

A Novel Validated UHPLC Method for the Estimation of Rosuvastatin and Its Complete Impurity Profile in Tablet Formulations
 , ,
, ,
Abstract
:1. Introduction
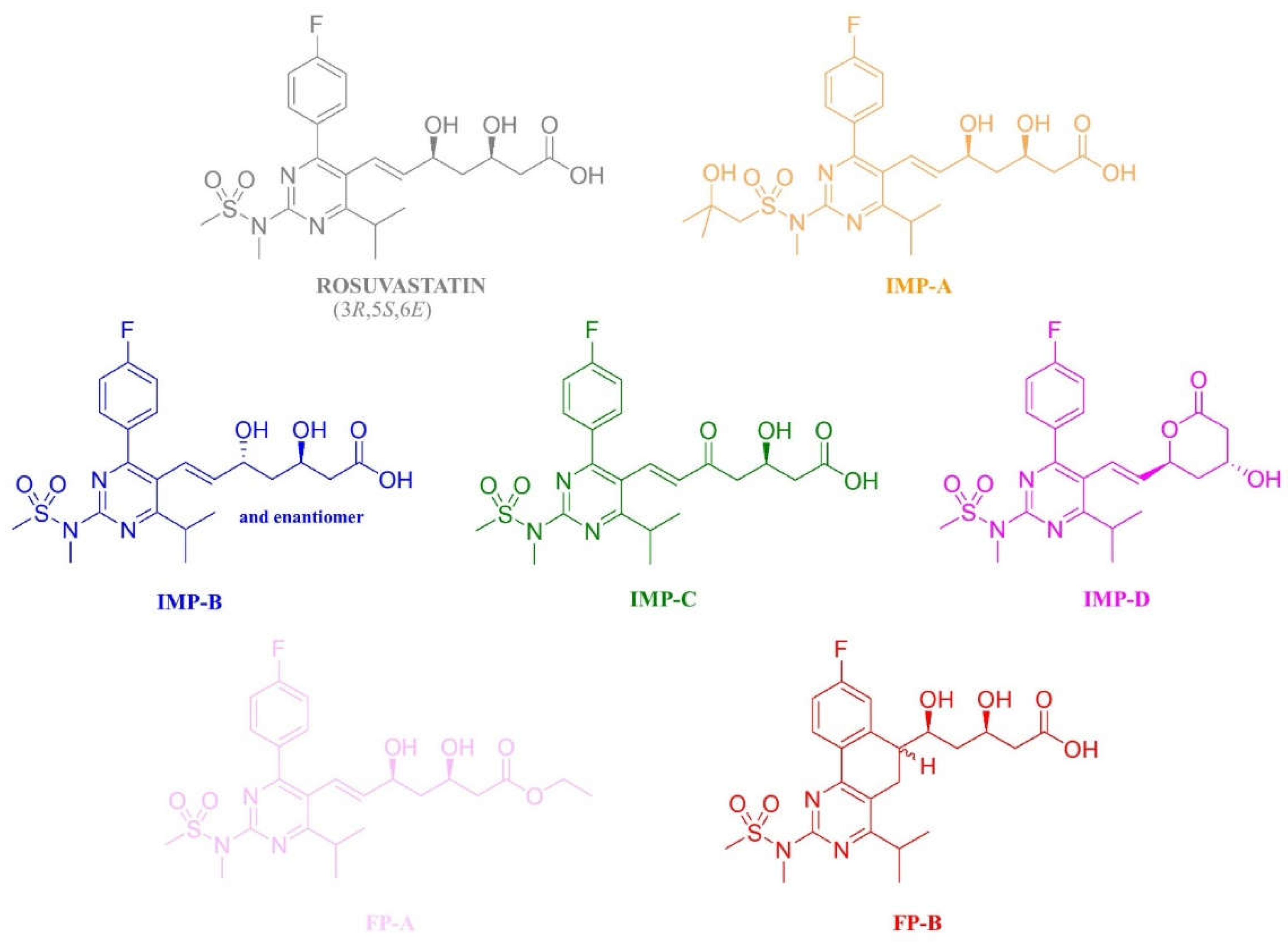
2. Results and Discussion
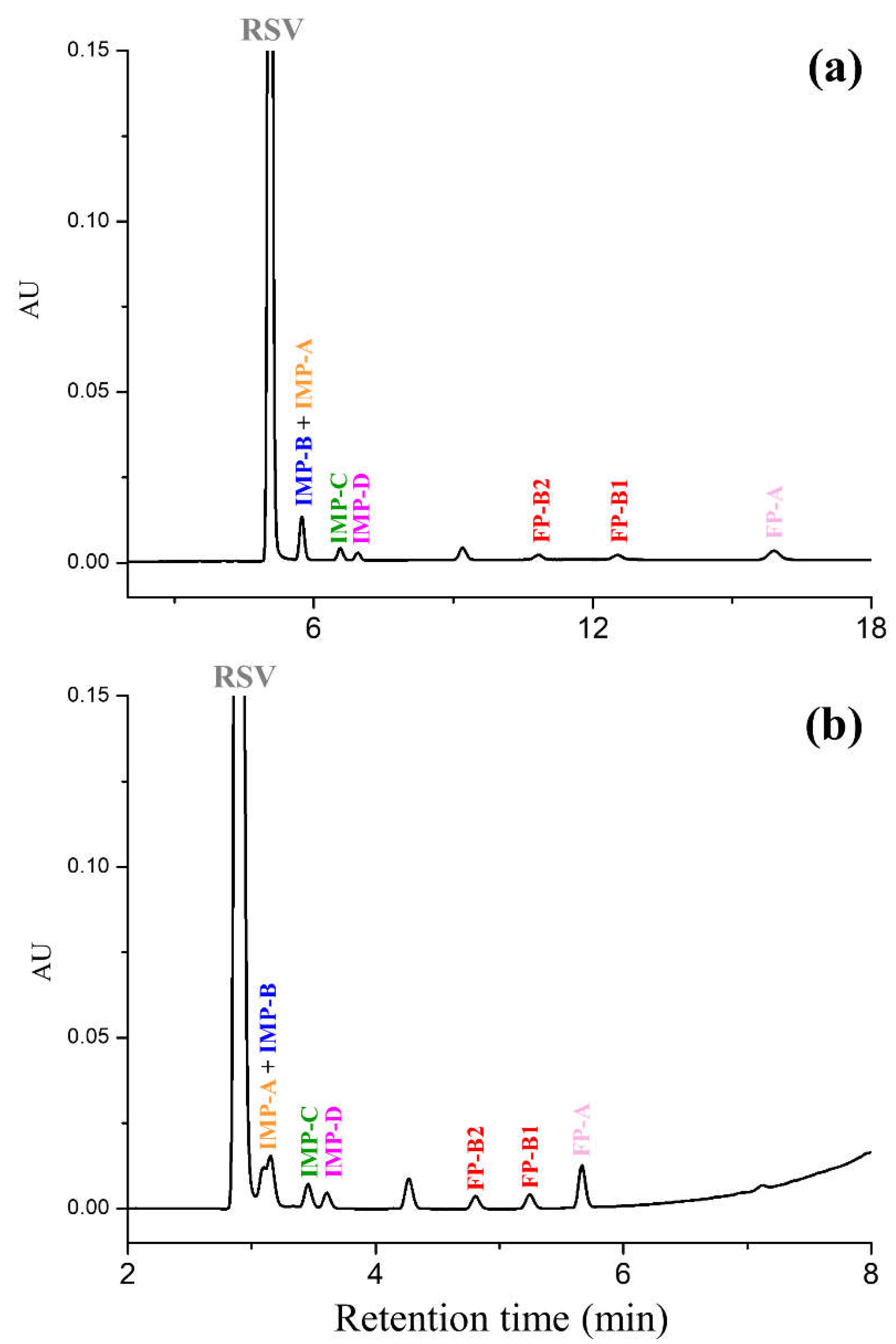
2.1. UHPLC Separation under Reversed-Phase Conditions
- (i)
- The TFA percentage was reduced from 0.1 to 0.025% with the purpose of preventing the potential on-column degradation of RSV.
- (ii)
- The methanol percentage was reduced from 50 to 45%, this change gave rise to a partial resolution of the critical pair IMP-B/IMP-A (Rs = 1.24) and an increase in the analysis time to 36 min.
- (iii)
- The column temperature and flow rate were increased from 40 to 55 °C and 0.3 to 0.5 mL/min, respectively. Under these conditions, all impurities were baseline separated from each other and eluted before 15 min.
- (i)
- Absence of buffer in the mobile phase; as a consequence, the risk of formation of precipitates in the column due to the poor solubility of salts in the organic modifier is overcome;
- (ii)
- Rapid elution of all the substances, both active substances and impurities, present in the multicomponent sample;
- (iii)
- High selectivity with excellent chromatographic resolution between adjacent peaks.
2.2. Method Validation
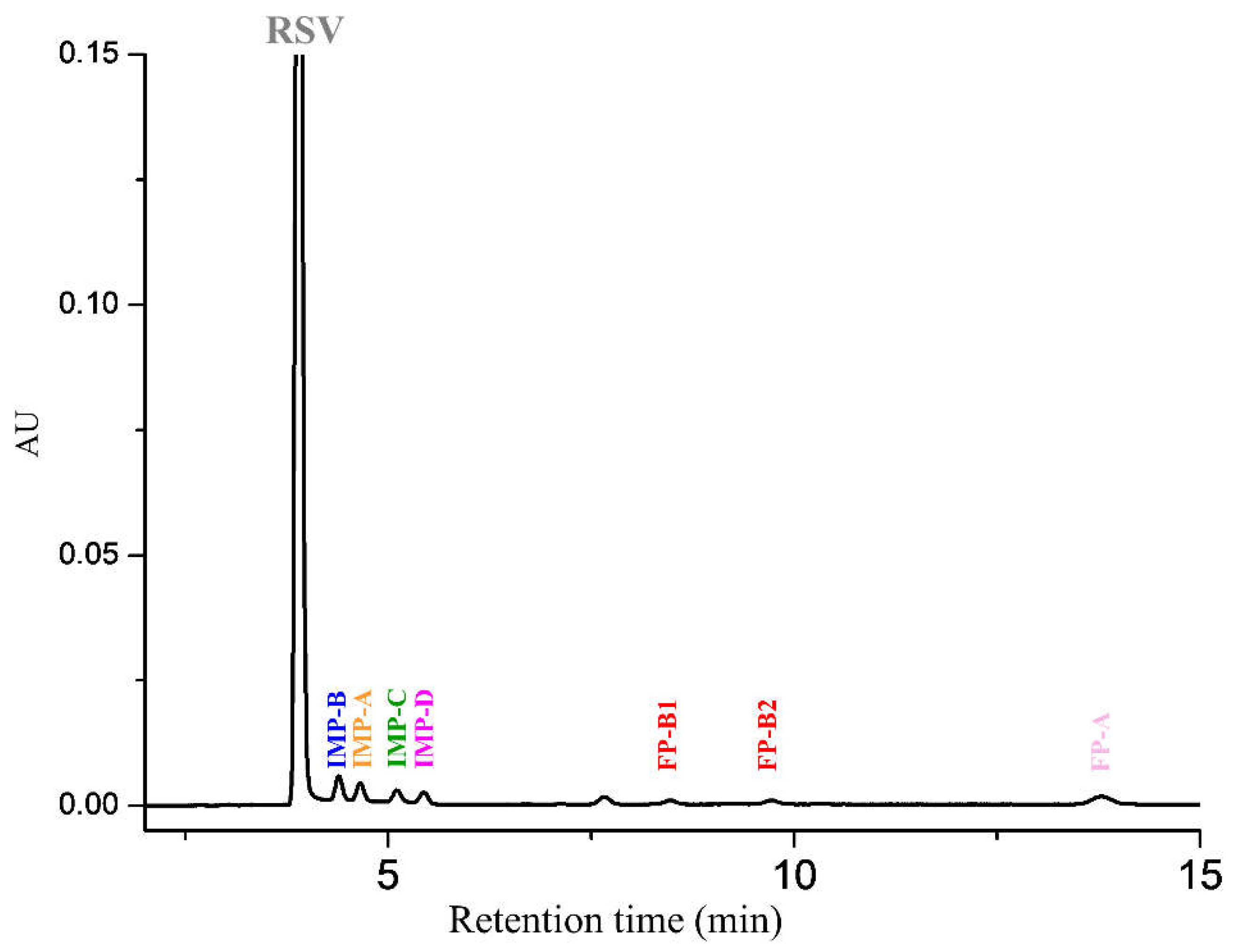
2.2.1. System Suitability
2.2.2. Linearity
2.2.3. Limits of Detection and Quantitation
2.2.4. Accuracy
2.2.5. Precision and Repeatability
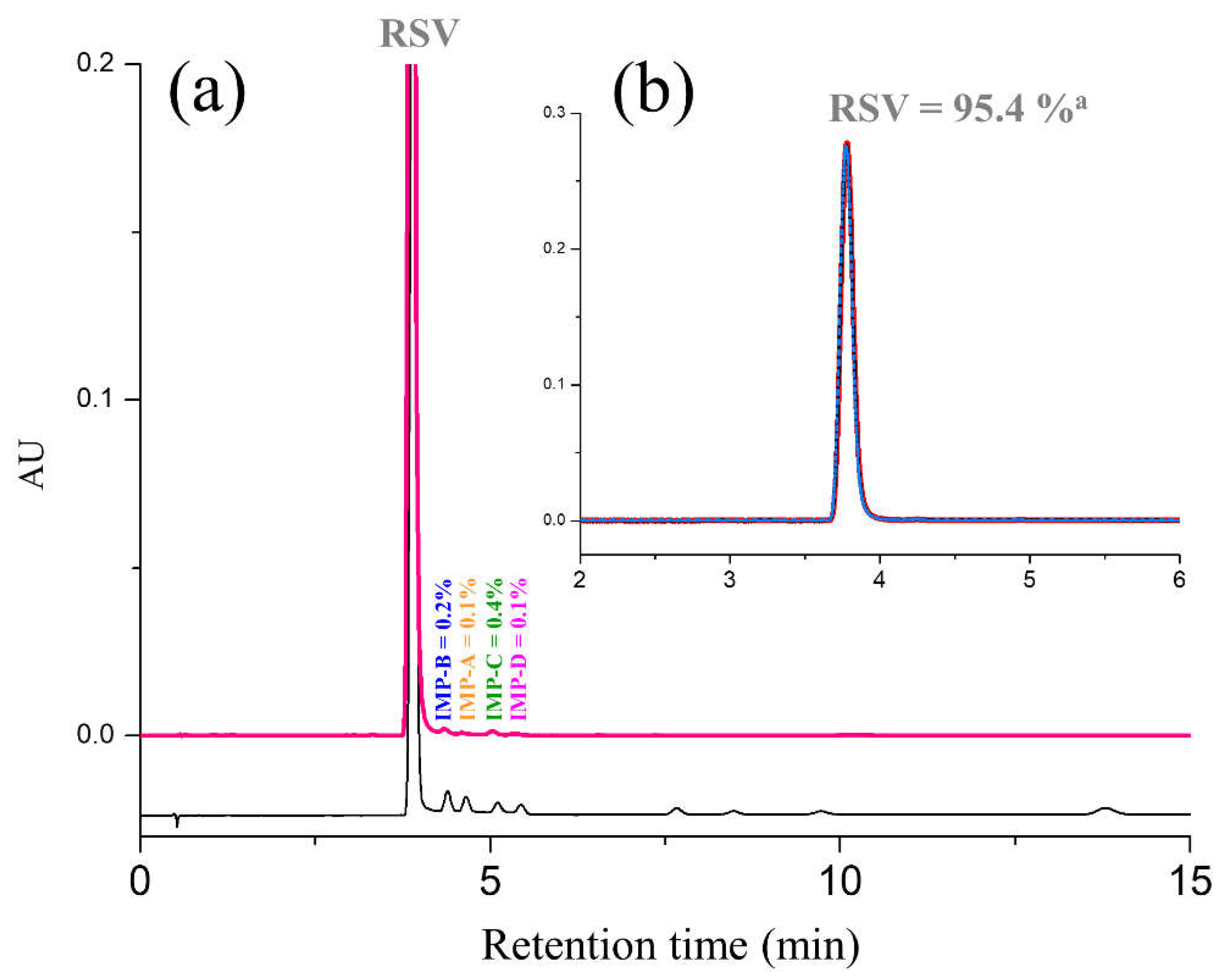
2.3. Determination of RSV and Impurity Contents in Commercial Tablets
3. Materials and Methods
3.1. Chemical and Reagents
3.2. Instruments
3.3. Synthesis of the Impurity FP-A
3.4. Method Validation
3.4.1. HPLC Operating Conditions
3.4.2. Specificity
3.4.3. Sample Solutions to Determine the Content of Rosuvastatin and Its Impurities
3.4.4. Standard Stock Solutions
3.4.5. Linearity
3.4.6. Limits of Detection and Quantitation
3.4.7. Precision
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jain, D.; Basniwal, P.K. Forced degradation and impurity profiling: Recent trends in analytical perspectives. J. Pharm. Biomed. Anal. 2013, 86, 11–35. [Google Scholar] [CrossRef] [PubMed]
- Patole, S.; Gosar, A.; Shaik, T. Impurities characterization in pharmaceuticals: A review. Int. J. Pharm. Res. 2019, 15, 46–64. [Google Scholar]
- Wang, Y.; Ai, F.; Ng, S.C.; Tan, T.T.Y. Sub-2 μm porous silica materials for enhanced separation performance in liquid chromatography. J. Chromatogr. A 2012, 1228, 99–109. [Google Scholar] [CrossRef]
- Swartz, M.E. UPLCTM: An introduction and review. J. Liq. Chromatogr. Relat. Technol. 2005, 28, 1253–1263. [Google Scholar] [CrossRef]
- Kątny, M.; Frankowski, M. Impurities in drug products and active pharmaceutical ingredients. Crit. Rev. Anal. Chem. 2016, 47, 187–193. [Google Scholar] [CrossRef]
- Thompson, R.B. Foundations for blockbuster drugs in federally sponsored research. FASEB J. 2001, 15, 1671–1676. [Google Scholar] [CrossRef]
- Pinal-Fernandez, I.; Casal-Dominguez, M.; Mammen, A.L. Statins: Pros and cons. Med. Clin. 2018, 150, 398–402. [Google Scholar] [CrossRef]
- Scott, L.J.; Curran, M.P.; Figgitt, D.P. Rosuvastatin: A review of its use in the management of dyslipidemia. Am. J. Cardiovasc. Drugs. 2004, 4, 117–138. [Google Scholar] [CrossRef]
- The European Pharmacopoeia, Monograph: Rosuvastatin Tablets 01/2022:3008. Available online: https://www.edqm.eu/en/-/monograph-on-rosuvastatin-tablets-3008-to-be-published-in-ph-eur-10.1 (accessed on 1 December 2022).
- Kishore, C.R.P.; Krishna Mohan, G.V. Structural identification and estimation of Rosuvastatin calcium related impurities in Rosuvastatin calcium tablet dosage form. Anal. Chem. Res. 2017, 12, 17–27. [Google Scholar] [CrossRef]
- Yulianita, R.; Sopyan, I.; Muchtaridi, M. Forced degradation study of statins: A review. Int. J. App. Pharm. 2018, 10, 38–42. [Google Scholar] [CrossRef]
- Mehta, T.N.; Patel, A.K.; Kulkarni, G.M.; Suubbaiah, G. Determination of rosuvastatin in the presence of its degradation products by a stability-indicating LC method. J. AOAC Int. 2005, 4, 1142–1147. [Google Scholar] [CrossRef]
- Reddy, G.V.R.; Reddy, B.V.; Haque, S.W.; Gautam, H.D.; Kumar, P.; Kumar, A.P.; Park, J.H. Development and validation of a stability-indicating UPLC method for rosuvastatin and its related impurities in pharmaceutical dosage forms. Quim. Nova 2011, 34, 250–255. [Google Scholar] [CrossRef] [Green Version]
- Trivedi, H.K.; Patel, M.C. Development and validation of a stability-indicating RP-UPLC method for determination of rosuvastatin and related substances in pharmaceutical dosage form. Sci. Pharm. 2012, 80, 393–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakrajšek, J.; Bevc-Černilec, K.; Bohanec, S.; Urleb, U. Optimization of UPLC method for simultaneous determination of rosuvastatin and rosuvastatin degradation products. Acta Chim. Slov. 2017, 64, 968–979. [Google Scholar] [CrossRef] [Green Version]
- Elbordiny, H.S.; Elonsy, S.M.; Daabees, H.G.; Belal, T.S. Implementation of two sustainable chromatographic methods for the simultaneous micro-quantitation and impurity profiling of metformin and rosuvastatin in recently approved fixed dose pills: Greenness and whiteness studies. Sustain. Chem. Pharm. 2022, 30, 100885. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Impurity | RRT | Rs | RRF |
|---|---|---|---|
| RSV | 1.00 | ||
| IMP-B | 1.13 | 3.23 | 0.87 |
| IMP-A | 1.19 | 1.60 | 0.72 |
| IMP-C | 1.31 | 2.56 | 0.46 |
| IMP-D | 1.39 | 1.76 | 0.95 |
| FP-B1 | 2.17 | 9.59 | 0.66 |
| FP-B2 | 2.49 | 3.73 | 0.66 |
| FP-A | 3.54 | 9.61 | 0.88 |
| Injection | Peak Area | Retention Time | Peak Tailing | Theoretical Plats |
|---|---|---|---|---|
| 1 | 1,689,234 | 3.954 | 1.09 | 11,407 |
| 2 | 1,694,889 | 3.945 | 1.09 | 11,326 |
| 3 | 1,689,344 | 3.963 | 1.09 | 11,289 |
| 4 | 1,693,483 | 3.945 | 1.08 | 11,397 |
| 5 | 1,689,940 | 3.93 | 1.09 | 11,333 |
| 6 | 1,686,928 | 3.935 | 1.09 | 11,280 |
| RSD(%) | 0.18 | 0.31 | ||
| 1 | 79,897 | 3.94 | 1.07 | 11,386 |
| 2 | 79,195 | 3.94 | 1.06 | 11,669 |
| 3 | 79,755 | 3.94 | 1.08 | 11,356 |
| 4 | 79,189 | 3.92 | 1.07 | 11,354 |
| 5 | 79,708 | 3.92 | 1.06 | 11,548 |
| 6 | 79,460 | 3.93 | 1.07 | 11,586 |
| RSD (%) | 0.38 | 0.26 |
| Amount Added | %Recovery | RSD% a |
|---|---|---|
| 0.06% (LOQ) | 100.92 | 0.93 |
| 0.5% | 100.45 | 0.34 |
| 80 | 100.81 | 0.51 |
| 100 | 100.77 | 0.62 |
| 120 | 99.25 | 0.68 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mammone, F.R.; Zanitti, L.; Puxeddu, M.; La Regina, G.; Silvestri, R.; Borioni, A.; Cirilli, R. A Novel Validated UHPLC Method for the Estimation of Rosuvastatin and Its Complete Impurity Profile in Tablet Formulations. Molecules 2023, 28, 431. https://doi.org/10.3390/molecules28010431
Mammone FR, Zanitti L, Puxeddu M, La Regina G, Silvestri R, Borioni A, Cirilli R. A Novel Validated UHPLC Method for the Estimation of Rosuvastatin and Its Complete Impurity Profile in Tablet Formulations. Molecules. 2023; 28(1):431. https://doi.org/10.3390/molecules28010431
Chicago/Turabian StyleMammone, Francesca Romana, Leo Zanitti, Michela Puxeddu, Giuseppe La Regina, Romano Silvestri, Anna Borioni, and Roberto Cirilli. 2023. "A Novel Validated UHPLC Method for the Estimation of Rosuvastatin and Its Complete Impurity Profile in Tablet Formulations" Molecules 28, no. 1: 431. https://doi.org/10.3390/molecules28010431