
Release of Volatile Cyclopentanone Derivatives from Imidazolidin-4-One Profragrances in a Fabric Softener Application

Abstract
:
1. Introduction
2. Results and Discussion
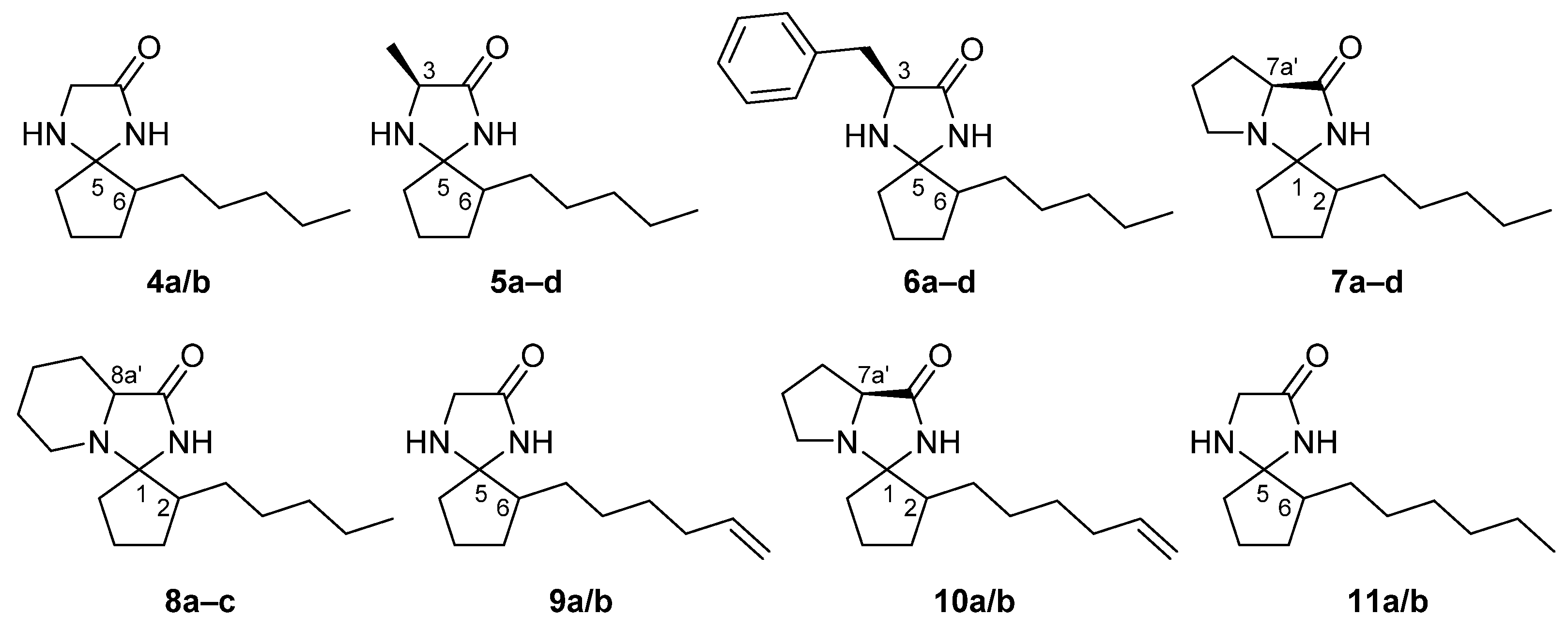
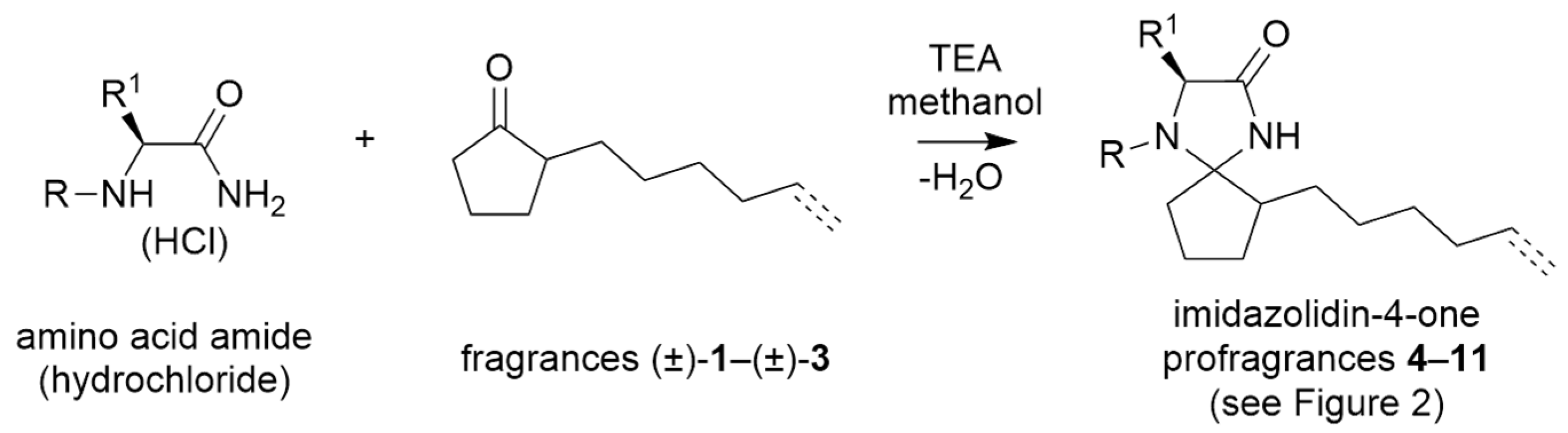
2.1. Synthesis and Characterisation of Imidazolidin-4-One-Based Profragrances
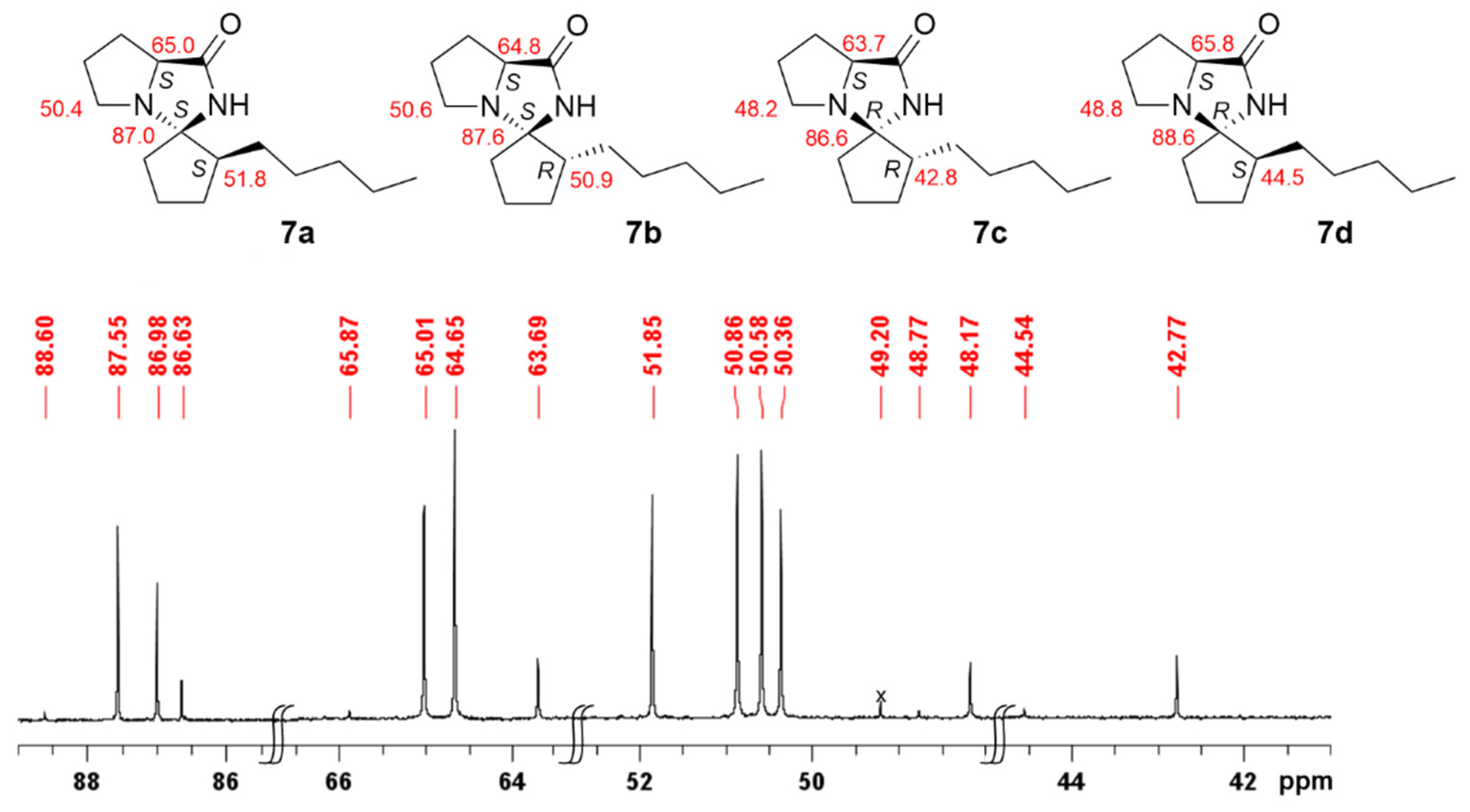
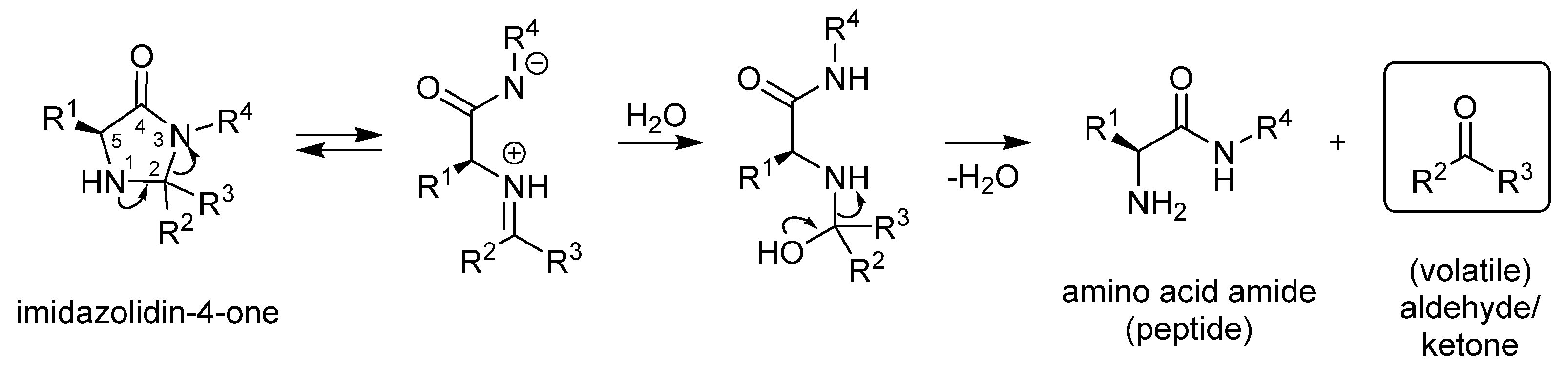
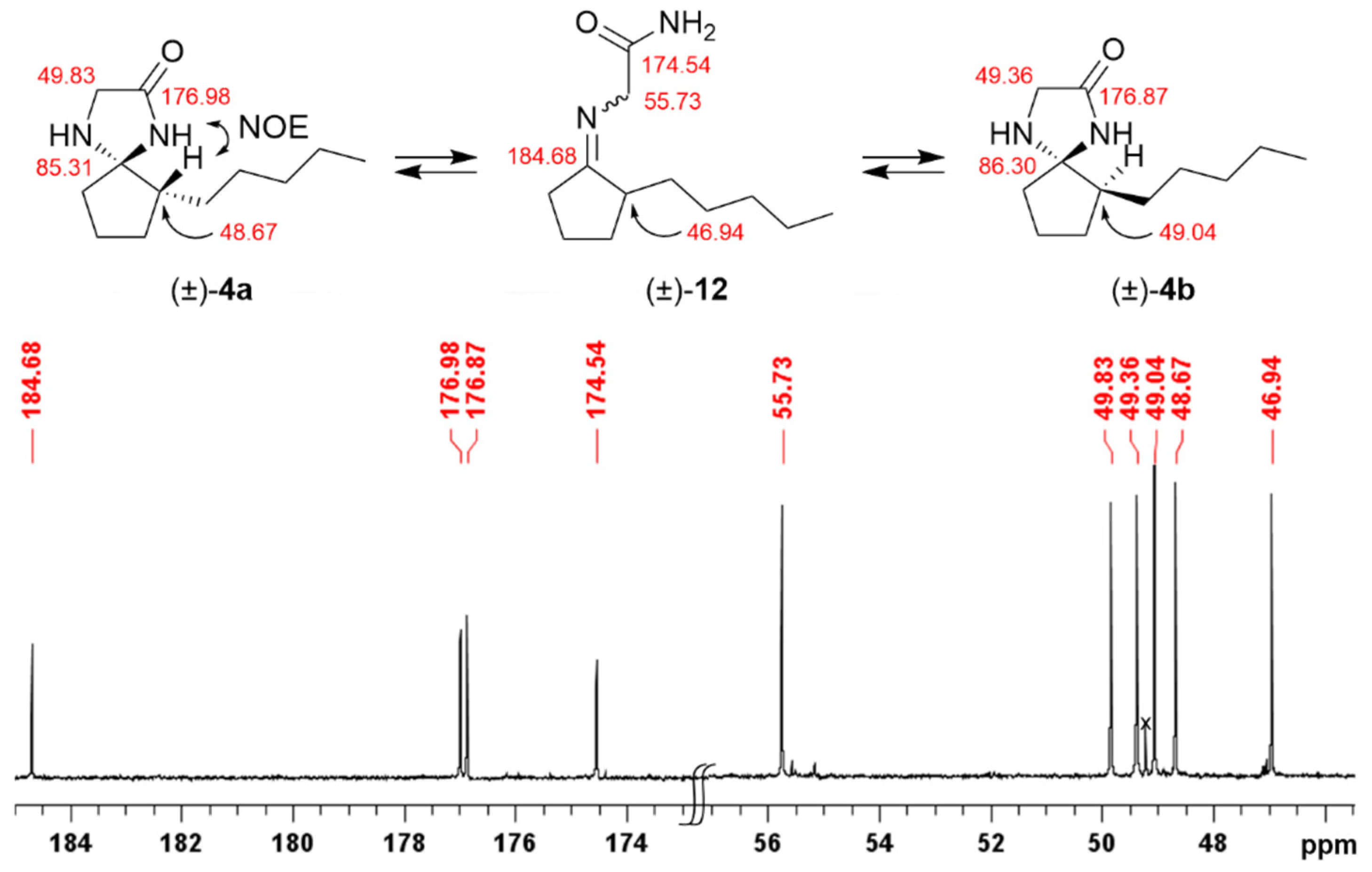
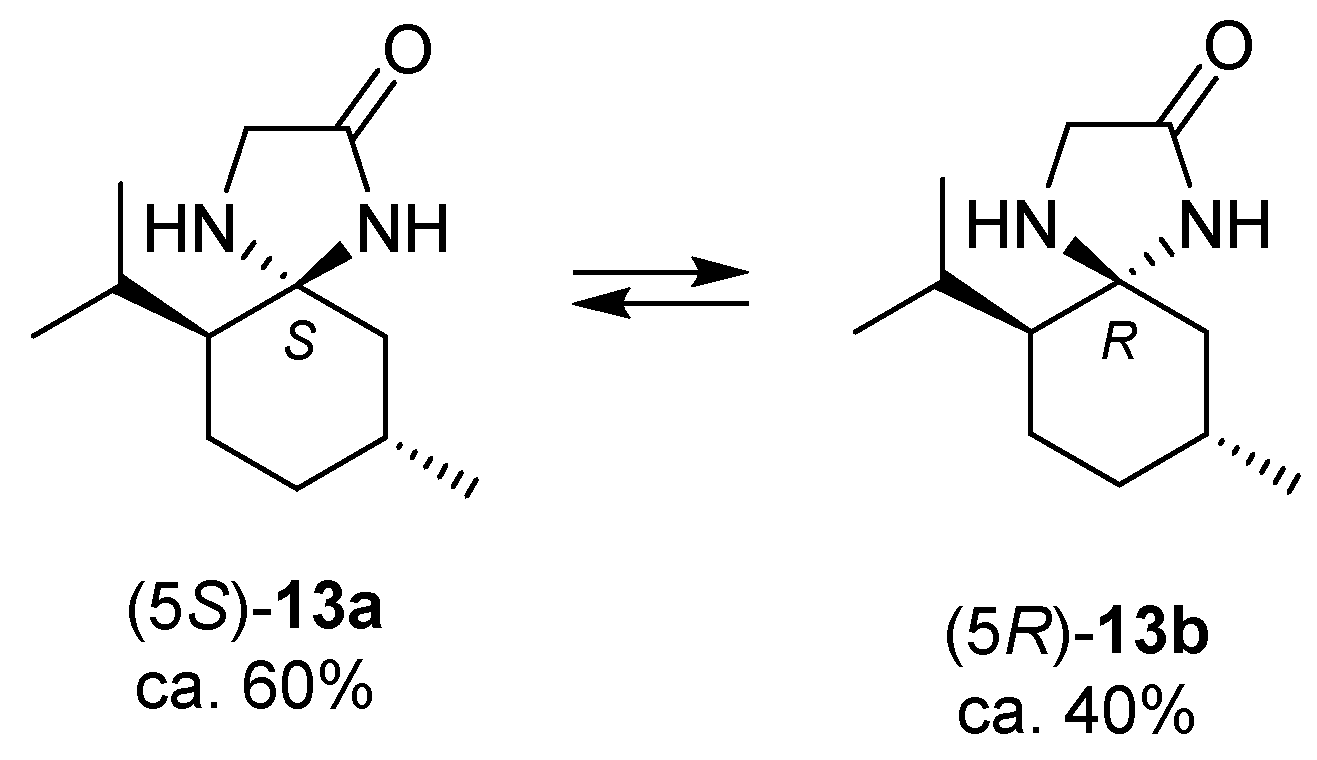
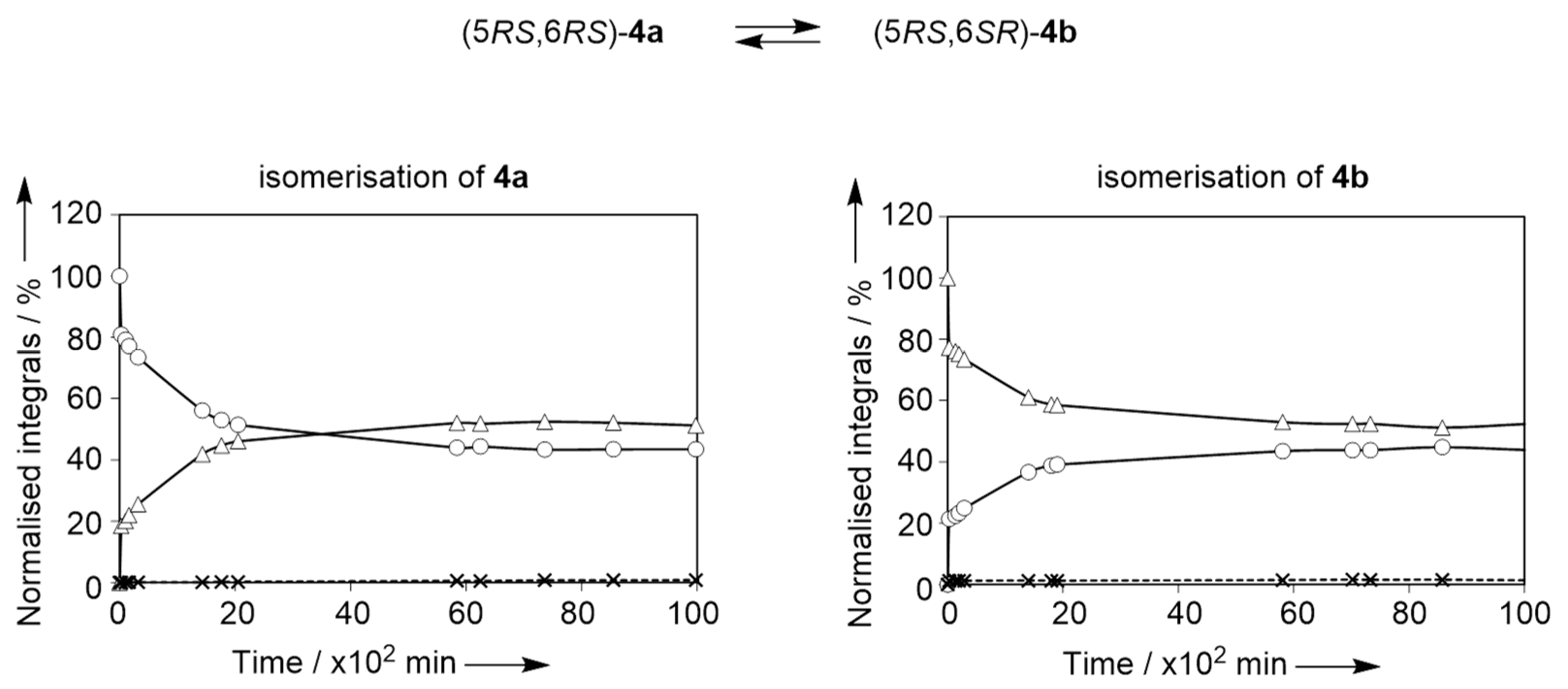
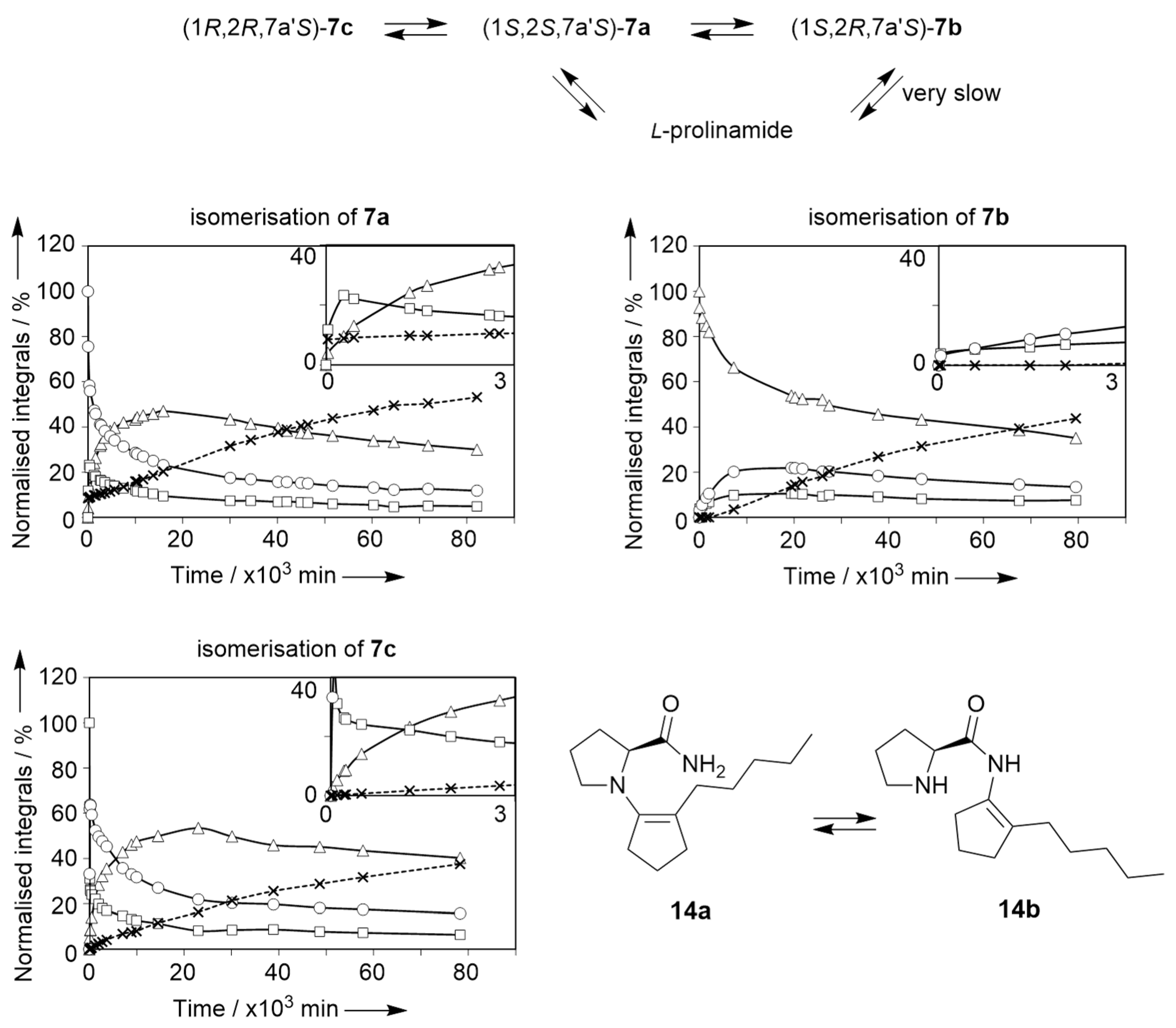
2.2. Isomerisation of Profragrances 4a/b and 7a–c in Methanol
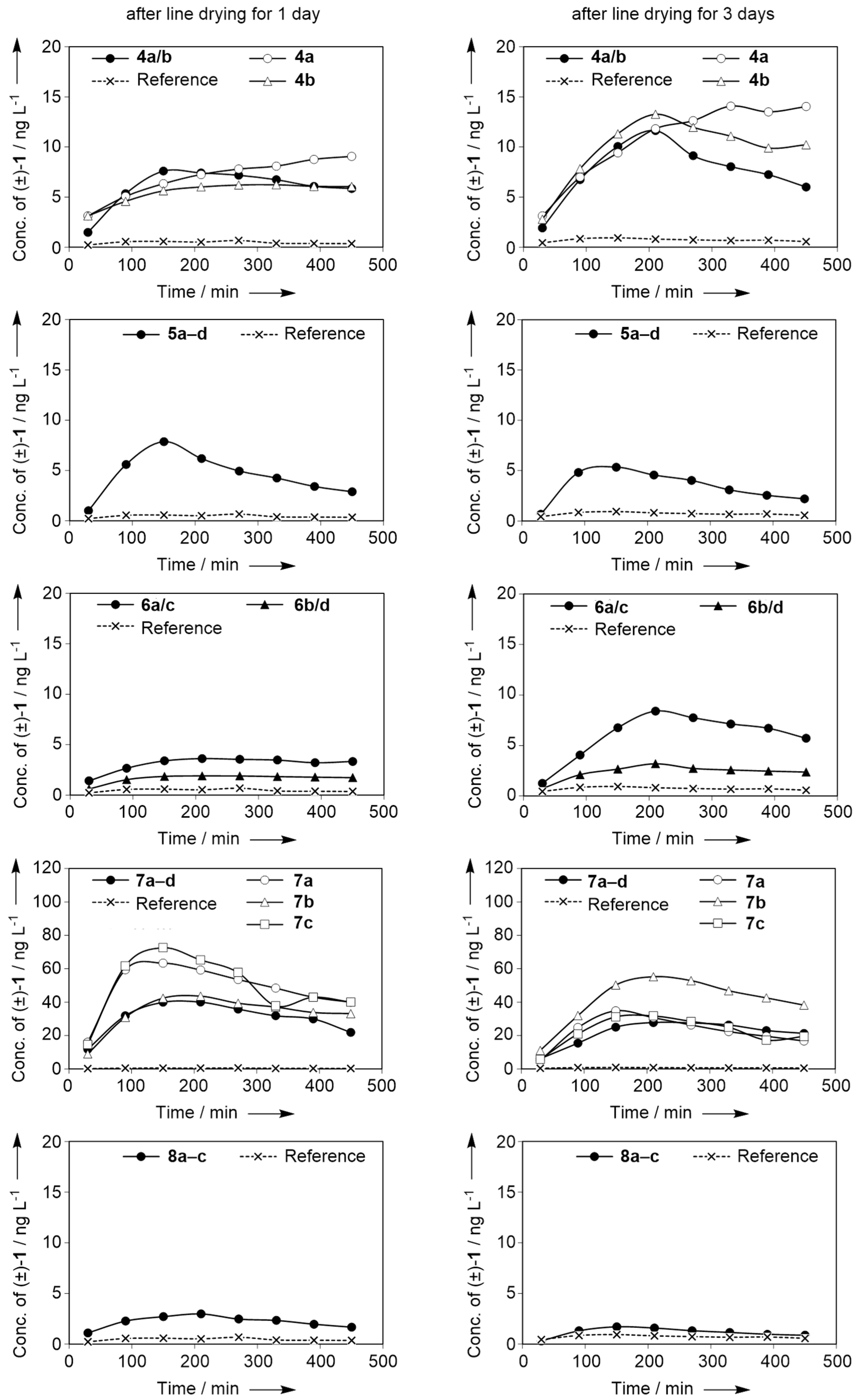
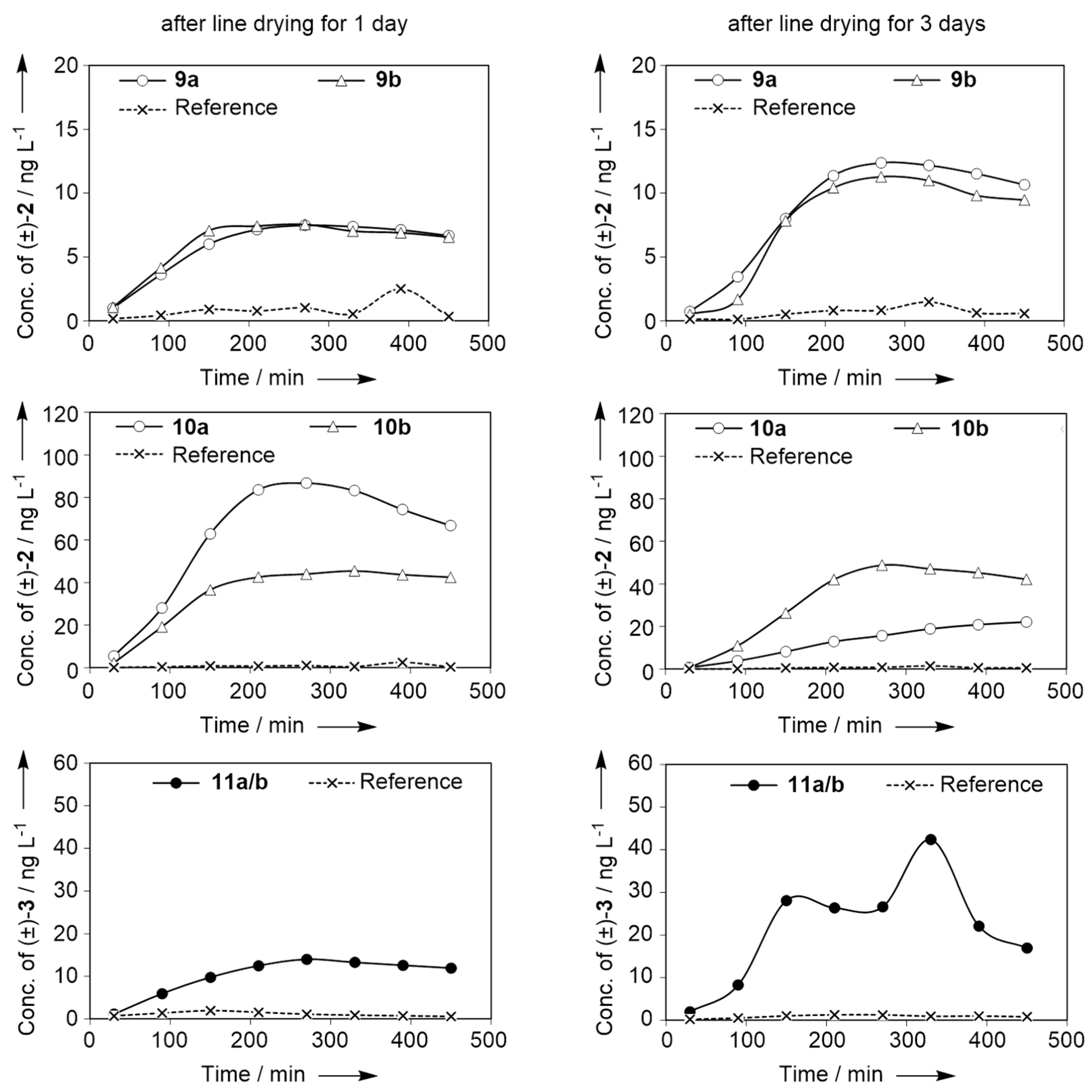
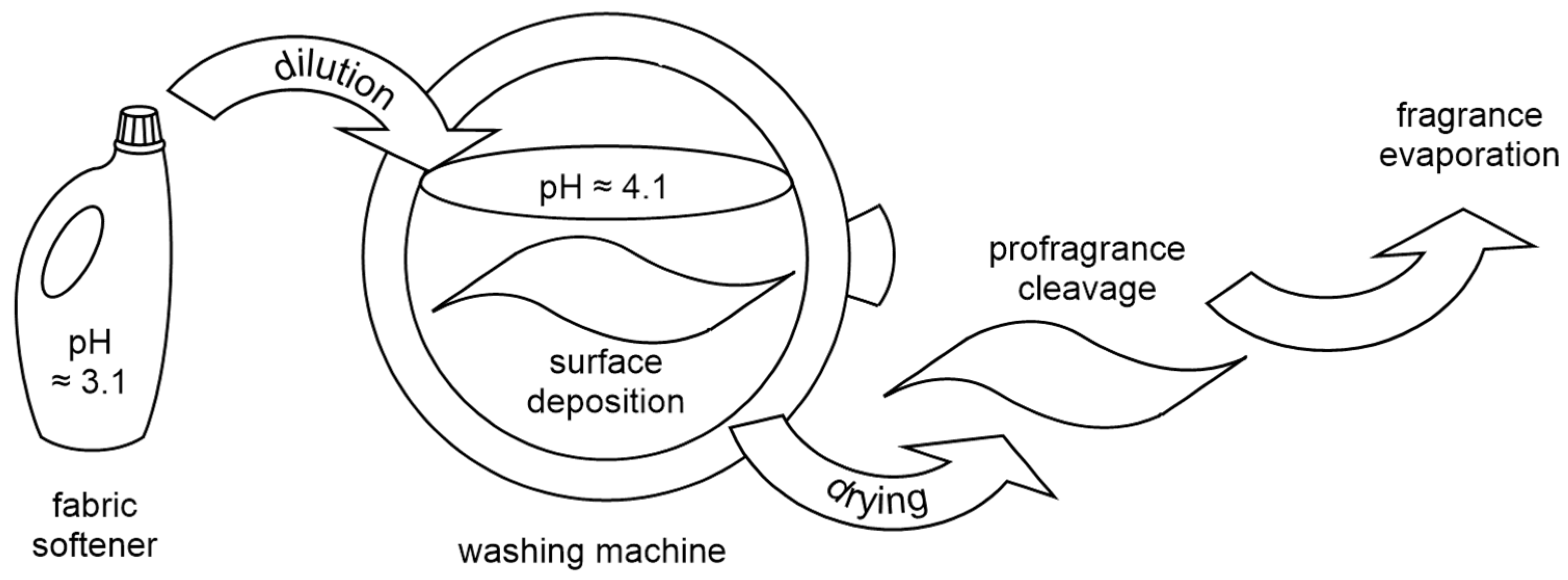
2.3. Dynamic Headspace Analysis on Dry Cotton after a Typical Fabric Softening Application
3. Materials and Methods
3.1. Synthesis and Characterisation of Imidazolidin-4-One Derivatives 4–11 [39]
3.1.1. Synthesis of 6-pentyl-1,4-diazaspiro[4.4]nonan-2-one (4a/b)
3.1.2. Synthesis of (3S)-3-methyl-6-pentyl-1,4-diazaspiro[4.4]nonan-2-one (5a–d)
3.1.3. Synthesis of (3S)-3-benzyl-6-pentyl-1,4-diazaspiro[4.4]nonan-2-one (6a–d)
3.1.4. Synthesis of (7a′S)-2-pentyltetrahydrospiro[cyclopentane-1,3′-pyrrolo[1,2-c]imidazol]-1′(2′H)-one (7a–d)
3.1.5. Synthesis of 2-pentyltetrahydro-2′H-spiro[cyclopentane-1,3′-imidazo[1,5-a]pyridin]-1′(5′H)-one (8a–d)
3.1.6. Synthesis of 6-(hex-5-enyl)-1,4-diazaspiro[4.4]nonan-2-one (9a/b)
3.1.7. Synthesis of (7a′S)-2-(hex-5-en-1-yl)tetrahydrospiro[cyclopentane-1,3′-pyrrolo[1,2-c]imidazol]-1′(2′H)-one (10a/b)
3.1.8. Synthesis of 6-hexyl-1,4-diazaspiro[4.4]nonan-2-one (11a/b)
3.2. Isomerisations in Methanol
3.3. Dynamic Headspace Measurements on Dry Cotton Sheets
3.3.1. Fabric Softening Procedure
3.3.2. Dynamic Headspace Sampling
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Herz, R.S.; Larsson, M.; Trujillo, R.; Casola, M.C.; Ahmed, F.K.; Lipe, S.; Brashear, M.E. A three-factor benefits framework for understanding consumer preference for scented household products: Psychological interactions and implications for future development. Cognit. Res. Princ. Implic. 2022, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Milotic, D. The impact of fragrance on consumer choice. J. Consum. Behav. 2003, 3, 179–191. [Google Scholar] [CrossRef]
- He, L.; Hu, J.; Deng, W. Preparation and application of flavor and fragrance capsules. Polym. Chem. 2018, 9, 4926–4946. [Google Scholar] [CrossRef]
- Kaur, R.; Kukkar, D.; Bhardwaj, S.K.; Kim, K.-H.; Deep, A. Potential use of polymers and their complexes as media for storage and delivery of fragrances. J. Control. Release 2018, 285, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Bruyninckx, K.; Dusselier, M. Sustainable chemistry considerations for the encapsulation of volatile compounds in laundry-type applications. ACS Sustain. Chem. Eng. 2019, 7, 8041–8054. [Google Scholar] [CrossRef]
- Perinelli, D.R.; Palmieri, G.F.; Cespi, M.; Bonacucina, G. Encapsulation of flavours and fragrances into polymeric capsules and cyclodextrins inclusion complexes: An update. Molecules 2020, 25, 5878. [Google Scholar] [CrossRef]
- Wei, M.; Pan, X.; Rong, L.; Dong, A.; He, Y.; Song, X.; Li, J. Polymer carriers for controlled fragrance release. Mater. Res. Express 2020, 7, 082001. [Google Scholar] [CrossRef]
- Tian, Q.; Zhou, W.; Cai, Q.; Ma, G.; Lian, G. Concepts, processing, and recent developments in encapsulating essential oils. Chin. J. Chem. Eng. 2021, 30, 255–271. [Google Scholar] [CrossRef]
- Mamusa, M.; Resta, C.; Sofroniou, C.; Baglioni, P. Encapsulation of volatile compounds in liquid media: Fragrances, flavors, and essential oils in commercial formulations. Adv. Colloid Interface Sci. 2021, 298, 102544. [Google Scholar] [CrossRef]
- Xiao, Z.; Sun, P.; Liu, H.; Zhao, Q.; Niu, Y.; Zhao, D. Stimulus responsive microcapsules and their aromatic applications. J. Control. Release 2022, 351, 198–214. [Google Scholar] [CrossRef]
- Zaitoon, A.; Luo, X.; Lim, L.-T. Triggered and controlled release of active gaseous/volatile compounds for active packaging applications of agri-food products: A review. Compr. Rev. Food Sci. Food Saf. 2022, 21, 541–579. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, A. Controlled release of volatiles under mild reaction conditions: From nature to everyday products. Angew. Chem. Int. Ed. 2007, 46, 5836–5863. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, A. Profragrances and properfumes. In The Chemistry and Biology of Volatiles; Herrmann, A., Ed.; John Wiley & Sons: Chichester, UK, 2010; pp. 333–362. [Google Scholar] [CrossRef]
- Herrmann, A. Profragrance chemistry as an interdisciplinary research area and key technology for fragrance delivery. Chimia 2017, 71, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, A. Controlled release of volatile compounds using the Norrish type II reaction. Photochemistry 2019, 46, 242–264. [Google Scholar] [CrossRef]
- Liang, Z.; Fang, Z.; Pai, A.; Luo, J.; Gan, R.; Gao, Y.; Lu, J.; Zhang, P. Glycosidically bound aroma precursors in fruits: A comprehensive review. Crit. Rev. Food Sci. Nutr. 2022, 62, 215–243. [Google Scholar] [CrossRef]
- Blackmore, T.R.; Thompson, P.E. Imidazolidin-4-ones: Their syntheses and applications. Heterocycles 2011, 83, 1953–1975. [Google Scholar] [CrossRef]
- Swain, S.P.; Mohanty, S. Imidazolidinones and imidazolidine-2,4-diones as antiviral agents. ChemMedChem 2019, 14, 291–302. [Google Scholar] [CrossRef]
- Klixbüll, U.; Bundgaard, H. Prodrugs as drug delivery systems. XXX. 4-Imidazolidinones as potential bioreversible derivatives for the α-aminoamide moiety in peptides. Int. J. Pharm. 1984, 20, 273–284. [Google Scholar] [CrossRef]
- Bak, A.; Fich, M.; Larsen, B.D.; Frokjaer, S.; Friis, G.J. N-Terminal 4-imidazolidinone prodrugs of Leu-enkephalin: Synthesis, chemical and enzymatic stability studies. Eur. J. Pharm. Sci. 1999, 7, 317–323. [Google Scholar] [CrossRef]
- Rasmussen, G.J.; Bundgaard, H. Prodrugs of peptides. 15. 4-Imidazolidinone prodrug derivatives of enkephalins to prevent aminopeptidase-catalyzed metabolism in plasma and absorptive mucosae. Int. J. Pharm. 1991, 76, 113–122. [Google Scholar] [CrossRef]
- Rasmussen, G.J.; Bundgaard, H. Prodrugs of peptides. 10. Protection of di- and tripeptides against aminopeptidase by formation of bioreversible 4-imidazolidinone derivatives. Int. J. Pharm. 1991, 71, 45–53. [Google Scholar] [CrossRef]
- Chambel, P.; Capela, R.; Lopes, F.; Iley, J.; Morais, J.; Gouveia, L.; Gomes, J.R.B.; Gomes, P.; Moreira, R. Reactivity of imidazolidin-4-one derivatives of primaquine: Implications for prodrug design. Tetrahedron 2006, 62, 9883–9891. [Google Scholar] [CrossRef] [Green Version]
- Vale, N.; Nogueira, F.; do Rosário, V.E.; Gomes, P.; Moreira, R. Primaquine dipeptide derivatives bearing an imidazolidin-4-one moiety at the N-terminus as potential antimalarial prodrugs. Eur. J. Med. Chem. 2009, 44, 2506–2516. [Google Scholar] [CrossRef] [Green Version]
- Weng Larsen, S.; Sidenius, M.; Ankersen, M.; Larsen, C. Kinetics of degradation of 4-imidazolidinone prodrug types obtained from reacting prilocaine with formaldehyde and acetaldehyde. Eur. J. Pharm. Sci. 2003, 20, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Durbin, A.K.; Rydon, H.N. The equilibrium between the antibiotics hetacillin and ampicillin in solution. J. Chem. Soc. D Chem. Commun. 1970, 1249–1250. [Google Scholar] [CrossRef]
- Tsuji, A.; Yamana, T. Kinetic approach to the development in β-lactam antibiotics. II. Prodrug. (1). Simultaneous determination of hetacillin and ampicillin, and its application to the stability of hetacillin in aqueous solution. Chem. Pharm. Bull. 1974, 22, 2434–2443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klixbüll, U.; Bundgaard, H. Kinetics of reversible reactions of ampicillin with various aldehydes and ketones with formation of 4-imidazolidinones. Int. J. Pharm. 1985, 23, 163–173. [Google Scholar] [CrossRef]
- Gomes, P.; Araújo, M.J.; Rodrigues, M.; Vale, N.; Azevedo, Z.; Iley, J.; Chambel, P.; Morais, J.; Moreira, R. Synthesis of imidazolidin-4-one and 1H-imidazo[2,1-a]isoindole-2,5(3H,9bH)-dione derivatives of primaquine: Scope and limitations. Tetrahedron 2004, 60, 5551–5562. [Google Scholar] [CrossRef] [Green Version]
- Giorgioni, G.; Claudi, F.; Ruggieri, S.; Ricciutelli, M.; Palmieri, G.F.; Di Stefano, A.; Sozio, P.; Cerasa, L.S.; Chiavaroli, A.; Ferrante, C.; et al. Design, synthesis, and preliminary pharmacological evaluation of new imidazolinones as L-DOPA prodrugs. Bioorg. Med. Chem. 2010, 18, 1834–1843. [Google Scholar] [CrossRef]
- Friedli, F.E.; Koehle, H.J.; Fender, M.; Watts, M.; Keys, R.; Frank, P.; Toney, C.J.; Doerr, M. Upgrading triethanolamine esterquat performance to new levels. J. Surfactants Deterg. 2002, 5, 211–216. [Google Scholar] [CrossRef]
- Mishra, S.; Tyagi, V.K. Ester quats: The novel class of cationic fabric softeners. J. Oleo Sci. 2007, 56, 269–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escher, S.D.; Oliveros, E. A quantitative study of factors that influence the substantivity of fragrance chemicals on laundered and dried fabrics. J. Am. Oil Chem. Soc. 1994, 71, 31–40. [Google Scholar] [CrossRef]
- Obendorf, S.K.; Liu, H.; Tan, K.; Leonard, M.J.; Young, T.J.; Incorvia, M.J. Adsorption of aroma chemicals on cotton fabric in different aqueous environments. J. Surfactants Deterg. 2009, 12, 43–58. [Google Scholar] [CrossRef]
- Trachsel, A.; Buchs, B.; Godin, G.; Crochet, A.; Fromm, K.M.; Herrmann, A. Preparation of imidazolidin-4-ones and their evaluation as hydrolytically cleavable precursors for the slow release of bioactive volatile carbonyl derivatives. Eur. J. Org. Chem. 2012, 2837–2854. [Google Scholar] [CrossRef] [Green Version]
- Surburg, H.; Panten, J. Common Fragrance and Flavor Materials—Preparation, Properties and Uses, 6th ed.; Wiley-VCH: Weinheim, Germany, 2016. [Google Scholar]
- Coulomb, J.; Demole, E. (to Firmenich SA). Cyclopentanone compounds. PCT Int. Patent Appl. WO 2020/078892, 23 April 2020. [Google Scholar]
- Arctander, S. Perfume and Flavor Chemicals; Published by the Author: Montclair, NJ, USA, 1969. [Google Scholar]
- Herrmann, A.; Lamboley, S. (to Firmenich SA). Compounds for providing a long-lasting odor. PCT Int. Patent Appl. WO 2021/209396, 21 October 2021. [Google Scholar]
- Koch, W.; Holthausen, M.C. A Chemist’s Guide to Density Functional Theory, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Sholl, D.S.; Steckel, J.A. Density Functional Theory—A Practical Introduction; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Schrödinger Release 2022-3: MacroModel; Schrödinger, LLC: New York, NY, USA, 2021.
- Spartan’18; Wavefunction Inc.: Irvine, CA, USA, 2018.
- Godin, G.; Levrand, B.; Trachsel, A.; Lehn, J.-M.; Herrmann, A. Reversible formation of aminals: A new strategy to control the release of bioactive volatiles from dynamic mixtures. Chem. Commun. 2010, 46, 3125–3127. [Google Scholar] [CrossRef]
- Buchs, B.; Godin, G.; Trachsel, A.; de Saint-Laumer, J.-Y.; Lehn, J.-M.; Herrmann, A. Reversible aminal formation: Controlling the evaporation of bioactive volatiles by dynamic combinatorial/covalent chemistry. Eur. J. Org. Chem. 2011, 681–695. [Google Scholar] [CrossRef]
- Herrmann, A. Dynamic mixtures: Challenges and opportunities for the amplification and sensing of scents. Chem. Eur. J. 2012, 18, 8568–8577. [Google Scholar] [CrossRef]
- Rouseff, R.L.; Cadwallader, K.R. (Eds.) Headspace Analysis of Food and Flavors: Theory and Practice; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2001. [Google Scholar] [CrossRef]
- Rubiolo, P.; Sgorbini, B.; Liberto, E.; Cordero, C.; Bicchi, C. Analysis of the plant volatile fraction. In The Chemistry and Biology of Volatiles; Herrmann, A., Ed.; John Wiley & Sons: Chichester, UK, 2010; pp. 49–93. [Google Scholar] [CrossRef]
- Lamboley, S.; Trachsel, A.; Herrmann, A. Polystyrene-based 2-oxoacetates for the light-induced release of fragrances under realistic application conditions. Macromol. Chem. Phys. 2020, 221, 2000196. [Google Scholar] [CrossRef]















{kind=link}
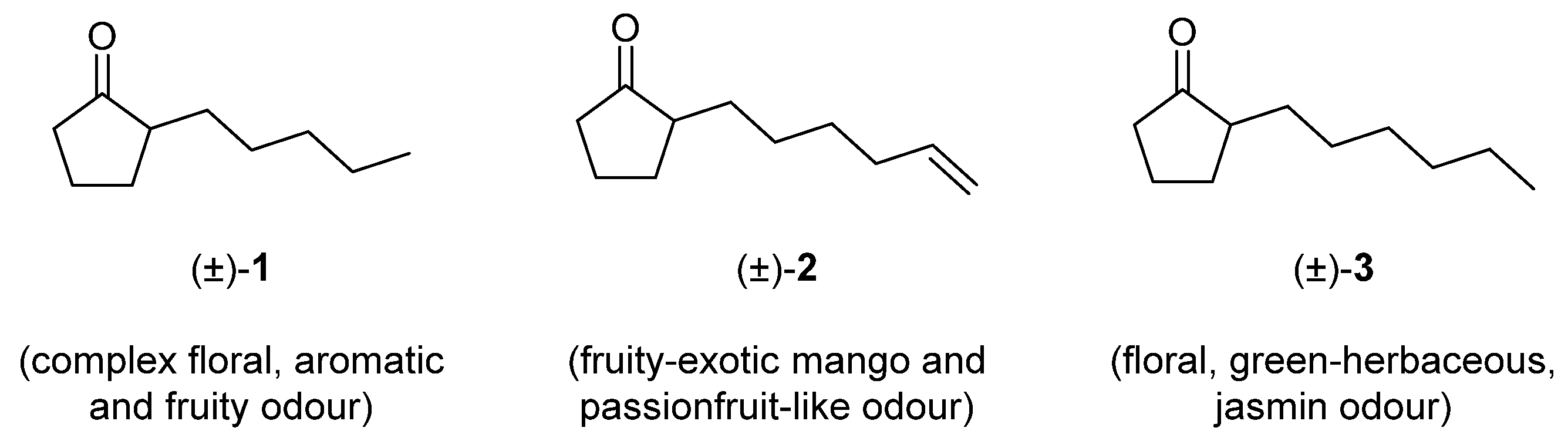{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Stereochemistry | Relative Energy [kJ] | Boltzmann Calc. [%] | Experimental Ratio [%] | Recorded 13C NMR Shift [ppm] |
|---|---|---|---|---|---|
| 7a | (1S,2S,7a′S) | 1.90 | 31 | 36 | 87.1 |
| 7b | (1S,2R,7a′S) | 0.00 | 66 | 52 | 87.7 |
| 7c | (1R,2R,7a′S) | 7.78 | 3 | 10 | 86.7 |
| 7d | (1R,2S,7a′S) | 10.26 | 1 | 2 | 88.6 |
| (±)-8a | (1RS,2RS,8a′SR) | 0.00 | 91 | 71 | 86.0 |
| (±)-8b | (1SR,2SR,8a′SR) | 7.74 | 4 | 10 | 85.0 |
| (±)-8c | (1RS,2SR,8a′SR) | 6.98 | 5 | 19 | 86.1 |
| (±)-8d | (1SR,2RS,8a′SR) | 22.91 | 0.01 | n.d. | n.d. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lamboley, S.; Vuichoud, B.; de Saint Laumer, J.-Y.; Herrmann, A. Release of Volatile Cyclopentanone Derivatives from Imidazolidin-4-One Profragrances in a Fabric Softener Application. Molecules 2023, 28, 382. https://doi.org/10.3390/molecules28010382
Lamboley S, Vuichoud B, de Saint Laumer J-Y, Herrmann A. Release of Volatile Cyclopentanone Derivatives from Imidazolidin-4-One Profragrances in a Fabric Softener Application. Molecules. 2023; 28(1):382. https://doi.org/10.3390/molecules28010382
Chicago/Turabian StyleLamboley, Serge, Basile Vuichoud, Jean-Yves de Saint Laumer, and Andreas Herrmann. 2023. "Release of Volatile Cyclopentanone Derivatives from Imidazolidin-4-One Profragrances in a Fabric Softener Application" Molecules 28, no. 1: 382. https://doi.org/10.3390/molecules28010382