
Supersaturation-Based Drug Delivery Systems: Strategy for Bioavailability Enhancement of Poorly Water-Soluble Drugs

 ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Supersaturated Drug Delivery Systems
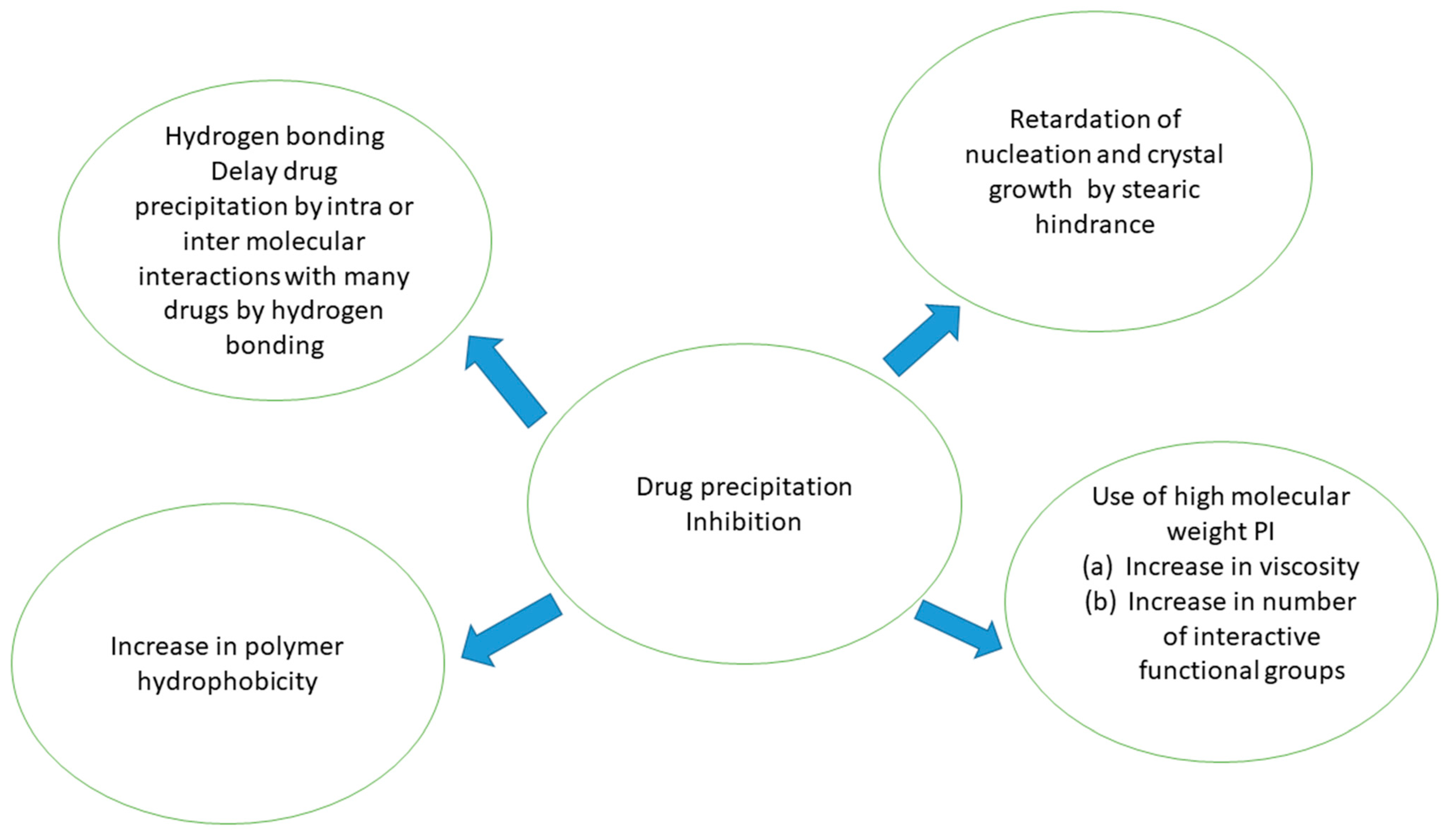
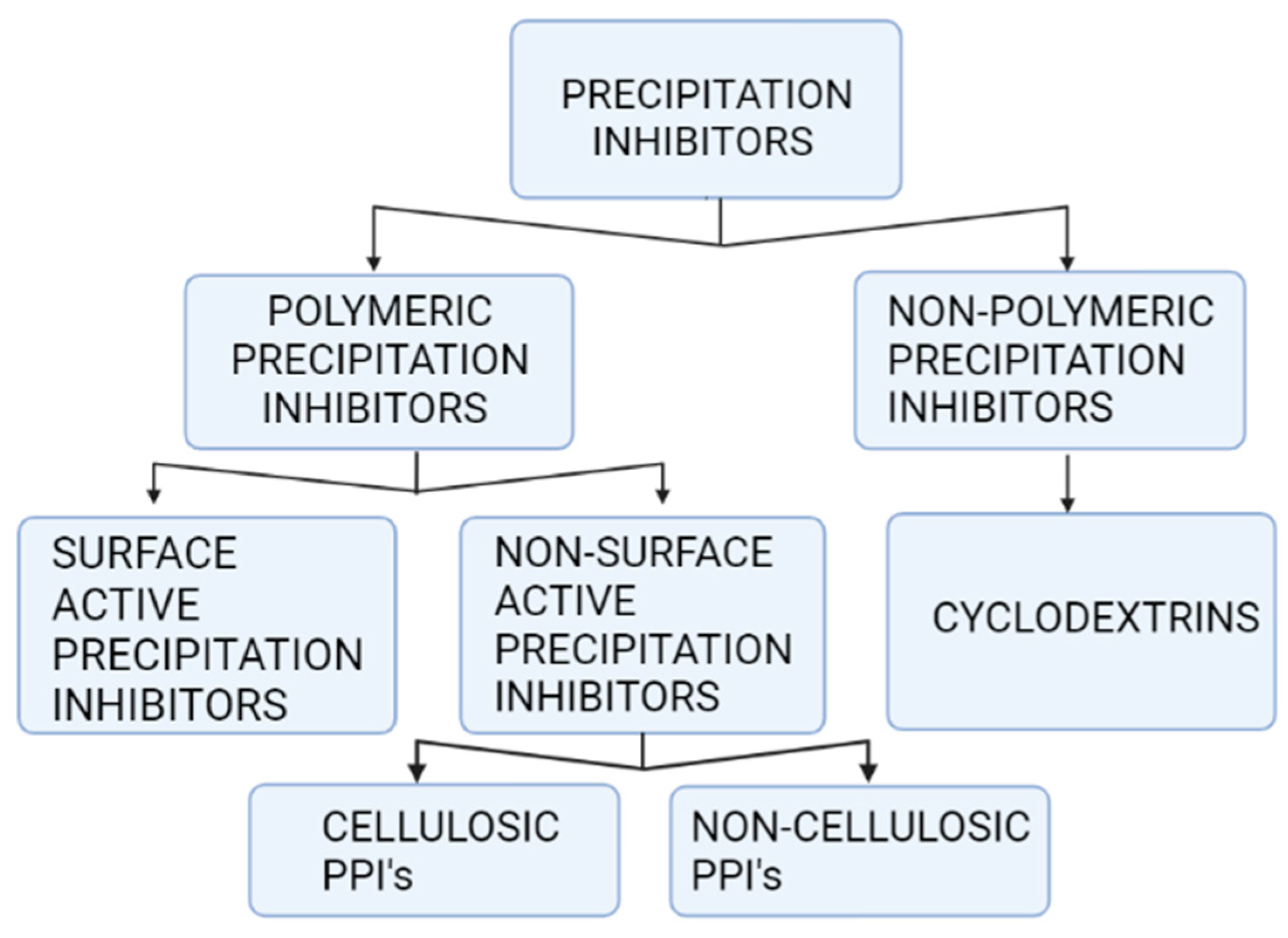
2.1. Precipitation Inhibitors

{kind=link}
{kind=link}
{kind=link}
| S.NO | Formulation (Spring Form) | Precipitation Inhibitors (Parachute) | Model Drug | In-Vivo and In-Vitro Performance (PK) | Reference |
|---|---|---|---|---|---|
| 1. | Solid dispersion | PEG 6000, PVP, HPMC | Tacrolimus | HPMC When compared to crystalline powder administration, there was a 10 fold increase in Cmax and AUC. | [14] |
| 2. | self-emulsifying drug delivery systems (SEDDS) | PEG 6000, PVP, HPMC | Paciltaxel | HPMC resulted in a 20-fold increase in Cmax and 10-fold increase in oral bioavailability | [15] |
| 3. | solid dispersions | HPMC, HPMCAS L, M, H SOL | Candesartan Cilexetil | HPMCAS M presented good anti-precipitation efficacy in both media, reaching higher AUC maintainind drug supersaturation for up to 120 min | [16] |
| 4. | Soild Dispersion | SOL, SLS, P188, PS20 | Chlorthalidone | SOL-SLS complex impacted positively release and Physical stability of chlorthalidone | [17] |
| 5. | Solid dispersion | SA, SLS | All SDs, demonstrated no drug recrystallization after 34 months of storage exception being those prepared with SA alone or SA-SLS at high drug loading | [18] | |
| 6. | Solid dispersion | P188, F127, SDS, HS15, ST and TPGS | Lacidipine | Nearly 3.3 and 3.7-fold increase in Cmax and AUC (0–∞) respectively was attained with formulation based on LCDP/SOL/SDS | [19] |
| 7. | Amorphous Solid dispersion | Eudragit EPO | Trimethoprin and sulfhmethooxazole | The 70% polymer formulation was able to produce and sustain the supersaturated phase of both compounds for 24 h. When compared to the combination of agents, improved antimicrobial effect was observed. | [20] |
| 8. | Amorphous Solid dispersion | hydroxypropylmethylcellulose acetate succinate (HPMCAS) type M | Candesartan cilexetil | Reduced the desupersaturation of both drugs | [21] |
| 9. | Amorphous Solid dispersion | Saccharin (SAC) | Griseofulvin | AUC increased 20% in comparison to conventional formulation | [22] |
| 10. | Soild dispersion | HPMC | Magnolol | Increased the bioavailability (the relative bioavailability was 213.69% | [23] |
| 11. | SEDDS | PEG 400, Tween 80, Miglyol 812 N | Carbamazepin | When compared to the commercial formulation, 200 mg of dosage resulted in 6.7 and 5.9 times larger increases in Cmax AUC, respectively. | [24] |
| 12. | SEDDS | HPMC-E5 PVP-12PF | Celecoxib | When comparison to solution and conventional capsule formulations, excellent IVIVC and Human PK was observed. | [25] |
| 13. | SEDDS | Soluplus | In comparison to drug powder, there was a 2.34-fold increase in Cmax and a 4.82-fold rise in AUC. | [26] | |
| 14. | SEDDS | Soluplus, PVP VA64, poloxamer 407, PEG 6000 | Celecoxib | PI effect of Soluplus is greater than PEG 6000 PVP, VA64, poloxamer 407 & PEG 6000 | [27] |
| 15. | SEDDS | Eudragit E PO | Curcumin | A 50 mg/kg dose of PI resulted in a 1.22 and 53.14-fold enhancement in absorption in rabbits when compared to the aqueous phase and standard SEDDS without PI, respectively. | [28] |
| 16. | SEDDS | Polyvinylpyrrolidone (PVP), hydroxypropyl methyl cellulose (HPMC) | Curcumin | The increased concentration-dependent effect was observed for PVP-K30 when used as PI in comparison to PVP-K90 without PI &HPMC. | [29] |
| 17. | SEDDS | HPMC K100 | Docetaxel | When SD rats were given a dose of 10 mg/kg, their AUC jumped by around 8.77 times which was 1.45-fold higher than the increases seen with the powder medication and traditional SEDDS without PI. | [30] |
| 18. | SEDDS | HPMC (5%, w/w) | Ginger extract | SD rats were given a dosage of 100 mg/kg in experimental model of animals, 6-gingerol and 8-gingerol had three time the antioxidant activity (BA) of the unformulated extract, i.e., control rats. | [31] |
| 19. | SEDDS | HPMC-E5 (5%, w/w) | Glipizide | AUC (2.7-fold) and Cmax (3.4-fold) were found to be increased in Himalayan rabbits when solid su-SEDDS were administered at a dose of 1 mg/kg as compared to the standard drug. | [32] |
| 20. | SEDDS | Poloxamer, HPMC | Griseofulvin | Aqueous suspension showed three-fold less permeability through the intestinal tract of Wister rats when given a dose of 1 mL at a concentration of 0.05 mg/mL (0.05 mg/mL). | [33] |
| 21. | SEEDS | HPMC, PEG 4000, PVP-K17 | Indirubin | When compared to the SEDDS without PI, the chemical exhibited better oral absorption and relative BA [129.5%] when delivered in vivo to SD rats at such a dose frequency of 2.58 mg/kg. | [34] |
| 22. | SEDDS | HPMC-E5LV | Paclitaxel | Compared to the Taxol® formulation and the standard SEDDS, the SD rats administered optimised formulation. At a dosage of 10 mg/kg, the Cmax and AUC were ten-fold and twenty-fold higher, respectively. | [35] |
| 23. | SEDDS | HPMC-E15LV | Resveratrol | After 20 mg/kg administration to Wistar rats, the su-SEDDS demonstrated a 1.33-fold increase in AUC compared to standard SEDDS lacking PI. | [36] |
| 24. | SEDDS | HPMC-E50LV | Silybin | SD rats were given a dosage of 533 mg/kg, which resulted in a 3-fold increase in AUC compared to the usual SEDDS without HPMC in vivo. | [37] |
| 25. | SEDDS | Poloxamer 407, Poloxamer 407 > HPCD, Eudragit L100 HPMCP | Silymarin | Using a dosage of 28 mg/kg of silybinvsLegalon® (a commercialized product) and a 76% BA of su-SEDDS concentration, silybin was evaluated in vivo in rabbits. | [38] |
| 26. | SEDDS | Soluplus, HPMC, PVP | Tacrolimus | As with conventional SEDDS, the Area under curve and Cmax of su-SEDDS at 1 dose of 5 mg/kg in SD rats were equivalent or larger than conventional SEDDS at the same dosage. | [39] |
| 27. | SEDDS | Poloxamer 407 | Valsartan | Using a dosage of 10 mg/kg, the medication was put to the test in SD rats. AUC ranges between about 177 and 198%when compared to API and Diovan®, a commercial product. | [40] |
| 28. | SNEEDS | HPMC, PVP, PVP/VA, and Soluplus® | Aprepitant | Increased dissolution rate of the drug due to enhanced solubility | [41] |
| 29. | SEDDS | HPMC E5 | Quercetin | improved AUC and Cmax values in comparison to conventional SEDDS | [42] |
| 30. | SNEDDS | HPMC | Albendazolum | Enhancement in the solubility and oral bioavailability | [43] |
| 31. | SNEDS | Poloxamer 407 (P 407), Eudragit® L100-55 (Eu), Kolliphor® HS15 (KHS15), Kolliphor® RH40 (K RH40), vitamin E TPGS (vit E TPGS) & Soluplus® | Cinnarizin | 2.7-fold increase in AUC 0–24 h | [44] |
| 32. | SEDDS | Cremophor RH40 & Macrogol 200 | Cepharanthine | Relative bioavailability was 203.46% | [45] |
| 33. | SNEDS | Polyoxyethylene (80) sorbitan monooleate (Tween® 80), d-α-tocopherol polyethylene glycol 1000 succinate (d-TPGS, Tocophersolan) | Celecoxib & fenofibrate | SNEDDS development in a short time with manageable resources | [46] |
| 34. | SMEDDS | PVP | Biphenyl dimethyl dicarboxylate | Significantly increased the Cmax and AUC | [47] |
| 35. | HPM C, HPMCA, SPV Pluronic F108, | Venetoclax | In vivo exposure of venetoclax was achieved | [48] |
Precipitation Inhibitors’ Influence on Supersaturation
2.2. Supersaturated Drug Delivery Systems (SDDSs) (Spring Form)
2.2.1. Solid Dispersion-Based Supersaturated Drug Delivery Systems (SDDSs)
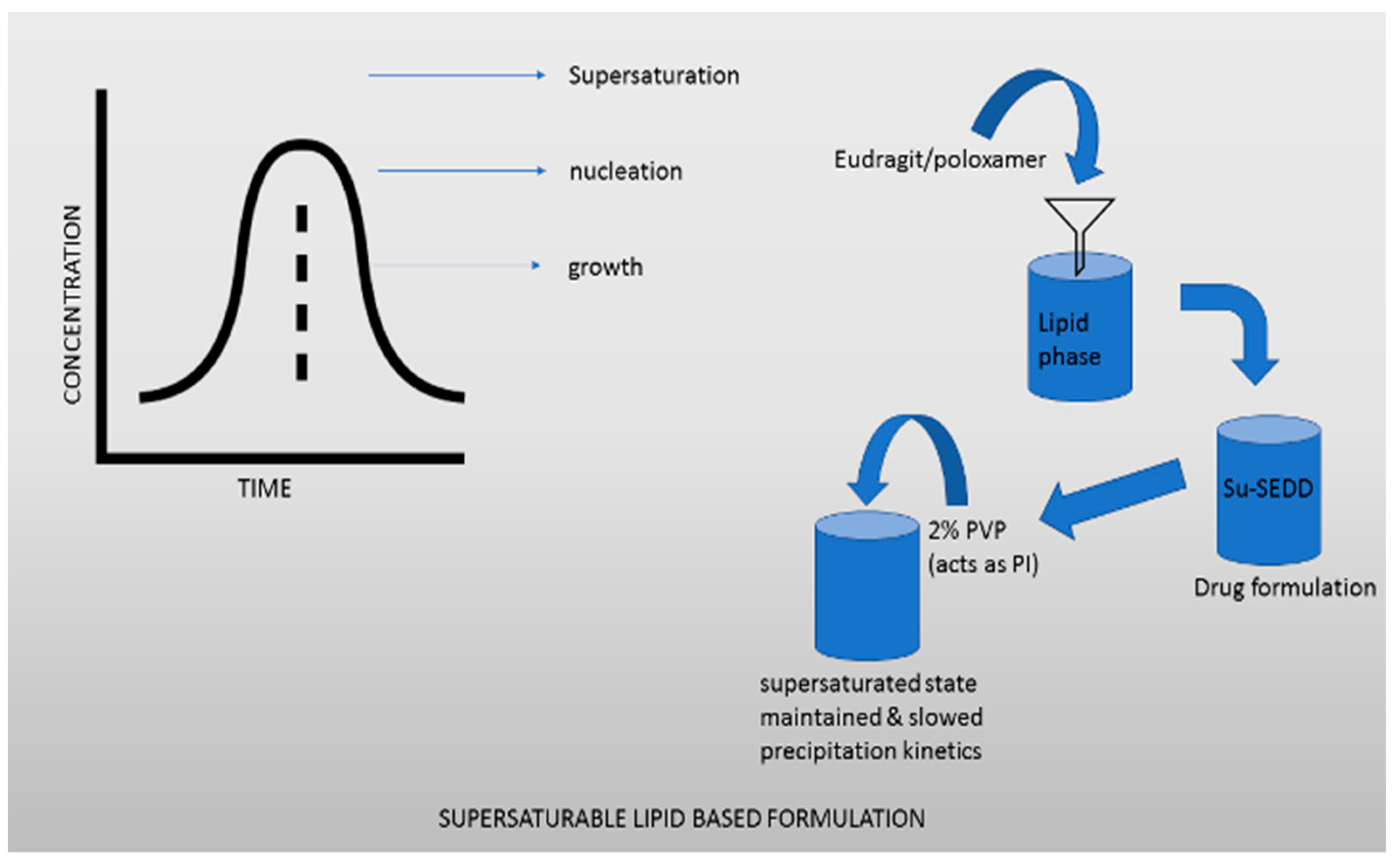
2.2.2. Supersaturable Lipid-Based Formulations
2.2.3. Transdermal Drug Delivery Systems (TDDSs)
3. Solid Dispersion-Based Supersaturated Drug Delivery Systems (SDDSs) vs. Supersaturable Lipid-Based Formulations
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brouwers, J.; Brewster, M.E.; Augustijns, P. Supersaturating drug delivery systems: The answer to solubility-limited oral bioavailability? J. Pharm. Sci. 2009, 98, 2549–2572. [Google Scholar] [CrossRef]
- Guzmán, H.R.; Tawa, M.; Zhang, Z.; Ratanabanangkoon, P.; Shaw, P.; Gardner, C.R.; Chen, H.; Moreau, J.P.; Almarsson, Ö.; Remenar, J.F. Combined use of crystalline salt forms and precipitation inhibitors to improve oral absorption of celecoxib from solid oral formulations. J. Pharm. Sci. 2007, 96, 2686–2702. [Google Scholar] [CrossRef]
- Franco, P.; De Marco, I. Nanoparticles and Nanocrystals by Supercritical CO2-Assisted Techniques for Pharmaceutical Applications: A Review. Appl. Sci. 2021, 11, 1476. [Google Scholar] [CrossRef]
- Bhatt, P.; Trehan, S.; Inamdar, N.; Mourya, V.K.; Misra, A. Polymers in Drug Delivery: An Update in Applications of Polymers in Drug Delivery; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Park, H.; Ha, E.S.; Kim, M.S. Current status of supersaturable self-emulsifying drug delivery systems. Pharmaceutics 2020, 12, 365. [Google Scholar] [CrossRef] [Green Version]
- Do Thi, T.; Van Speybroeck, M.; Barillaro, V.; Martens, J.; Annaert, P.; Augustijns, P.; Van Humbeeck, J.; Vermant, J.; Van den Mooter, G. Formulate-ability of ten compounds with different physicochemical profiles in SMEDDS. Eur. J. Pharm. Sci. 2009, 38, 479–488. [Google Scholar] [CrossRef]
- Anby, M.U.; Williams, H.D.; McIntosh, M.; Benameur, H.; Edwards, G.A.; Pouton, C.W.; Porter, C.J. Lipid digestion as a trigger for supersaturation: Evaluation of the impact of supersaturation stabilization on the in vitro and in vivo performance of self-emulsifying drug delivery systems. Mol. Pharm. 2012, 9, 2063–2079. [Google Scholar] [CrossRef]
- Warren, D.B.; Benameur, H.; Porter, C.J.; Pouton, C.W. Using polymeric precipitation inhibitors to improve the absorption of poorly water-soluble drugs: A mechanistic basis for utility. J. Drug Target. 2010, 18, 704–731. [Google Scholar] [CrossRef]
- Li, P.; Hynes, S.R.; Haefele, T.F.; Pudipeddi, M.; Royce, A.E.; Serajuddin, A.T. Development of clinical dosage forms for a poorly water-soluble drug II: Formulation and characterization of a novel solid microemulsion preconcentrate system for oral delivery of a poorly water-soluble drug. J. Pharm. Sci. 2009, 98, 1750–1764. [Google Scholar] [CrossRef]
- Serajuddin, A.T.; Li, P.; Haefele, T. Development of lipid-based drug delivery systems for poorly water-soluble drugs as viable oral dosage forms—Present status and future prospects. Am. Pharm. Rev. 2008, 11, 34–42. [Google Scholar]
- Dannenfelser, R.M.; He, H.; Joshi, Y.; Bateman, S.; Serajuddin, A.T. Development of clinical dosage forms for a poorly water-soluble drug I: Application of polyethylene glycol–polysorbate 80 solid dispersion carrier system. J. Pharm. Sci. 2004, 93, 1165–1175. [Google Scholar] [CrossRef]
- Warren, D.B.; Bergström, C.A.S.; Benameur, H.; Porter, C.J.S.; Pouton, C.W. Evaluation of the structural determinants of polymeric precipitation inhibitors using solvent shift methods and principle component analysis. Mol. Pharm. 2013, 10, 2823–2848. [Google Scholar] [CrossRef]
- Suys, E.J.; Chalmers, D.K.; Pouton, C.W.; Porter, C.J. Polymeric precipitation inhibitors promote fenofibrate supersaturation and enhance drug absorption from a type IV lipid-based formulation. Mol. Pharm. 2018, 15, 2355–2371. [Google Scholar] [CrossRef]
- Chauhan, H.; Hui-Gu, C.; Atef, E. Correlating the behavior of polymers in solution as precipitation inhibitor to its amorphous stabilization ability in solid dispersions. J. Pharm. Sci. 2013, 102, 1924–1935. [Google Scholar] [CrossRef]
- Usui, F.; Maeda, K.; Kusai, A.; Nishimura, K.; Yamamoto, K. Inhibitory effects of water-soluble polymers on precipitation of RS-8359. Int. J. Pharm. 1997, 154, 59–66. [Google Scholar] [CrossRef]
- Tang, B.; Cheng, G.; Gu, J.C.; Xu, C.H. Development of solid self-emulsifying drug delivery systems: Preparation techniques and dosage forms. Drug Discov. Today 2008, 13, 606–612. [Google Scholar] [CrossRef]
- Herpin, M.J.; Smyth, H.D. Super-heated aqueous particle engineering (SHAPE): A novel method for the micronization of poorly water-soluble drugs. J. Pharm. Investig. 2018, 48, 135–142. [Google Scholar] [CrossRef]
- Hajjar, B.; Zier, K.I.; Khalid, N.; Azarmi, S.; Löbenberg, R. Evaluation of a microemulsion-based gel formulation for topical drug delivery of diclofenac sodium. J. Pharm. Investig. 2018, 8, 351–362. [Google Scholar] [CrossRef]
- Singh, D.; Bedi, N.; Tiwary, A.K. Enhancing solubility of poorly aqueous soluble drugs: Critical appraisal of techniques. J. Pharm. Investig. 2018, 48, 509–526. [Google Scholar] [CrossRef]
- Dokania, S.; Joshi, A.K. Self-microemulsifying drug delivery system (SMEDDS)–challenges and road ahead. Drug Deliv. 2015, 22, 675–690. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Zhang, W.; Jin, Y.; Quan, D.Q. Studies on preparation of carbamazepine (CBZ) supersaturatable self-microemulsifying (S-SMEDDS) formulation and relative bioavailability in beagle dogs. Pharm. Dev. Technol. 2011, 16, 415–421. [Google Scholar] [CrossRef]
- Shi, Y.; Gao, P.; Gong, Y.; Ping, H. Application of a biphasic test for characterization of in vitro drug release of immediate release formulations of celecoxib and its relevance to in vivo absorption. Mol. Pharm. 2010, 7, 1458–1465. [Google Scholar] [CrossRef]
- Chavan, R.B.; Modi, S.R.; Bansal, A.K. Role of solid carriers in pharmaceutical performance of solid supersaturable SEDDS of celecoxib. Int. J. Pharm. 2015, 495, 374–384. [Google Scholar] [CrossRef]
- Pinto, J.M.O.; Leão, A.F.; Bazzo, G.C.; Mendes, C.; Madureira, L.M.P.; Caramori, G.F.; Parreira, R.L.T.; Stulzer, H.K. Supersaturating drug delivery systems containing fixed-dose combination of two antihypertensive drugs: Formulation, in vitro evaluation and molecular metadynamics simulations. Eur. J. Pharm. Sci. 2021, 163, 105860. [Google Scholar] [CrossRef]
- França, M.T.; Marcos, M.T.; Costa, P.F.A. Eutectic mixture and amorphous solid dispersion: Two different supersaturating drug delivery system strategies to improve griseofulvin release using saccharin. Int. J. Pharm. 2022, 615, 121–498. [Google Scholar] [CrossRef]
- Zhao, J.; Gao, P.; Mu, C. Preparation and Evaluation of Novel Supersaturated Solid Dispersion of Magnolol Theme: Advancements in Amorphous Solid Dispersions to Improve Bioavailability. AAPS PharmSciTech 2022, 23, 97. [Google Scholar] [CrossRef]
- Song, W.H.; Park, J.H.; Yeom, D.W.; Ahn, B.K.; Lee, K.M.; Lee, S.G.; Woo, H.S.; Choi, Y.W. Enhanced dissolution of celecoxib by supersaturating self-emulsifying drug delivery system (S-SEDDS) formulation. Arch. Pharm. Res. 2013, 36, 69–78. [Google Scholar] [CrossRef]
- Jaisamut, P.; Wiwattanawongsa, K.; Graidist, P.; Sangsen, Y.; Wiwattanapatapee, R. Enhanced oral bioavailability of curcumin using a supersaturatable self-microemulsifying system incorporating a hydrophilic polymer; in vitro and in vivo investigations. AAPS PharmSciTech 2018, 19, 730–740. [Google Scholar] [CrossRef]
- Gosangari, S.; Dyakonov, T. Enhanced dissolution performance of curcumin with the use of supersaturatable formulations. Pharm. Dev. Technol. 2013, 18, 475–480. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, C.; Zheng, J.; Chen, Z.; Shi, Q.; Liu, H. Development of a solid supersaturatable self-emulsifying drug delivery system of docetaxel with improved dissolution and bioavailability. Biol. Pharm. Bull. 2011, 34, 278–286. [Google Scholar] [CrossRef] [Green Version]
- Ogino, M.; Yakushiji, K.; Suzuki, H.; Shiokawa, K.; Kikuchi, H.; Seto, Y.; Sato, H.; Onoue, S. Enhanced pharmacokinetic behavior and hepatoprotective function of ginger extract-loaded supersaturable self-emulsifying drug delivery systems. J. Funct. Foods 2018, 40, 156–163. [Google Scholar] [CrossRef]
- Dash, R.N.; Mohammed, H.; Humaira, T.; Reddy, A.V. Solid supersaturatable self-nanoemulsifying drug delivery systems for improved dissolution, absorption and pharmacodynamic effects of glipizide. J. Drug Deliv. Sci. Technol. 2015, 28, 28–36. [Google Scholar] [CrossRef]
- Zadeha, B.S.M.; Salimi, A.; Aminib, R. Novel Super Saturated Self-Emulsifying System for Oral Delivery of Griseofulvin: Design, Preparation and ex-vivo Intestinal Permeability. J. Rep. Pharm. Sci. 2017, 6, 180–190. [Google Scholar]
- Chen, Z.Q.; Liu, Y.; Zhao, J.-H.; Wang, L.; Feng, N.-P. Improved oral bioavailability of poorly water-soluble indirubin by a supersaturatable self-microemulsifying drug delivery system. Int. J. Nanomed. 2012, 7, 1115. [Google Scholar]
- Gao, P.; Rush, B.D.; Pfund, W.P.; Huang, T.; Bauer, J.M.; Morozowich, W.; Kuo, M.S.; Hageman, M.J. Development of a supersaturable SEDDS (S-SEDDS) formulation of paclitaxel with improved oral bioavailability. J. Pharm. Sci. 2003, 92, 2386–2398. [Google Scholar] [CrossRef]
- Singh, G.; Pai, R.S. In vitro and in vivo performance of supersaturable self-nanoemulsifying system of trans-resveratrol. Artif. Cells Nanomed. Biotechnol. 2016, 44, 510–516. [Google Scholar] [CrossRef]
- Wei, Y.; Ye, X.; Shang, X.; Peng, X.; Bao, Q.; Liu, M.; Guo, M.; Li, F. Enhanced oral bioavailability of silybin by a supersaturatable self-emulsifying drug delivery system (S-SEDDS). Colloids Surf. A Physicochem. Eng. Asp. 2012, 396, 22–28. [Google Scholar] [CrossRef]
- Tung, N.T.; Tran, C.S.; Nguyen, H.A.; Nguyen, T.D.; Chi, S.-C.; Pham, D.V.; Bui, Q.D.; Ho, X.H. Formulation and biopharmaceutical evaluation of supersaturatable self-nanoemulsifying drug delivery systems containing silymarin. Int. J. Pharm. 2019, 555, 63–76. [Google Scholar] [CrossRef]
- Lee, D.R.; Ho, M.J.; Jung, H.J.; Cho, H.R.; Park, J.S.; Yoon, S.-H.; Choi, Y.S.; Choi, Y.W.; Oh, C.-H.; Kang, M.J. Enhanced dissolution and oral absorption of tacrolimus by supersaturable self-emulsifying drug delivery system. Int. J. Nanomed. 2016, 11, 1109. [Google Scholar]
- Shin, D.J.; Chae, B.R.; Goo, Y.T.; Yoon, H.Y.; Kim, C.H.; Sohn, S.I.; Oh, D.; Lee, A.; Song, S.H.; Choi, Y.W. Improved Dissolution and Oral Bioavailability of Valsartan Using a Solidified Supersaturable Self-Microemulsifying Drug Delivery System Containing Gelucire® 44/14. Pharmaceutics 2019, 11, 58. [Google Scholar] [CrossRef] [Green Version]
- Nazlı, H.; Mesut, B.; Özsoy, Y. In Vitro Evaluation of a Solid Supersaturated Self Nanoemulsifying Drug Delivery System (Super-SNEDDS) of Aprepitant for Enhanced Solubility. Pharmaceuticals 2021, 14, 1089. [Google Scholar] [CrossRef]
- Sirvi, A.; Kuche, K.; Chaudhari, D.; Ghadi, R.; Date, T.; Katiyar, S.S.; Jain, S. Supersaturable self-emulsifying drug delivery system: A strategy for improving the loading and oral bioavailability of quercetin. J. Drug Deliv. Sci. Technol. 2022, 71, 103289. [Google Scholar] [CrossRef]
- Cavalu, S.; Bisboaca, S.; Mates, I.M.; Pasca, P.M.; Laslo, V.; Costea, T.; Fritea, L.; Vicas, S. Novel Formulation Based on Chitosan-Arabic Gum Nanoparticles Entrapping Propolis Extract Production, physico-chemical and structural characterization. Rev. Chim. 2018, 69, 3756–3760. [Google Scholar] [CrossRef]
- Lie, A.R.; Griffin, B.T.; Vetzoni, M.; Kuentz, M.; Kolakovic, R.; Prudic-Paus, A.; Malash, A.; Bohets, H.; Herman, J.; Holm, R. Exploring precipitation inhibitors to improve in vivo absorption of cinnarizine from supersaturated lipid-based drug delivery systems. Eur. J. Pharm. Sci. 2021, 159, 105691. [Google Scholar]
- Gao, P.; Jiang, Z.; Luo, Q. Preparation and Evaluation of Self-emulsifying Drug Delivery System (SEDDS) of Cepharanthine. AAPS PharmSciTech 2021, 22, 245. [Google Scholar] [CrossRef]
- Schmied, F.P.; Bernhardt, A.; Engel, A. A Customized Screening Tool Approach for the Development of a Self-Nanoemulsifying Drug Delivery System (SNEDDS). AAPS PharmSciTech 2022, 23, 39. [Google Scholar] [CrossRef]
- Jiang, Q.; Wang, T.; Li, G. Evaluation on a supersaturatable self-microemulsifying (s-smdds) formulation of biphenyl dimethyl dicarboxylate (BDD) in Vitro and in Vivo. Food Sci. Technol. 2022. [Google Scholar] [CrossRef]
- Ma, X.; Williams, R.O. Polymeric nanomedicines for poorly soluble drugs in oral delivery systems: An update. J. Pharm. Investig. 2018, 48, 61–75. [Google Scholar]
- Patel, D. Kinetics and Mechanisms of Crystal Growth Inhibition of Indomethacin by Model Precipitation Inhibitors; University of Kentucky: Lexington, KY, USA, 2015. [Google Scholar]
- Phaechamud, T.; Lertsuphotvanit, N.; Issarayungyuen, P.; Chantadee, T. Design, fabrication and characterization of xanthan gum/liquid-loaded porous natural rubber film. J. Pharm. Investig. 2019, 49, 149–160. [Google Scholar] [CrossRef]
- Dias, M.M.; Raghavan, S.L.; Pellett, M.A.; Hadgraft, J. The effect of β-cyclodextrins on the permeation of diclofenac from supersaturated solutions. Int. J. Pharm. 2003, 263, 173–181. [Google Scholar] [CrossRef]
- Iervolino, M.; Raghavan, S.L.; Hadgraft, J. Membrane penetration enhancement of ibuprofen using supersaturation. Int. J. Pharm. 2000, 198, 229–238. [Google Scholar] [CrossRef]
- Brewster, M.E.; Vandecruys, R.; Peeters, J.; Neeskens, P.; Verreck, G.; Loftsson, T. Comparative interaction of 2-hydroxypropyl-β-cyclodextrin and sulfobutylether-β-cyclodextrin with itraconazole: Phase-solubility behavior and stabilization of supersaturated drug solutions. Eur. J. Pharm. Sci. 2008, 34, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Vogensen, S.B.; Brewster, M.E.; Konráðsdóttir, F. Effects of cyclodextrins on drug delivery through biological membranes. J. Pharm. Sci. 2007, 96, 2532–2546. [Google Scholar] [CrossRef] [PubMed]
- Amin, O.M.; Ammar, A.; Eladawy, S.A. Febuxostat loaded β-cyclodextrin based nanosponge tablet: An in vitro and in vivo evaluation. Eur. J. Pharm. Sci. 2020, 50, 399–411. [Google Scholar] [CrossRef]
- Wu, Z.; Tucker, I.G.; Razzak, M.; Yang, L.; McSporran, K.; Medlicott, N.J. Absorption and tissue tolerance of ricobendazole in the presence of hydroxypropyl-β-cyclodextrin following subcutaneous injection in sheep. Int. J. Pharm. 2010, 397, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Curatolo, W.; Nightingale, J.A.; Herbig, S.M. Utility of hydroxypropylmethylcellulose acetate succinate (HPMCAS) for initiation and maintenance of drug supersaturation in the GI milieu. Pharm. Res. 2009, 26, 1419–1431. [Google Scholar] [CrossRef]
- Cho, W.; Kim, M.S.; Kim, J.S.; Park, J.; Park, H.J.; Cha, K.H.; Park, J.S.; Hwang, H.J. Optimized formulation of solid self-microemulsifying sirolimus delivery systems. Int. J. Nanomed. 2013, 8, 1673–1682. [Google Scholar]
- Pouton, C.W. Formulation of poorly water-soluble drugs for oral administration: Physicochemical and physiological issues and the lipid formulation classification system. Eur. J. Pharm. Sci. 2006, 29, 278–287. [Google Scholar] [CrossRef]
- Amara, S.; Bourlieu, C.; Humbert, L.; Rainteau, D.; Carrière, F. Variations in gastrointestinal lipases, pH and bile acid levels with food intake, age and diseases: Possible impact on oral lipid-based drug delivery systems. Adv. Drug Deliv. Rev. 2019, 142, 3–15. [Google Scholar] [CrossRef]
- Schram, C.J.; Beaudoin, S.P.; Taylor, L.S. Impact of polymer conformation on the crystal growth inhibition of a poorly water-soluble drug in aqueous solution. Langmuir 2015, 31, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Bevernage, J.; Forier, T.; Brouwers, J.; Tack, J.; Annaert, P.; Augustijns, P. Excipient-mediated supersaturation stabilization in human intestinal fluids. Mol. Pharm. 2011, 8, 564–570. [Google Scholar] [CrossRef]
- Boyd, B.J.; Bergström, C.A.; Vinarov, Z.; Kuentz, M.; Brouwers, J.; Augustijns, P.; Brandl, M.; Bernkop-Schnürch, A.; Shrestha, N.; Préat, V.; et al. Successful oral delivery of poorly water-soluble drugs both depends on the intraluminal behavior of drugs and of appropriate advanced drug delivery systems. Eur. J. Pharm. Sci. 2019, 137, 104967. [Google Scholar] [CrossRef] [PubMed]
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, C.J.; Trevaskis, N.L.; Charman, W.N. Lipids and lipid-based formulations: Optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug Discov. 2007, 6, 231–248. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.; Sangamwar, A.T. Stabilizing supersaturated drug-delivery system through mechanism of nucleation and crystal growth inhibition of drugs. Ther. Deliv. 2018, 9, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Danafar, H.; Rostamizadeh, K.; Hamidi, M. Polylactide/poly (ethylene glycol)/polylactide triblock copolymer micelles as carrier for delivery of hydrophilic and hydrophobic drugs: A comparison study. J. Pharm. Investig. 2018, 48, 381–391. [Google Scholar] [CrossRef]
- Berthelsen, R.; Klitgaard, M.; Rades, T.; Müllertz, A. In vitro digestion models to evaluate lipid-based drug delivery systems; present status and current trends. Adv. Drug Deliv. Rev. 2019, 142, 35–49. [Google Scholar] [CrossRef]
- Fong, S.Y.; Bauer-Brandl, A.; Brandl, M. Oral bioavailability enhancement through supersaturation: An update and meta-analysis. Expert Opin. Drug Deliv. 2017, 14, 403–426. [Google Scholar] [CrossRef]
- Schittny, A.; Huwyler, J.; Puchkov, M. Mechanisms of increased bioavailability through amorphous solid dispersions: A review. Drug Deliv. 2020, 27, 110–127. [Google Scholar] [CrossRef]
- Lipp, R. The innovator pipeline: Bioavailability challenges and advanced oral drug delivery opportunities. Am. Pharm. Rev. 2013, 16, 14–16. [Google Scholar]
- Waring, M.J.; Arrowsmith, J.; Leach, A.R.; Leeson, P.D.; Mandrell, S.; Owen, R.M.; Pairaudeau, G.; Pennie, W.D.; Pickett, S.D.; Wang, J.; et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 2015, 14, 475–486. [Google Scholar] [CrossRef]
- Padden, B.E.; Miller, J.M.; Robbins, T.; Prasad, L.; Spence, J.K.; LaFountaine, J. Formulation development-amorphous solid dispersions as enabling formulations for discovery and early development. Am. Pharm. Rev. 2011, 14, 66. [Google Scholar]
- Zhang, J.; Han, R.; Chen, W.; Zhang, W.; Li, Y.; Ji, Y.; Chen, L.; Pan, H.; Yang, X.; Pan, W.; et al. Analysis of the literature and patents on solid dispersions from 1980 to 2015. Molecules 2018, 23, 1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Xia, D.; Zhu, Q.; Zhu, C.; Chen, D.; Gan, Y. Supersaturated polymeric micelles for oral cyclosporine A delivery. Eur. J. Pharm. Biopharm. 2013, 85, 1325–1336. [Google Scholar] [CrossRef]
- Fule, R.; Dhamecha, D.; Maniruzzaman, M.; Khale, A.; Amin, P. Development of hot melt co-formulated antimalarial solid dispersion system in fixed dose form (ARLUMELT): Evaluating amorphous state and in vivo performance. Int. J. Pharm. 2015, 496, 137–156. [Google Scholar] [CrossRef] [PubMed]
- Fule, R.; Paithankar, V.; Amin, P. Hot melt extrusion based solid solution approach: Exploring polymer comparison, physicochemical characterization and in-vivo evaluation. Int. J. Pharm. 2016, 499, 280–294. [Google Scholar] [CrossRef]
- Agrawal, A.M.; Dudhedia, M.S.; Zimny, E. Hot melt extrusion: Development of an amorphous solid dispersion for an insoluble drug from mini-scale to clinical scale. AAPS PharmSciTech 2016, 17, 133–147. [Google Scholar] [CrossRef]
- Kate, L.; Gokarna, V.; Borhade, V.; Prabhu, P.; Deshpande, V.; Pathak, S.; Sharma, S.; Patravale, V. Bioavailability enhancement of atovaquone using hot melt extrusion technology. Eur. J. Pharm. Sci. 2016, 86, 103–114. [Google Scholar] [CrossRef]
- Mitra, A.; Li, L.; Marsac, P.; Marks, B.; Liu, Z.; Brown, C. Impact of polymer type on bioperformance and physical stability of hot melt extruded formulations of a poorly water-soluble drug. Int. J. Pharm. 2016, 505, 107–114. [Google Scholar] [CrossRef]
- Xia, D.; Yu, H.; Tao, J.; Zeng, J.; Zhu, Q.; Zhu, C.; Gan, Y. Supersaturated polymeric micelles for oral cyclosporine A delivery: The role of Soluplus–sodium dodecyl sulfate complex. Colloids Surf. B Biointerfaces 2016, 141, 301–310. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Luo, Y.; Yao, Q.; Zhong, Y.; Tian, B.; Tang, X. Extruded Soluplus/SIM as an oral delivery system: Characterization, interactions, in vitro and in vivo evaluations. Drug Deliv. 2016, 23, 1902–1911. [Google Scholar]
- Knopp, M.M.; Wendelboe, J.; Holm, R.; Rades, T. Effect of amorphous phase separation and crystallization on the in vitro and in vivo performance of an amorphous solid dispersion. Eur. J. Pharm. Biopharm. 2018, 130, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Liu, Z.; Chen, Y.; Chen, Z.; Chen, H.; Pui, Y.; Qian, F. Oral bioavailability enhancement of β-lapachone, a poorly soluble fast crystallizer, by cocrystal, amorphous solid dispersion, and crystalline solid dispersion. Eur. J. Pharm. Biopharm. 2018, 124, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Six, K.; Daems, T.; de Hoon, J.; Van Hecken, A.; Depre, M.; Bouche, M.P.; Prinsen, P.; Verreck, G.; Peeters, J.; Brewster, M.E.; et al. Clinical study of solid dispersions of itraconazole prepared by hot-stage extrusion. Eur. J. Pharm. Sci. 2005, 24, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.; Herzog, M.; König, S.; Storch, C.H.; Ketabi-Kiyanvash, N.; Haefeli, W.E. Induction of multiple drug transporters by efavirenz. J. Pharmacol. Sci. 2009, 109, 242–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moes, J.J.; Koolen, S.L.; Huitema, A.D.; Schellens, J.H.; Beijnen, J.H.; Nuijen, B. Pharmaceutical development and preliminary clinical testing of an oral solid dispersion formulation of docetaxel (ModraDoc001). Int. J. Pharm. 2011, 420, 244–250. [Google Scholar] [CrossRef]
- Krishna, G.; Ma, L.; Martinho, M.; Preston, R.A.; O’Mara, E. A new solid oral tablet formulation of posaconazole: A randomized clinical trial to investigate rising single-and multiple-dose pharmacokinetics and safety in healthy volunteers. J. Antimicrob. Chemother. 2012, 67, 2725–2730. [Google Scholar] [CrossRef] [Green Version]
- Marchetti, S.; Stuurman, F.; Koolen, S.; Moes, J.; Hendrikx, J.; Thijssen, B.; Huitema, A.D.; Nuijen, B.; Rosing, H.; Keessen, M.; et al. Phase I study of weekly oral docetaxel (ModraDoc001) plus ritonavir in patients with advanced solid tumors. J. Clin. Oncol. 2012, 30, 2550. [Google Scholar] [CrossRef]
- Zayed, R.; Kamel, A.O.; Shukr, M.; El-Shamy, A.E. An in vitro and in vivo comparative study of directly compressed solid dispersions and freeze dried sildenafil citrate sublingual tablets for management of pulmonary arterial hypertension. Acta Pharm. 2012, 62, 411–432. [Google Scholar] [CrossRef] [Green Version]
- Othman, A.A.; Cheskin, H.; Locke, C.; Nothaft, W.; Dutta, S. A Phase 1 Study to Evaluate the Bioavailability and Food Effect of 2 Solid Dispersion Formulations of the TRPV1 Antagonist ABT_102, Relative to the Oral Solution Formulation, in Healthy Human Volunteers. Clin. Pharmacol. Drug Dev. 2012, 1, 24–31. [Google Scholar] [CrossRef]
- Othman, A.A.; Nothaft, W.; Awni, W.M.; Dutta, S. Pharmacokinetics of the TRPV1 antagonist ABT_102 in healthy human volunteers: Population analysis of data from 3 phase 1 trials. J. Clin. Pharmacol. 2012, 52, 1028–1041. [Google Scholar] [CrossRef]
- Moes, J.; Koolen, S.; Huitema, A.; Schellens, J.; Beijnen, J.; Nuijen, B. Development of an oral solid dispersion formulation for use in low-dose metronomic chemotherapy of paclitaxel. Eur. J. Pharm. Biopharm. 2013, 83, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Prasannaraju, Y.; Harini Chowdary, V.; Jayasri, V.; Asuntha, G.; Kishore Kumar, N.; V. Ramana Murthy, K.; Nair, R. Bioavailability and pharmacokinetic studies of rofecoxib solid dispersion. Curr. Drug Deliv. 2013, 10, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Schittny, A.; Philipp-Bauer, S.; Detampel, P.; Huwyler, J.; Puchkov, M. Mechanistic insights into effect of surfactants on oral bioavailability of amorphous solid dispersions. J. Control. Release 2020, 320, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Gong, W.; Wang, Y.; Shan, L.; Li, Y.; Gao, C. Bioavailability Improvement Strategies for Poorly Water-Soluble Drugs Based on the Supersaturation Mechanism: An Update. J. Pharm. Pharm. Sci. 2016, 19, 208–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, 1074–1082. [Google Scholar] [CrossRef]
- Newman, A. (Ed.) Pharmaceutical Amorphous Solid Dispersions; John Wiley & Sons: Hoboken, NJ, USA, 2015. [Google Scholar]
- Park, K. Drug release mechanisms from amorphous solid dispersions. J. Control. Release 2015, 211, 171. [Google Scholar] [CrossRef]
- Tho, I.; Liepold, B.; Rosenberg, J.; Maegerlein, M.; Brandl, M.; Fricker, G. Formation of nano/micro-dispersions with improved dissolution properties upon dispersion of ritonavir melt extrudate in aqueous media. Eur. J. Pharm. Sci. 2010, 40, 25–32. [Google Scholar] [CrossRef]
- Ashwathy, P.; Anto, A.T.; Sudheesh, M.S. A mechanistic review on the dissolution phase behavior and supersaturation stabilization of amorphous solid dispersions. Drug Dev. Ind. Pharm. 2021, 47, 1–11. [Google Scholar] [CrossRef]
- Taylor, L.S.; Zhang, G.G. Physical chemistry of supersaturated solutions and implications for oral absorption. Adv. Drug Deliv. Rev. 2016, 101, 122–142. [Google Scholar] [CrossRef]
- Kanzer, J.; Hupfeld, S.; Vasskog, T.; Tho, I.; Hölig, P.; Mägerlein, M.; Fricker, G.; Brandl, M. In situ formation of nanoparticles upon dispersion of melt extrudate formulations in aqueous medium assessed by asymmetrical flow field-flow fractionation. J. Pharm. Biomed. Anal. 2010, 53, 359–365. [Google Scholar] [CrossRef]
- Frank, K.J.; Westedt, U.; Rosenblatt, K.M.; Hölig, P.; Rosenberg, J.; Mägerlein, M.; Fricker, G.; Brandl, M. The amorphous solid dispersion of the poorly soluble ABT-102 forms nano/microparticulate structures in aqueous medium: Impact on solubility. Int. J. Nanomed. 2012, 7, 5757. [Google Scholar]
- Ueda, K.; Higashi, K.; Moribe, K. Mechanistic elucidation of formation of drug-rich amorphous nanodroplets by dissolution of the solid dispersion formulation. Int. J. Pharm. 2019, 561, 82–92. [Google Scholar] [CrossRef]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Understanding the generation and maintenance of supersaturation during the dissolution of amorphous solid dispersions using modulated DSC and 1H NMR. Int. J. Pharm. 2018, 536, 414–425. [Google Scholar] [CrossRef]
- Cavalu, S.; Banica, F.; Gruian, C.; Vanea, E.; Goller, G.; Simon, V. Microscopic and spectroscopic investigation of bioactive glasses for antibiotic controlled release. J. Mol. Struct. 2013, 1040, 47–52. [Google Scholar] [CrossRef]
- Ueda, K.; Higashi, K.; Yamamoto, K.; Moribe, K. Equilibrium state at supersaturated drug concentration achieved by hydroxypropyl methylcellulose acetate succinate: Molecular characterization using (1)H NMR technique. Mol. Pharm. 2015, 12, 1096–1104. [Google Scholar] [CrossRef]
- Ueda, K.; Higashi, K.; Moribe, K. Direct NMR monitoring of phase separation behavior of highly supersaturated nifedipine solution stabilized with hypromellose derivatives. Mol. Pharm. 2017, 14, 2314–2322. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, S.; Wang, S.; Liu, C.; Su, C.; Hageman, M.; Hussain, M.; Haskell, R.; Stefanski, K.; Qian, F. Sodium lauryl sulfate competitively interacts with HPMC-AS and consequently reduces oral bioavailability of posaconazole/HPMC-AS amorphous solid dispersion. Mol. Pharm. 2016, 13, 2787–2795. [Google Scholar] [CrossRef]
- Szafraniec, J.; Antosik, A.; Knapik-Kowalczuk, J.; Chmiel, K.; Kurek, M.; Gawlak, K.; Paluch, M.; Jachowicz, R. Enhanced dissolution of solid dispersions containing bicalutamide subjected to mechanical stress. Int. J. Pharm. 2018, 542, 18–26. [Google Scholar] [CrossRef]
- Raina, S.A.; Van Eerdenbrugh, B.; Alonzo, D.E.; Mo, H.; Zhang, G.G.; Gao, Y.; Taylor, L.S. Trends in the precipitation and crystallization behavior of supersaturated aqueous solutions of poorly water-soluble drugs assessed using synchrotron radiation. J. Pharm. Sci. 2015, 104, 1981–1992. [Google Scholar] [CrossRef]
- Guan, J.; Jin, L.; Liu, Q.; Xu, H.; Wu, H.; Zhang, X.; Mao, S. Exploration of supersaturable lacidipine ternary amorphous solid dispersion for enhanced dissolution and in vivo absorption. Eur. J. Pharm. Sci. 2019, 139, 105043. [Google Scholar] [CrossRef]
- Baird, J.A.; Van Eerdenbrugh, B.; Taylor, L.S. A classification system to assess the crystallization tendency of organic molecules from undercooled melts. J. Pharm. Sci. 2010, 99, 3787–3806. [Google Scholar] [CrossRef] [PubMed]
- Van Eerdenbrugh, B.; Baird, J.A.; Taylor, L.S. Crystallization tendency of active pharmaceutical ingredients following rapid solvent evaporation—Classification and comparison with crystallization tendency from under cooled melts. J. Pharm. Sci. 2010, 99, 3826–3838. [Google Scholar] [CrossRef] [PubMed]
- Mahlin, D.; Ponnambalam, S.; Heidarian Höckerfelt, M.; Bergström, C.A. Toward in silico prediction of glass-forming ability from molecular structure alone: A screening tool in early drug development. Mol. Pharm. 2011, 8, 498–506. [Google Scholar] [CrossRef]
- Gao, P.; Shi, Y. Characterization of supersaturatable formulations for improved absorption of poorly soluble drugs. AAPS J. 2012, 14, 703–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Dai, W.G. Drug precipitation inhibitors in supersaturable formulations. Int. J. Pharm. 2013, 453, 36–43. [Google Scholar] [CrossRef]
- Kojima, T.; Higashi, K.; Suzuki, T.; Tomono, K.; Moribe, K.; Yamamoto, K. Stabilization of a supersaturated solution of mefenamic acid from a solid dispersion with EUDRAGIT® EPO. Pharm. Res. 2012, 29, 2777–2791. [Google Scholar] [CrossRef] [PubMed]
- Mosquera-Giraldo, L.I.; Borca, C.H.; Parker, A.S.; Dong, Y.; Edgar, K.J.; Beaudoin, S.P.; Slipchenko, L.V.; Taylor, L.S. Crystallization inhibition properties of cellulose esters and ethers for a group of chemically diverse drugs: Experimental and computational insight. Biomacromolecules 2018, 19, 4593–4606. [Google Scholar] [CrossRef]
- Ilevbare, G.A.; Liu, H.; Edgar, K.J.; Taylor, L.S. Understanding Polymer Properties Important for Crystal Growth Inhibition, Impact of Chemically Diverse Polymers on Solution Crystal Growth of Ritonavir. Cryst. Growth Des. 2012, 12, 3133–3143. [Google Scholar] [CrossRef]
- Brough, C.; Williams, R.O., 3rd. Amorphous solid dispersions and nano-crystal technologies for poorly water-soluble drug delivery. Int. J. Pharm. 2013, 453, 157–166. [Google Scholar] [CrossRef]
- Raina, S.A.; Zhang, G.G.; Alonzo, D.E.; Wu, J.; Zhu, D.; Catron, N.D.; Gao, Y.; Taylor, L.S. Enhancements and limits in drug membrane transport using supersaturated solutions of poorly water-soluble drugs. J. Pharm. Sci. 2014, 103, 2736–2748. [Google Scholar] [CrossRef]
- Williams, H.D.; Trevaskis, N.L.; Yeap, Y.Y.; Anby, M.U.; Pouton, C.W.; Porter, C.J. Lipid-based formulations and drug supersaturation: Harnessing the unique benefits of the lipid digestion/absorption pathway. Pharm. Res. 2013, 30, 2976–2992. [Google Scholar] [CrossRef]
- Suys, E.J.A.; Brundel, D.H.S.; Chalmers, D.K.; Pouton, C.W.; Porter, C.J.H. Interaction with biliary and pancreatic fluids drives supersaturation and drug absorption from lipid-based formulations of low (saquinavir) and high (fenofibrate) permeability poorly soluble drugs. J. Control. Release 2021, 331, 45–61. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Quan, D.Q. Assessment of a supersaturated self-microemulsifying formulation of biphenyl dimethyl dicarboxylate in vitro and in vivo. Chin. Pharm. J. 2011, 46, 600–604. [Google Scholar]
- Desai, S.; Disouza, J.; Sable, A.; Hosmani, A. Development of orodispersible tablet of atorvastatin calcium using hot melt extrusion. Drug Deliv. Lett. 2015, 5, 19–30. [Google Scholar] [CrossRef]
- Charkoftaki, G.; Dokoumetzidis, A.; Valsami, G.; Macheras, P. Supersaturated dissolution data and their interpretation: The TPGS–carbamazepine model case. J. Pharm. Pharmacol. 2011, 63, 352–361. [Google Scholar] [CrossRef]
- Ganesh, M.; Jeon, U.J.; Ubaidulla, U.; Hemalatha, P.; Saravanakumar, A.; Peng, M.M.; Jang, H.T. Chitosan cocrystals embedded alginate beads for enhancing the solubility and bioavailability of aceclofenac. Int. J. Biol. Macromol. 2015, 74, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Kubota, N.; Yokota, M.; Mullin, J.W. The combined influence of supersaturation and impurity concentration on crystal growth. J. Cryst. Growth 2000, 212, 480–488. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Katare, O.P.; Singh, B. Development of optimized supersaturable self-nanoemulsifying systems of ezetimibe: Effect of polymers and efflux transporters. Expert Opin. Drug Deliv. 2014, 11, 479–492. [Google Scholar] [CrossRef]
- Kim, M.S.; Kim, J.S.; Park, H.J.; Cho, W.K.; Cha, K.H.; Hwang, S.J. Enhanced bioavailability of sirolimus via preparation of solid dispersion nanoparticles using a supercritical antisolvent process. Int. J. Nanomed. 2011, 6, 2997. [Google Scholar]
- Miller, M.A.; DiNunzio, J.; Matteucci, M.E.; Ludher, B.S.; Williams, R.O.; Johnston, K.P. Flocculated amorphous itraconazole nanoparticles for enhanced in vitro supersaturation and in vivo bioavailability. Drug Dev. Ind. Pharm. 2012, 38, 557–570. [Google Scholar] [CrossRef]
- Van Speybroeck, M.; Mellaerts, R.; Martens, J.A.; Annaert, P.; Van den Mooter, G.; Augustijns, P. Ordered Mesoporous Silica for the Delivery of Poorly Soluble Drugs; Springer: Boston, MA, USA, 2011; pp. 203–219. [Google Scholar]
- Gao, P.; Akrami, A.; Alvarez, F.; Hu, J.; Li, L.; Ma, C.; Surapaneni, S. Characterization and optimization of AMG 517 supersaturatable self-emulsifying drug delivery system (S-SEDDS) for improved oral absorption. J. Pharm. Sci. 2009, 98, 516–528. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shi, Q.; Chen, Z.; Zheng, J.; Xu, H.; Li, J.; Liu, H. Preparation and characterization of emulsified solid dispersions containing docetaxel. Arch. Pharm. Res. 2011, 34, 1909–1917. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, S.; Katare, O.P.; Singh, B. Optimized self nano-emulsifying systems of ezetimibe with enhanced bioavailability potential using long chain and medium chain triglycerides. Colloids Surf. B Biointerfaces 2012, 100, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Craig, H.; Hayward, T. Oxygen supersaturation in the ocean: Biological versus physical contributions. Science 1987, 235, 199–202. [Google Scholar] [CrossRef]
- Jog, R.; Burgess, D.J. Pharmaceutical Amorphous Nanoparticles. J. Pharm. Sci. 2017, 106, 39–65. [Google Scholar] [CrossRef] [Green Version]
- Quan, G.; Wu, Q.; Zhang, X.; Zhan, Z.; Zhou, C.; Chen, B.; Zhang, Z.; Li, G.; Pan, X.; Wu, C. Enhancing in vitro dissolution and in vivo bioavailability of fenofibrate by solid self-emulsifying matrix combined with SBA-15 mesoporous silica. Colloids Surf. B 2016, 141, 476–482. [Google Scholar] [CrossRef]
- Matteucci, M.E.; Paguio, J.C.; Miller, M.A.; Williams 3rd, R.O.; Johnston, K.P. Highly supersaturated solutions from dissolution of amorphous itraconazole microparticles at pH 6.8. Mol. Pharm. 2009, 6, 375–385. [Google Scholar] [CrossRef]
- Maleki, A.; Kettiger, H.; Schoubben, A.; Rosenholm, J.M.; Ambrogi, V.; Hamidi, M. Mesoporous silica materials: From physico-chemical properties to enhanced dissolution of poorly water-soluble drugs. J. Control. Release 2017, 262, 329–347. [Google Scholar] [CrossRef]
- Wang, F.L.; Ji, H.M.; Zhu, J.Y.; Xu, G.J.; Guan, Y.Z.; Chen, Y.J. Penetration enhancement effect of turpentine oil on transdermal film of ketorolac. Trop. J. Pharm. Res. 2015, 14, 1341–1348. [Google Scholar] [CrossRef] [Green Version]
- Coldman, M.F.; Poulsen, B.J.; Higuchi, T. Enhancement of percutaneous absorption by the use of volatile: Nonvolatile systems as vehicles. J. Pharm. Sci. 1969, 58, 1098–1102. [Google Scholar] [CrossRef]
- Gupta, J.; Felner, E.I.; Prausnitz, M.R. Minimally invasive insulin delivery in subjects with type 1 diabetes using hollow microneedles. Diabetes Technol. Ther. 2009, 11, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Morrow, D.I.; McCarron, P.A.; Woolfson, A.D.; Donnelly, R.F. Innovative strategies for enhancing topical and transdermal drug delivery. Open Drug Deliv. J. 2007, 1, 1. [Google Scholar] [CrossRef]
- Poulsen, B.J.; Young, E.; Coquilla, V.; Katz, M. Effect of topical vehicle composition on the in vitro release of fluocinolone acetonide and its acetate ester. J. Pharm. Sci. 1968, 57, 928–933. [Google Scholar] [CrossRef]
- Cilurzo, F.; Minghetti, P.; Casiraghi, A.; Tosi, L.; Pagani, S.; Montanari, L. Polymethacrylates as crystallization inhibitors in monolayer transdermal patches containing ibuprofen. Eur. J. Pharm. Biopharm. 2005, 60, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Miere, F.; Vicas, S.I.; Timar, A.V.; Ganea, M.; Zdrinca, M.; Cavalu, S.; Fritea, L.; Vicas, L.; Muresan, M.; Pallag, A.; et al. Preparation and Characterization of Two Different Liposomal Formulations with Bioactive Natural Extract for Multiple Applications. Processes 2021, 9, 432. [Google Scholar] [CrossRef]
- Löffelmann, M.; Mersmann, A. How to measure supersaturation? Chem. Eng. Sci. 2002, 57, 4301–4310. [Google Scholar] [CrossRef]
- Boskey, A.L.; Posner, A.S. Formation of hydroxyapatite at low supersaturation. J. Phys. Chem. 1976, 80, 40–45. [Google Scholar] [CrossRef]
- Femenia-Font, A.; Padula, C.; Marra, F.; Balaguer-Fernandez, C.; Merino, V.; Lopez-Castellano, A.; Nicoli, S.; Santi, P. Bioadhesive monolayer film for the in vitro transdermal delivery of sumatriptan. J. Pharm. Sci. 2006, 95, 1561–1569. [Google Scholar] [CrossRef]
- Barichello, J.M.; Handa, H.; Kisyuku, M.; Shibata, T.; Ishida, T.; Kiwada, H. Inducing effect of liposomalization on the transdermal delivery of hydrocortisone: Creation of a drug supersaturated state. J. Control. Release 2006, 115, 94–102. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, A.; Arora, K.; Mohapatra, H.; Sindhu, R.K.; Bulzan, M.; Cavalu, S.; Paneshar, G.; Elansary, H.O.; El-Sabrout, A.M.; Mahmoud, E.A.; et al. Supersaturation-Based Drug Delivery Systems: Strategy for Bioavailability Enhancement of Poorly Water-Soluble Drugs. Molecules 2022, 27, 2969. https://doi.org/10.3390/molecules27092969
Sharma A, Arora K, Mohapatra H, Sindhu RK, Bulzan M, Cavalu S, Paneshar G, Elansary HO, El-Sabrout AM, Mahmoud EA, et al. Supersaturation-Based Drug Delivery Systems: Strategy for Bioavailability Enhancement of Poorly Water-Soluble Drugs. Molecules. 2022; 27(9):2969. https://doi.org/10.3390/molecules27092969
Chicago/Turabian StyleSharma, Arvind, Kanika Arora, Harapriya Mohapatra, Rakesh K. Sindhu, Madalin Bulzan, Simona Cavalu, Gulsheen Paneshar, Hosam O. Elansary, Ahmed M. El-Sabrout, Eman A. Mahmoud, and et al. 2022. "Supersaturation-Based Drug Delivery Systems: Strategy for Bioavailability Enhancement of Poorly Water-Soluble Drugs" Molecules 27, no. 9: 2969. https://doi.org/10.3390/molecules27092969