
Coconut Oil Alleviates the Oxidative Stress-Mediated Inflammatory Response via Regulating the MAPK Pathway in Particulate Matter-Stimulated Alveolar Macrophages


Abstract
:1. Introduction
2. Materials and Methods
2.1. APM Generation
2.2. Preparation of APM, DEP, and Coconut Oil
2.3. Measurement of Organic Carbon/Elemental Carbon (OC/EC)
2.4. Cell Culture and APM, DEP, Coconut Oil, and N-Acetylcysteine (NAC) Treatments
2.5. Cell Viability Assay
2.6. Measurement of Reactive Oxygen Species (ROS) Level
2.7. Intracellular Glutathione (GSH) and Nicotinamide Adenine Dinucleotide Phosphate (NADP+)/Reduced Form (NADPH) Levels
2.8. Measurement of Cytokine Levels
2.9. RNA Extraction and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.10. Western Blotting Analyses
2.11. Statistical Analysis
3. Results
3.1. Characterization of DEP and APM
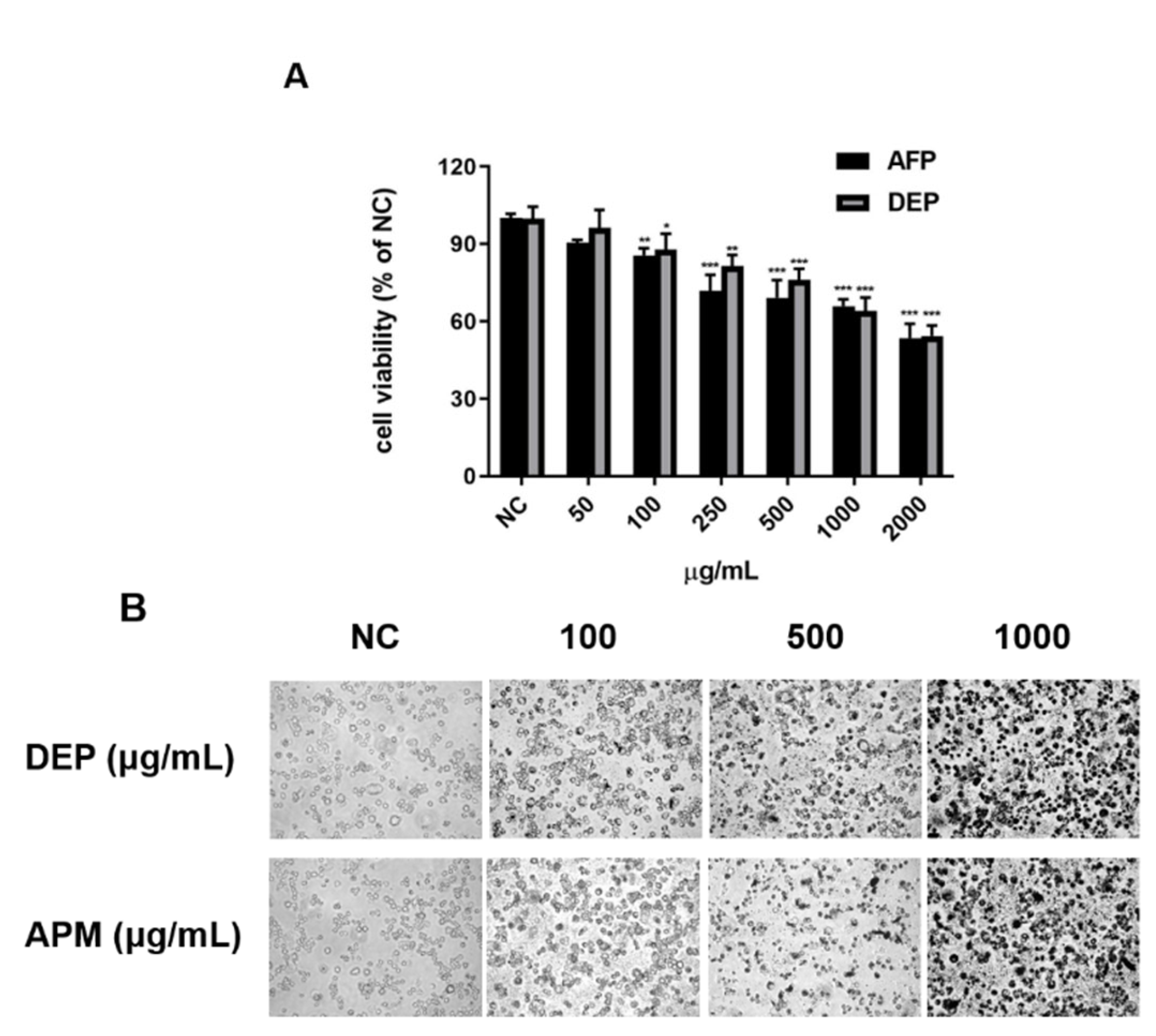
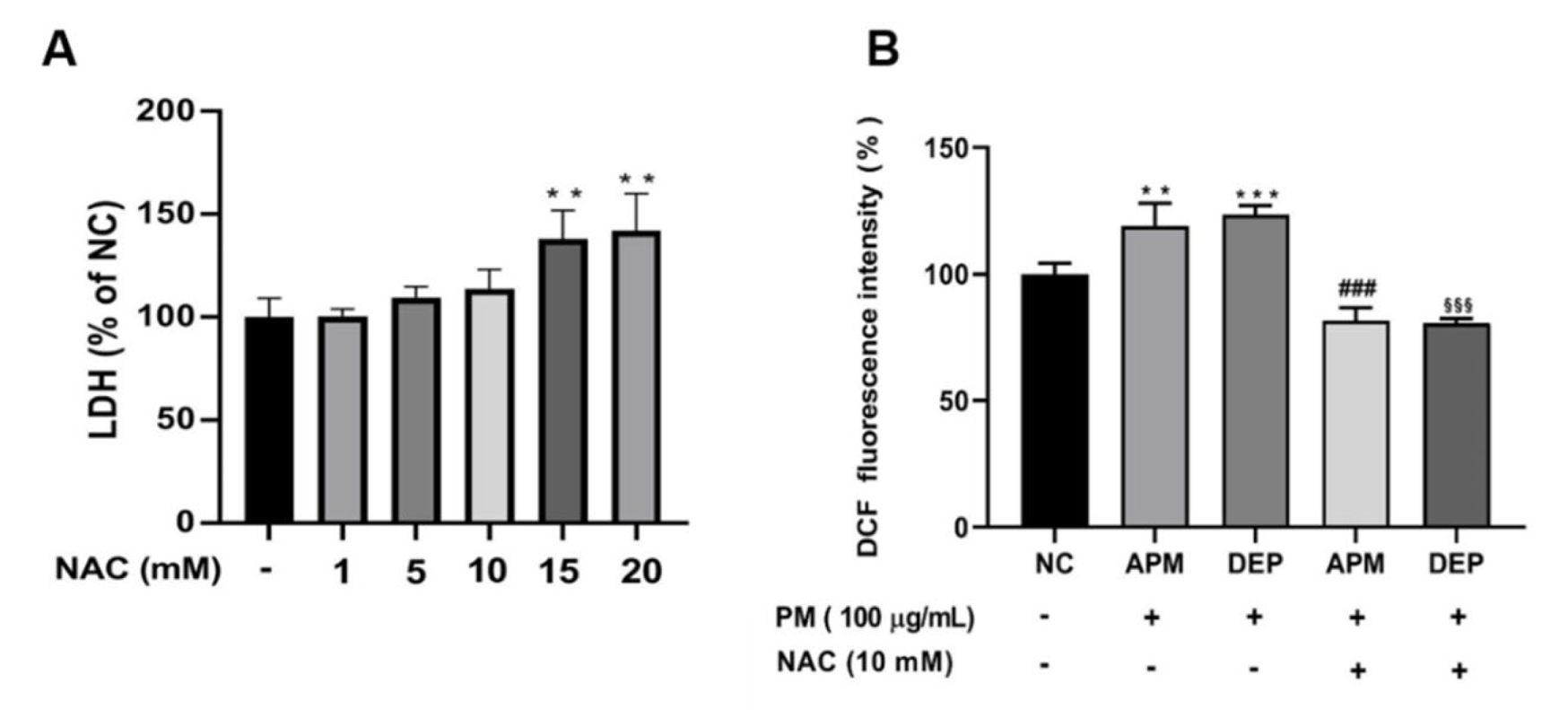
3.2. PM Induces Oxidative Stress-Mediated Cell Damage in AMs
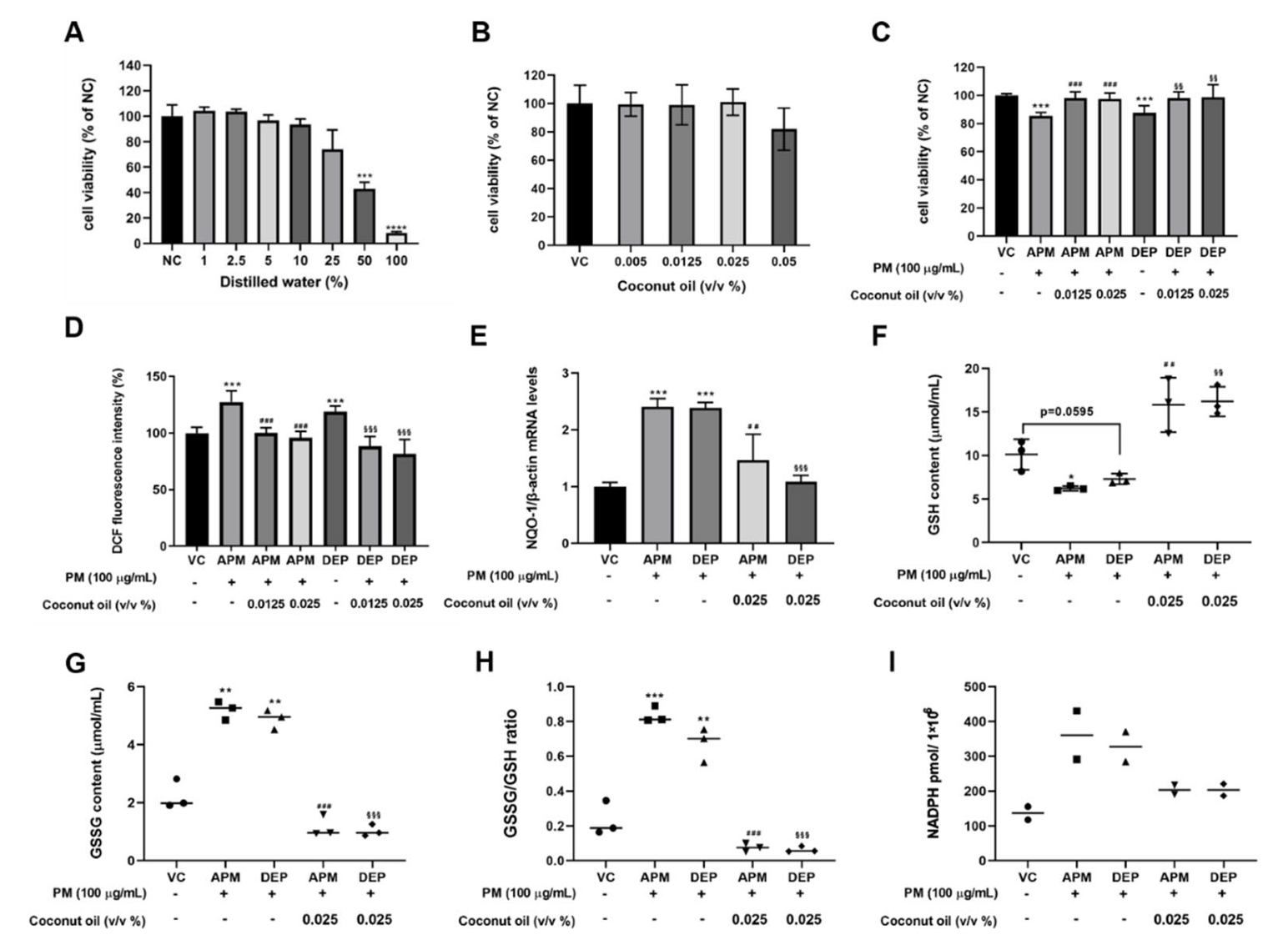
3.3. Effect of Coconut Oil on Cytotoxicity and Oxidative Stress in PM-Stimulated AMs
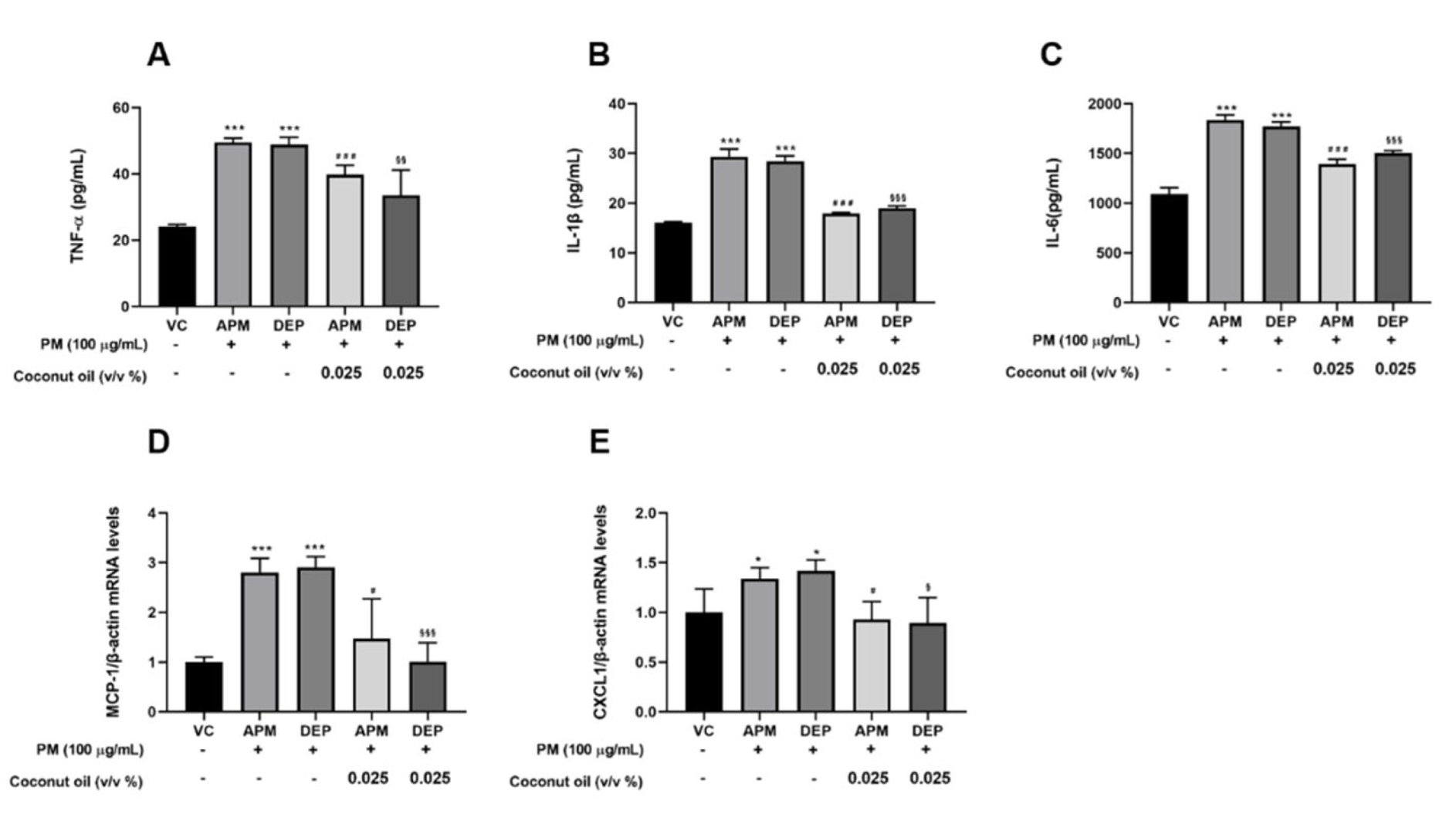
3.4. Coconut Oil Suppresses Inflammatory Genes and Protein Expression in PM-Stimulated AMs
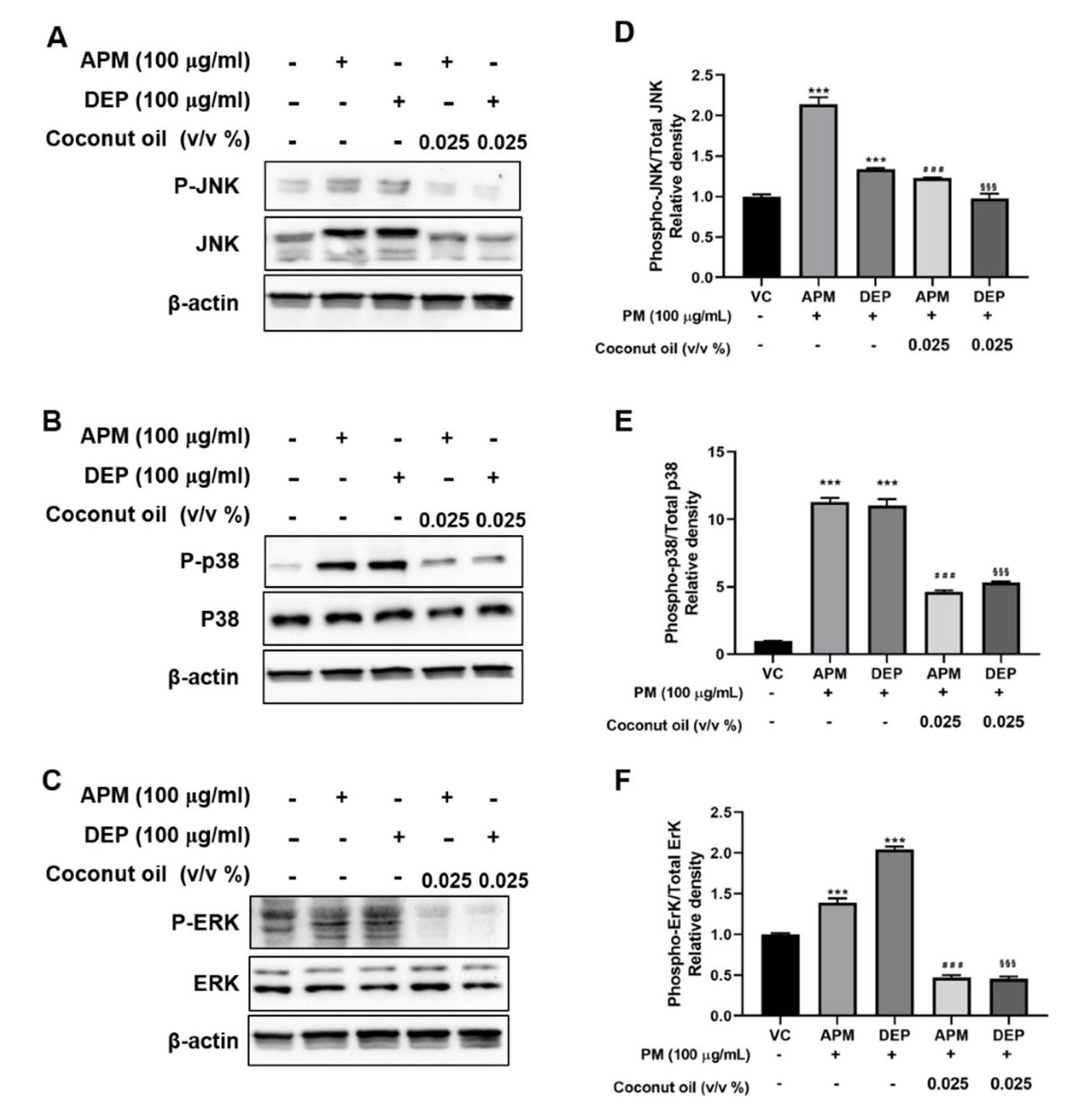
3.5. Coconut Oil Attenuates PM-Induced MAPK Signaling Activation in AMs
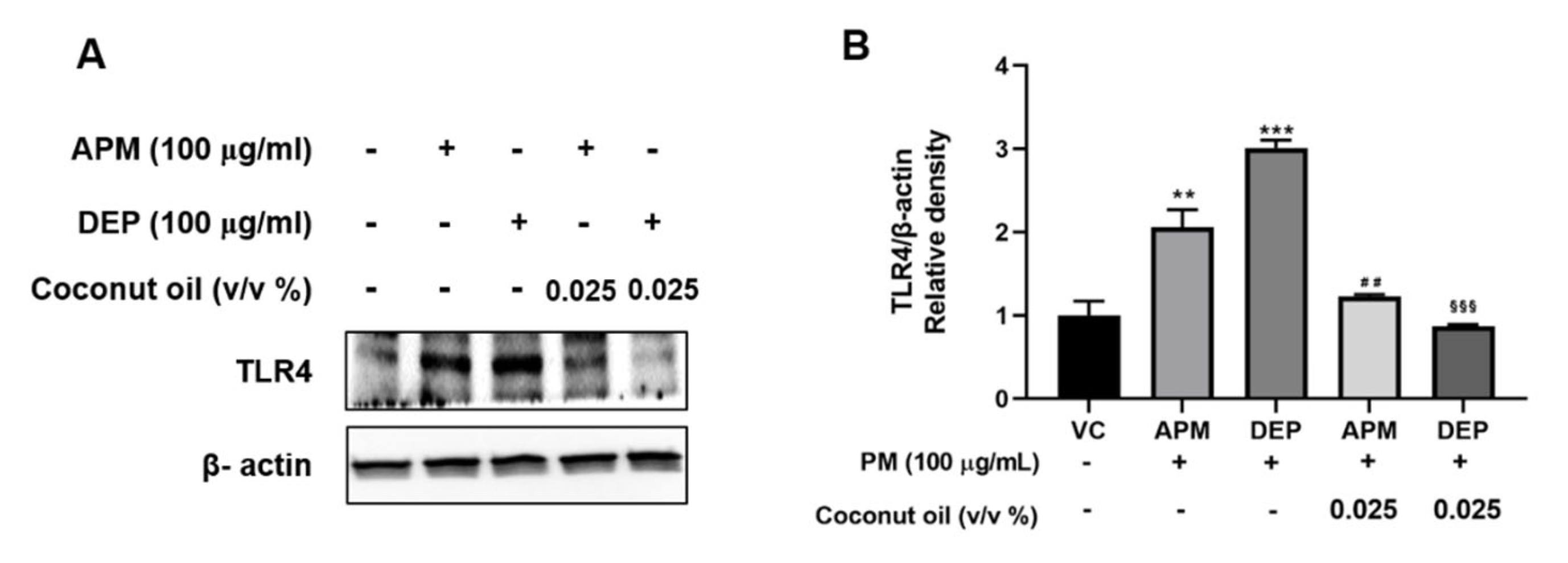
3.6. Coconut Oil Inhibits PM-Induced TLR4 Activation in AMs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Li, J.; Liu, H.; Lv, Z.; Zhao, R.; Deng, F.; Wang, C.; Qin, A.; Yang, X. Estimation of PM2.5 mortality burden in China with new exposure estimation and local concentration-response function. Environ. Pollut. 2018, 243, 1710–1718. [Google Scholar] [CrossRef] [PubMed]
- Gilmour, J.M.M.I.; Duvall, R.M.; Dailey, L.; Daniels, M.; Boykin, E.; Cho, S.-H.; Doerfler, D.; Gordon, T.; Devlin, R.B. Comparative toxicity of size-fractionated airborne particulate matter obtained from different cities in the United States. Inhal. Toxicol. 2007, 19 (Suppl. 1), 7–16. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Jin, H.; Wang, H.; Yao, Y.; Aniagu, S.; Tong, J.; Jiang, Y. Aryl hydrocarbon receptor mediates the cardiac developmental toxicity of EOM from PM2.5 in P19 embryonic carcinoma cells. Chemosphere 2018, 216, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Coccini, T.; Barni, S.; Mustarelli, P.; Locatelli, C.; Roda, E. One-month persistence of inflammation and alteration of fibrotic marker and cytoskeletal proteins in rat kidney after Cd-doped silica nanoparticle instillation. Toxicol. Lett. 2015, 232, 449–457. [Google Scholar] [CrossRef]
- Dong, G.-H. Perspective for Future Research Direction about Health Impact of Ambient Air Pollution in China. Adv. Exp. Med. Biol. 2017, 1017, 263–268. [Google Scholar]
- Bowe, B.; Xie, Y.; Li, T.; Yan, Y.; Xian, H.; Al-Aly, Z. Particulate Matter Air Pollution and the Risk of Incident CKD and Progression to ESRD. J. Am. Soc. Nephrol. 2018, 29, 218–230. [Google Scholar] [CrossRef] [Green Version]
- Stone, V.; Miller, M.R.; Clift, M.J.D.; Elder, A.; Mills, N.L.; Moller, P.; Schins, R.P.F.; Vogel, U.; Kreyling, W.G.; Jensen, K.A.; et al. Nanomaterials Versus Ambient Ultrafine Particles: An Opportunity to Exchange Toxicology Knowledge. Env. Health Perspect. 2017, 125, 106002. [Google Scholar] [CrossRef]
- Karottki, D.G.; Spilak, M.; Frederiksen, M.; Andersen, Z.J.; Madsen, A.M.; Ketzel, M.; Massling, A.; Gunnarsen, L.; Møller, P.; Loft, S. Indoor and Outdoor Exposure to Ultrafine, Fine and Microbiologically Derived Particulate Matter Related to Cardiovascular and Respiratory Effects in a Panel of Elderly Urban Citizens. Int. J. Environ. Res. Public Health 2015, 12, 1667–1686. [Google Scholar] [CrossRef]
- Kim, D.I.; Song, M.K.; Kim, S.H.; Park, C.Y.; Lee, K. TF-343 Alleviates Diesel Exhaust Particulate-Induced Lung Inflammation via Modulation of Nuclear Factor-kappaB Signaling. J. Immunol. Res. 2019, 2019, 8315845. [Google Scholar] [CrossRef] [Green Version]
- Li, C.-P.; Qin, G.; Shi, R.-Z.; Zhang, M.-S.; Lv, J.-Y. Ginsenoside Rg1 reduces toxicity of PM2.5 on human umbilical vein endothelial cells by upregulating intracellular antioxidative state. Environ. Toxicol. Pharmacol. 2013, 35, 21–29. [Google Scholar] [CrossRef]
- Oberdorster, G.; Ferin, J.; Gelein, R.; Soderholm, S.C.; Finkelstein, J. Role of the Alveolar Macrophage in Lung Injury: Studies with Ultrafine Particles. Environ. Health Perspect. 1992, 97, 193. [Google Scholar]
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef]
- Rylance, J.; Fullerton, D.G.; Scriven, J.; Aljurayyan, A.N.; Mzinza, D.; Barrett, S.; Wright, A.K.A.; Wootton, D.G.; Glennie, S.J.; Baple, K.; et al. Household Air Pollution Causes Dose-Dependent Inflammation and Altered Phagocytosis in Human Macrophages. Am. J. Respir. Cell Mol. Biol. 2015, 52, 584–593. [Google Scholar] [CrossRef] [Green Version]
- Jia, J.; Sun, Y.; Hu, Z.; Li, Y.; Ruan, X. Propofol inhibits the release of interleukin-6, 8 and tumor necrosis factor-alpha correlating with high-mobility group box 1 expression in lipopolysaccharides-stimulated RAW 264.7 cells. BMC Anesthesiol. 2017, 17, 148. [Google Scholar] [CrossRef] [Green Version]
- Guo, E.Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. RK/MAPK signalling pathway and tumorigenesis. Exp Ther Med. 2020, 19, 1997–2007. [Google Scholar]
- Wang, J.; Huang, J.; Wang, L.; Chen, C.; Yang, D.; Jin, M.; Bai, C.; Song, Y. Urban particulate matter triggers lung inflammation via the ROS-MAPK-NF-ĸB signaling pathway. J. Thorac. Dis. 2017, 9, 4398–4412. [Google Scholar] [CrossRef] [Green Version]
- Na, H.G.; Kim, Y.D.; Choi, Y.S.; Bae, C.H.; Song, S.Y. Diesel exhaust particles elevate MUC5AC and MUC5B expression via the TLR4-mediated activation of ERK1/2, p38 MAPK, and NF-kappaB signaling pathways in human airway epithelial cells. Biochem. Biophys. Res. Commun. 2019, 512, 53–59. [Google Scholar] [CrossRef]
- Kim, D.I.; Song, M.-K.; Kim, H.-I.; Han, K.M.; Lee, K. Diesel Exhaust Particulates Induce Neutrophilic Lung Inflammation by Modulating Endoplasmic Reticulum Stress-Mediated CXCL1/KC Expression in Alveolar Macrophages. Molecules 2020, 25, 6046. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhao, Y.; Wang, Q.; Chen, H.; Zhou, X. Fine particulate matter exposure promotes M2 macrophage polarization through inhibiting histone deacetylase 2 in the pathogenesis of chronic obstructive pulmonary disease. Ann. Transl. Med. 2020, 8, 1303. [Google Scholar] [CrossRef]
- Kyung, S.Y.; Jeong, S.H. Particulate-Matter Related Respiratory Diseases. Tuberc. Respir. Dis. 2020, 83, 116–121. [Google Scholar] [CrossRef]
- Mohamed, F.R. Fruit Oils: Chemistry and Functionality. In Oils From Fruit Nuts, Coconut (Cocos nucifera) Oil; Osman, A., Ed.; E-Publishing Inc.: Cham, Switzerland, 2019; pp. 209–221. [Google Scholar]
- Dia, V.P.; Garcia, V.V.; Mabesa, R.C. Comparative physiological characteristics of virgin coconut oil produced by different methods. Philipp. Agric. Sci. Philipp. 2005, 88, 462–475. [Google Scholar]
- DebMandal, M.; Mandal, S. Coconut (Cocos nucifera L.: Arecaceae): In health promotion and disease prevention. Asian Pac. J. Trop. Med. 2011, 4, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Kamariah, A.A.L.; Rosmawati, A.; Ching, M.G.W.; Sivapragasam, M.D.A.A.; Tan, C.P.; Lai, O.M. Physico-chemical and quality characteristics of virgin coconut oil—A Malaysian survey. J. Trop. Agric. Food Sci. 2008, 36, 239–248. [Google Scholar]
- Winarsi, H.H.; Purwanto, A. Virgin Coconut Oil (VCO) Enriched with Zn as Immunostimulator for Vaginal Candidiasis Patient. HAYATI J. Biosci. 2008, 15, 135–139. [Google Scholar] [CrossRef] [Green Version]
- Intahphuak, S.; Khonsung, P.; Panthong, A. Anti-inflammatory, analgesic, and antipyretic activities of virgin coconut oil. Pharm. Biol. 2010, 48, 151–157. [Google Scholar] [CrossRef] [Green Version]
- Nevin, K.G.; Rajamohan, T. Effect of topical application of virgin coconut oil on skin components and antioxidant status during dermal wound healing in young rats. Ski. Pharm. Physiol. 2010, 23, 290–297. [Google Scholar] [CrossRef]
- Choi, E.H.; Kang, J.I.; Cho, J.Y.; Lee, S.H.; Kim, T.S.; Yeo, I.H.; Chun, H.S. Supplementation of standardized lipid-soluble extract from maca (Lepidium meyenii) increases swimming endurance capacity in rats. J. Funct. Foods 2012, 4, 568–573. [Google Scholar] [CrossRef]
- Chandrashekar, P.; Lokesh, B.R.; Krishna, A.G.G. Hypolipidemic effect of blends of coconut oil with soybean oil or sunflower oil in experimental rats. Food Chem. 2010, 123, 728–733. [Google Scholar] [CrossRef]
- Vasconcelos, L.H.C.; Silva, M.; Costa, A.C.; de Oliveira, G.A.; de Souza, I.L.L.; Righetti, R.F.; Queiroga, F.R.; Cardoso, G.A.; Silva, A.S.; da Silva, M.P.; et al. Virgin Coconut Oil Supplementation Prevents Airway Hyperreactivity of Guinea Pigs with Chronic Allergic Lung Inflammation by Antioxidant Mechanism. Oxid. Med. Cell Longev. 2020, 2020, 5148503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, Y.T.; Youn, J.S.; Moon, H.G.; Chen, X.Y.; Kim, D.I.; Cho, W.H.; Lee, K.-H.; Joen, K.-J. Relationship between Cytotoxicity and Surface Oxidation of Artificial Black Carbon. Nanomaterials 2021, 11, 1455. [Google Scholar] [CrossRef] [PubMed]
- Hwangbo, M.C.S.; Moon, C. Effect of oil particle size on dispersion stability in oil in water emulsion. Part. Aerosol Res. 2017, 13, 133–139. [Google Scholar]
- Birch, M.E.; Cary, R.A. Elemental Carbon-Based Method for Monitoring Occupational Exposures to Particulate Diesel Exhaust. Aerosol Sci. Technol. 1996, 25, 221–241. [Google Scholar] [CrossRef]
- Liu, Y.; Peterson, D.A.; Kimura, H.; Schubert, D. Mechanism of cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction. J. Neurochem. 1997, 69, 581–593. [Google Scholar] [CrossRef]
- Afzal, M.; Afzal, A.; Jones, A.; Armstrong, D.; Afzal, M.; Afzal, A.; Jones, A.; Armstrong, D. A Rapid Method for the Quantification of GSH and GSSG in Biological Samples. In Oxidative Stress Biomarkers and Antioxidant Protocols; Armstrong, D., Ed.; Humana Press: Totova, NJ, USA, 2002. [Google Scholar]
- Robinson, R.K.; Birrell, M.A.; Adcock, J.J.; Wortley, M.A.; Dubuis, E.D.; Chen, S.; McGilvery, C.M.; Hu, S.; Shaffer, M.S.P.; Bonvini, S.J.; et al. Mechanistic link between diesel exhaust particles and respiratory reflexes. J. Allergy. Clin. Immunol. 2018, 141, 1074–1084. [Google Scholar] [CrossRef] [Green Version]
- Le, K.C.; Lefumeux, C.; Pino, T. Differential Raman backscattering cross sections of black carbon nanoparticles. Sci. Rep. 2017, 7, 17124. [Google Scholar] [CrossRef] [Green Version]
- Qi, Z.H.; Zhang, Y.H.; Chen, Z.F.; Yang, C.; Song, Y.Y.; Liao, X.L.; Li, W.Q.; Tsang, S.Y.; Liu, G.G.; Cai, W.Z. Chemical identity and cardiovascular toxicity of hydrophobic organic components in PM2.5 Ecotoxicol. Environ. Saf. 2020, 201, 110827. [Google Scholar] [CrossRef]
- Janssen, N.A.H.; Gerlofs-Nijland, M.E.; Lanki, T.; Salonen, R.O.; Cassee, F.; Brunekreef, B.; Hoek, G.; Fischer, P.; Krzyzanowski, M. Health Effects of Black Carbon; WHO Regional Office for Europe: Copenhagen, Denmark, 2012. [Google Scholar]
- Kitz, K.; Windischhofer, W.; Leis, H.J.; Huber, E.; Kollroser, M.; Malle, E. 15-Deoxy-Delta12,14-prostaglandin J2 induces Cox-2 expression in human osteosarcoma cells through MAPK and EGFR activation involving reactive oxygen species. Free Radic. Biol. Med. 2011, 50, 854–865. [Google Scholar] [CrossRef]
- Totlandsdal, A.I.; Cassee, F.R.; Schwarze, P.; Refsnes, M.; Lag, M. Diesel exhaust particles induce CYP1A1 and pro-inflammatory responses via differential pathways in human bronchial epithelial cells. Part Fibre. Toxicol. 2010, 7, 41. [Google Scholar] [CrossRef] [Green Version]
- Nunes, C.; Pereira, A.M.; Morais-Almeida, M. Asthma costs and social impact. Asthma. Res. Pract. 2017, 3, 1. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Huang, L.; Gao, S.; Gao, S.; Wang, L. Measurements of PM10 and PM2.5 in urban area of Nanjing, China and the assessment of pulmonary deposition of particle mass. Chemosphere 2002, 48, 689–695. [Google Scholar] [CrossRef]
- Cavanagh, J.A.; Trought, K.; Brown, L.; Duggan, S. Exploratory investigation of the chemical characteristics and relative toxicity of ambient air particulates from two New Zealand cities. Sci. Total Environ. 2009, 407, 5007–5018. [Google Scholar] [CrossRef]
- Huang, X.; Shi, X.; Zhou, J.; Li, S.; Zhang, L.; Zhao, H. The activation of antioxidant and apoptosis pathways involved in damage of human proximal tubule epithelial cells by PM2.5 exposure. Environ. Sci. Europe. 2020, 32, 2. [Google Scholar] [CrossRef] [Green Version]
- Seriani, R.; Mde, S.J.; de Toledo, A.C.; Martins, M.A.; Seckler, M.; Alencar, A.M.; Negri, E.M.; Silva, L.F.F.; Mauad, T.; Saldiva, P.H.N.; et al. Diesel exhaust particulates affect cell signaling, mucin profiles, and apoptosis in trachea explants of Balb/C mice. Environ. Toxicol. 2015, 30, 1297–1308. [Google Scholar] [CrossRef]
- Hashimoto, S.; Gon, Y.; Takeshita, I.; Matsumoto, K.; Jibiki, I.; Takizawa, H.; Kudoh, S.; Horie, T. Diesel exhaust particles activate p38 MAP kinase to produce interleukin 8 and RANTES by human bronchial epithelial cells and N-acetylcysteine attenuates p38 MAP kinase activation. Am. J. Respir. Crit. Care. Med. 2000, 161, 280–285. [Google Scholar] [CrossRef] [Green Version]
- Nemmar, A.; Al-Salam, S.; Zia, S.; Dhanasekaran, S.; Shudadevi, M.; Ali, B.H. Time-course effects of systemically administered diesel exhaust particles in rats. Toxicol. Lett. 2010, 194, 58–65. [Google Scholar] [CrossRef]
- De Filippo, K.; Henderson, R.B.; Laschinger, M.; Hogg, N. Neutrophil chemokines KC and macrophage-inflammatory protein-2 are newly synthesized by tissue macrophages using distinct TLR signaling pathways. J. Immunol. 2008, 180, 4308–4315. [Google Scholar] [CrossRef] [Green Version]
- Hussein, M.S.M.M.; Abdel-Shafy, I. A review on polycyclic aromatic hydrocarbons: Source, environmental impact, effect on human health and remediation. Egypt. J. Pet. 2016, 25, 107–123. [Google Scholar]
- Gurbani, S.K.B.D.; Kumar, A.; Pandey, A.K.; Ana, G.R.E.E.; Verma, A.; Khan, A.H.; Patel, D.K.; Mudiam, M.K.R.; Jain, S.K.; Roy, R.; et al. Polycyclic aromatic hydrocarbons and their quinones modulate the metabolic profile and induce DNA damage in human alveolar and bronchiolar cells. Int. J. Hyg. Environ. Health 2013, 216, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Xia, Y.; Niu, P.; Jiang, L.; Duan, J.; Yu, Y.; Zhou, X.; Li, Y.; Sun, Z. Silica nanoparticles induce oxidative stress, inflammation, and endothelial dysfunction in vitro via activation of the MAPK/Nrf2 pathway and nuclear factor-kappaB signaling. Int. J. Nanomed. 2015, 10, 1463–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, H. Toxico-pharmacological perspective of the Nrf2-Keap1 defense system against oxidative stress in kidney diseases. Biochem. Pharmacol. 2013, 85, 865–872. [Google Scholar] [CrossRef]
- Varma, S.R.; Sivaprakasam, T.O.; Arumugam, I.; Dilip, N.; Raghuraman, M.; Pavan, K.B.; Rafiq, M.; Paramesh, R. In vitro anti-inflammatory and skin protective properties of Virgin coconut oil. J. Tradit. Complement. Med. 2019, 9, 5–14. [Google Scholar] [CrossRef]
- Allard, B.; Panariti, A.; Martin, J.M. Alveolar macrophages in the resolution of inflammation, tissue repair, and tolerance to infection. Front. Immunol. 2018, 9, 1777. [Google Scholar] [CrossRef] [Green Version]
- Hiraiwa, K.; Stephan, F.; Eeden, v. Contribution of lung macrophages to the inflammatory responses induced by exposure to air pollutants. Mediat. Inflamm. 2013, 2013, 619523. [Google Scholar] [CrossRef] [Green Version]
- Mauderly, J.; Hoyer, M. Health Assessment Document for Diesel Engine Exhaust; National Center for Environmental Assessment, Environmental Protection Agency: Washington, DC, USA, 2002; p. 669. [Google Scholar]
- Turner, J.; Hernandez, M.; Snawder, J.E.; Handorean, A.; McCabe, K.M. A toxicology suite adapted for comparing parallel toxicity responses of model human lung cells to diesel exhaust particles and their extracts. Aerosol. Sci. Technol. 2015, 49, 599–610. [Google Scholar] [CrossRef] [Green Version]
- Roh, J.L.; Jang, H.; Kim, E.H.; Shin, D. Targeting of the Glutathione, Thioredoxin, and Nrf2 Antioxidant Systems in Head and Neck Cancer. Antioxid Redox Signal. 2017, 27, 106–114. [Google Scholar] [CrossRef]
- Katharine, B.; Stefania, S.; Miller, F., Jr.; Krause, K.-H. Reactive oxygen species: From health to disease. Swiss Med. Wkly. 2012, 142, 13659. [Google Scholar]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in Inflammatory Disease. Int. J. Mol. Sci. 2019, 20, 6008. [Google Scholar] [CrossRef] [Green Version]
- Ling, S.H.; Eeden, S.F.v. Particulate matter air pollution exposure: Role in the development and exacerbation of chronic obstructive pulmonary disease. Int. J. Chron. Obstruct. Pulmon. Dis. 2009, 4, 233–243. [Google Scholar] [CrossRef] [Green Version]
- Espinosa-Diez, C.; Miguel, V.; Mennerich, D.; Kietzmann, T.; Sanchez-Perez, P.; Cadenas, S.; Lamas, S. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015, 6, 183–197. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Schwarze, P.E.; Totlandsdal, A.I.; Lag, M.; Refsnes, M.; Holme, J.A.; Ovrevik, J. Inflammation-related effects of diesel engine exhaust particles: Studies on lung cells in vitro. Biomed Res. Int. 2013, 2013, 685142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pushpakumar, S.; Ren, L.; Kundu, S.; Gamon, A.; Tyagi, S.C.; Sen, U. Toll-like Receptor 4 Deficiency Reduces Oxidative Stress and Macrophage Mediated Inflammation in Hypertensive Kidney. Sci. Rep. 2017, 7, 6349. [Google Scholar] [CrossRef] [PubMed]
- Jasek-Gajda, E.; Jurkowska, H.; Jasinska, M.; Lis, G.J. Targeting the MAPK/ERK and PI3K/AKT Signaling Pathways Affects NRF2, Trx and GSH Antioxidant Systems in Leukemia Cells. Antioxidants 2020, 9, 633. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward Primer | Reverse Primer |
|---|---|---|
| CXCL–1 | 5′-AACCGAAGTCATAGCCACAC-3′ | 5′-GTTGGATTTGTCACTGTTCAGC-3′ |
| MCP–1 | 5′-TGATCCCAATGAGTAGGCTGGAG-3′ | 5′-ATGTCTGGACCCATTCCTTCTTG-3′ |
| NQO–1 | 5′-GGTGATATTTCAGTTCCCATTG-3′ | 5′-ACTCTCTCAAACCAGCCTTTC-3′ |
| β–Actin | 5′-GGCACCACACCT TCTACAATG-3′ | 5′-GGGGTGTTGAAGGTCTCAAAC-3′ |
| DEP (a) | APM | |
|---|---|---|
| Particle size (nm) | mostly <600 nm [36] | approximately 30 nm [31] |
| Pore diameter (nm) | 4−35 nm (b) | approximately 30 nm [31] |
| Morphology | small irregular agglomerates and roughly spherical [36] | spherical and irregular carbonaceous particles [31] |
| Raman spectrum | D peak: 1340, G peak: 1600 [37] | D peak: 1340, G peak: 1600 [31] |
| OC/EC ratio values (c) | 1.9 ± 0.001 | 2.2 ± 0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Kim, D.I.; Moon, H.-G.; Chu, M.; Lee, K. Coconut Oil Alleviates the Oxidative Stress-Mediated Inflammatory Response via Regulating the MAPK Pathway in Particulate Matter-Stimulated Alveolar Macrophages. Molecules 2022, 27, 2898. https://doi.org/10.3390/molecules27092898
Chen X, Kim DI, Moon H-G, Chu M, Lee K. Coconut Oil Alleviates the Oxidative Stress-Mediated Inflammatory Response via Regulating the MAPK Pathway in Particulate Matter-Stimulated Alveolar Macrophages. Molecules. 2022; 27(9):2898. https://doi.org/10.3390/molecules27092898
Chicago/Turabian StyleChen, Xinyu, Dong Im Kim, Hi-Gyu Moon, Minchul Chu, and Kyuhong Lee. 2022. "Coconut Oil Alleviates the Oxidative Stress-Mediated Inflammatory Response via Regulating the MAPK Pathway in Particulate Matter-Stimulated Alveolar Macrophages" Molecules 27, no. 9: 2898. https://doi.org/10.3390/molecules27092898