
Stable Carbon Isotope Analysis of Hexachlorocyclohexanes by Liquid–Liquid Extraction Gas Chromatography Isotope Ratio Mass Spectrometry: Method Evaluation and Applications

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
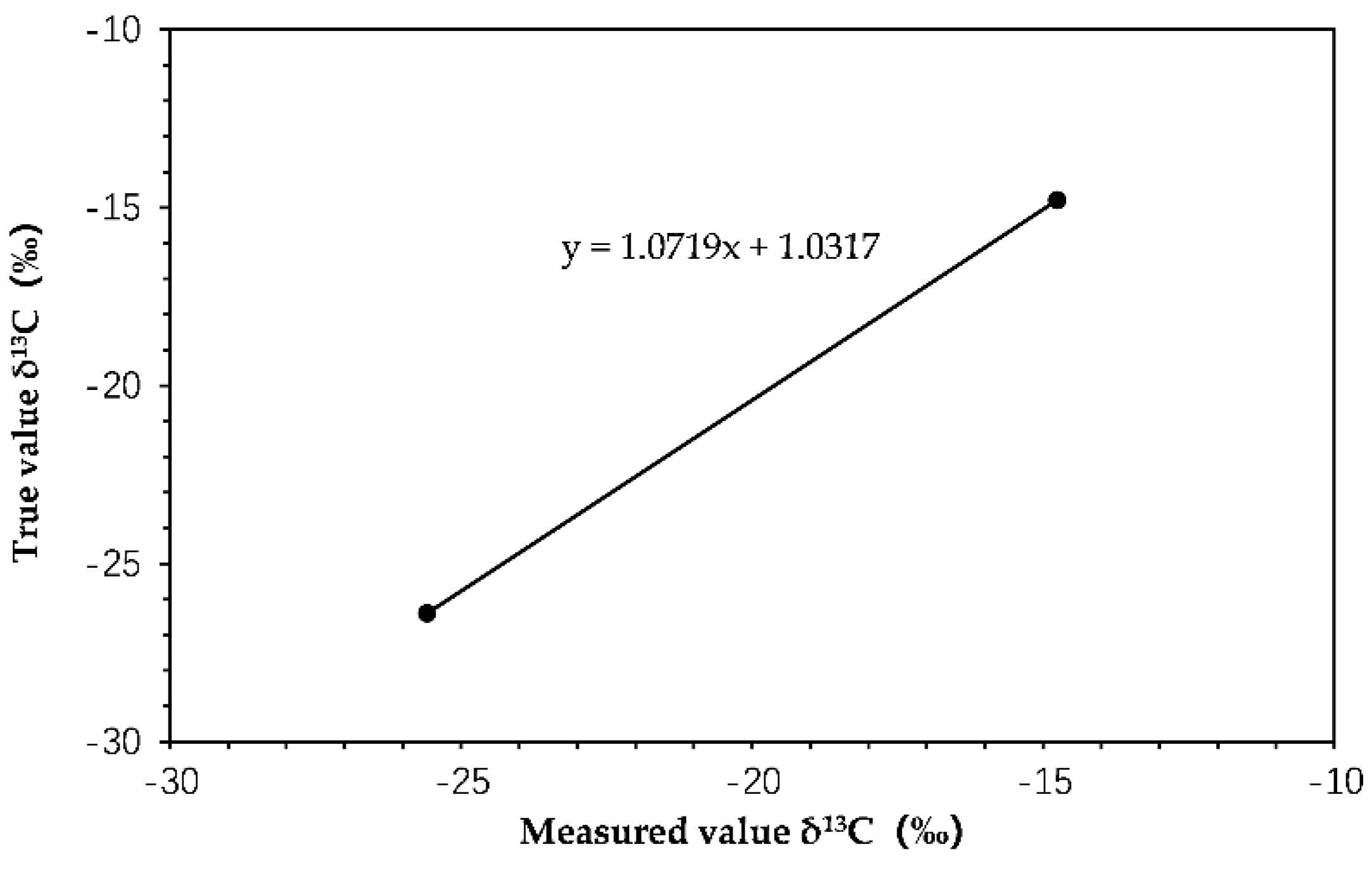
2.1. Calibration of Isotope Ratios of HCHs Reference Samples
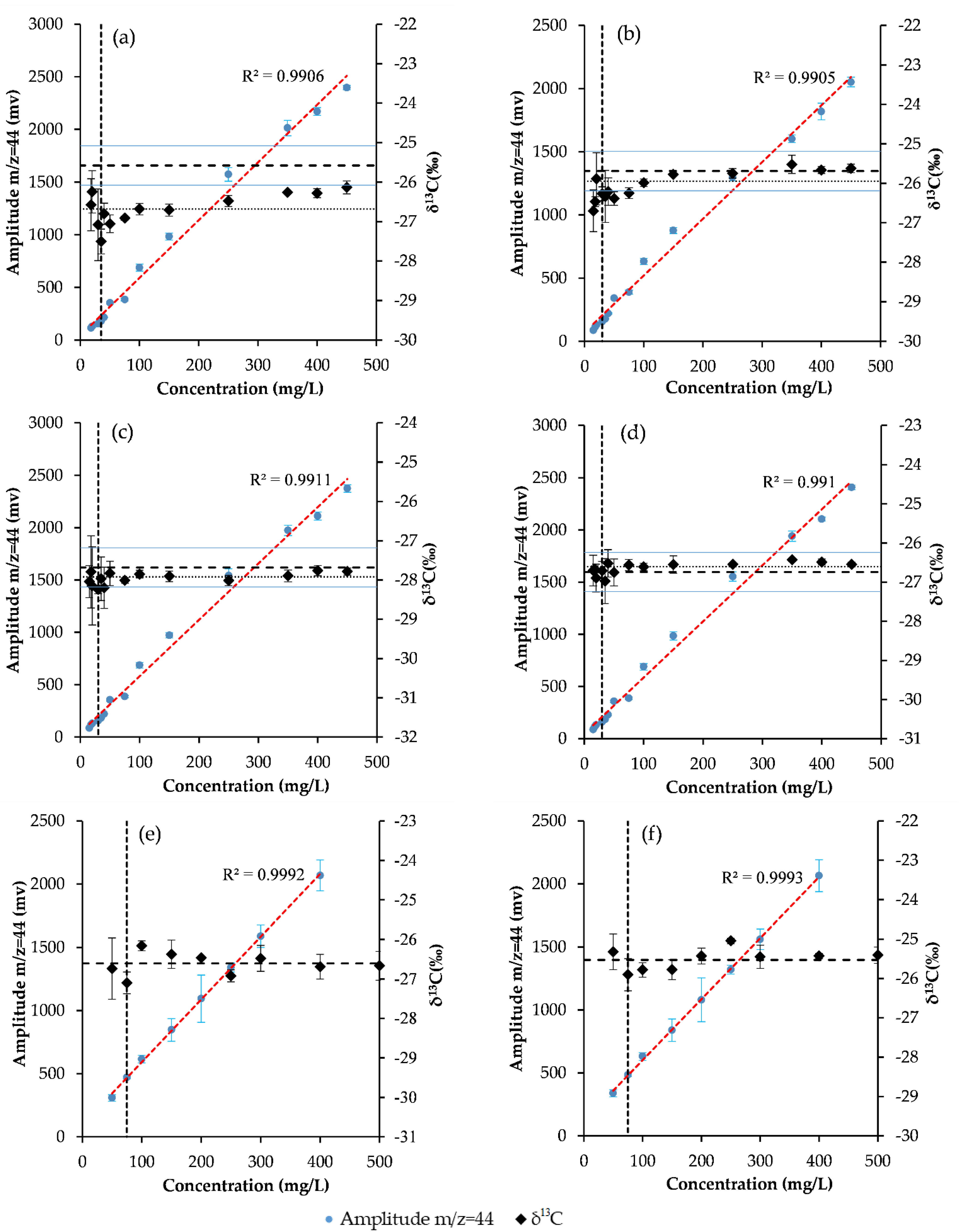
2.2. Resolution of the HCHs and Determination of Their Carbon Isotope Ratios
2.3. Applications
2.3.1. Source Identification
2.3.2. Identification Degradation of HCHs in Groundwater from Contaminated Site
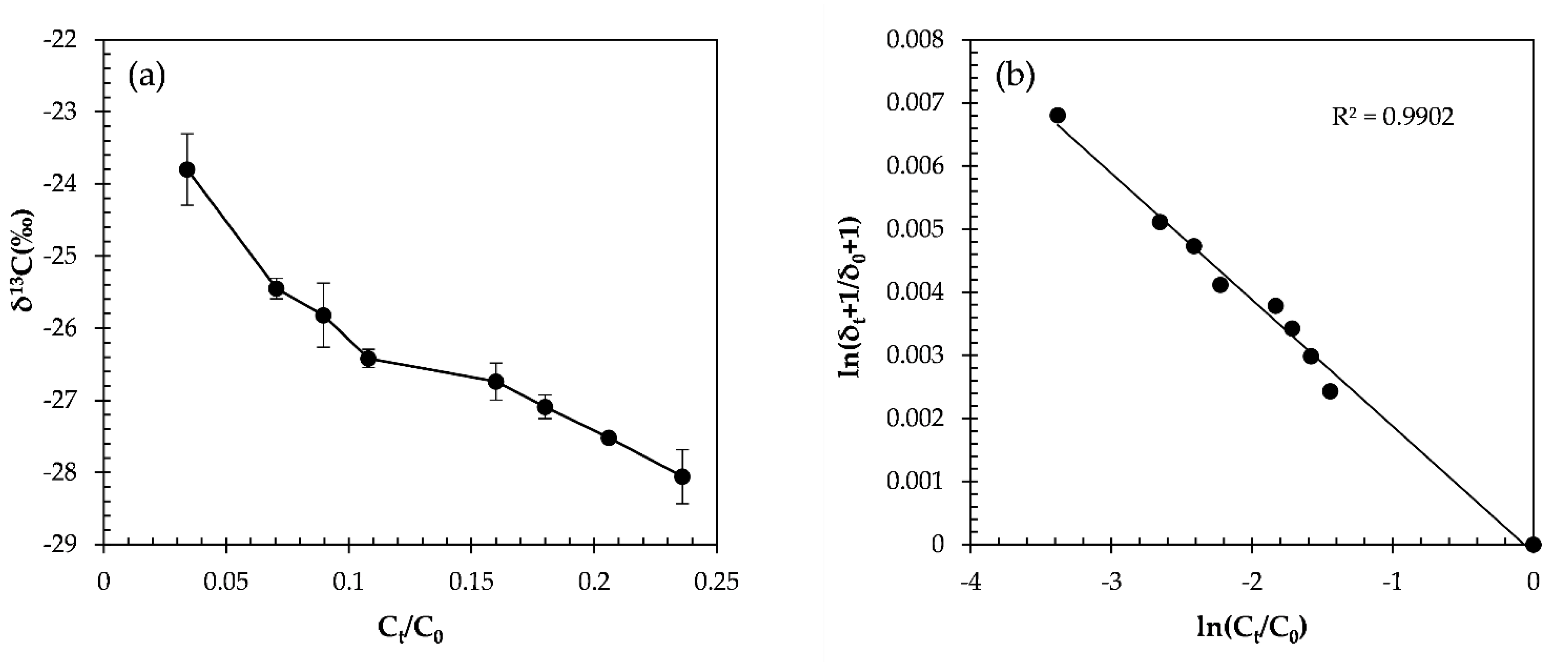
2.3.3. The α-HCH Oxidation by the CaO2/Fe(II) Fenton System
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. EA-IRMS Measurement for Determination of Reference Values
3.3. GC-IRMS Conditions for Carbon Isotope Analysis
3.4. GC-ECD for the HCHs Testing
3.5. Experimental Method
3.5.1. Standard Solution
3.5.2. Sample Pre-Concentration
3.6. Instrumental Method Detection Limits
3.7. Groundwater Samples
3.8. Oxidation Experiment
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Ivdra, N.; Herrero-Martín, S.; Fischer, A. Validation of user- and environmentally friendly extraction and clean-up methods for compound-specific stable carbon isotope analysis of organochlorine pesticides and their metabolites in soils. J. Chromatogr. A 2014, 1355, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Vijgen, J.; Abhilash, P.C.; Li, Y.F.; Lal, R.; Forter, M.; Torres, J.; Singh, N.; Yunus, M.; Tian, C.; Schäffer, A. Hexachlorocyclohexane (HCH) as new Stockholm Convention POPs--a global perspective on the management of Lindane and its waste isomers. Environ. Sci. Pollut. Res. Int. 2011, 18, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Niu, L.; Qiu, J.; Zhou, Y.; Lu, H.; Liu, W. Stereoselective accumulations of hexachlorocyclohexanes (HCHs) are correlated with Sphingomonas spp. in agricultural soils across China. Environ. Pollut. 2018, 240, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.Y.; Liu, W.J.; Xu, Y.S.; Zhao, Y.Z.; Cai, C.Y.; Liu, Y.; Wang, X.; Xiong, G.N.; Tao, S.; Liu, W.X. Organochlorine pesticides in ambient air from the littoral cities of northern China: Spatial distribution, seasonal variation, source apportionment and cancer risk assessment. Sci. Total Environ. 2019, 652, 163–176. [Google Scholar] [CrossRef]
- Pan, H.; Lei, H.; He, X.; Xi, B.; Xu, Q. Spatial distribution of organochlorine and organophosphorus pesticides in soil-groundwater systems and their associated risks in the middle reaches of the Yangtze River Basin. Environ. Geochem. Health 2019, 41, 1833–1845. [Google Scholar] [CrossRef]
- Liu, B.; Li, Y.; Ma, J.; Huang, L.; Chen, L. Detection of semi-volatile organic compounds (SVOCs) in surface water, soil, and groundwater in a chemical industrial park in Eastern China. Water Sci. Technol. 2016, 73, 1175–1189. [Google Scholar] [CrossRef]
- Wu, C.; Luo, Y.; Gui, T.; Huang, Y. Concentrations and potential health hazards of organochlorine pesticides in shallow groundwater of Taihu Lake region, China. Sci. Total Environ. 2014, 470–471, 1047–1055. [Google Scholar] [CrossRef]
- Elsner, M.; Jochmann, M.A.; Hofstetter, T.B.; Hunkeler, D.; Bernstein, A.; Schmidt, T.C.; Schimmelmann, A. Current challenges in compound-specific stable isotope analysis of environmental organic contaminants. Anal. Bioanal. Chem. 2012, 403, 2471–2491. [Google Scholar] [CrossRef]
- Fischer, A.; Manefield, M.; Bombach, P. Application of stable isotope tools for evaluating natural and stimulated biodegradation of organic pollutants in field studies. Curr. Opin. Biotechnol. 2016, 41, 99–107. [Google Scholar] [CrossRef]
- Nijenhuis, I.; Renpenning, J.; Kümmel, S.; Richnow, H.H.; Gehre, M. Recent advances in multi-element compound-specific stable isotope analysis of organohalides: Achievements, challenges and prospects for assessing environmental sources and transformation. Trends Environ. Anal. Chem. 2016, 11, 1–8. [Google Scholar] [CrossRef]
- Badea, S.-L.; Danet, A.-F. Enantioselective stable isotope analysis (ESIA)—A new concept to evaluate the environmental fate of chiral organic contaminants. Sci. Total Environ. 2015, 514, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Badea, S.-L.; Vogt, C.; Weber, S.; Danet, A.-F.; Richnow, H.-H. Stable Isotope Fractionation of γ-Hexachlorocyclohexane (Lindane) during Reductive Dechlorination by Two Strains of Sulfate-Reducing Bacteria. Environ. Sci. Technol. 2009, 43, 3155–3161. [Google Scholar] [CrossRef] [PubMed]
- Badea, S.-L.; Vogt, C.; Gehre, M.; Fischer, A.; Danet, A.-F.; Richnow, H.-H. Development of an enantiomer-specific stable carbon isotope analysis (ESIA) method for assessing the fate of α-hexachlorocyclo-hexane in the environment. Rapid Commun. Mass Spectrom. 2011, 25, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Bashir, S.; Fischer, A.; Nijenhuis, I.; Richnow, H.-H. Enantioselective Carbon Stable Isotope Fractionation of Hexachlorocyclohexane during Aerobic Biodegradation by Sphingobium spp. Environ. Sci. Technol. 2013, 47, 11432–11439. [Google Scholar] [CrossRef]
- Zhang, N.; Bashir, S.; Qin, J.; Schindelka, J.; Fischer, A.; Nijenhuis, I.; Herrmann, H.; Wick, L.Y.; Richnow, H.H. Compound specific stable isotope analysis (CSIA) to characterize transformation mechanisms of α-hexachlorocyclohexane. J. Hazard. Mater. 2014, 280, 750–757. [Google Scholar] [CrossRef]
- Bashir, S.; Hitzfeld, K.L.; Gehre, M.; Richnow, H.H.; Fischer, A. Evaluating degradation of hexachlorcyclohexane (HCH) isomers within a contaminated aquifer using compound-specific stable carbon isotope analysis (CSIA). Water Res. 2015, 71, 187–196. [Google Scholar] [CrossRef]
- Chartrand, M.; Passeport, E.; Rose, C.; Lacrampe-Couloume, G.; Bidleman, T.F.; Jantunen, L.M.; Sherwood Lollar, B. Compound specific isotope analysis of hexachlorocyclohexane isomers: A method for source fingerprinting and field investigation of in situ biodegradation. Rapid Commun. Mass Spectrom. 2015, 29, 505–514. [Google Scholar] [CrossRef]
- Liu, Y.; Bashir, S.; Stollberg, R.; Trabitzsch, R.; Weiß, H.; Paschke, H.; Nijenhuis, I.; Richnow, H.-H. Compound Specific and Enantioselective Stable Isotope Analysis as Tools to Monitor Transformation of Hexachlorocyclohexane (HCH) in a Complex Aquifer System. Environ. Sci. Technol. 2017, 51, 8909–8916. [Google Scholar] [CrossRef]
- Ivdra, N.; Fischer, A.; Herrero-Martin, S.; Giunta, T.; Bonifacie, M.; Richnow, H.-H. Carbon, Hydrogen and Chlorine Stable Isotope Fingerprinting for Forensic Investigations of Hexachlorocyclohexanes. Environ. Sci. Technol. 2017, 51, 446–454. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, L.; Kohli, P.; Kumar, R.; Stryhanyuk, H.; Nijenhuis, I.; Lal, R.; Richnow, H.-H. Enantiomer and Carbon Isotope Fractionation of α-Hexachlorocyclohexane by Sphingobium indicum Strain B90A and the Corresponding Enzymes. Environ. Sci. Technol. 2019, 53, 8715–8724. [Google Scholar] [CrossRef]
- Schilling, I.E.; Bopp, C.E.; Lal, R.; Kohler, H.-P.E.; Hofstetter, T.B. Assessing Aerobic Biotransformation of Hexachlorocyclohexane Isomers by Compound-Specific Isotope Analysis. Environ. Sci. Technol. 2019, 53, 7419–7431. [Google Scholar] [CrossRef] [PubMed]
- Schilling, I.E.; Hess, R.; Bolotin, J.; Lal, R.; Hofstetter, T.B.; Kohler, H.-P.E. Kinetic Isotope Effects of the Enzymatic Transformation of γ-Hexachlorocyclohexane by the Lindane Dehydrochlorinase Variants LinA1 and LinA2. Environ. Sci. Technol. 2019, 53, 2353–2363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Kümmel, S.; Yao, J.; Nijenhuis, I.; Richnow, H.-H. Dual C–Cl isotope analysis for characterizing the anaerobic transformation of α, β, γ, and δ-hexachlorocyclohexane in contaminated aquifers. Water Res. 2020, 184, 116128. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.; Renpenning, J.; Nijenhuis, I.; Richnow, H.-H. Dual C–Cl Isotope Analysis for Characterizing the Reductive Dechlorination of α- and γ-Hexachlorocyclohexane by Two Dehalococcoides mccartyi Strains and an Enrichment Culture. Environ. Sci. Technol. 2020, 54, 7250–7260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, S.; Gui, J.; Li, X.; Qi, G. Compound-Specific Chlorine Isotope Analysis of Organochlorine Pesticides by Gas Chromatography-Negative Chemical Ionization Mass Spectrometry. J. Anal. Methods Chem. 2021, 2021, 8874679. [Google Scholar] [CrossRef]
- Badea, S.-L.; Stegarus, D.-I.; Niculescu, V.-C.; Enache, S.; Soare, A.; Ionete, R.-E.; Gori, D.; Höhener, P. Dehalogenation of α-hexachlorocyclohexane by iron sulfide nanoparticles: Study of reaction mechanism with stable carbon isotopes and pH variations. Sci. Total Environ. 2021, 801, 149672. [Google Scholar] [CrossRef]
- Ai, G.; Sun, T.; Dong, X. Gas chromatography/isotope ratio mass spectrometry: Analysis of methanol, ethanol and acetic acid by direct injection of aqueous alcoholic and acetic acid samples. Rapid Commun. Mass Spectrom. 2014, 28, 1674–1682. [Google Scholar] [CrossRef]
- Cheng, G.; Gao, S.; Gao, Y.; Yu, Z.; Peng, P.a. Compound-specific stable carbon isotope analysis of hexabromocyclododecane diastereoisomers using gas chromatography/isotope ratio mass spectrometry. Rapid Commun. Mass Spectrom. 2019, 33, 1318–1323. [Google Scholar] [CrossRef]
- Neves, L.A.; Rodrigues, J.M.; Daroda, R.J.; Silva, P.R.M.; Ferreira, A.A.; Aranda, D.A.G.; Eberlin, M.N.; Fasciotti, M. The influence of different referencing methods on the accuracy of δ13C value measurement of ethanol fuel by gas chromatography/combustion/isotope ratio mass spectrometry. Rapid Commun. Mass Spectrom. 2015, 29, 1938–1946. [Google Scholar] [CrossRef]
- Jochmann, M.A.; Blessing, M.; Haderlein, S.B.; Schmidt, T.C. A new approach to determine method detection limits for compound-specific isotope analysis of volatile organic compounds. Rapid Commun. Mass Spectrom. 2006, 20, 3639–3648. [Google Scholar] [CrossRef]
- Liu, X.; Wu, L.; Kümmel, S.; Richnow, H.H. Stable isotope fractionation associated with the synthesis of hexachlorocyclohexane isomers for characterizing sources. Chemosphere 2022, 296, 133938. [Google Scholar] [CrossRef] [PubMed]
- Coplen, T.B. Guidelines and recommended terms for expression of stable-isotope-ratio and gas-ratio measurement results. Rapid Commun. Mass Spectrom. 2011, 25, 2538–2560. [Google Scholar] [CrossRef] [PubMed]
- Coplen, T.B.; Brand, W.A.; Gehre, M.; Gröning, M.; Meijer, H.A.J.; Toman, B.; Verkouteren, R.M. After two decades a second anchor for the VPDB δ13C scale. Rapid Commun. Mass Spectrom. 2006, 20, 3165–3166. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Methods | Isotope | Scale | Contaminants | Applications | References |
|---|---|---|---|---|---|
| CSIA | C | Batch | γ-HCH | Biotic degradation identification | [12] |
| C | Field | α-, β-, γ-HCH | Source identification; in situ biodegradation identification | [17] | |
| C | Field | α-, β-, γ-, δ-HCH | In situ biodegradation quantification | [16] | |
| C, H, Cl | Batch | α-, β-, γ-, δ-HCH | Source identification | [19] | |
| C, H | Batch | γ-HCH | Biotic degradation identification | [22] | |
| C, Cl | Field | α-, β-, γ-, δ-HCH | In situ biodegradation identification | [23] | |
| C, Cl | Batch | α-HCH | Abiotic degradation identification | [26] | |
| Cl | Batch, field | α-, β-, γ-, δ-HCH | Source identification; in situ biodegradation identification | [25] | |
| CSIA, ESIA | C | Batch, field | α-HCH | Source identification; in situ biodegradation identification | [13] |
| C | Batch | α-HCH | Biotic degradation identification | [14] | |
| C | Batch | α-HCH | Abiotic degradation identification | [15] | |
| C | Field | α-, β-, γ-, δ-HCH | In situ biodegradation quantification | [18] | |
| C, H | Batch | α-, β-, γ-, δ-HCH | Biotic degradation identification | [21] | |
| C | Batch | α-HCH | Biotic degradation identification | [20] | |
| C | Batch | α-, γ-HCH | Biotic degradation identification | [24] |
| Compounds | EA-IRMS/‰ 2 | GC-IRMS/‰ | Amplitude Mass 44 at MDL (mV) 2 | |||
|---|---|---|---|---|---|---|
| Pure Chemicals 2 | Liquid–Liquid Extraction (mg/L) | |||||
| 200 2 | 300 2 | 400 2 | ||||
| α-HCH | −25.58 ± 0.02 | −26.23 ± 0.25 | −26.05 ± 0.17 | −26.21 ± 0.15 | −26.19 ± 0.28 | 181 ± 2 |
| β-HCH | −25.69 ± 0.05 | −25.90 ± 0.31 | −25.90 ± 0.21 | −25.90 ± 0.28 | −25.90 ± 0.30 | 265 ± 3 |
| γ-HCH | −27.68 ± 0.03 | −27.93 ± 0.31 | −27.98 ± 0.32 | −27.73 ± 0.25 | −27.69 ± 0.21 | 177 ± 2 |
| δ-HCH | −26.74 ± 0.03 | −26.84 ± 0.25 | −26.64 ± 0.22 | −26.63 ± 0.15 | −26.68 ± 0.18 | 176 ± 1 |
| (−)α-HCH | n.d.1 | −26.61 ± 0.39 | −26.73 ± 0.46 | −26.58 ± 0.39 | −26.90 ± 0.05 | 469 ± 8 |
| (+)α-HCH | n.d.1 | −25.44 ± 0.46 | −25.44 ± 0.33 | −25.46 ± 0.14 | −25.40 ± 0.13 | 338 ± 23 |
| Sample ID | δ13C/‰ (n = 3) | |||||
|---|---|---|---|---|---|---|
| α-HCH | β-HCH | γ-HCH | δ-HCH | (−)α-HCH | (+)α-HCH | |
| A | −24.94 ± 0.18 | −25.61 ± 0.20 | −27.08 ± 0.24 | −25.62 ± 0.21 | −26.61 ± 0.39 | −25.44 ± 0.46 |
| B | −24.63 ± 0.18 | −25.30 ± 0.09 | −26.86 ± 0.10 | −29.56 ± 0.17 | −26.30 ± 0.19 | −25.41 ± 0.22 |
| C | −26.13 ± 0.20 | −25.94 ± 0.10 | −27.64 ± 0.17 | −35.52 ± 0.16 | - | - |
| Sample ID | δ13C/‰ (n = 3) | |||||
|---|---|---|---|---|---|---|
| α-HCH | β-HCH | γ-HCH | δ-HCH | (−)α-HCH | (+)α-HCH | |
| D1 | −25.63 ± 0.18 | −26.30 ± 0.09 | −25.86 ± 0.10 | −22.56 ± 0.13 | −25.61 ± 0.39 | −26.74 ± 0.46 |
| D2 | −22.34 ± 0.18 | −24.51 ± 0.20 | −24.05 ± 0.21 | −25.74 ± 0.31 | −23.30 ± 0.19 | −22.36 ± 0.32 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, C.; Wang, Y.; Xia, Y.; Liu, H.; Cheng, W.; Xie, Y.; Yang, Y. Stable Carbon Isotope Analysis of Hexachlorocyclohexanes by Liquid–Liquid Extraction Gas Chromatography Isotope Ratio Mass Spectrometry: Method Evaluation and Applications. Molecules 2022, 27, 2874. https://doi.org/10.3390/molecules27092874
Gao C, Wang Y, Xia Y, Liu H, Cheng W, Xie Y, Yang Y. Stable Carbon Isotope Analysis of Hexachlorocyclohexanes by Liquid–Liquid Extraction Gas Chromatography Isotope Ratio Mass Spectrometry: Method Evaluation and Applications. Molecules. 2022; 27(9):2874. https://doi.org/10.3390/molecules27092874
Chicago/Turabian StyleGao, Cuiping, Yunlong Wang, Yu Xia, Haixian Liu, Weiguo Cheng, Yi Xie, and Yuesuo Yang. 2022. "Stable Carbon Isotope Analysis of Hexachlorocyclohexanes by Liquid–Liquid Extraction Gas Chromatography Isotope Ratio Mass Spectrometry: Method Evaluation and Applications" Molecules 27, no. 9: 2874. https://doi.org/10.3390/molecules27092874