
Diels–Alder Adducts of Morphinan-6,8-Dienes and Their Transformations †

 , , , and
, , , and
Abstract
:1. Introduction
2. Synthesis of 6,14-Ethenomorphinans
2.1. Diels–Alder Reactions
2.1.1. Nomenclature and Stereochemistry
2.1.2. Reactions of Δ6,8-morphinandienes
Thebaine
N17-Substituted Northebaine Derivatives
Morphinandienes with An Opened E-Ring
6-Demethoxythebaine
6-Halo-, 7-Halomorphinandienes and Pseudohalo Analogues of Thebaine
5β-Alkylthebaines
Substituted 5β-methyl-6-demethoxythebaine Derivatives
6-O-Alkylmorphinandienes
Heteroring-Fused Morphinandienes
Miscellaneous Cycloadducts
2.2. Grignard Addition
2.3. Orvinols
2.3.1. Synthesis of Orvinols
Diprenorphine, Buprenorphine, Etorphine, and Their Analogues
2.3.2. Synthesis of 7β-substituted 6,14-ethenomorphinans
Separation of 7α- and 7β-substituted 6,14-ethenomorphinan Derivatives
Isomerization
Synthesis of 7β-analogues of Diprenorphine, Buprenorphine, and Etorphine
2.3.3. On the 6,14-etheno Bridge Functionalized Analogues
2.3.4. Synthesis of Thevinone Analogues by Methylation of Its Enolate
2.3.5. N-demethylation and O-demethylation of 6,14-ethenomorphinans
N-demethylation
3-O-Demethylation
6-O-Demethylation
2.4. Hetero Diels–Alder (HDA) Reactions
2.4.1. Nitrosocarbonyl Dienophiles
2.4.2. Azadienophiles
2.4.3. Thebaine HDA Adducts for the Synthesis of Oripavine and Heroin Vaccine Haptens
2.5. Acetylenic Dienophiles
2.6. 7α-Amino and 7α-Aminomethyl Derivatives and Their Cyclic Analogues
2.7. Nepenthone Derivatives and 7α-phenyl-6,14-ethenomorphinan Analogues
“The Zeus’s daughter Helen thought of something else.Into the mixing-bowl from which they drank their wineshe slipped a drug, heart’s-ease, dissolving anger,magic to make us all forget our pains.No one who drank it deeply, mulled in wine,could let a tear roll down his cheeks that day,not even if his mother should die, his father die,not even if right before his eyes some enemy brought downa brother or darling son with a sharp bronze blade.”(Homer, Odyssey, 4, 219–227) (translation: Fagles, R.)
2.8. 1-Substituted 6,14-ethenomorphinans
3. Radiochemistry
3.1. Tritium Labelled Orvinol Derivatives
3.2. [125I]Iodinated Orvinols
3.3. Carbon-11 Labeled Orvinol Derivatives for PET
3.4. 18F-Labeled 6,14-ethenomorphinans
4. Conclusions and Outlook
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Presley, C.C.; Lindsley, C.W. Dark classics in chemical neuroscience: Opium, a historical perspective. ACS Chem. Neurosci. 2018, 9, 2503–2518. [Google Scholar] [CrossRef] [PubMed]
- Devereaux, A.L.; Mercer, S.L.; Cunningham, C.W. Dark classics in chemical neuroscience: Morphine. ACS Chem. Neurosci. 2018, 9, 2395–2407. [Google Scholar] [CrossRef] [PubMed]
- Wicks, C.; Hudlicky, T.; Rinner, U. Chapter Two—Morphine alkaloids: History, biology, and synthesis. In The Alkaloids: Chemistry and Biology; Knölker, H.-J., Ed.; Academic Press: Cambridge, MA, USA, 2021; Volume 86, pp. 145–342. ISBN 978-0-12-824618-4. [Google Scholar]
- Bolser, D.C. Central Mechanisms II: Pharmacology of brainstem pathways. In Pharmacology and Therapeutics of Cough, Handbook of Experimental Pharmacology; Chung, K.F., Widdicombe, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; Volume 187. [Google Scholar]
- Eddy, N.B.; Friebel, H.; Hahn, K.J.; Halbach, H. Codeine and its alternates for pain and cough relief. 3. The antitussive action of codeine-mechanism, methodology and evaluation. Bull. World Health Org. 1969, 40, 425–454. [Google Scholar] [PubMed]
- Cardinale, G.J.; Donnerer, J.; Finck, A.D.; Kantrowitz, J.D.; Oka, K.; Spector, S. Morphine and codeine are endogenous components of human cerebrospinal fluid. Life Sci. 1987, 40, 301–306. [Google Scholar] [CrossRef]
- Zhang, X.-Y.; Dou, Y.-N.; Yuan, L.; Li, Q.; Zhu, Y.-J.; Wang, M.; Sun, Y.-G. Different neuronal populations mediate inflammatory pain analgesia by exogenous and endogenous opioids. eLife 2020, 9, e55289. [Google Scholar] [CrossRef] [PubMed]
- Cumming, P.; Marton, J.; Lilius, T.O.; Olberg, D.E.; Rominger, A. A Survey of Molecular Imaging of Opioid Receptors. Molecules 2019, 24, 4190. [Google Scholar] [CrossRef] [Green Version]
- Szántay, C.; Dörnyei, G.; Blaskó, G. The morphine alkaloids. In The Alkaloids: Chemistry and Physiology; Cordell, G.A., Brossi, A., Eds.; Academic Press Inc.: New York, NY, USA; London, UK, 1988; Volume 45, pp. 128–232. [Google Scholar]
- Hosztafi, S., III. Chemistry-Biochemistry of Poppy, 1. Chemical Structures of Alkaloids. In Poppy, The Genus Papaver; Bernáth, J., Ed.; Harwood Academic Publishers: Amsterdam, The Netherlands, 1998; Volume 3, pp. 105–159. [Google Scholar]
- Sandermann, W. Diën-Anlagerungsverbindunden des Thebains. Ber. Dtsch. Chem. Ges. 1938, 71, 648–650. [Google Scholar] [CrossRef]
- Schöpf, C.; von Gottberg, K.; Petri, W. Über Thebain-maleinsäureanhydrid, Thebainchinon, Thebainhydrochinon und dessen Säreumlagerungsprodukt, das Flavothebaon. Justus Liebigs Ann. Chem. 1938, 536, 216–257. [Google Scholar] [CrossRef]
- Gulland, J.M.; Robinson, R. Constitution of codeine and thebaine. Mem. Proc. Manchester Lit. Phil. Soc. 1925, 69, 79–86. [Google Scholar]
- Diels, O.; Alder, K. Synthesen in der hydroaromatischen Reihe, I. Mitteilung: Anlagerungen von „Di-en“-kohlenwasserstoffen. Justus Liebigs Ann. Chem. 1928, 460, 98–122. [Google Scholar] [CrossRef]
- Kanevskaya, S.I.; Mitryagina, S.F. Investigations in the field of thebaine II. Study of the product of condensing thebaine with acroleine. J. Gen. Chem. (USSR) 1947, 17, 1203–1207. [Google Scholar]
- Bentley, K.W.; Thomas, A.F. The morphine–thebaine group of alkaloids. Part VI. The condensation of thebaine with dienophils. J. Chem. Soc. 1956, 1863–1867. [Google Scholar] [CrossRef]
- Bentley, K.W.; Hardy, D.G. New potent analgesics in morphine series. Proc. Chem. Soc. 1963, 220. [Google Scholar]
- Bentley, K.W. The morphine alkaloids. In The Alkaloids: Chemistry and Physiology; Manske, R.H.F., Ed.; Academic Press Inc.: New York, NY, USA; London, UK, 1971; Volume 13. [Google Scholar]
- Casy, A.F.; Parfitt, R.T. Diels-Alder adducts of thebaine. In Opioid Analgesics. Chemistry and Receptors; Plenum Press: New York, NY, USA, 1986; pp. 69–84. [Google Scholar]
- Massotte, D.; Kieffer, B.L. A molecular basis for opiate action. Essays Biochem. 1998, 33, 65–67. [Google Scholar]
- Benyhe, S.; Zádor, F.; Ötvös, F. Biochemistry of opioid (morphine) receptors: Binding, structure and molecular modelling. Acta Biol. Szeged 2015, 59 (Suppl. S1), 17–37. [Google Scholar]
- Borsodi, A.; Bruchas, M.; Caló, G.; Chavkin, C.; Christie, M.J.; Civelli, O.; Connor, M.; Cox, B.M.; Devi, L.A.; Evans, C.; et al. Opioid receptors (version 2019.4) in the IUPHAR/BPS Guide to Pharmacology Database. IUPHAR/BPS GtoPdb CITE 2019, 4. [Google Scholar] [CrossRef]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the mu-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Granier, S.; Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Weis, W.I.; Kobilka, B.K. Structure of the delta-opioid receptor bound to naltrindole. Nature 2012, 485, 400–404. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Wacker, D.; Mileni, M.; Katritch, V.; Han, G.W.; Vardy, E.; Liu, W.; Thompson, A.A.; Huang, X.-P.; Carroll, F.I.; et al. Structure of the human κ-opioid receptor in complex with JDTic. Nature 2012, 485, 327–332. [Google Scholar] [CrossRef]
- Thompson, A.A.; Liu, W.; Chun, E.; Katritch, V.; Wu, H.; Vardy, E.; Huang, X.-P.; Trapella, C.; Guerrini, R.; Calo, G.; et al. Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature 2012, 485, 395–399. [Google Scholar] [CrossRef]
- Lever, J.R. PET and SPECT imaging of the opioid system: Receptors, radioligands and avenues for drug discovery and development. Curr. Pharm. Des. 2007, 13, 33–49. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, G.; Willoch, F. Imaging of opioid receptors in the central nervous system. Brain 2008, 131, 1171–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dannals, R.F. Positron emission tomography radioligands for the opioid system. J. Label. Compd. Radiopharm. 2013, 56, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Üçeyler, N.; Buchholz, H.-G.; Kewenig, S.; Ament, S.-J.; Birklein, F.; Schreckenberger, M.; Sommer, C. Cortical binding potential of opioid receptors in patients with fibromyalgia syndrome and reduced systemic interleukin-4 levels—A pilot study. Front. Neurosci. 2020, 14, 512. [Google Scholar] [CrossRef] [PubMed]
- Kling, M.A.; Carson, R.E.; Borg, L.; Zametkin, A.; Matochik, J.A.; Schluger, J.; Herscovitch, P.; Rice, K.C.; Ho, A.; Eckelman, W.C.; et al. Opioid receptor imaging with positron emission tomography and [18F]cyclofoxy in long-term, methadone-treated former heroin addicts. J. Pharm. Exp. Ther. 2000, 295, 1070–1076. [Google Scholar]
- Kantonen, T.; Pekkarinen, L.; Karjalainen, T.; Bucci, M.; Kalliokoski, K.; Haaparanta-Solin, M.; Aarnio, R.; Dickens, A.M.; von Eyken, A.; Laitinen, K.; et al. Obesity risk is associated with altered cerebral glucose metabolism and decreased μ-opioid and CB1 receptor availability. Int. J. Obes. 2022, 46, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, H.K.; Tuominen, L.; Tuulari, J.J.; Hirvonen, J.; Parkkola, R.; Helin, S.; Salminen, P.; Nuutila, P.; Nummenmaa, L. Obesity is associated with decreased mu-opioid but unaltered dopamine D2 receptor availability in the brain. J. Neurosci. 2015, 35, 3959–3965. [Google Scholar] [CrossRef] [Green Version]
- Majuri, J.; Joutsa, J.; Johansson, J.; Voon, V.; Alakurtti, K.; Parkkola, R.; Lahti, T.; Alho, H.; Hirvonen, J.; Arponen, E.; et al. Dopamine and opioid neurotransmission in behavioral addictions: A comparative PET study in pathological gambling and binge eating. Neuropsychopharmacology 2017, 42, 1169–1177. [Google Scholar] [CrossRef] [Green Version]
- Vijay, A.; Cavallo, D.; Goldberg, A.; de Laat, B.; Nabulsi, N.; Huang, Y.; Krishnan-Sarin, S.; Morris, E.D. PET imaging reveals lower kappa opioid receptor availability in alcoholics but no effect of age. Neuropsychopharmacology 2018, 43, 2539–2547. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, M.L.; Wulf, H.A.; Browne, C.A.; Lucki, I. Chapter 1—Opioid modulation of cognitive impairment in depression. In Progress in Brain Research; Elsevier: Amsterdam, The Netherlands, 2018; Volume 239, pp. 1–48. [Google Scholar]
- Saanijoki, T.; Tuominen, L.; Tuulari, J.J.; Nummenmaa, L.; Arponen, E.; Kalliokoski, K.; Hirvonen, J. Opioid release after high-intensity interval training in healthy human subjects. Neuropsychopharmacology 2018, 43, 246–254. [Google Scholar] [CrossRef] [Green Version]
- Berényi, S.; Csutorás, C.; Sipos, A. Recent developments in the chemistry of thebaine and its transformation products as pharmacological targets. Curr. Med. Chem. 2009, 16, 3215–3242. [Google Scholar] [CrossRef] [PubMed]
- Hosztafi, S. Recent Advances in the Chemistry of Oripavine and Its Derivatives. Adv. Biosci. Biotechnol. 2014, 5, 704–717. [Google Scholar] [CrossRef] [Green Version]
- Maat, L.; Peters, J.A.; Prazeres, M.A. Diels-Alder reaction of thebaine via N-formylnorthebaine with nitroethene; reduction of the nitro group in 7α-nitroetheno isomorphinans (Chemistry of Opium Alkaloids, part XX). Recl. Trav. Chim. Pays-Bas 1985, 104, 205–208. [Google Scholar] [CrossRef]
- Fleischhacker, W.; Richter, B. 14,17-Ethano-norcodeinone. Monatsh. Chem. 1992, 123, 837–848. [Google Scholar] [CrossRef]
- Maurer, P.J.; Rapoport, H. Nitrogen-bridged conformationally constrained etorphine analogs. Synthesis and biological evaluation. J. Med. Chem. 1987, 30, 2016–2026. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.W.; Rushworth, W.I. Novel analgesics and molecular rearrangements in the morphine–thebaine group. Part XVII. Compounds related to the thebaine–maleic anhydride adduct. J. Chem. Soc. C 1970, 4, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Barton, J.W.; Coop, A.; Lewis, J.W. Diels-Alder reactions of thebaines with cycloalkenones; Lithium tetrafluoroborate as a novel diels-alder catalyst. Tetrahedron Lett. 1993, 34, 6777–6778. [Google Scholar] [CrossRef]
- Sandulenko, I.V.; Semenova, D.V.; Zelentsova, M.V.; Moiseev, S.K.; Koldobskii, A.B.; Peregudov, A.S.; Bushmarinov, I.S.; Kalinin, V.N. Reactions of thebaine derivatives with trifluoroacetyl acetylenes: [4+2]-addition solely. J. Fluor. Chem. 2016, 189, 7–12. [Google Scholar] [CrossRef]
- Bentley, K.W.; Robinson, R. Some notes on the reduction of thebaine and related topic. Experientia 1950, 6, 353–354. [Google Scholar] [CrossRef]
- Hayakawa, K.; Fujii, I.; Kanematsu, K. Addition reaction of thebaine and related compounds with acetylenic dienophiles: The structure-reactivity relationship. J. Org. Chem. 1983, 48, 166–173. [Google Scholar] [CrossRef]
- Ghosh, A.C.; Portlock, D.E.; Dalzell, H.C.; Malmberg, C.; Herlihy, P.; Razdan, R.K.; Duax, W.L.; Smith, G.D. Novel opiates and antagonists. Part 7. Diels-Alder reaction of beta-dihydrothebaine and its 4-phenyl ether with methyl vinyl ketone: Synthesis of 6,14-exo-ethenomorphinans. J. Org. Chem. 1983, 48, 4137–4139. [Google Scholar] [CrossRef]
- Linders, J.T.M.; Kokje, J.P.; Overhand, M.; Lie, T.S.; Maat, L. Diels-Alder reaction of 6-demethoxy-β-dihydrothebaine with methyl vinyl ketone using microwave heating; preparation and pharmacology of 3-hydroxy-α,α, 17-trimethyl-6β,14β- ethenomorphinan-7β-methanol, a novel deoxygenated diprenorphine analogue (Chemistry of Opium Alkaloids, part XXV). Recl. Trav. Chim. Pays-Bas 1988, 107, 449–454. [Google Scholar]
- Linders, J.T.M.; Lipman, P.J.L.; Saly, E.; Lie, T.S.; Maat, L. Diels-Alder Adducts From 4-O-Acetyl-6-Demethoxy-N-Formyl-β-Dihydro-N-Northebaine With Methyl Vinyl Ketone and Nitroethene (Chemistry of Opium Alkaloids, Part XXIX). Bull. Soc. Chim. Belg. 1989, 98, 257–264. [Google Scholar] [CrossRef]
- Knipmeyer, L.L.; Rapoport, H. Analgesics of the 6,14-ethenomorphinan type. 6-deoxy-7-alpha-orvinols and 6-deoxy-8-alpha-orvinols. J. Med. Chem. 1985, 28, 461–466. [Google Scholar] [CrossRef]
- Berényi, S.; Makleit, S.; Bognár, R.; Tegdes, A. Conversions of tosyl and mesyl derivatives of the morphine group XXII. Some nucleophilic substitution reactions of 6-O-mesylneopine. Acta Chim. Acad. Sci. Hung. 1980, 103, 365–369. [Google Scholar] [CrossRef]
- Berényi, S.; Hosztafi, S.; Makleit, S.; Molnár, I. Preparation of demethoxyoripavine and its conversion into N-substituted-N-demethylapomorphine derivatives. Acta Chim. Hung. 1983, 113, 51–60. [Google Scholar]
- Linders, J.T.M.; Briel, P.; Fog, E.; Lie, T.S.; Maat, L. Preparation of 6-demethoxy-N-formyl-N-northebaine and its Diels-Alder reactions with methyl vinyl ketone and nitroethene; novel 8-nitro-substituted 6α,14α-ethenoisomorphinans and 6β,14β-ethenomorphinans (Chemistry of Opium Alkaloids, part XXVIII). Recl. Trav. Chim. Pays-Bas 1989, 108, 268–274. [Google Scholar] [CrossRef]
- Berényi, S.; Hosztafi, S.; Makleit, S. A new efficient method for the preparation of 2-fluoro-N-propylnorapomorphine. J. Chem. Soc. Perkin Trans. 1 1992, 20, 2693–2694. [Google Scholar] [CrossRef]
- Berényi, S.; Makleit, S.; Rantal, F. Conversion of tosyl and mesyl derivatives of the morphine group, XXIV. Reactions of morphine derivatives containing double allylic system. Acta Chim. Hung. 1985, 120, 171–174. [Google Scholar]
- Berényi, S.; Sepsi, Á.; Gyulai, S.; Szilágyi, L. Synthesis of sulfur-containing morphinane dienes. Synth. Commun. 1995, 25, 3307–3314. [Google Scholar] [CrossRef]
- Csutorás, C.; Berényi, S.; Czakó, B.; Makleit, S. Syntheses and transformations of novel nitrogen and sulfur containing morphinanedienes. Monatsh. Chem. 1997, 128, 1267–1273. [Google Scholar] [CrossRef]
- Simon, C.; Berényi, S.; Makleit, S.; Fekete, V. Conversions of tosyl and mesyl derivatives of the morphine group, XXV. Studies of the nucleophilic substitution reactions of 6-0-mesyl-7.alpha.-chloro(bromo)neopine. Acta Chim. Hung. 1987, 124, 497–503. [Google Scholar]
- Boden, R.M.; Gates, M.; Ho, S.P.; Sundararaman, P. Derivatives of the thebaine anion. 1. Structure of metopon. A direct demonstration. J. Org. Chem. 1982, 47, 1347–1349. [Google Scholar] [CrossRef]
- Woudenberg, R.H.; Oosterhoff, B.E.; Lie, T.S.; Maat, L. Synthesis of 5β-alkylthebaines, 5β-alkyl-10α-ethylthebaines and 5β,10α, 10β-triethylthebaine; Diels-Alder reactions to 5β-alkyl- and 5β-alkyl-10α-ethyl-6α, 14α-ethenoisomorphinans (Chemistry of Opium Alkaloids, Part XXXVI). Recl. Trav. Chim. Pays-Bas 1992, 111, 119–125. [Google Scholar] [CrossRef]
- Woudenberg, R.H.; Lie, T.S.; Maat, L. Reactions of thebaine and 6-demethoxythebaine anions with carbonyl compounds; novel Diels-Alder adducts from 5β-substituted thebaines (Chemistry of Opium Alkaloids, Part XXXIX). Recl. Trav. Chim. Pays-Bas 1993, 112, 557–564. [Google Scholar] [CrossRef]
- Chen, W.; Wu, H.; Bernard, D.; Metcalf, M.D.; Deschamps, J.R.; Flippen-Anderson, J.L.; MacKerell, A.D.; Coop, A. Rearrangement of 5-trimethylsilylthebaine on treatment with L-Selectride: An efficient synthesis of (+)-bractazonine. J. Org. Chem. 2003, 68, 1929–1932. [Google Scholar] [CrossRef]
- Woudenberg, R.H.; Piet, D.P.; Sinnema, A.; Lie, T.S.; Maat, L. Synthesis of 5β-methyl-6-demethoxythebaine and its Diels-Alder reaction to 6α,14α-ethenoisomorphinans and 6β,14β-ethenomorphinans (Chemistry of Opium Alkaloids, Part XXXV. Recl. Trav. Chim. Pays-Bas 1991, 110, 405–413. [Google Scholar] [CrossRef]
- Woudenberg, R.H.; Maat, L. Synthesis and biological activity of new etorphine analogues from 7-chloro-6-demethoxythebaine and 7-chloro-5β-methyl-6-demethoxythebaine (Chemistry of Opium Alkaloids, part XXXVII). Recl. Trav. Chim. Pays-Bas 1993, 112, 113–122. [Google Scholar] [CrossRef]
- Woudenberg, R.H.; Meuzelaar, G.J.; Maat, L. Synthesis of 7-substituted 6-demethoxythebaines and Diels-Alder reaction of 7-methoxy-5β-methyl-6-demethoxythebaine (chemistry of opium alkaloids, part XLI). Recl. Trav. Chim. Pays-Bas 1993, 112, 578–583. [Google Scholar] [CrossRef]
- Czakó, B.; Marton, J.; Berényi, S.; Gach, K.; Fichna, J.; Storr, M.; Tóth, G.; Sipos, A.; Janecka, A. Synthesis and opioid activity of novel 6-substituted-6-demethoxy-ethenomorphinans. Bioorg. Med. Chem. 2010, 18, 3535–3542. [Google Scholar] [CrossRef]
- Sipos, A.; Skaliczki, T.; Berényi, S.; Antus, S. Thiazole constrained analogues of the thevinones: Synthesis and structure. Magn. Reson. Chem. 2009, 47, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Pindur, U.; Keilhofer, D. New studies and reinvestigation on [4+2] cycloadditions of (-)-thebaine: Asymmetrical Diels-Alder reactions with a conformationally fixed chiral diene. Liebigs Ann. Chem. 1993, 9, 947–953. [Google Scholar] [CrossRef]
- Pindur, U.; Keilhofer, D.; Schollmeyer, D. New Diels-Alder reactions for (-)-thebaine and first X-ray crystallographic structure analyses of the cycloadducts. Z. Naturforsch. B 1994, 49, 272–279. [Google Scholar] [CrossRef]
- Linders, J.T.M.; Maat, L. Face selectivity of the Diels-Alder reaction of thebaine-like morphinandienes, A computational approach (Chemistry of opium alkaloids, Part XXXI). Bull. Soc. Chim. Belg. 1989, 98, 265–276. [Google Scholar] [CrossRef]
- Linders, J.T.M.; Lie, T.S.; Maat, L. Synthesis and preliminary pharmacology of the rigid dehydroxylated etorphine analogue 4,5α-epoxy-α,α,17-trimethyl-6α,14α-etheno-isomorphinan-7α-methanol (Chemistry of Opium Alkaloids, part XXVI). Bull. Soc. Chim. Belg. 1988, 97, 463–467. [Google Scholar] [CrossRef]
- Maat, L. Novel thebainelike morphinan-dienes and their Diels-Alder adducts. In National Institute on Drug Abuse, Research Monograph Series, Drugs of Abuse: Chemistry, Pharmacology, Immunology, and AIDS; Pham, P.T.K., Rice, K.C., Eds.; U.S. DHHS, Public Health Service, Alcohol, Drug Abuse, and Mental Health Administration, NIDA: Rockville, MD, USA, 1990; pp. 35–49. [Google Scholar]
- Bentley, K.W.; Hardy, D.G. Novel analgesics and molecular rearrangements in the morphine-thebaine group I. Ketones derived from 6,14-endo-ethenotetrahydrtothebaine. J. Am. Chem. Soc. 1967, 89, 3267–3273. [Google Scholar] [CrossRef]
- Crabbendam, P.R.; Lie, T.S.; Linders, J.T.M.; Maat, L. Synthesis of 6, 14-ethenoisomorphinans and 6, 14-ethenomorphinans based on Diels-Alder adducts of 6-demethoxythebaine and 6-demethoxy-β-dihydrothebaine; pharmacology of the isomorphinans (Chemistry of Opium Alkaloids, Part XIX). Recl. Trav. Chim. Pays-Bas 1984, 103, 296–300. [Google Scholar] [CrossRef]
- Marton, J.; Garadnay, S.; Szabó, Z.; Hosztafi, S.; Makleit, S. Isomerization reactions of 7-substituted 6,14-bridged thebaine derivatives (Bentley compounds). Acta Chem. Scand. 1998, 52, 1234–1238. [Google Scholar] [CrossRef] [Green Version]
- Bentley, K.W.; Ball, J.C. Acid-catalyzed rearrangements in the nepenthone series. J. Org. Chem. 1958, 23, 1720–1725. [Google Scholar] [CrossRef]
- Linders, J.T.M.; Prazeres, M.A.; Lie, T.S.; Maat, L. A 1H and 13C NMR study of N-formylmorphinans and their 6,14-bridged derivatives; Comparison with N-Me and N-H analogues. Magn. Reson. Chem. 1989, 27, 980–986. [Google Scholar] [CrossRef]
- Coop, A.; Grivas, K.; Husbands, S.M.; Lewis, J.W. Methylation of the enolates of thevinone and some analogues. Tetrahedron 1995, 51, 9681–9698. [Google Scholar] [CrossRef]
- Prazeres, M.A.; Peters, J.A.; Linders, J.T.M.; Maat, L. 4,5α-epoxy-3-methoxy-8β-nitro-6β, 14β-ethenomorphinan, a novel type Diels-Alder adduct from 6-demethoxy-17-formylnorthebaine and nitroethene (Chemistry of Opium Alkaloids, part XXIII). Recl. Trav. Chim. Pays-Bas 1986, 105, 554–555. [Google Scholar] [CrossRef]
- Berényi, S.; Tóth, Z.; Sepsi, Á.; Zékány, A.; Gyulai, S.; Makleit, S. Morphine alkaloids, Part 131. Synthesis and biological evaluation of some halogenated 6,14-ethenomorphinan derivatives. Med. Chem. Res. 1994, 5, 26–32. [Google Scholar]
- Marton, J.; Szabó, Z.; Csorvássy, I.; Simon, C.; Hosztafi, S.; Makleit, S. Reaction of morphinan-6,8-dienes with azadienophiles. Tetrahedron 1996, 52, 2449–2464. [Google Scholar] [CrossRef]
- Woudenberg, R.H.; Lie, T.S.; Maat, L. Rigid etheno-bridged metopon analogues from Diels-Alder reaction of 5-methylthebaine. (Chemistry of Opium Alkaloids, Part XXXII). Recl. Trav. Chim. Pays-Bas 1990, 109, 353–357. [Google Scholar] [CrossRef]
- Woudenberg, R.H.; Lie, T.S.; Maat, L. Chemistry of opium alkaloids. 38. Synthesis of rigid morphinans doubly bridged at ring C (Chemistry of Opium Alkaloids. Part 38.). J. Org. Chem. 1993, 58, 6139–6142. [Google Scholar] [CrossRef]
- Chen, W.; Strahan, G.D.; Parrish, D.A.; Deschamps, J.R.; Coop, A. Studies into the Diels-Alder reactions of 5-trimethylsilylthebaine. Tetrahedron Lett. 2005, 46, 131–133. [Google Scholar] [CrossRef]
- Seki, I. Studies on the morphine alkaloids and its related compounds. XVII. One-step preparations of enol ether and pyrrolidinyl dienamine of normorphinone derivatives. Chem. Pharm. Bull. 1970, 18, 671–676. [Google Scholar] [CrossRef] [Green Version]
- Berényi, S.; Makleit, S.; Szilágyi, L. Conversions of tosyl and mesyl derivatives of the morphine group, XXIII. Preparation of new 6-substituted-6-demethoxythebaine derivatives. Acta Chim. Hung. 1984, 117, 307–312. [Google Scholar] [CrossRef]
- Zhang, A.; van Vliet, S.; Neumeyer, J.L. Synthesis of aminothiazole derived morphinans. Tetrahedron Lett. 2003, 44, 6459–6462. [Google Scholar] [CrossRef]
- Zhang, A.; Xiong, W.; Hilbert, J.E.; DeVita, E.K.; Bidlack, J.M.; Neumeyer, J.L. 2-Aminothiazole-derived opioids. Bioisosteric replacement of phenols. J. Med. Chem. 2004, 47, 1886–1888. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Yan, Z.; Sromek, A.; Knapp, B.I.; Scrimale, T.; Bidlack, J.M.; Neumeyer, J.L. Aminothiazolomorphinans with Mixed kappa and mu Opioid Activity. J. Med. Chem. 2011, 54, 1903–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Görlitzer, K.; Schumann, R. Darstellung und Charackterisierung von Bromierungsprodukten des Oxycodons. Pharmazie 1992, 47, 893–897. [Google Scholar]
- Görlitzer, K.; Schumann, R. Zur Reaktion von Dihydrocodeinhydrogentartrat mit Brom in essigsauer Lösung. Pharmazie 1993, 48, 525–527. [Google Scholar]
- Tóth, M.; Gyulai, Z.; Berényi, S.; Sipos, A. Synthesis and transformation of thiazolomorphinanedienes. Lett. Org. Chem. 2007, 4, 539–543. [Google Scholar] [CrossRef]
- Hromatka, O.; Knollmüller, M.; Sengstschmid, G. Über eine neue Diels-Alder-Reaktion am Thebain. Monatsh. Chem. 1968, 99, 1662–1665. [Google Scholar] [CrossRef]
- Hromatka, O.; Sengstschmid, G.; Eichinger, K. Thebain-Maleinimid-und Thebain-N-substituierte Maleinimid-Addukte. Monatsh. Chem. 1971, 102, 1015–1021. [Google Scholar] [CrossRef]
- Coop, A.; Grivas, K.; Husbands, S.M.; Lewis, J.W.; Porter, J. Ring constrained analogues of the thevinones; Diels-Alder reactions of thebaines with 1-indenone and methylene cycloalkanones. Tetrahedron Lett. 1995, 36, 1689–1692. [Google Scholar] [CrossRef]
- Bentley, K.W.; Hardy, D.G.; Meek, B. Novel analgesics and molecular rearrangements in the morphine-thebaine group. II. Alcohols derived from 6,14-endo-etheno- and 6,14-endo-ethanotetrahydrothebaine. J. Am. Chem. Soc. 1967, 89, 3273–3280. [Google Scholar] [CrossRef]
- Marton, J.; Schoultz, B.W.; Hjørnevik, T.; Drzezga, A.; Yousefi, B.H.; Wester, H.-J.; Willoch, F.; Henriksen, G. Synthesis and evaluation of a full-agonist orvinol for PET-Imaging of opioid receptors: [11C]PEO. J. Med. Chem. 2009, 52, 5586–5589. [Google Scholar] [CrossRef]
- Bentley, K.W.; Hardy, D.G.; Crocker, H.P.; Haddlesey, D.I.; Mayor, P.A. Novel analgesics and molecular rearrangements in the morphine-thebaine group. VI. Base-catalyzed rearrangements in the 6,14-endo-ethenotetrahydrothebaine series. J. Am. Chem. Soc. 1967, 89, 3312–3321. [Google Scholar] [CrossRef] [PubMed]
- Marton, J.; Sipos, A.; Henriksen, G.; Cumming, P.; Berényi, S.; Schmitt, B.M.; Szabó, Z. NMR Analysis of a series of 6,14-ethenomorphinan derivatives as PET precursors and reference substances. ChemistrySelect 2021, 6, 5994–6005. [Google Scholar] [CrossRef]
- Marton, J.; Szabó, Z.; Garadnay, S.; Miklós, S.; Makleit, S. Studies on the synthesis of beta-thevinone derivatives. Tetrahedron 1998, 54, 9143–9152. [Google Scholar] [CrossRef]
- Marton, J.; Szabó, Z.; Hosztafi, S. Herstellung von 6,14 Ethenomorphinan-Derivaten. Liebigs Ann. Chem. 1993, 8, 915–919. [Google Scholar] [CrossRef]
- Marton, J.; Hosztafi, S.; Berényi, S.; Simon, C.; Makleit, S. Herstellung von 6,14-Ethenomorphinan- Derivaten. Monatsh. Chem. 1994, 125, 1229–1239. [Google Scholar] [CrossRef]
- Marton, J.; Miklós, S.; Hosztafi, S.; Makleit, S. Synthesis of N-substituted 7-beta-diprenorphine derivatives. Synth. Commun. 1995, 25, 829–848. [Google Scholar] [CrossRef]
- Kosten, T.R.; George, T.P. The neurobiology of opioid dependence: Implications for treatment. Sci. Pract. Perspect. 2002, 1, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.X.; Zhu, Y.C.; Jin, W.Q.; Chen, X.J.; Chen, J.; Ji, R.Y.; Chi, Z.Q. Stereoisomers of N-[1-hydroxy-(2-phenylethyl)-3-methyl-4-piperidyl]-N-phenylpropanamide: Synthesis, stereochemistry, analgesic activity, and opioid receptor binding characteristics. J. Med. Chem. 1995, 38, 3652–3659. [Google Scholar] [CrossRef]
- Carliss, R.D.; Keefer, J.F.; Perschke, S.; Welch, S.; Rich, T.C.; Weissman, A.D. Receptor reserve reflects differential intrinsic efficacy associated with opioid diastereomers. Pharmacol. Biochem. Behav. 2009, 92, 495–502. [Google Scholar] [CrossRef]
- Contet, C.; Kieffer, B.L.; Befort, K. Mu opioid receptor: A gateway to drug addiction. Curr. Opin. Neurobiol. 2004, 14, 370–378. [Google Scholar] [CrossRef]
- Shippenberg, T.S.; LeFevour, A.; Chefer, V.I. Targeting endogenous mu- and delta-opioid receptor systems for the treatment of drug addiction. CNS Neurol. Disord. Drug Targets 2008, 7, 442–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marton, J.; Simon, C.; Hosztafi, S.; Szabó, Z.; Márki, Á.; Borsodi, A.; Makleit, S. New nepenthone and thevinone derivatives. Bioorg. Med. Chem. 1997, 5, 369–382. [Google Scholar] [CrossRef]
- Bentley, K.W.; Hardy, D.G. Novel analgesics and molecular rearrangements in the morphine-thebaine group. III. Alcohols of the 6,14-endo-ethenotetrahydrooripavine series and derived analogs of N-allylnormorphine and -norcodeine. J. Am. Chem. Soc. 1967, 89, 3281–3292. [Google Scholar] [CrossRef] [PubMed]
- Fulmor, W.; Lancaster, J.E.; Morton, G.O.; Brown, J.J.; Howell, C.F.; Nora, C.T.; Hardy, R.A. Nuclear magnetic resonance studies in the 6, 14-endo-ethenotetrahydrothebaine series. J. Am. Chem. Soc. 1967, 89, 3322–3330. [Google Scholar] [CrossRef] [PubMed]
- Biyashev, D.; Garadnay, S.; Marton, J.; Makleit, S.; Borsodi, A.; Benyhe, S. Biochemical characterization of newly developed beta-etorphine and beta-dihydroetorphine derivatives. Eur. J. Pharm. 2002, 442, 23–27. [Google Scholar] [CrossRef]
- Kumar, V.; Ridzwan, I.E.; Grivas, K.; Lewis, J.W.; Clark, M.J.; Meurice, C.; Jimenez-Gomez, C.; Pogozheva, I.; Mosberg, H.; Traynor, J.R.; et al. Selectively promiscuous opioid ligands: Discovery of high affinity/low efficacy opioid ligands with substantial nociceptin opioid peptide receptor affinity. J. Med. Chem. 2014, 57, 4049–4057. [Google Scholar] [CrossRef]
- Konowalaowa, R.; Yunussoff, S.; Orechoff, A. Über Alkaloide der Papaver-Arten, I. Mitteil.: Alkaloide von Papaver Armeniacum und Papaver orientale. Ber. Dtsch. Chem. Ges. 1935, 68, 2158–2163. [Google Scholar] [CrossRef]
- Lewis, J.W.; Husbands, S.M. The Orvinols and Related Opioids—High Affinity Ligands with Diverse Efficacy Profiles. Curr. Pharm. Des. 2004, 10, 717–732. [Google Scholar]
- Herz, A.; Höllt, V. Receptor occupation and pharmacological activity as demonstrated in opiates. Arzneim.-Forsch./Drug. Res. 1977, 27, 1865–1867. [Google Scholar]
- Lutfy, K.; Cowan, A. Buprenorphine: A unique drug with complex pharmacology. Curr. Neuropharm. 2004, 2, 395–402. [Google Scholar] [CrossRef]
- Husbands, S.H. Buprenorphine and related orvinols. In Research and Development of Opioid-Related Ligands; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2013; pp. 127–144. [Google Scholar]
- Kyzer, J.L.; Wenthur, C.J. Classics in Chemical Neuroscience: Buprenorphine. ACS Chem. Neurosci. 2020, 11, 1385–1399. [Google Scholar] [CrossRef] [PubMed]
- Heel, R.C.; Brogden, R.N.; Speight, T.M.; Avery, G.S. Buprenorphine: A review of its pharmacological properties and therapeutic efficacy. Drugs 1979, 17, 81–110. [Google Scholar] [CrossRef] [PubMed]
- Wnendt, S.; Krüger, T.; Janocha, E.; Hildebrandt, D.; Englberger, W. Agonistic effect of buprenorphine in a nociceptin/OFQ receptor triggered reporter gene assay. Mol. Pharmacol. 1999, 56, 334–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mijele, D.; Njoroge, M.; Kaitho, T. Surgical treatment of an umbilical hernia in a free-ranging sub-adult African elephant in Samburu National Reserve, Kenya. Vet. Med. Res. Rep. 2015, 6, 165–170. [Google Scholar]
- Takemori, A.E.; Hayashi, G.; Smits, S.E. Studies on the quantitative antagonism of analgesics by naloxone and diprenorphine. Eur. J. Pharmacol. 1972, 20, 85–92. [Google Scholar] [CrossRef]
- Ohmori, S.; Morimoto, Y. Dihydroetorphine: A potent analgesic: Pharmacology, toxicology, pharmacokinetics, and clinical effects. CNS Drug Rev. 2002, 8, 391–404. [Google Scholar] [CrossRef] [Green Version]
- Olofsen, E.; Boom, M.; Sarton, E.; van Velzen, M.; Baily, P.; Smith, K.J.; Oksche, A.; Dahan, A.; Niesters, M. Analgesic and respiratory depressant effects of R-dihydroetorphine: A pharmacokinetic-pharmacodynamic analysis in healthy male volunteers. Anesthesiology 2019, 131, 1327–1339. [Google Scholar] [CrossRef]
- Lewis, J.W.; Bentley, K.W.; Cowan, A. Narcotic analgesics and antagonists. Ann. Rev. Pharmacol. 1971, 11, 241–270. [Google Scholar] [CrossRef]
- Bentley, K.W.; Lewis, J.W. The relationship between structure and activity in the 6,14-endoethenotetrahydrothebaine series of analgesics. In Agonist and Antagonist Actions of Narcotic Analgesic Drugs; Kosterlitz, H.W., Collier, H.O.J., Villarreal, J.E., Eds.; University Park Press Baltimore: London, UK; Tokyo, Japan, 1972. [Google Scholar]
- Liu, H.; Zhong, B.-H.; Liu, C.-H.; Wu, B.; Gong, Z.-H. Synthesis, crystal structural and pharmacological study of N-cyclopropylmethyl-7α-[(R)-1-hydroxy-1-methyl-3-(thien-2-yl)propyl]-6,14-endoethanotetrahydronororipavine. Acta Chim. Slov. 2005, 52, 80–85. [Google Scholar]
- Khroyan, T.V.; Polgar, W.E.; Cami-Kobeci, G.; Husbands, S.M.; Zaveri, N.T.; Toll, L. The first universal opioid ligand, (2S)-2-[(5R,6R,7R,14S)-N-cyclopropylmethyl-4,5-epoxy-6,14-ethano-3-hydroxy-6-methoxymorphinan-7-yl]-3,3-dimethylpentan-2-ol (BU08028): Characterization of the in vitro profile and in vivo behavioral effects in mouse models of acute pain and cocaine-induced reward. J. Pharm. Exp. Ther. 2011, 336, 952–961. [Google Scholar]
- Cueva, J.P.; Roche, C.; Ostovar, M.; Kumar, V.; Clark, M.J.; Hillhouse, T.M.; Lewis, J.W.; Traynor, J.R.; Husbands, S.M. C7β-Methyl Analogues of the Orvinols: The discovery of kappa opioid antagonists with nociceptin/orphanin FQ peptide (NOP) receptor partial agonism and low, or zero efficacy at mu opioid receptors. J. Med. Chem. 2015, 58, 4242–4249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almatroudi, A.; Ostovar, M.; Bailey, C.P.; Husbands, S.M.; Bailey, S.J. Antidepressantlike effects of BU10119, a novel buprenorphine analogue with mixed κ/μ receptor antagonist properties, in mice. Brit. J. Pharmacol. 2018, 175, 2869–2880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillhouse, T.M.; Olson, K.M.; Hallahan, J.E.; Rysztak, L.G.; Sears, B.F.; Meurice, C.; Ostovar, M.; Koppenhaver, P.O.; West, J.L.; Jutkiewicz, E.M.; et al. The buprenorphine analogue BU10119 attenuates drug-primed and stress-induced cocaine reinstatement in mice. J. Pharm. Exp. Ther. 2021, 378, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Werner, L.; Machara, A.; Adams, D.R.; Cox, D.P.; Hudlicky, T. Synthesis of buprenorphine from oripavine via N-demethylation of oripavine quaternary salts. J. Org. Chem. 2011, 76, 4628–4634. [Google Scholar] [CrossRef] [PubMed]
- Machara, A.; Werner, L.; Endoma-Arias, M.A.; Cox, D.P.; Hudlicky, T. Improved synthesis of buprenorphine from thebaine and/or oripavine via Palladium-catalyzed N-demethylation/acylation and/or concomitant O-demethylation. Adv. Synth. Catal. 2012, 354, 613–626. [Google Scholar] [CrossRef]
- Hudlicky, T. Recent advances in process development for opiate-derived pharmaceutical agents. Can. J. Chem. 2015, 93, 492–501. [Google Scholar] [CrossRef]
- Von Braun, J. Die Einwirkung von Bromcyan auf tertiäre Amine. Berichte 1900, 33, 1438–1452. [Google Scholar] [CrossRef]
- McHugh, R.K.; Fulciniti, F.; Mashhoon, Y.; Weiss, R.D. Cue-induced craving to paraphernalia and drug images in opioid dependence. Am. J. Addict. 2016, 25, 105–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castleman, P.; Szwabowski, G.; Bowman, D.; Cole, J.; Parrill, A.L.; Baker, D.L. Ligand-based G protein coupled receptor pharmacophore modeling: Assessing the role of ligand function in model development. J. Mol. Graph. Model. 2022, 111, 108107. [Google Scholar] [CrossRef]
- Puls, K.; Schmidhammer, H.; Wolber, G.; Spetea, M. Mechanistic characterization of the pharmacological profile of HS-731, a peripherally acting opioid analgesic, at the µ-, δ-, κ-Opioid and nociceptin receptors. Molecules 2022, 27, 919. [Google Scholar] [CrossRef]
- Bickel, W.K.; Amass, L. Buprenorphine treatment of opioid dependence: A review. Exp. Clin. Psychopharm. 1995, 3, 447–489. [Google Scholar] [CrossRef]
- Subasinghe, K.R.; Jackson, W.R.; Young, J.F.; Papanastasiou, M.; Jarrott, B. Non-CNS acting opiates bearing guanidino substituents. Aust. J. Chem. 2004, 57, 427–438. [Google Scholar] [CrossRef]
- Park, H.S.; Lee, H.L.; Kim, Y.H.; Park, J.K.; Zvartau, E.E.; Lee, H. A highly selective κ-opioid receptor agonist with low addictive potential and dependence liability. Bioorg. Med. Chem. Lett. 2006, 16, 3609–3613. [Google Scholar] [CrossRef] [PubMed]
- Cone, E.J.; Gorodetzky, C.W.; Darwin, W.D.; Buchwald, W.F. Stability of the 6,14-endo-ethanotetrahydrooripavine analgesics: Acid-catalyzed rearrangement of buprenorphine. J. Pharm. Sci. 1984, 73, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Husbands, S.M.; Breeden, S.W.; Grivas, K.; Lewis, J.W. Ring constrained analogues of the orvinols: The furanomorphides. Bioorg. Med. Chem. Lett. 1999, 9, 831–834. [Google Scholar] [CrossRef]
- Greedy, B.M.; Bradbury, F.; Thomas, M.P.; Grivas, K.; Cami-Kobeci, G.; Archambeau, A.; Bosse, K.; Clark, M.J.; Aceto, M.; Lewis, J.W.; et al. Orvinols with mixed kappa/mu opioid receptor agonist activity. J. Med. Chem. 2013, 56, 3207–3216. [Google Scholar] [CrossRef] [Green Version]
- Boura, A.L.; Fitzgerald, A.E. The pharmacology of N-(cyclopropylmethyl)-19-isopentylnororvinol hydrochloride. A potent and long lasting central depressant. Br. J. Pharmacol. 1966, 26, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Ben Haddou, T.; Malfacini, D.; Calo, G.; Aceto, M.D.; Harris, L.S.; Traynor, J.R.; Coop, A.; Schmidhammer, H.; Spetea, M. Exploring pharmacological activities and signaling of morphinans substituted in position 6 as potent agonists interacting with the mu opioid receptor. Mol. Pain 2014, 10, 1744–8069. [Google Scholar] [CrossRef] [Green Version]
- Hasanpour, Z.; Salehi, P.; Bunch, L.; Khoramjouy, M.; Bararjanian, M.; Staerk, D.; Faizi, M. Semi-synthesis of Novel Buprenorphine Derivatives and their Anti-nociceptive Properties and Dependency Potential. Can. J. Chem. 2021, 99, 910–919. [Google Scholar] [CrossRef]
- McCamley, K.; Ripper, J.A.; Singer, R.D.; Scammells, P.J. Efficient N-demethylation of opiate alkaloids using a modified nonclassical Polonovski reaction. J. Org. Chem. 2003, 68, 9847–9850. [Google Scholar] [CrossRef]
- Zhu, L.; Brassard, C.J.; Zhang, X.; Guha, P.M.; Clark, R.J. On the mechanism of copper(I)-catalyzed azide–alkyne cycloaddition. Chem. Rec. 2016, 16, 1501–1517. [Google Scholar] [CrossRef] [PubMed]
- Bentley, K.W.; Ball, J.C. Base-catalyzed rearrangements in the nepenthone series. J. Org. Chem. 1958, 23, 1725–1730. [Google Scholar] [CrossRef]
- Bentley, K.W.; Hardy, D.G.; Meek, B. Novel analgesics and molecular rearrangements in the morphine-thebaine group. IV. Acid-catalyzed rearrangements of alcohols of the 6,14-endo-ethenotetrahydrothebaine series. J. Am. Chem. Soc. 1967, 89, 3293–3303. [Google Scholar] [CrossRef] [PubMed]
- Garadnay, S.; Marton, J.; Makleit, S. Acid-catalyzed isomerization of alpha-thevinone and alpha-dihydrothevinone into beta-thevinone and beta-dihydrothevinone. ACH-Models Chem. 2000, 137, 789–792. [Google Scholar]
- Derrick, I.; Coop, A.; Al-Mousawi, S.M.; Husbands, S.M.; Lewis, J.W. Perchloric acid induced epimerisation of the thevinones: An improved synthesis of 7beta-dihydrothevinones. Tetrahedron Lett. 2000, 41, 7571–7576. [Google Scholar] [CrossRef]
- Reisch, H.A. 7-beta-Substituted 6-alpha,14-alpha-Ethenomorphinans and 7-beta-Substituted 6-alpha,14-alpha-Ethanomorphinans. (Rhodes Technologies, Coventry, RI, US). U.S. Patent 9,834,562, 2017. [Google Scholar]
- Chen, W.; Parrish, D.; Deschamps, J.; Coop, A. Functionalization of the 6,14-bridge of the orvinols. 1. Preparation and Diels-Alder reaction of 7-phenylsilylthebaines. Helv. Chim. Acta 2005, 88, 822–829. [Google Scholar] [CrossRef]
- Wu, H.; Bernard, D.; Chen, W.; Strahan, G.D.; Deschamps, J.R.; Parrish, D.A.; Lewis, J.W.; MacKerell, A.D.; Coop, A. Functionalization of the 6,14-Bridge of the orvinols. 2. Preparation of 18- and 19-hydroxyl-substituted thevinols and their treatment with benzyl bromide. J. Org. Chem. 2005, 70, 1907–1910. [Google Scholar] [CrossRef]
- Wu, H.; Smith, T.A.; Huang, H.; Wang, J.B.; Deschamps, J.R.; Coop, A. Functionalization of the 6,14-bridge of the orvinols. Part 3: Preparation and pharmacological evaluation of 18- and 19-hydroxyl substituted orvinols. Bioorg. Med. Chem. Lett. 2007, 17, 4829–4831. [Google Scholar] [CrossRef] [Green Version]
- Uff, B.C.; Mallard, A.S.; Davis, J.A.; Henson, R. NMR Spectra and Stereochemistry of some 7-Substituted 6,14-Bridged Thebaine Derivatives. Magn. Reson. Chem. 1985, 23, 454–459. [Google Scholar] [CrossRef]
- Olofson, R.A.; Martz, J.T.; Senet, J.P.; Piteau, M.; Malfroot, T. A new reagent for the selective, high-yield N-dealkylation of tertiary amines: Improved syntheses of naltrexone and nalbuphine. J. Org. Chem. 1984, 49, 2081–2082. [Google Scholar] [CrossRef]
- Hosztafi, S. Reactions of azodicarboxylic esters with amines. Sci. Pharm. 1987, 55, 61–75. [Google Scholar]
- Kok, G.B.; Scammells, P.J. Further investigations into the N-demethylation of oripavine using iron and stainless steel. Org. Biomol. Chem. 2011, 9, 1008–1011. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Long, J.-D.; Qian, Y.-Y.; Long, Y.; Xu, X.-J.; Wang, Y.-J.; Shen, Q.; Wang, Z.-N.; Yang, X.-C.; Xiao, L.; et al. The pharmacological heterogeneity of nepenthone analogs in conferring highly selective and potent kappa-opioid agonistic activities. ACS Chem. Neurosci. 2017, 8, 766–776. [Google Scholar] [CrossRef]
- Kok, G.B.; Pye, C.C.; Singer, R.D.; Scammells, P.J. Two-Step Iron(0)-Mediated N-Demethylation of N-Methyl Alkaloids. J. Org. Chem. 2010, 75, 4806–4811. [Google Scholar] [CrossRef] [PubMed]
- Neumeyer, J.L.; Zhang, B.; Zhang, T.; Sromek, A.W.; Knapp, B.I.; Cohen, D.J.; Bidlack, J.M. Synthesis, binding affinity, and functional in vitro activity of 3-benzylaminomorphinan and 3-benzylaminomorphine ligands at opioid receptors. J. Med. Chem. 2012, 55, 3878–3890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decker, M.; Si, Y.-G.; Knapp, B.I.; Bidlack, J.M.; Neumeyer, J.L. Synthesis and opioid receptor binding affinities of 2-substituted and 3-aminomorphinans: Ligands for mu, kappa, and delta opioid receptors. J. Med. Chem. 2010, 53, 402–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kugita, H.; Takeda, M.; Inoue, H. Synthesis of B/C trans-fused morphine structures—III: Synthesis of B/C trans-morpine. Tetrahedron 1969, 25, 1851–1862. [Google Scholar] [CrossRef]
- Lawson, J.A.; DeGraw, J.I. An improved method for O-demethylation of codeine. J. Med. Chem. 1977, 20, 165–166. [Google Scholar] [CrossRef]
- Andre, J.-D.; Dormoy, J.-R.; Heymes, A. O-Demethylation of opioid derivatives with methane sulfonic acid/methionine: Application to the synthesis of naloxone and analogues. Synth. Commun. 1992, 22, 2313–2327. [Google Scholar] [CrossRef]
- Hilscher, J.C. Notiz zur Reduktion von 17α-Äthinylöstradiolen mit Aluminiumalkylen. Chem. Ber. 1976, 109, 1208–1210. [Google Scholar] [CrossRef]
- Coop, A.; Lewis, J.W.; Rice, K.C. Direct and simple O-demethylation of thebaine to oripavine. J. Org. Chem. 1996, 61, 6774. [Google Scholar] [CrossRef] [PubMed]
- Coop, A.; Janetka, J.W.; Lewis, J.W.; Rice, K.C. L-Selectride as a general reagent for the O-demethylation and N-decarbomethoxylation of opium alkaloids and derivatives. J. Org. Chem. 1998, 63, 4392–4396. [Google Scholar] [CrossRef]
- Bentley, K.W.; Hardy, D.G.; Meek, B.; Taylor, J.B.; Brown, J.J.; Morton, G.O. Novel analgesics and molecular rearrangements in the morphine-thebaine group. Part X. Further acid-catalysed rearrangements of alcohols in the 6,14-endo-ethenotetrahydrothebaine series. J. Chem. Soc. C 1969, 18, 2229–2232. [Google Scholar] [CrossRef] [PubMed]
- Hutchins, C.W.; Rapoport, H. Analgesics of the orvinol type. 19-Deoxy and 6,20-epoxy derivatives. J. Med. Chem. 1984, 27, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Coop, A.; Norton, C.L.; Berzetei-Gurske, I.; Burnside, J.; Toll, L.; Husbands, S.M.; Lewis, J.W. Structural determinants of opioid activity in the orvinols and related structures: Ethers of orvinol and isoorvinol. J. Med. Chem. 2000, 43, 1852–1857. [Google Scholar] [CrossRef] [PubMed]
- Marton, J.; Henriksen, G. Design and synthesis of an 18F-labeled version of phenylethyl orvinol ([18F]FE-PEO) for PET-imaging of opioid receptors. Molecules 2012, 17, 11554–11569. [Google Scholar] [CrossRef] [Green Version]
- Hutchins, C.W.; Cooper, G.K.; Puerro, S.; Rapoport, H. 6-Demethoxythebaine and its conversion to analgesics of the 6,14-ethenomorphinan type. J. Med. Chem. 1981, 24, 773–777. [Google Scholar] [CrossRef]
- Lessor, R.A.; Bajwa, B.S.; Rice, K.C.; Jacobson, A.E.; Streaty, R.A.; Klee, W.A.; Smith, C.B.; Aceto, M.D.; May, E.L.; Harris, L.S. Probes for narcotic receptor mediated phenomena. 13. Potential irreversible narcotic antagonist-based ligands derived from 6,14-endo-ethenotetrahydrooripavine with 7-(methoxyfumaroyl)amino, (bromoacetyl)amino, or isothiocyanate electrophiles: Chemistry, biochemistry, and pharmacology. J. Med. Chem. 1986, 29, 2136–2141. [Google Scholar]
- Rennison, D.; Neal, A.P.; Cami-Kobeci, G.; Aceto, M.D.; Martinez-Bermejo, F.; Lewis, J.W.; Husbands, S.M. Cinnamoyl derivatives of 7α-aminomethyl-6,14-endo-ethanotetrahydrothebaine and 7α-aminomethyl-6,14-endo-ethanotetrahydrooripavine and related opioid ligands. J. Med. Chem. 2007, 50, 5176–5182. [Google Scholar] [CrossRef] [Green Version]
- Machara, A.; Hudlicky, T. Advances in N-and O-demethylation of opiates. In Targets in Heterocyclic Systems, Chemistry and Properties; Attanasi, O.A., Merino, P., Spinelli, D., Eds.; Societa Chimica Italiana: Roma, Italy, 2016; Volume 20, pp. 113–138. [Google Scholar]
- Kopcho, J.J.; Schaeffer, J.C. Selective O-demethylation of 7.alpha.-(aminomethyl)6,14-endo-ethenotetrahydrothebaine. J. Org. Chem. 1986, 51, 1620–1622. [Google Scholar] [CrossRef]
- Lever, J.R.; Dannals, R.F.; Wilson, A.A.; Ravert, H.T.; Wagner, H.N., Jr. Synthesis of carbon-11 labeled diprenorphine: A radioligand for positron emission tomographic studies of opiate receptors. Tetrahedron Lett. 1987, 28, 4015–4018. [Google Scholar] [CrossRef]
- Lever, J.R.; Mazza, S.M.; Dannals, R.F.; Ravert, H.T.; Wilson, A.A.; Wagner, H.N., Jr. Facile synthesis of [11C]buprenorphine for positron emission tomographic studies of opioid receptors. Int. J. Radiat. Appl. Instrum. Part A Appl. Radiat. Isot. 1990, 41, 745–752. [Google Scholar] [CrossRef]
- Luthra, S.K.; Brady, F.; Turton, D.R.; Brown, D.J.; Dowsett, K.; Waters, S.L.; Jones, A.K.P.; Matthews, R.W.; Crowder, J.C. Automated radiosyntheses of [6-O-methyl-11C]diprenorphine and [6-O-methyl-11C]buprenorphine from 3-O-trityl protected precursors. Appl. Radiat. Isot. 1994, 45, 857–873. [Google Scholar] [CrossRef]
- Breeden, S.W.; Coop, A.; Husbands, S.M.; Lewis, J.W. 6-O-Demethylation of the thevinols with lithium aluminum hydride: Selective demethylation of a tertiary alkyl methyl ether in the presence of an aryl methyl ether. Helv. Chim. Acta 1999, 82, 1978–1980. [Google Scholar] [CrossRef]
- Shults, E.E.; Shakirov, M.M.; Tolstikov, G.A.; Kalinin, V.N.; Schmidhammer, H. Thebaine adducts with maleimides. Synthesis and transformations. Russ. J. Org. Chem. 2005, 41, 1132–1144. [Google Scholar] [CrossRef]
- Finke, A.O.; Ravaeva, M.Y.; Krasnov, V.I.; Cheretaev, I.V.; Chuyan, E.N.; Baev, D.S.; Shults, E.E. Cross-coupling-cyclocondensation reaction sequence to access a library of ring-C bridged pyrimidino-tetrahydrothebaines and pyrimidinotetrahydrooripavines. ChemistrySelect 2021, 6, 7391–7397. [Google Scholar] [CrossRef]
- Cami-Kobeci, G.; Polgar, W.E.; Khroyan, T.V.; Toll, L.; Husbands, S.M. Structural determinants of opioid and NOP receptor activity in aerivatives of buprenorphine. J. Med. Chem. 2011, 54, 6531–6537. [Google Scholar] [CrossRef] [Green Version]
- Szűcs, E.; Marton, J.; Szabó, Z.; Hosztafi, S.; Kékesi, G.; Tuboly, G.; Bánki, L.; Horváth, G.; Szabó, P.T.; Tömböly, C.; et al. Synthesis, biochemical, pharmacological characterization and in silico profile modelling of highly potent opioid orvinol and thevinol derivatives. Eur. J. Med. Chem. 2020, 191, 112145. [Google Scholar] [CrossRef]
- Kirby, G.W.; Sweeny, J.G. Nitrosocarbonyl compounds as intermediates in the oxidative cleavage of hydroxamic acids. J. Chem. Soc. Chem. Commun. 1973, 19, 704–705. [Google Scholar] [CrossRef]
- Kirby, G.W.; Sweeny, J.G. Formation and dienophilic reactions of transient C-nitrosocarbonyl compounds. J. Chem. Soc. Perkin Trans. 1 1981, 3250–3254. [Google Scholar] [CrossRef]
- Kirby, G.W.; Mackinnon, J.W.M.; Sharma, R.P. Reaction of alkoxycarbonylnitrenes with dimethyl sulphoxide; formation of transient nitrosoformates. Tetrahedron Lett. 1977, 18, 215–218. [Google Scholar] [CrossRef]
- Kirby, G.W.; Bentley, K.W.; Horsewood, P.; Singh, S. Cyclo-adducts of thebaine with nitrosoarenes. J. Chem. Soc. Perkin Trans. 1 1979, 3064–3066. [Google Scholar] [CrossRef]
- Schwab, L.S. 14-(Arylhydroxyamino)codeinones and derivatives as analgetics and antagonists. J. Med. Chem. 1980, 23, 698–702. [Google Scholar] [CrossRef] [PubMed]
- Corrie, J.E.T.; Kirby, G.W.; Mackinnon, J.W.M. Reactions of transient C-nitrosocarbonyl compounds with dienes, mono-olefins, and nucleophiles. J. Chem. Soc. Perkin Trans. 1 1985, 883–886. [Google Scholar] [CrossRef]
- Kirby, G.W.; McGuigan, H.; Mackinnon, J.W.M.; McLean, D.; Sharma, R.P. Formation and reactions of C-nitrosoformate esters a new class of transient dienophiles. J. Chem. Soc. Perkin Trans. 1 1985, 1437–1442. [Google Scholar] [CrossRef]
- Sheldrake, G.N.; Soissons, N. Selective opening of ring C in the morphine skeleton by an unexpected cleavage of the C5−C6 bond in cycloadducts of thebaine and acyl nitroso compounds. J. Org. Chem. 2006, 71, 789–791. [Google Scholar] [CrossRef]
- Li, F.; Yang, B.; Miller, M.J.; Zajicek, J.; Noll, B.C.; Möllmann, U.; Dahse, H.-M.; Miller, P.A. Iminonitroso Diels-Alder reactions for efficient derivatization and functionalization of complex diene-containing natural products. Org. Lett. 2007, 9, 2923–2926. [Google Scholar] [CrossRef]
- Kirby, G.W.; McLean, D. An efficient synthesis of 14-beta-aminocodeinone fro thebaine. J. Chem. Soc. Perkin Trans. 1 1985, 1442–1445. [Google Scholar]
- Révész, L.; Siegel, R.A.; Buescher, H.-H.; Marko, M.; Maurer, R.; Meigel, H. Design and synthesis of novel opiate antagonists with LH-stimulating properties. Helv. Chim. Acta 1990, 73, 326–336. [Google Scholar] [CrossRef]
- Kirby, G.W.; Sclare, A.D. Synthesis of 20-alkyl-8-thiathevinols, opiate agonists derived from 8-thiathevinone, the cycloadduct of thebaine and 2-oxopropanethial. J. Chem. Soc. Perkin Trans. 1 1991, 10, 2329–2338. [Google Scholar] [CrossRef]
- Jeong, I.H.; Kim, Y.S.; Cho, K.Y.; Kim, K.J. Unexpected effect of fluorine in Diels-Alder reaction of 2-fluoroacrolein with thebaine. Bull. Korean Chem. Soc. 1990, 11, 178–179. [Google Scholar]
- Jeong, I.H.; Kim, Y.S.; Cho, K.Y.; Kim, K.J. Hetero Diels-Alder reaction of thebaine with perfluoroaldehydes and chemical transformation of their adducts. Bull. Korean Chem. Soc. 1991, 12, 125–126. [Google Scholar] [CrossRef]
- Merz, H.; Pook, K.-H. Reaktionen des Thebains mit Azodicarbonsäureestern. Tetrahedron 1970, 26, 1727–1741. [Google Scholar] [CrossRef]
- Hosztafi, S.; Tímár, T.; Makleit, S. Morfin alkaloidok N-demetilezése III. Magy. Kém. Foly. 1985, 91, 126–130. [Google Scholar]
- Hromatka, O.; Sengstschmid, G. Diels-Alder-Reaktionen am Thebain mit cyclischen Azodienophilen. Monatsh. Chem. 1971, 102, 1022–1027. [Google Scholar] [CrossRef]
- Giger, R.; Rubinstein, R.; Ginsburg, D. Synthesis and reactions of the Diels-Alder adduct of thebaine with 4-phenyl-1,2,4-triazoline-3,5-dione. Tetrahedron 1973, 29, 2387–2391. [Google Scholar] [CrossRef]
- Makleit, S.; Radics, L.; Bognár, R.; Mile, T.; Oláh, É. Conversions of tosyl and mesyl derivatives of the morphine group, X. 14-Hydroxymorpine derivatives, I. Azido and amino derivatives. Acta Chim. Acad. Sci. Hung. 1972, 74, 99–113. [Google Scholar]
- Barton, D.H.R.; Lusinchi, X.; Ramírez, J.S. Improved synthesis of 1,2,4-triazoline-3,5-dione derivatives of ergosterol and a new method for their reconversion to ergosterol. Tetrahedron Lett. 1983, 24, 2995–2998. [Google Scholar] [CrossRef]
- Murphy, B.; Šnajdr, I.; Machara, A.; Endoma-Arias, M.A.A.; Stamatatos, T.C.; Cox, D.P.; Hudlický, T. Conversion of thebaine to oripavine and other useful intermediates for the semisynthesis of opiate-derived agents: Synthesis of hydromorphone. Adv. Synth. Catal. 2014, 356, 2679–2687. [Google Scholar] [CrossRef]
- Gutman, E.S.; Irvin, T.C.; Morgan, J.B.; Barrientos, R.C.; Torres, O.B.; Beck, Z.; Matyas, G.R.; Jacobson, A.E.; Rice, K.C. Synthesis and immunological effects of C14-linked 4, 5-epoxymorphinan analogues as novel heroin vaccine haptens. RSC Chem. Biol. 2021, 2, 835–842. [Google Scholar] [CrossRef]
- Ziaks, T.J.; Hwang, C.S. Is it possible to design a clinically viable heroin vaccine? The progress and pitfalls. Expert Opin. Drug Discov. 2022, 17, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Stowe, G.N.; Schlosburg, J.E.; Vendruscolo, L.F.; Edwards, S.; Misra, K.K.; Schulteis, G.; Zakhari, J.S.; Koob, G.F.; Janda, K.D. Developing a vaccine against multiple psychoactive targets: A case study of heroin. CNS Neurol. Disor. Drug Targets 2011, 10, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Lee, J.C.; Eubanks, L.M.; Ellis, B.; Zhou, B.; Janda, K.D. Improvements on a chemically contiguous hapten for a vaccine to address fentanyl-contaminated heroin. Bioorg. Med. Chem. 2021, 41, 116225. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, H.; Sheldrick, P. The Diels-Alder reaction with thebaine. Thermal rearrangement of some adducts from acetylenic dienophiles. J. Am. Chem. Soc. 1963, 85, 1636–1642. [Google Scholar] [CrossRef]
- Hayakawa, K.; Motohiro, S.; Fujii, I.; Kanematsu, K. Novel addition reaction of thebaine with acetylenic dienophiles: Construction of a new morphine skeleton. J. Am. Chem. Soc. 1981, 103, 4605–4606. [Google Scholar] [CrossRef]
- Singh, A.; Archer, S.; Hoogsteen, K.; Hirshfield, J. Thebaine and acetylenic dienophiles. J. Org. Chem. 1983, 48, 173–177. [Google Scholar] [CrossRef]
- Jeong, I.H.; Kim, Y.S.; Cho, K.Y.; Kim, K.J. Reaction of thebaine with trifluoromethyl substituted acetylenic dienophiles. Bull. Korean Chem. Soc. 1990, 11, 258–260. [Google Scholar]
- Sandulenko, I.V. Dual reactivity of the alkaloid thebaine and its derivatives towards acetylenes and the synthetic potential thereof. INEOS OPEN 2021, 4, 61–67. [Google Scholar] [CrossRef]
- Bentley, K.W.; Hardy, D.G.; Smith, A.C.B. Novel analgesics and molecular rearrangements in the morphine–thebaine group. Part XII. Derivatives of 7-amino-6,14-endo-etheno-tetrahydrothebaine. J. Chem. Soc. C 1969, 18, 2235–2236. [Google Scholar] [CrossRef]
- Burke, T.R.; Bajwa, B.S.; Jacobson, A.E.; Rice, K.C.; Streaty, R.A.; Klee, W.A. Probes for narcotic receptor mediated phenomena. 7. Synthesis and pharmacological properties of irreversible ligands specific for mu or delta opiate receptors. J. Med. Chem. 1984, 27, 1570–1574. [Google Scholar] [CrossRef]
- Klein, P.; Nelson, W.L.; Yao, Y.-H.; Simon, E.J. Electrophilic. alpha.-methylene-.gamma.-lactone and isothiocyanate opioid ligands related to etorphine. J. Med. Chem. 1990, 33, 2286–2296. [Google Scholar] [CrossRef] [PubMed]
- Dortch-Carnes, J.; Potter, D.E. Bremazocine: A kappa-opioid agonist with potent analgesic and other pharmacologic properties. CNS Drug Rev. 2005, 11, 95–212. [Google Scholar] [CrossRef] [PubMed]
- Berényi, S.; Gulyás, G.; Batta, G.; Makleit, S. Synthesis of a new azatetracyclodecane ring system via intramolecular addition of azide on double bond. Org. Prep. Proced. Int. 1991, 23, 111–113. [Google Scholar] [CrossRef]
- Berényi, S.; Gulyás, G.; Batta, G.; Gunda, T.; Makleit, S. Steric factors in the azidolysis-thermolysis of some 5-tosyloxy methylbicyclo [2.2.2]oct-2-enes to yield 4-azatetracyclo[4.4.0.0.0]decanes. J. Chem. Soc. Perkin Trans. 1 1991, 5, 1139–1142. [Google Scholar] [CrossRef]
- Sepsi, Á.; Berényi, S.; Makleit, S.; Tóth, Z. Morphine Alkaloids, CXVII: Investigation of the Azidolysis of Tertiary Alcohols of Thebaine Derivatives with Bridged Ring C. Arch. Pharm. 1993, 326, 313–317. [Google Scholar] [CrossRef]
- Derrick, I.; Husbands, S.; Broadbear, J.; Traynor, J.; Woods, J.; Lewis, J. Cinnamoyl derivatives of 7α-amino- and 7α-(aminomethyl)-N-(cyclopropylmethyl)-6,14-endo-ethanotetrahydronororipavines are high-potency opioid antagonists. Helv. Chim. Acta 2000, 83, 3122–3130. [Google Scholar] [CrossRef]
- Fujii, H.; Narita, M.; Mizoguchi, H.; Murachi, M.; Tanaka, T.; Kawai, K.; Tseng, L.F.; Nagase, H. Drug design and synthesis of ε opioid receptor agonist: 17-(cyclopropylmethyl)-4,5α-epoxy-3,6β-dihydroxy-6,14-endoethenomorphinan-7α-(N-methyl-N-phenethyl)carboxamide (TAN-821) inducing antinociception mediated by putative ε opioid receptor. Bioorg. Med. Chem. 2004, 12, 4133–4145. [Google Scholar] [CrossRef]
- Portoghese, P.S. Bivalent ligands and the message-address concept in the design of selective opioid receptor antagonists. Trends Pharm. Sci. 1989, 10, 230–235. [Google Scholar] [CrossRef]
- Portoghese, P.S.; Sultana, M.; Takemori, A.E. Design of peptidomimetic delta-opioid receptor antagonists using the message address concept. J. Med. Chem. 1990, 33, 1714–1720. [Google Scholar] [CrossRef]
- Narita, M.; Tseng, L.F. Evidence for the existence of the β-endorphin-sensitive “ε-opioid receptor” in the brain: The mechanisms of ε-mediated antinociception. Jpn. J. Pharmacol. 1998, 76, 233–253. [Google Scholar] [CrossRef] [Green Version]
- Nock, B.; Giordano, A.L.; Moore, B.W.; Cicero, T.J. Properties of the putative epsilon opioid receptor: Identification in rat, guinea pig, cow, pig and chicken brain. J. Pharmacol. Exp. Ther. 1993, 264, 349–359. [Google Scholar] [PubMed]
- Contet, C.; Matifas, A.; Kieffer, B.L. No evidence for G-protein-coupled epsilon receptor in the brain of triple opioid receptor knockout mouse. Eur. J. Pharmacol. 2004, 492, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Narita, M.; Mizoguchi, H.; Hirokawa, J.; Kawai, K.; Tanaka, T.; Tseng, L.F.; Nagase, H. Rational drug design and synthesis of a selective ε opioid receptor antagonist on the basis of the accessory site concept. Bioorg. Med. Chem. Lett. 2004, 14, 4241–4243. [Google Scholar] [CrossRef] [PubMed]
- Nógrády, T. Medicinal Chemistry, A Biochemical Approach; Oxford University Press: New York, NY, USA, 1985; pp. 68–69. [Google Scholar]
- Fujii, H.; Nagase, H. Rational drug design of selective epsilon opioid receptor agonist TAN-821 and antagonist TAN-1014. Curr. Med. Chem. 2006, 13, 1109–1118. [Google Scholar] [CrossRef]
- Moynihan, H.A.; Derrick, I.; Broadbear, J.H.; Greedy, B.M.; Aceto, M.D.; Harris, L.S.; Purington, L.C.S.; Thomas, M.P.; Woods, J.H.; Traynor, J.R.; et al. Fumaroylamino-4,5-epoxymorphinans and related opioids with irreversible μ opioid receptor antagonist effects. J. Med. Chem. 2012, 55, 9868–9874. [Google Scholar] [CrossRef] [PubMed]
- Yavuz, S.; Yıldırır, Y. Synthesis and characterization of new 7-substituted 6,14-ethenomorphinane derivatives: N-{5-[(5α,7α)-4,5-epoxy-3,6-dimethoxy-17-methyl-6,14-ethenomorphinan-7-yl]-1,3,4-oxadiazol-2-yl}arenamines. Helv. Chim. Acta 2010, 93, 2406–2418. [Google Scholar] [CrossRef]
- Yildirir, Y.; Pamir, Ö.; Yavuz, S.; Dişli, A. Synthesis and characterization of new thebaine derivatives as potential opioid agonists and antagonists: 2-[N-(1H-tetrazol-5-yl)-6,14-endo-etheno-6,7,8,14-tetrahydrothebaine-7α-yl]-5-phenyl-1,3,4-oxadiazoles. J. Heterocyclic Chem. 2013, 50, E93–E99. [Google Scholar] [CrossRef]
- Yavuz, S.; Ünal, Y.; Pamir, Ö.; Yilmazer, D.; Kurtipek, Ö.; Kavutçu, M.; Arslan, M.; Ark, M.; Yildirir, Y. Synthesis and pharmacological evaluation of some novel thebaine derivatives: N-(tetrazol-1H-5-yl)-6,14-endoethenotetrahydrothebaine incorporating the 1,3,4-oxadiazole or the 1,3,4-thiadiazole moiety. Arch. Pharm. Pharm. Med. Chem. 2013, 346, 455–462. [Google Scholar] [CrossRef]
- Nagase, H.; Yamamoto, N. Morphinan Derivative. U.S. Patent 9,963,460, 8 May 2018. [Google Scholar]
- Lewis, J.W.; Readhead, M.J. Novel analgesics and molecular rearrangements in the morphine–thebaine group. Part XX. Reaction of 7β-cyano-6,14-endo-etheno-7α-methyl-6,7,8,14-tetrahydrothebaine with phenylmagnesium bromide. J. Chem. Soc. C 1971, 1161–1163. [Google Scholar] [CrossRef]
- Lewis, J.W.; Readhead, M.J.; Smith, A.C.B. Novel analgesics and molecular rearrangements in the morphine-thebaine group. XXVIII. Derivatives of 6,14-endo-etheno-7-Oxo-6,7,8,14-tetrahydrothebaine and 6,14-endo-etheno-6,7,8,14-tetrahydrothebaine. J. Med. Chem. 1973, 16, 9–12. [Google Scholar] [CrossRef]
- Bentley, K.W.; Bower, J.D.; Lewis, J.W. Novel analgesics and molecular rearrangements in the morphine–thebaine group. Part XVI. Some derivatives of 6,14-endo-etheno-7,8-dihydromorphine. J. Chem. Soc. C 1969, 19, 2569–2572. [Google Scholar] [CrossRef] [PubMed]
- Krüll, J.; Fehler, S.K.; Hofmann, L.; Nebel, N.; Maschauer, S.; Prante, O.; Gmeiner, P.; Lanig, H.; Hübner, H.; Heinrich, M.R. Synthesis, radiosynthesis and biological evaluation of buprenorphine-derived phenylazocarboxamides as novel mu-opioid receptor ligands. ChemMedChem 2020, 15, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Bentley, K.W.; Ball, J.C. Flavonepenthone. Chem. Ind. 1956, 47, 1428. [Google Scholar]
- Lewis, J.W.; Readhead, M.J.; Selby, I.A.; Smith, A.C.B.; Young, C.A. Novel analgesics and molecular rearrangements in the morphine–thebaine group. Part XIX. Further Diels–Alder adducts of thebaine. J. Chem. Soc. C 1971, 1158–1161. [Google Scholar] [CrossRef]
- Lewis, J.W.; Readhead, M.J. Novel analgesics and molecular rearrangements in the morphine–thebaine group. Part XXI. Alcohols derived from 7-methyl-epi-nepenthone. J. Chem. Soc. C 1971, 2296–2298. [Google Scholar] [CrossRef]
- Husbands, S.M.; Lewis, J.W. Morphinan cyclic imines and pyrrolidines containing a constrained phenyl group: High affinity opioid agonists. Bioorg. Med. Chem. Lett. 1995, 5, 2969–2974. [Google Scholar] [CrossRef]
- Childers, S.R.; Xiao, R.; Vogt, L.; Sim, L.J. Lack of evidence of kappa2-selective activation of G-proteins: Kappa opioid receptor stimulation of [35S] GTPgammaS binding in guinea pig brain. Biochem. Pharmacol. 1998, 56, 113–120. [Google Scholar] [CrossRef]
- Marton, J.; Szabó, Z.; Simon, C.; Hosztafi, S.; Makleit, S. Synthesis of new nepenthone derivatives. Liebigs Ann. Chem. 1996, 1653–1656. [Google Scholar] [CrossRef]
- Micskei, K.; Gyarmati, J.; Kovács, G.; Makleit, S.; Simon, C.; Szabó, Z.; Marton, J.; Hosztafi, S.; Reinke, H.; Drexler, H.-J. Reactions of nepenthone with chromium(II) reagents in neutral aqueous medium. Eur. J. Org. Chem. 1999, 1, 149–153. [Google Scholar] [CrossRef]
- Bermejo, F.M.; Husbands, S.M.; Lewis, J.W. Derivatives of flavonepenthone: Kappa opioid receptor selectivity in an N-methylmorphinan. Helv. Chim. Acta 1999, 82, 1721–1727. [Google Scholar] [CrossRef]
- MacDonald, R.; Heiner, J.; Villarreal, J.; Strote, J. Loperamide dependence and abuse. BMJ Case Rep. 2015, 2015, bcr2015209705. [Google Scholar] [CrossRef] [PubMed]
- Kamendulis, L.M.; Brzezinski, M.R.; Pindel, E.V.; Bosron, W.F.; Dean, R.A. Metabolism of cocaine and heroin is catalyzed by the same human liver carboxylesterases. J. Pharmacol. Exp. Ther. 1996, 279, 713–717. [Google Scholar] [PubMed]
- Qiao, Y.; Han, K.; Zhan, C.G. Fundamental reaction pathway and free energy profile for butyrylcholinesterase-catalyzed hydrolysis of heroin. Biochemistry 2013, 52, 6467–6479. [Google Scholar] [CrossRef] [Green Version]
- Ray, S.B.; Gupta, H.; Gupta, Y.K. Up-regulation of mu-opioid receptors in the spinal cord of morphine-tolerant rats. J. Biosci. 2004, 29, 51–56. [Google Scholar] [PubMed]
- Shichinohe, S.; Ozawa, H.; Saito, T.; Hashimoto, E.; Lang, C.; Riederer, P.; Takahata, N. Differential alteration of adenylyl cyclase subtypes I, II, and V/VI in postmortem human brains of heroin addicts. Alcohol. Clin. Exp. Res. 1998, 22 Pt 1, 84S–87S. [Google Scholar] [CrossRef] [PubMed]
- Manu, P.; Rogozea, L.M.; Shulman, M. Pharmacological management of heroin withdrawal syndrome: A century of expert opinions in cecil textbook of medicine. Am. J. Ther. 2022, 29, e193–e198. [Google Scholar] [CrossRef] [PubMed]
- Gerra, G.; Fantoma, A.; Zaimovic, A. Naltrexone and buprenorphine combination in the treatment of opioid dependence. J. Psychopharmacol. 2006, 20, 806–814. [Google Scholar] [CrossRef]
- Sandulenko, I.V.; Kovaleva, E.S.; Peregudov, A.S.; Kalinin, V.N.; Moiseev, S.K. 21,21,21-Trifluorothevinone: The straightest way to fluorinated thevinols and orvinols. ChemistrySelect 2016, 1, 1004–1005. [Google Scholar] [CrossRef]
- Eremin, D.B.; Kadentsev, V.I.; Sandulenko, I.V.; Moiseev, S.K. Tandem high-resolution electrospray ionization mass spectrometry of fluorinated thevinols and 18,19-dihydrothevinols. J. Anal. Chem. 2015, 70, 1561–1568. [Google Scholar] [CrossRef]
- Zelentsova, M.V.; Sandulenko, I.V.; Melnikova, E.K.; Moiseev, S.K. 19F NMR determination of the C20 absolute configuration of C21-fluorinated arylthevinols. Mendeleev Commun. 2022, 32, 97–99. [Google Scholar] [CrossRef]
- Sun, H.J.; Wang, Y.-H.; Yuan, C.-M.; Kong, L.-H.; Xu, X.-J.; Wang, Y.-J.; Wu, H.-H.; Lin, C.; Qian, Y.-Y.; Huang, H.-M.; et al. 7beta-Methyl substituent is a structural locus associated with activity cliff for nepenthone analogues. Bioorg. Med. Chem. 2018, 26, 4254–4263. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Wang, Y.; Zhang, M.; Wu, W.; Kong, L.; Ma, Y.; Xu, X.; Liu, X.; He, Q.; Qian, Y.; et al. Discovery of a highly selective and potent kappa-opioid receptor agonist from N-cyclopropylmethyl- 7alpha-phenyl-6,14-endoethanotetrahydronorthebaines with reduced central nervous system (CNS) side effects navigated by the message-address concept. J. Med. Chem. 2019, 62, 11054–11070. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, S.; Kong, L.; Ye, R.; Xiao, L.; Xu, X.; He, Q.; Wei, Y.; Li, Z.; Sun, H.; et al. Exploration of the SAR connection between morphinan- and arylacetamide-based kappa-opioid receptor (KOR) agonists using the strategy of bridging. ACS Chem. Neurosci. 2021, 12, 1018–1030. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Wei, Y.; Liu, X.; Ye, R.; Kong, L.; Li, Z.; Jiang, S.; Yu, L.; Chai, J.; Xie, Q.; et al. Discovery of an m-substituted N-cyclopropylmethyl-7α-phenyl-6,14-endoethanotetrahydronorthebaine as a selective, potent, and orally active kappa-opioid receptor agonist with an improved central nervous system safety profile. J. Med. Chem. 2021, 64, 12414–12433. [Google Scholar] [CrossRef] [PubMed]
- Bauman, V.T.; Shults, E.E.; Shakirov, M.M.; Tolstikov, G.A. Synthetic transformations of isoquinoline alkaloids. Synthesis of N′-substituted 1-alkynyl-7α, 8α-(2, 5-dioxopyrrolidino)-[3, 4-h]-6, 14-endo-ethenotetrahydrothebaines and their transformations. Russian J. Org. Chem. 2012, 48, 1473–1483. [Google Scholar] [CrossRef]
- Bauman, V.T.; Shults, E.E.; Shakirov, M.M.; Tolstikov, G.A. Synthetic Transformations of Isoquinoline Alkaloids. Catalytic Alkinylation of 6, 14-endo-Ethenotetrahydrothebaine and 6, 14-endo-Ethenodihydrothebaine Hydroquinone Derivatives. Chem. Sustain. Dev. 2007, 5, 529–534. [Google Scholar]
- Valenzano, K.J.; Miller, W.; Chen, Z.; Shan, S.; Crumley, G.; Victory, S.F.; Davies, E.; Huang, J.-C.; Allie, N.; Nolan, S.J.; et al. DiPOA ([8-(3,3-Diphenyl-propyl)-4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]dec-3-yl]-acetic acid), a novel, systemically available, and peripherally restricted mu-opioid agonist with antihyperalgesic activity: I. In vitro pharmacological characterization and pharmacokinetic properties. J. Pharmacol. Exp. Ther. 2004, 310, 783–792. [Google Scholar]
- Raynor, K.; Kong, H.; Chen, Y.; Yasuda, K.; Yu, L.; Bell, G.I.; Reisine, T. Pharmacological characterization of the cloned kappa-, delta-, and mu-opioid receptors. Mol. Pharmacol. 1994, 45, 330–334. [Google Scholar]
- Aceto, M.D.; Harris, L.S.; May, E.L. Dependence studies of new compounds in the rhesus monkey, rat and mouse. 1983. NIDA Res. Monogr. 1984, 49, 368–420. [Google Scholar]
- Schoultz, B.W.; Hjørnevik, T.; Reed, B.J.; Marton, J.; Coello, C.S.; Willoch, F.; Henriksen, G. Synthesis and evaluation of three structurally related 18F-labeled orvinols of different intrinsic activities: 6-O-[18F]Fluoroethyl-diprenorphine ([18F]FDPN), 6-O-[18F]fluoroethyl-buprenorphine ([18F]FBPN), and 6-O-[18F]fluoroethyl-phenethyl-orvinol ([18F]FPEO). J. Med. Chem. 2014, 57, 5464–5469. [Google Scholar]
- Maat, L.; Woudenberg, R.H.; Meuzelaar, G.J.; Linders, J.T.M. Chemistry of opium alkaloids. Part 44: Synthesis and opioid receptor binding profile of substituted ethenoisomorphinans and ethenomorphinans. Bioorg. Med. Chem. 1999, 7, 529–541. [Google Scholar] [CrossRef]
- Katsumata, S.; Minami, M.; Nakagawa, T.; Iwamura, T.; Satoh, M. Pharmacological study of dihydoetorphine in cloned mu- delta- and kappa opioid-receptors. Eur. J. Pharm. Mol. Pharm. Sect. 1995, 291, 367–373. [Google Scholar] [CrossRef]
- Smith, C.F.C. 16-Methyl-cyprenorphine (RX 8008M): A potent opioid antagonist with some delta selectivity. Life Sci. 1987, 40, 267–274. [Google Scholar] [CrossRef]
- Li, J.-X.; Becker, G.L.; Traynor, J.R.; Gong, Z.-H.; France, C.P. Thienorphine: Receptor binding and behavioral effects in rhesus monkeys. J. Pharm. Exp. Ther. 2007, 321, 227–236. [Google Scholar] [CrossRef]
- Lewis, J.; Husbands, S. Orvinol and Thevinol Derivatives Useful in the Treatment of Drug and Alcohol Abuse. U.S. Patent WO 2013/0007986 A1, 17 January 2013. [Google Scholar]
- Dannals, R.F.; Ravert, H.T.; Frost, J.J.; Wilson, A.A.; Burns, H.D.; Wagner, H.N. Radiosynthesis of an opiate receptor binding radiotracer: [11C]carfentanil. Int. J. Appl. Radiat. Isot. 1985, 36, 303–306. [Google Scholar] [CrossRef]
- Lever, J.R.; Kinter, C.M.; Ravert, H.T.; Musachio, J.L.; Mathews, W.B.; Dannals, R.F. Synthesis of N1′-([11C]methyl)naltrindole ([11C]MeNTI): A radioligand for positron emission tomographic studies of delta opioid receptors. J. Label. Compd. Radiopharm. 1995, 36, 137–145. [Google Scholar] [CrossRef]
- Carson, R.E.; Channing, M.A.; Blasberg, R.G.; Dunn, B.B.; Cohen, R.M.; Rice, K.C.; Herscovitch, P. Comparison of bolus and infusion methods for receptor quantitation: Application to [18F] cyclofoxy and positron emission tomography. J. Cereb. Blood Flow Metab. 1993, 13, 24–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talbot, P.S.; Narendran, R.; Butelman, E.R.; Huang, Y.; Ngo, K.; Slifstein, M.; Martinez, D.; Laruelle, M.; Hwang, D.-R. 11C-GR103545, a radiotracer for imaging kappa-opioid receptors in vivo with PET: Synthesis and evaluation in baboons. J. Nucl. Med. 2005, 46, 484–494. [Google Scholar]
- Hammers, A.; Lingford-Hughes, A. Opioid imaging. Neuroimag. Clin. N. Am. 2006, 16, 529–552. [Google Scholar] [CrossRef]
- Filer, C.N. Morphinan alkaloids labeled with tritium: Synthesis and applications. J. Label. Compd. Radiopharm. 2013, 56, 639–648. [Google Scholar] [CrossRef]
- Tóth, G.; Mallareddy, J.R. Tritiated opioid receptor ligands as radiotracers. Curr. Pharm. Design 2013, 19, 7461–7472. [Google Scholar] [CrossRef] [PubMed]
- van Waarde, A.; Absalom, A.R.; Visser, A.K.D.; Dierckx, R.A.J.O. Positron emission tomography (PET) imaging of opioid receptors. In PET and SPECT of Neurobiological Systems; Dierckx, R.A.J.O., Otte, A., de Vries, E.F.J., van Waarde, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 585–623. [Google Scholar]
- Haddlesey, D.I.; Lewis, J.W.; Mayor, P.A.; Young, G.R. Novel analgesics and molecular rearrangements in the morphine-thebaine group. Part XXIV. 15,16-Didehydro-6,14-endo-etheno-6,7,8,14-tetrahydro-thebaines and -oripavines. J. Chem. Soc. Perkin Trans. 1 1972, 872–874. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.W.; Rance, M.J.; Young, G.R. A procedure for preparing 3H-labeled tertiary amines. Synthesis of [3H]-6,14-endo-etheno-6,7,8,14-tetrahydrooripavine derivatives. J. Med. Chem. 1974, 17, 465–466. [Google Scholar] [CrossRef] [PubMed]
- Pert, C.B.; Kuhar, M.J.; Snyder, S.H. Opiate receptor: Autoradiographic localization in rat brain. Proc. Natl. Acad. Sci. USA 1976, 73, 3729–3733. [Google Scholar] [CrossRef] [Green Version]
- Herkenham, M.; Pert, C.B. Neurobiology in vitro autoradiography of opiate receptors in rat brain suggests loci of “opiatergic” pathways. Proc. Natl. Acad. Sci. USA 1980, 77, 5532–5536. [Google Scholar] [CrossRef] [Green Version]
- Ötvös, F.; Hosztafi, S.; Simon, C.; Tóth, G. Synthesis of high specific activity [15,16-3H2]buprenorphine. J. Labell. Compds. Radiopharm. 1995, 36, 79–83. [Google Scholar] [CrossRef]
- Debrabandere, L.; van Boven, M.; Daenens, P. Synthesis of iodobuprenorphine for use in radioimmunoassay. J. Labell. Compds. Radioharm. 1992, 31, 575–588. [Google Scholar] [CrossRef]
- Tafani, J.-A.M.; Francés, B.; Coulais, Y.; Méjan-Galzi, A.; Goeldner, M.; Hirth, C.; Guiraud, R.; Zajac, J.-M. In vivo binding of [125I]NH2-carfentanil to mu opioid receptors in mouse brain. Nucl. Med. Biol. 1994, 21, 231–238. [Google Scholar] [CrossRef]
- Musachio, J.L.; Lever, J.R. Vinylstannylated alkylating agents as prosthetic groups for radioiodination of small molecules: Design, synthesis and application to iodoallyl analogues of spiperone and diprenorphine. Bioconjugate Chem. 1992, 3, 167–175. [Google Scholar] [CrossRef]
- Lever, J.R.; Ilgin, N.; Musachio, J.L.; Scheffel, U.; Finley, P.A.; Flesher, J.E.; Natarajan, T.K.; Wagner, H.N., Jr.; Frost, J.J. Autoradiographic and SPECT imaging of cerebral opioid receptors with an iodine-123 labeled analogue of diprenorphine. Synapse 1998, 29, 172–182. [Google Scholar] [CrossRef]
- Wang, R.F.; Tafani, J.-A.M.; Bergon, M.; Tisnes, P.; Coulais, Y.; Zajac, J.M.; Guiraud, R. Synthesis and characterization of 7alpha-O-iodoallyl diprenorphine: A new ligand for potential SPECT imaging of opioid receptors. J. Labell. Compds. Radiopharm. 1995, 36, 611–623. [Google Scholar] [CrossRef]
- Wang, R.F.; Tafani, J.-A.M.; Francés, B.; Bergon, M.; Coulais, Y.; Zajac, J.M.; Guiraud, R. Evaluation of [125I]7-alpha-O-iodoallyl diprenorphine as a new potential SPECT opioid receptor imaging agent. Nucl. Med. Biol. 1997, 24, 553–558. [Google Scholar] [CrossRef]
- Maziere, M.; Godot, J.M.; Berger, G.; Prenant, C.; Comar, D. 11C-Labelled etorphine for in vivo studies of opiate receptors in brain. J. Radioanal. Chem. 1981, 62, 279–284. [Google Scholar] [CrossRef]
- Burns, H.D.; Lever, J.R.; Dannals, R.F.; Frost, J.J.; Wilson, A.A.; Ravert, H.T.; Subramanian, B.; Zeyman, S.E.; Langstrom, B.; Wagner, H.N., Jr. Synthesis of ligands for imaging opiate receptors by positron emission tomography: Carbon-11 labelled diprenorphine. J. Labell. Compds. Radiopharm. 1984, 22, 1167–1169. [Google Scholar]
- Luthra, S.K.; Pike, V.W.; Brady, F. The preparation of carbon-11 labelled diprenorphine: A new radioligand for the study of the opiate receptor system in vivo. J. Chem. Soc. Chem. Commun. 1985, 1423–1425. [Google Scholar] [CrossRef]
- Luthra, S.K.; Pike, V.W.; Brady, F.; Horlock, P.L.; Prenant, C.; Crouzel, C. Preparation of [11C]buprenorphine—A potential radioligand for the study of the opiate receptor system in vivo. Int. J. Radiat. Appl. Instrum. Part A Appl. Radiat. Isot. 1987, 38, 65–66. [Google Scholar] [CrossRef]
- Shiue, C.-Y.; Bai, L.-Q.; Teng, R.-R.; Arnett, C.D.; Dewey, S.L.; Wolf, A.P.; McPherson, D.W.; Fowler, J.S.; Logan, J.; Holland, M.J.; et al. A Comparison of the Brain Uptake of N-(Cyclopropyl[11C]methyl)-norbuprenorphine ([11C]buprenorphine) and N-(cyclopropyl[11C]methyl)-nordiprenorphine ([11C]diprenorphine) in baboon using PET. Int. J. Radiat. Appl. Instrum. Part B Nucl. Med. Biol. 1991, 18, 281–288. [Google Scholar] [CrossRef]
- Galynker, I.; Schlyer, D.J.; Dewey, S.L.; Fowler, J.S.; Logan, J.; Gatley, S.J.; MacGregor, R.R.; Ferrieri, R.A.; Holland, M.J.; Brodie, J.; et al. Opioid receptor imaging and displacement studies with [6-O-[11C]methyl]buprenorphine in baboon brain. Nucl. Med. Biol. 1996, 23, 325–331. [Google Scholar] [CrossRef]
- Koepp, M.J.; Richardson, M.P.; Brooks, D.J.; Duncan, J.S. Focal cortical release of endogenous opioids during reading-induced seizures. Lancet 1998, 352, 952–955. [Google Scholar] [CrossRef]
- Fairclough, M.; Prenant, C.; Brown, G.; McMahon, A.; Lowe, J.; Jones, A. The automated radiosynthesis and purification of the opioid receptor antagonist, [6-O-methyl-11C]diprenorphine on the GE TRACERlab FXFE radiochemistry module. J. Label. Compd. Radiopharm. 2014, 57, 388–396. [Google Scholar] [CrossRef]
- Marton, J.; Wester, H.J.; Willoch, F.; Henriksen, G. PET-Imaging of the Opioid Receptors with Labeled PEO Derivatives. In Proceedings of the NRC-Seventh International Conference on Nuclear and Radiochemistry, Budapest, Hungary, 24–29 August 2008; Hungarian Chemical Society: Budapest, Hungary, 2008; p. 241. [Google Scholar]
- Wester, H.-J.; Willoch, F.; Tölle, T.R.; Munz, F.; Herz, M.; Øye, I.; Schadrack, J.; Schwaiger, M.; Bartenstein, P. 6-O-(2-[18F]Fluoroethyl-6-O-desmethyldiprenorphine ([18F]DPN): Synthesis, biologic evaluation, and comparison with [11C]DPN in humans. J. Nucl. Med. 2000, 41, 1279–1286. [Google Scholar] [PubMed]
- Chesis, P.L.; Hwang, D.-R.; Welch, M.J. N-(3-[18F]Fluoropropyl)-N-nordiprenorphine: Synthesis and characterization of a new agent for imaging opioid receptors with positron emission tomography. J. Med. Chem. 1990, 33, 1482–1490. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.-Q.; Teng, R.-R.; Shiue, C.-Y.; Wolf, A.P.; Dewey, S.L.; Holland, M.J.; Simon, E.J. No-carrier-added (NCA) N-(3-[18F]fluoropropyl-N-norbuprenorphine and N-(3-[18F]fluoropropyl-N-nordiprenorphine—Synthesis distribution in mice and rats, and tomographic studies in baboon. Int. J. Radiat. Appl. Instr. Part B Nucl. Med. Chem. 1990, 17, 217–227. [Google Scholar] [CrossRef]
- Spilker, M.E.; Sprenger, T.; Valet, M.; Henriksen, G.; Wagner, K.; Wester, H.-J.; Toelle, T.R.; Boecker, H. Quantification of [18F]diprenorphine kinetics in the human brain with compartmental and non-compartmental modeling approaches. NeuroImage 2004, 22, 1523–1533. [Google Scholar] [CrossRef]
- Boecker, H.; Sprenger, T.; Henriksen, G.; Toelle, T.R.; Spilker, M.E. Optimal duration of PET studies with 18F-fluoroethyl-diprenorphine. J. Nucl. Med. 2005, 46, 2092–2096. [Google Scholar]
- Sprenger, T.; Valet, M.; Boecker, H.; Henriksen, G.; Spilker, M.E.; Willoch, F.; Wagner, K.J.; Wester, H.-J.; Toelle, T. R Opioidergic activation in the medial pain system after heat pain. Pain 2006, 122, 63–67. [Google Scholar] [CrossRef]
- Riss, P.J.; Hong, Y.T.; Marton, J.; Caprioli, D.; Williamson, D.J.; Ferrari, V.; Saigal, N.; Roth, B.L.; Henriksen, G.; Fryer, T.D.; et al. Synthesis and evaluation of 18F-FE-PEO in rodents: An 18F-labeled full agonist for opioid receptor imaging. J. Nucl. Med. 2013, 54, 299–305. [Google Scholar] [CrossRef] [Green Version]
- Schoultz, B.W.; Reed, B.J.; Marton, J.; Willoch, F.; Henriksen, G. A fully automated radiosynthesis of [18F]fluoroethyl-diprenorphine on a single module by use of SPE cartridges for preparation of high quality 2-[18F]fluoroethyl tosylate. Molecules 2013, 18, 7271–7278. [Google Scholar] [CrossRef] [Green Version]
- Willoch, F.; Hjornevik, T.; Lindbom, I.; Reed, B.J.; Coello, C.S. Common reference tissue kinetic model for comparison of three opioid receptor tracers in the rat brain. In Proceedings of the Ninth International Symposium on Functional Neuroreceptor Mapping of the Living Brain (NRM2012), Baltimore, MD, USA, 9–11 August 2012; pp. S155–S156. [Google Scholar]
- Sandulenko, I.V.; Ambartsumyan, A.A.; Moiseev, S.K. Fluorinated and [18F]fluorinated morphinan based opioid ligands. Org. Biomol. Chem. 2020, 18, 5533–5557. [Google Scholar] [CrossRef]
- Soyka, M. Transition from full mu opioid agonists to buprenorphine in opioid dependent patients—A critical review. Front. Pharmacol. 2021, 12, 2901. [Google Scholar] [CrossRef]
- Krenz, J.R.; Hayes, B.D.; Wakeman, S.E.; Martin, A.; Raja, A.S.; White, B.A.; Koehl, J.L. Continuation of outpatient buprenorphine therapy after dispensing Buprenorphine-Naloxone from the emergency department. Clin. Toxicol. 2022, 60, 429–432. [Google Scholar] [CrossRef] [PubMed]
- Webster, L.; Gudin, J.; Raffa, R.B.; Kuchera, J.; Rauck, R.; Fudin, J.; Adler, J.; Mallick-Searle, T. Understanding buprenorphine for use in chronic pain: Expert opinion. Pain Med. 2020, 21, 714–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, R.; Nash, M.; Peperzak, K.A.; Ziga, T.M.; Dinges, E.M.; Delgado, C.; Wu, J.; Terman, G.W.; Dale, R.C. Comparison between preoperative methadone and buprenorphine use on postoperative opioid requirement: A retrospective cohort study. Clin. J. Pain 2022, 38, 311–319. [Google Scholar] [CrossRef] [PubMed]





















































































































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
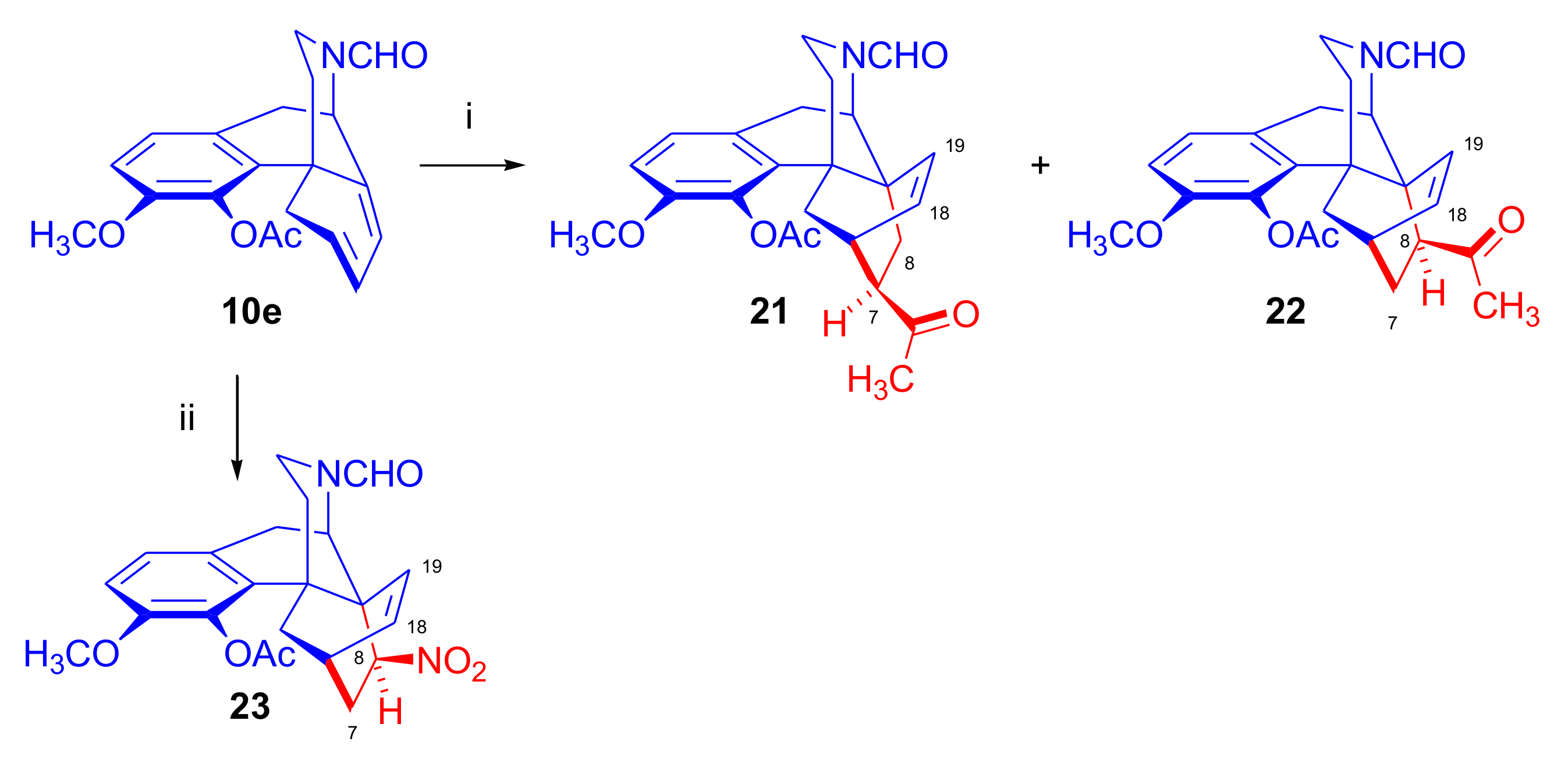{kind=link}
{kind=link}
{kind=link}
{kind=link}
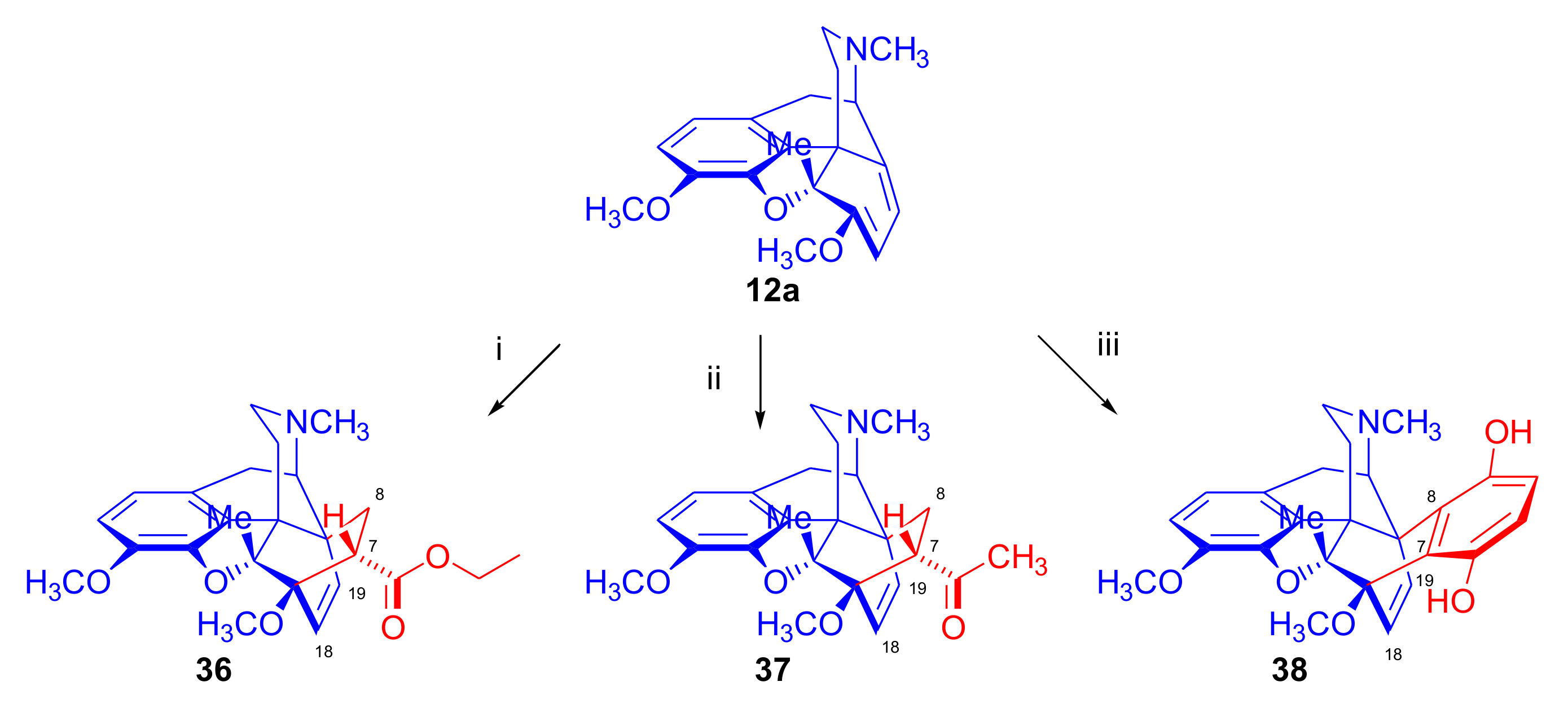{kind=link}
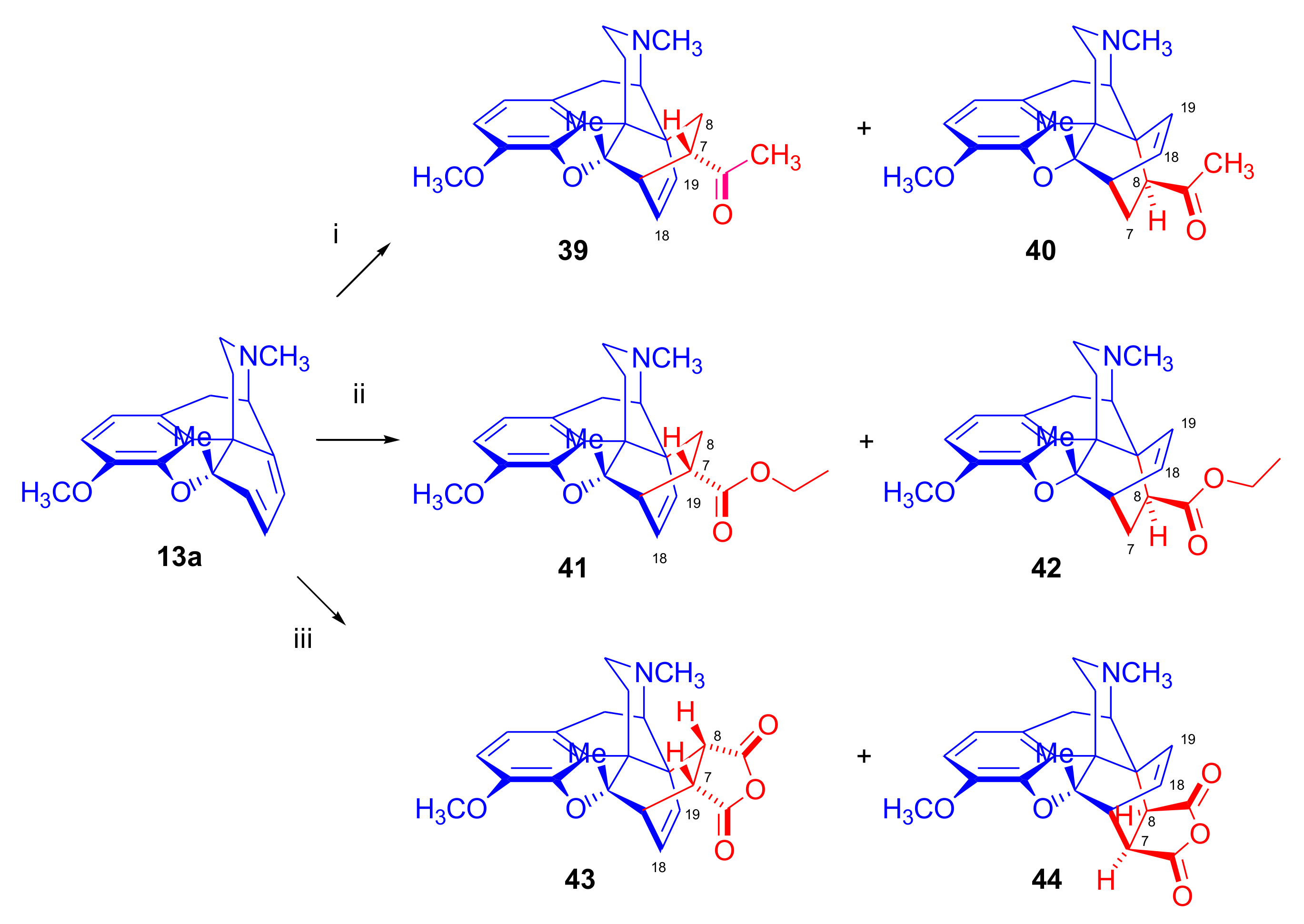{kind=link}
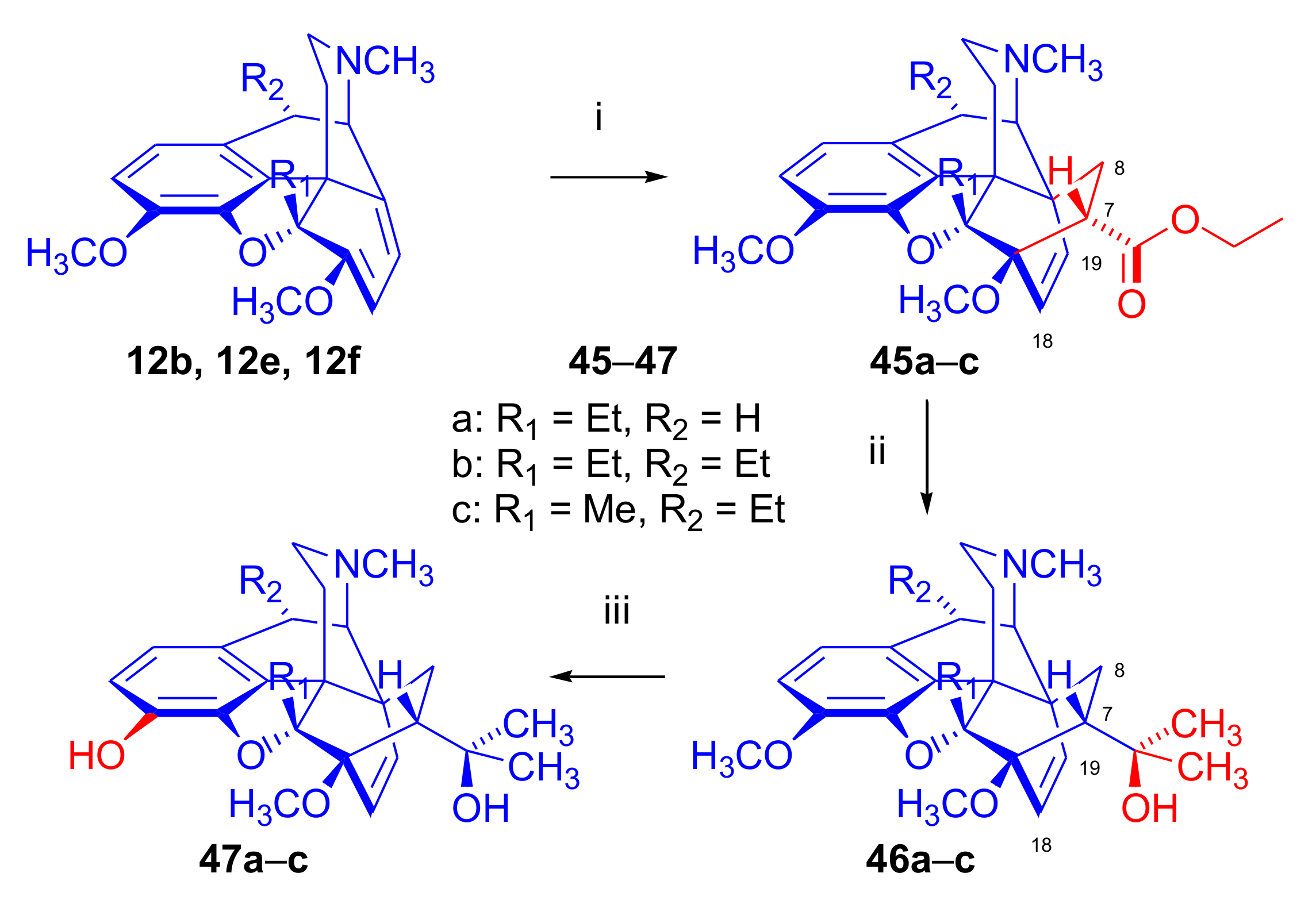{kind=link}
{kind=link}
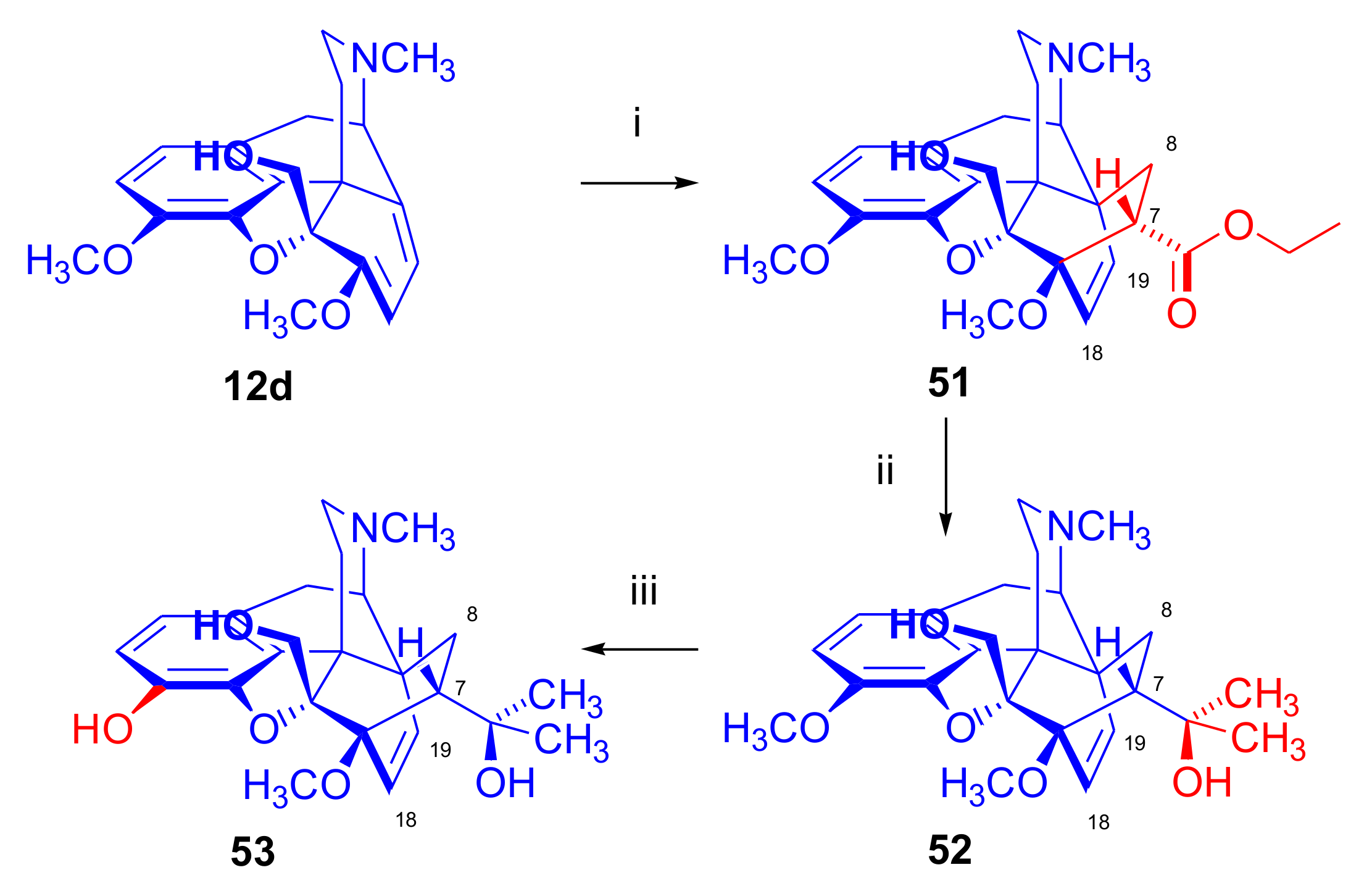{kind=link}
{kind=link}
{kind=link}
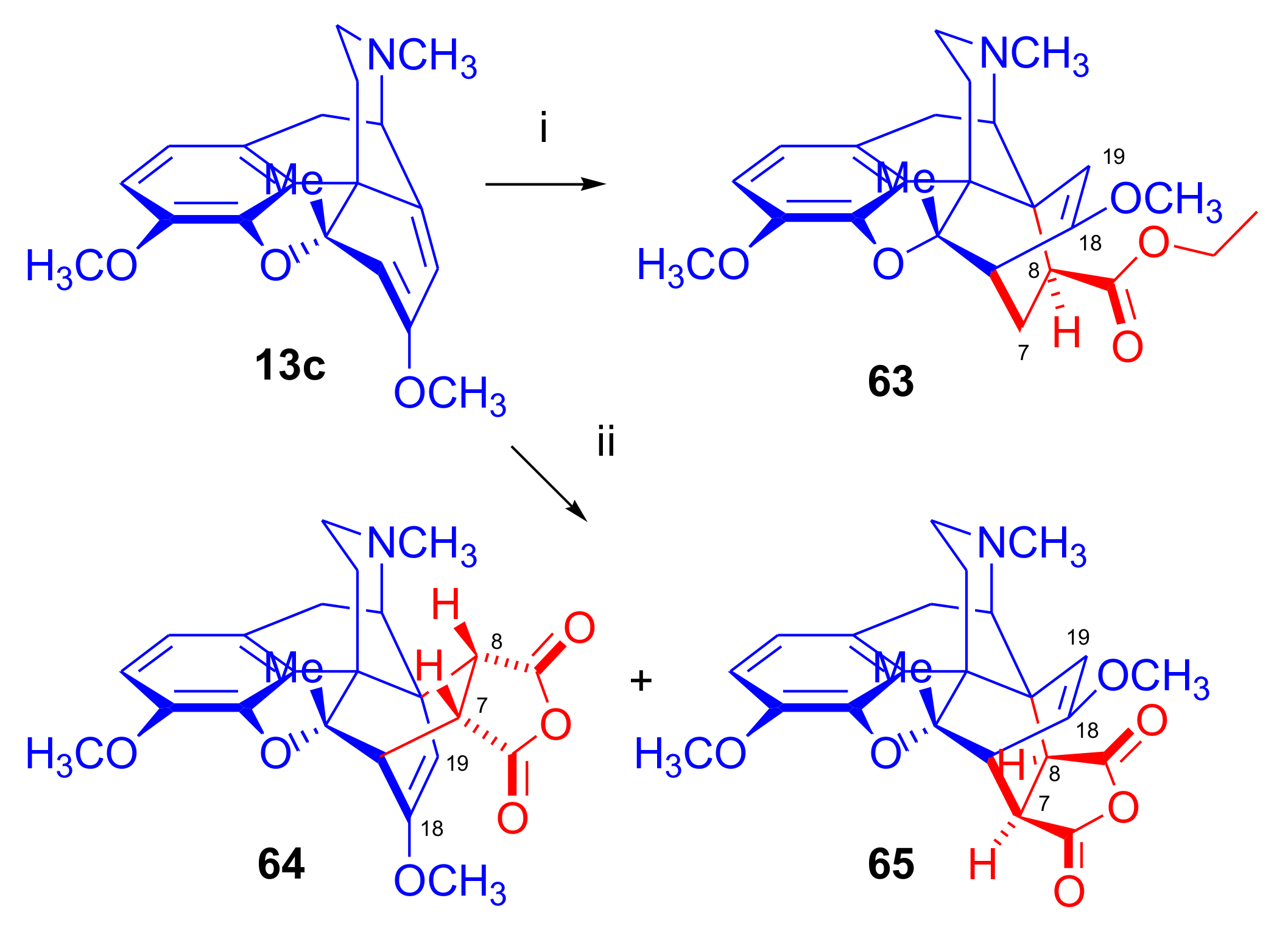{kind=link}
{kind=link}
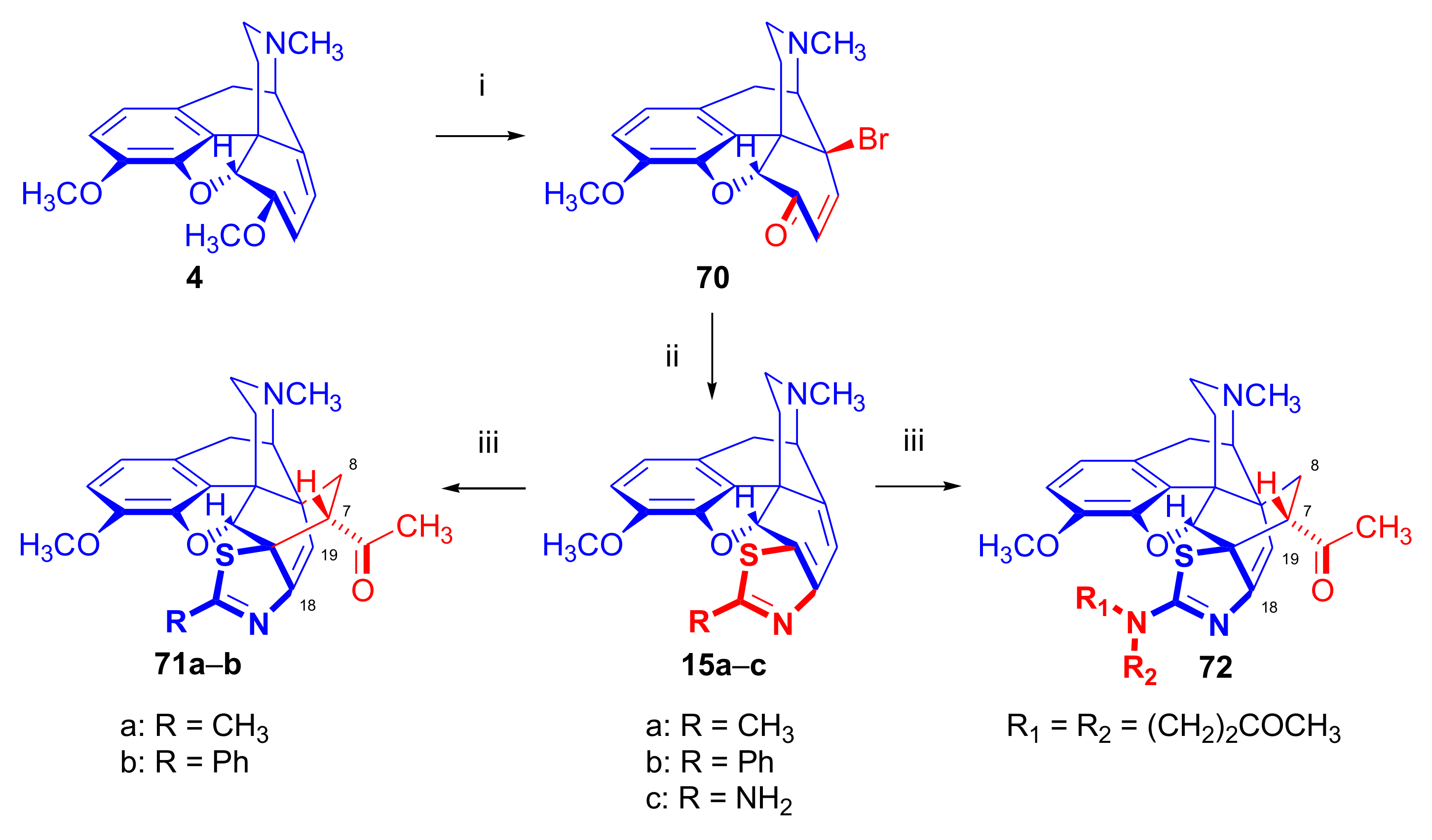{kind=link}
{kind=link}
{kind=link}
{kind=link}
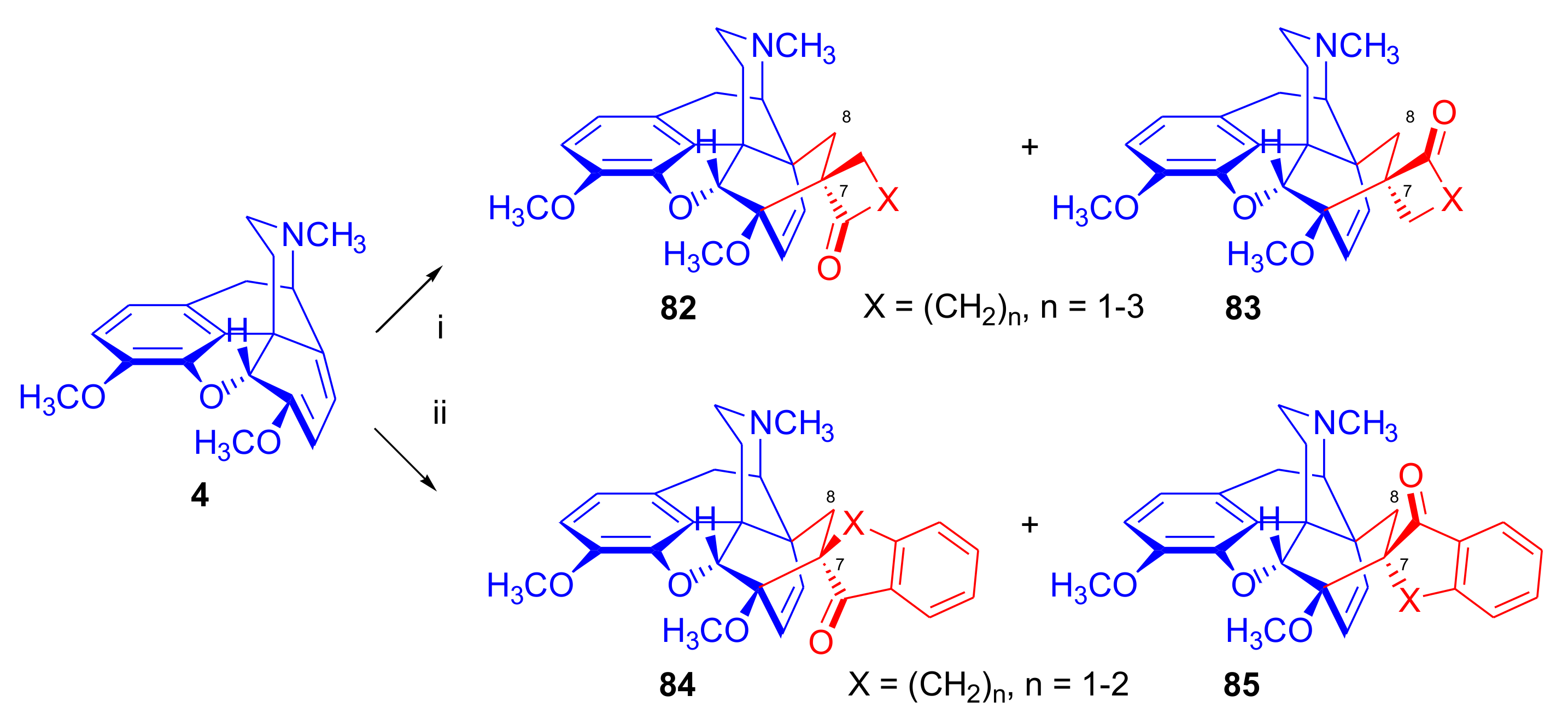{kind=link}
{kind=link}
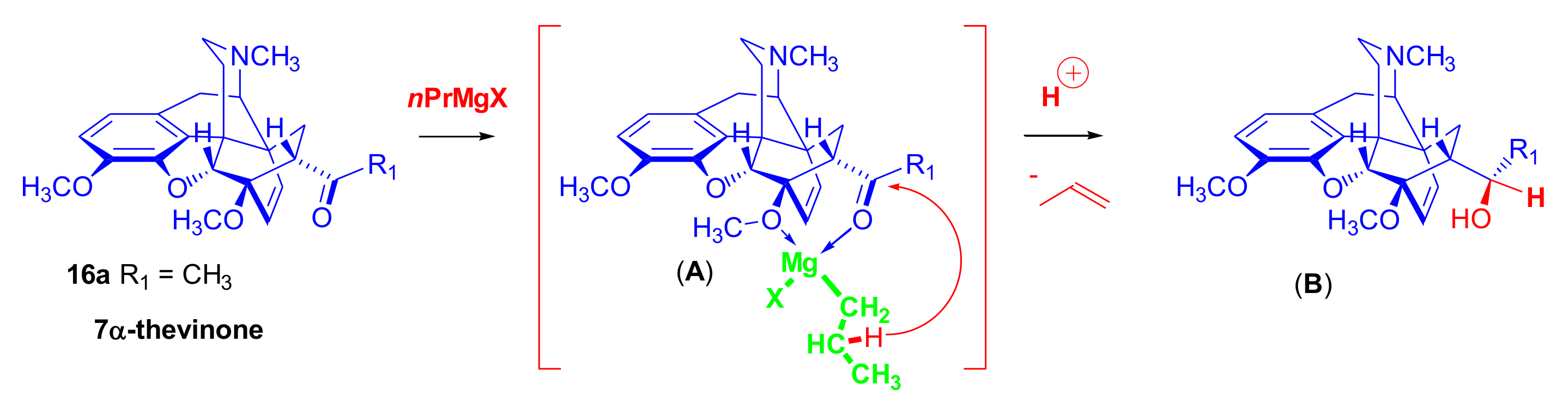{kind=link}
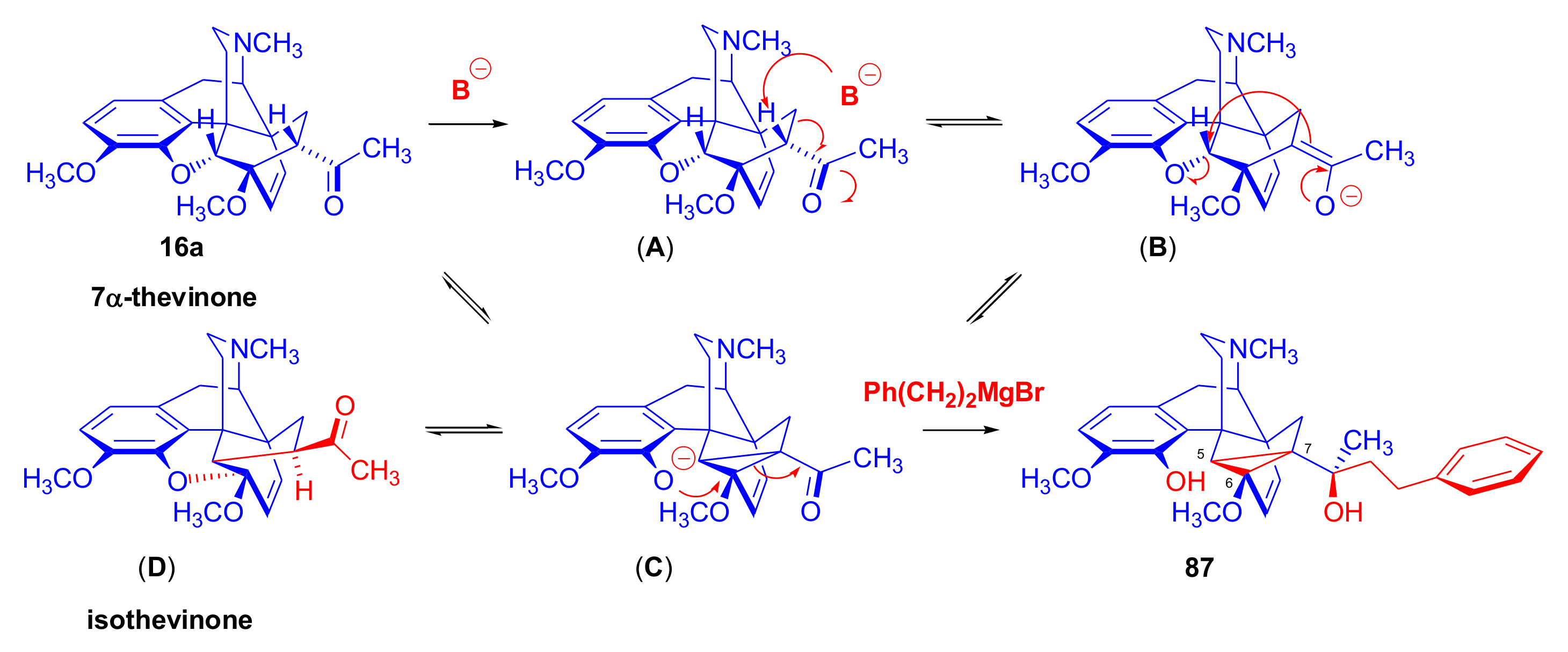{kind=link}
{kind=link}
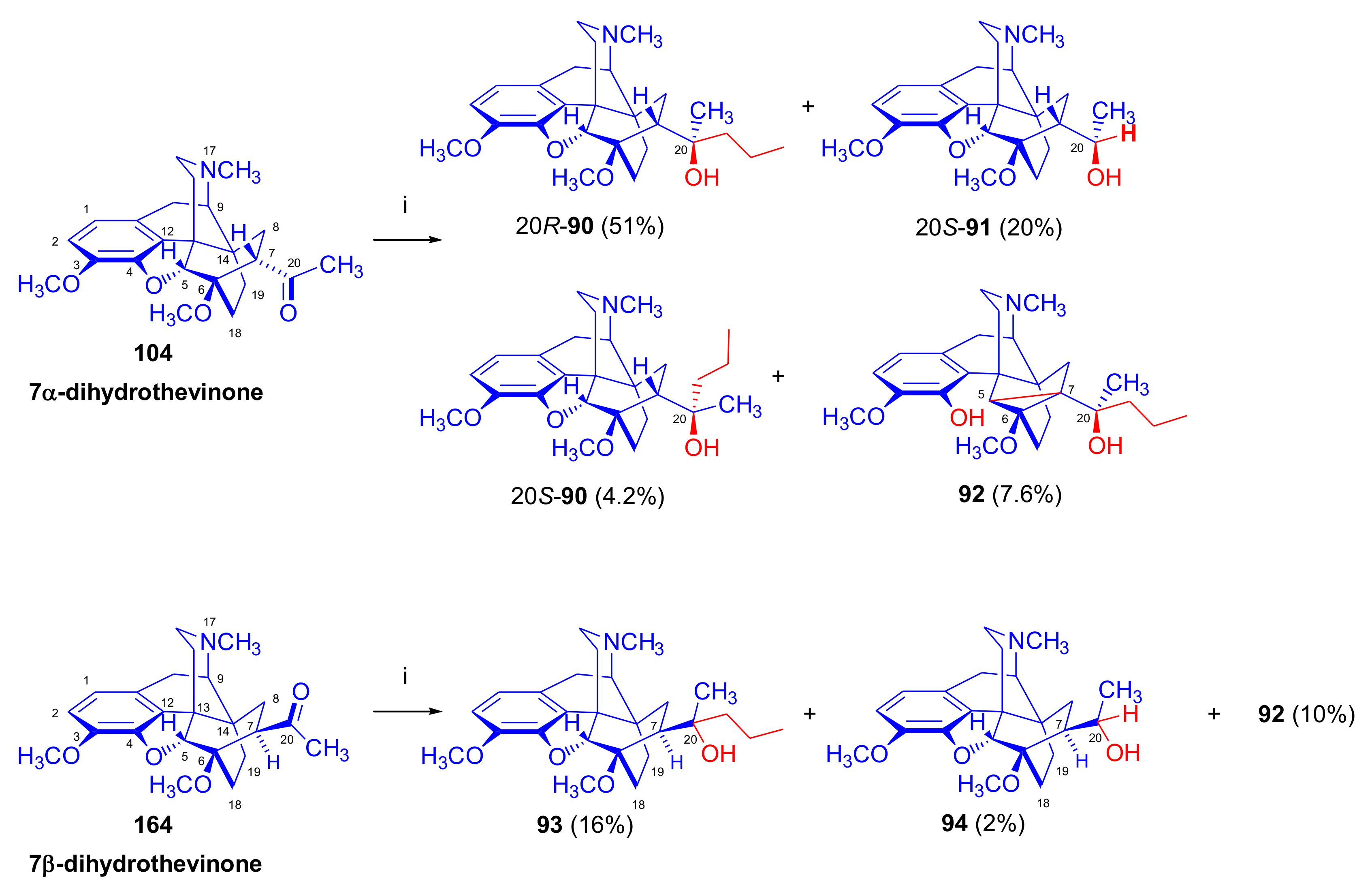{kind=link}
{kind=link}
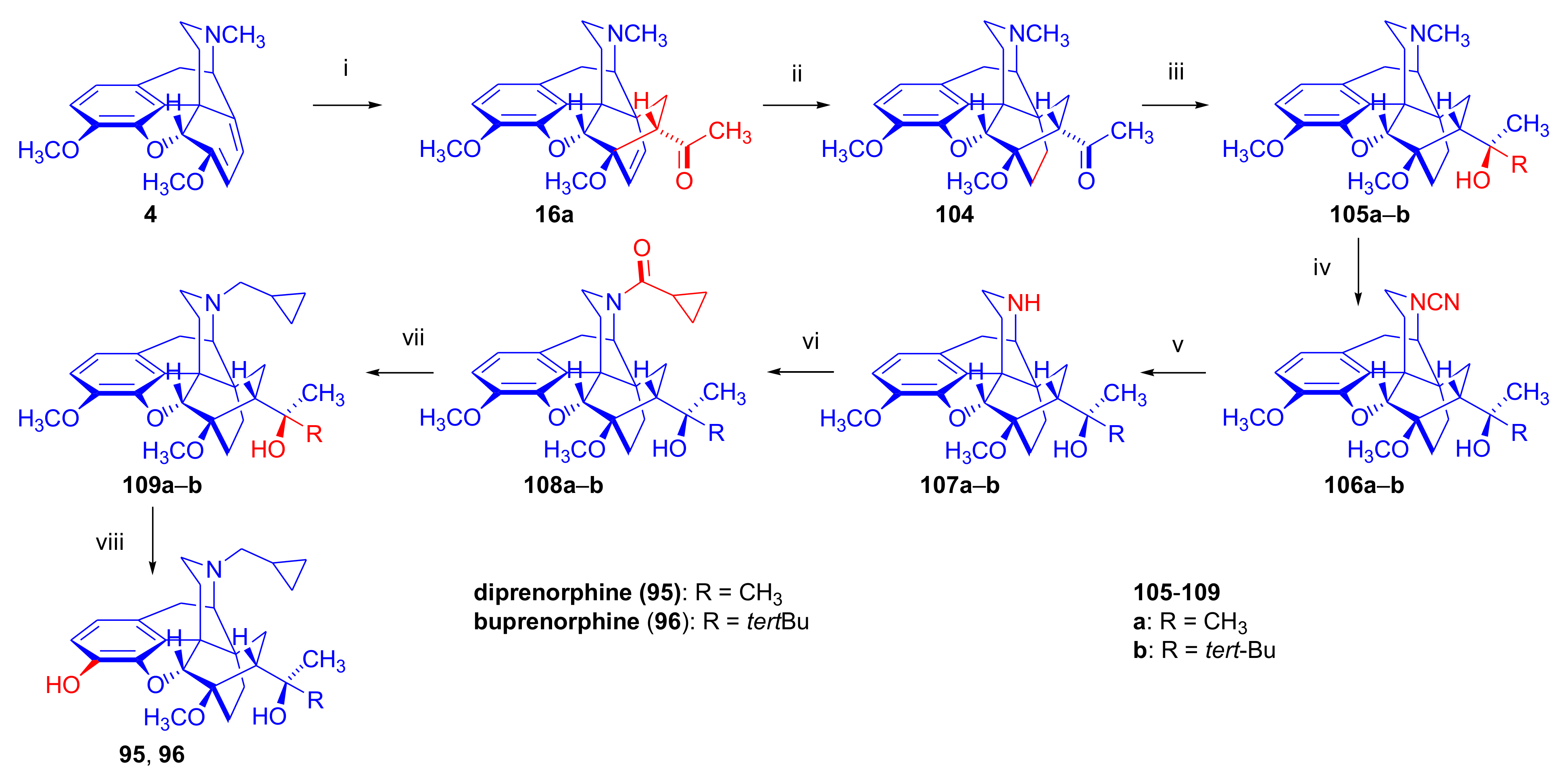{kind=link}
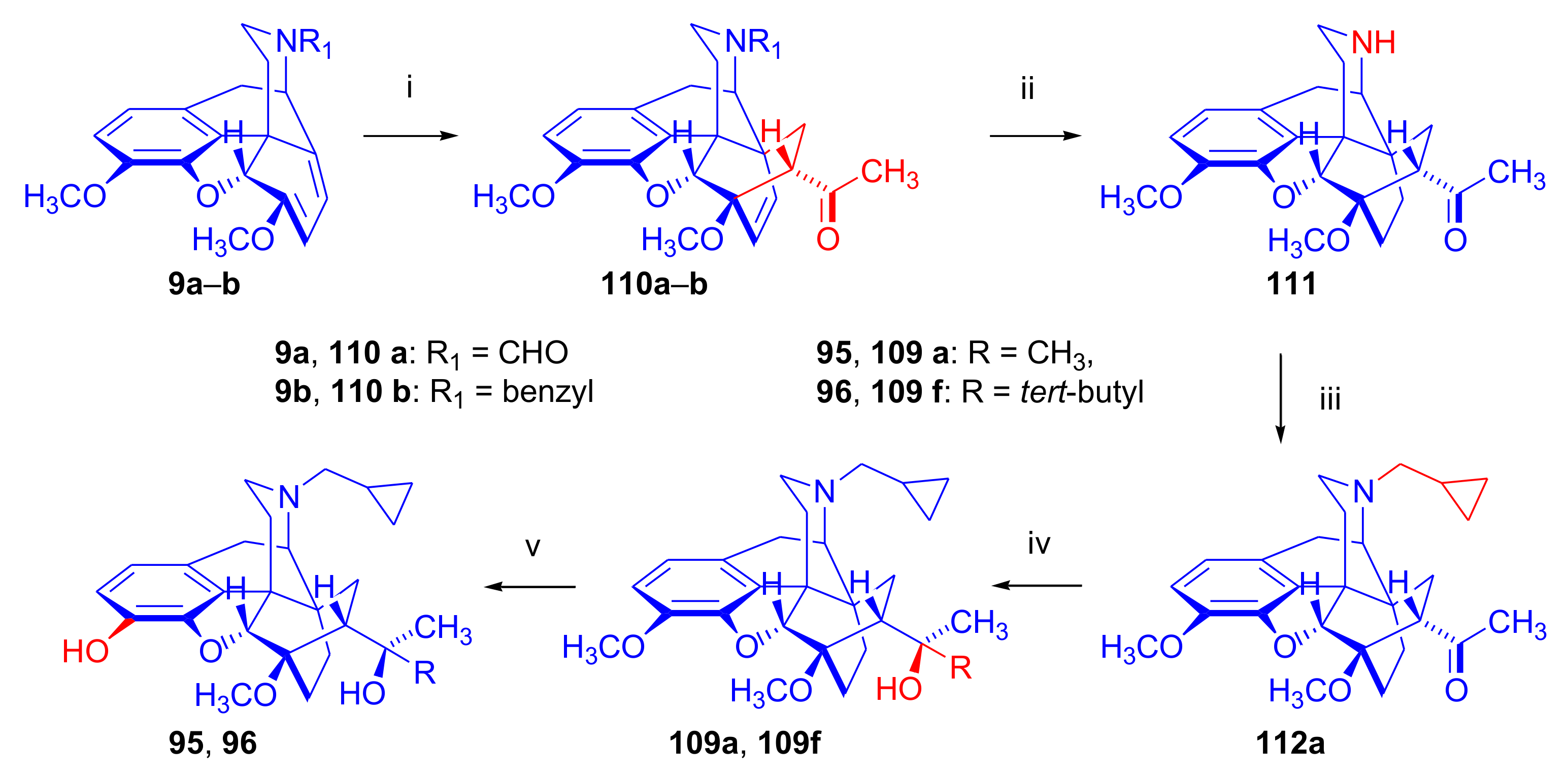{kind=link}
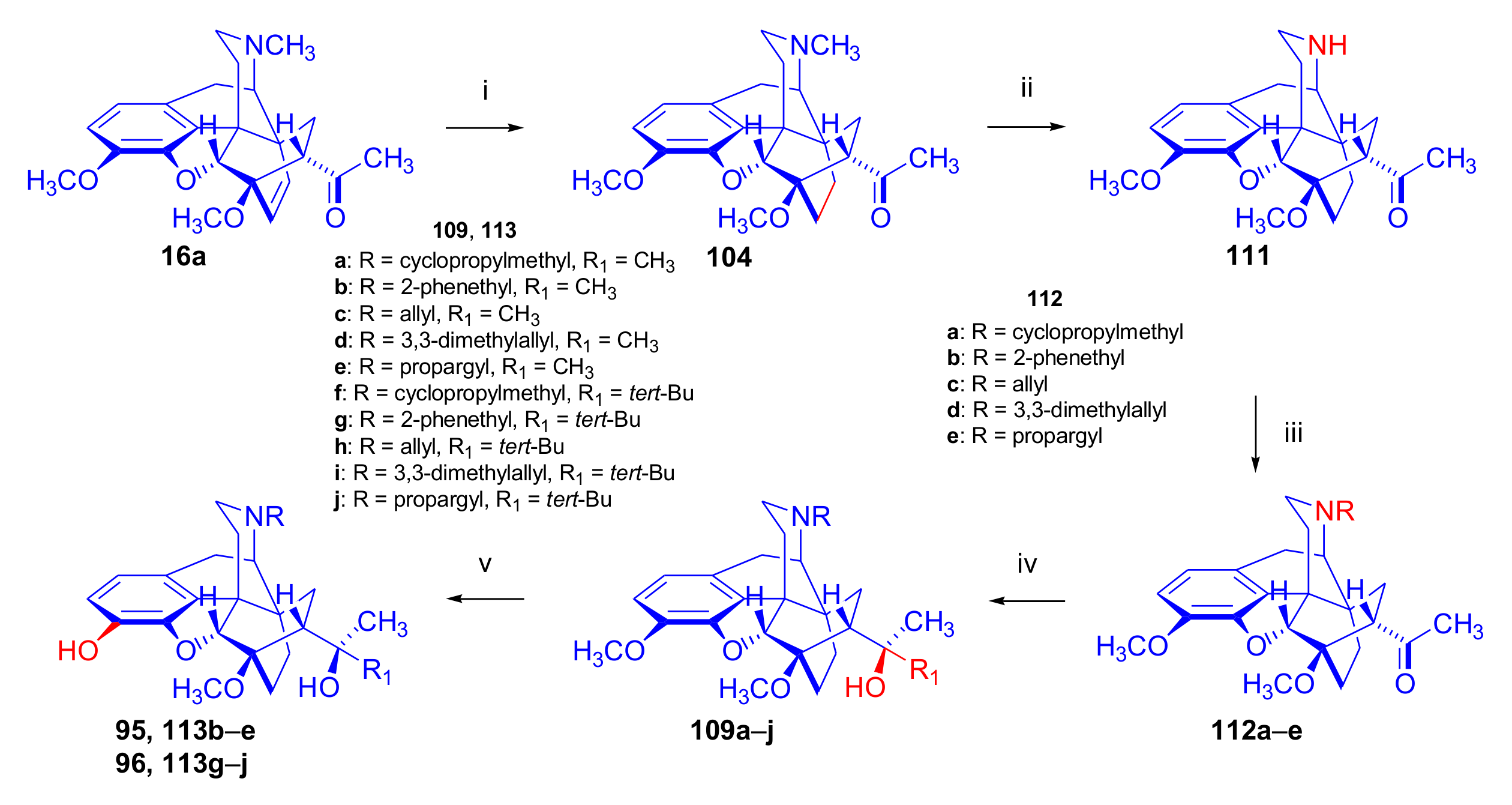{kind=link}
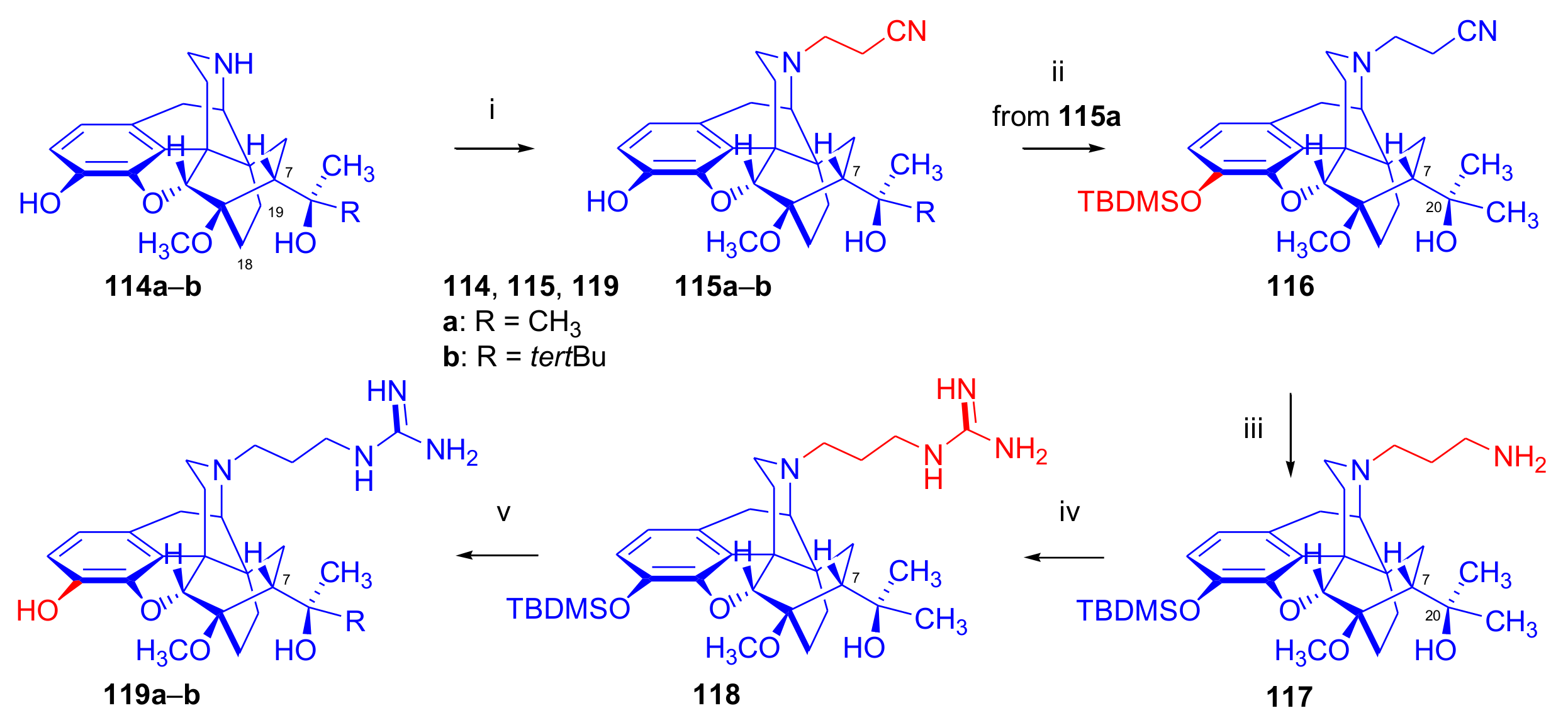{kind=link}
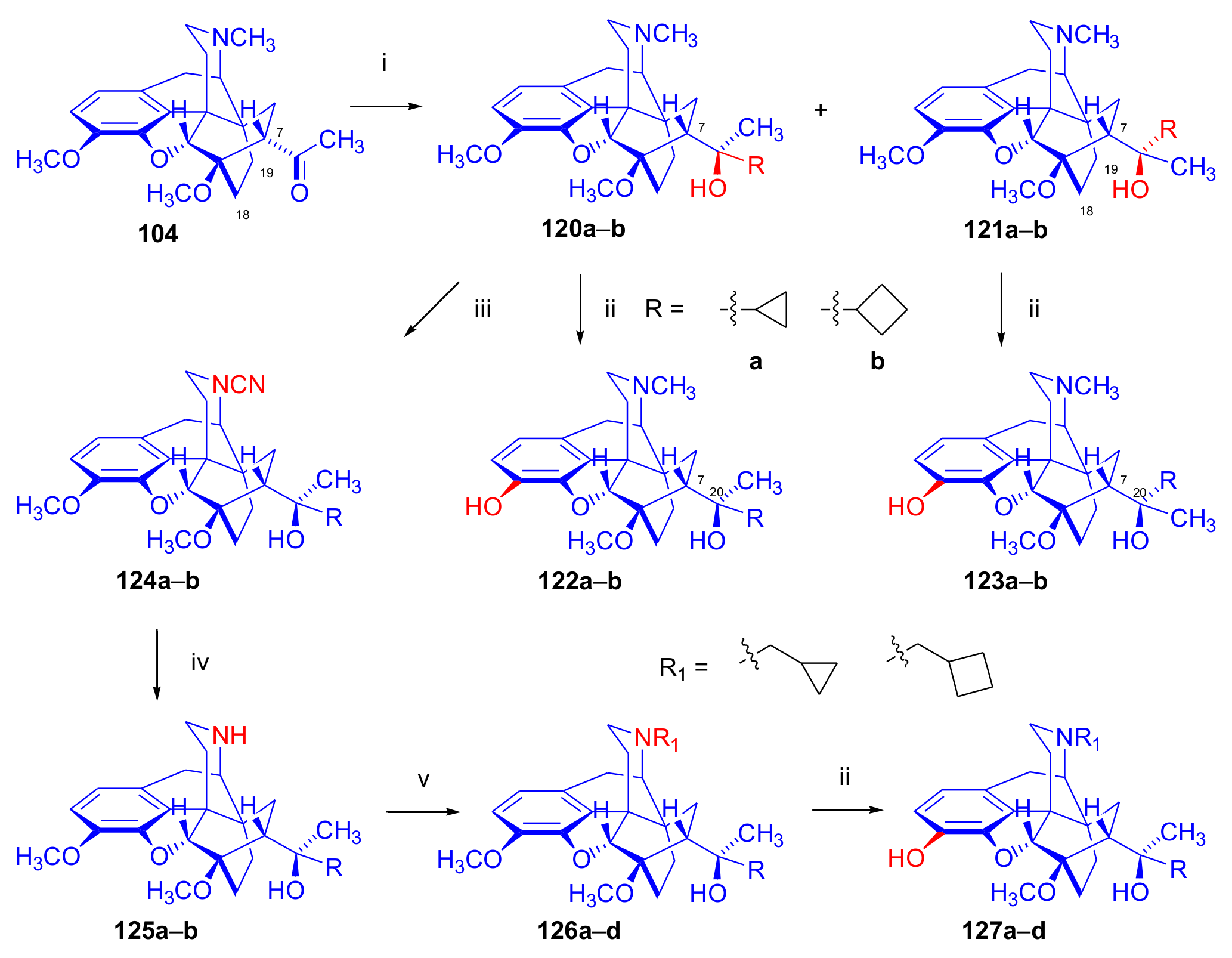{kind=link}
{kind=link}
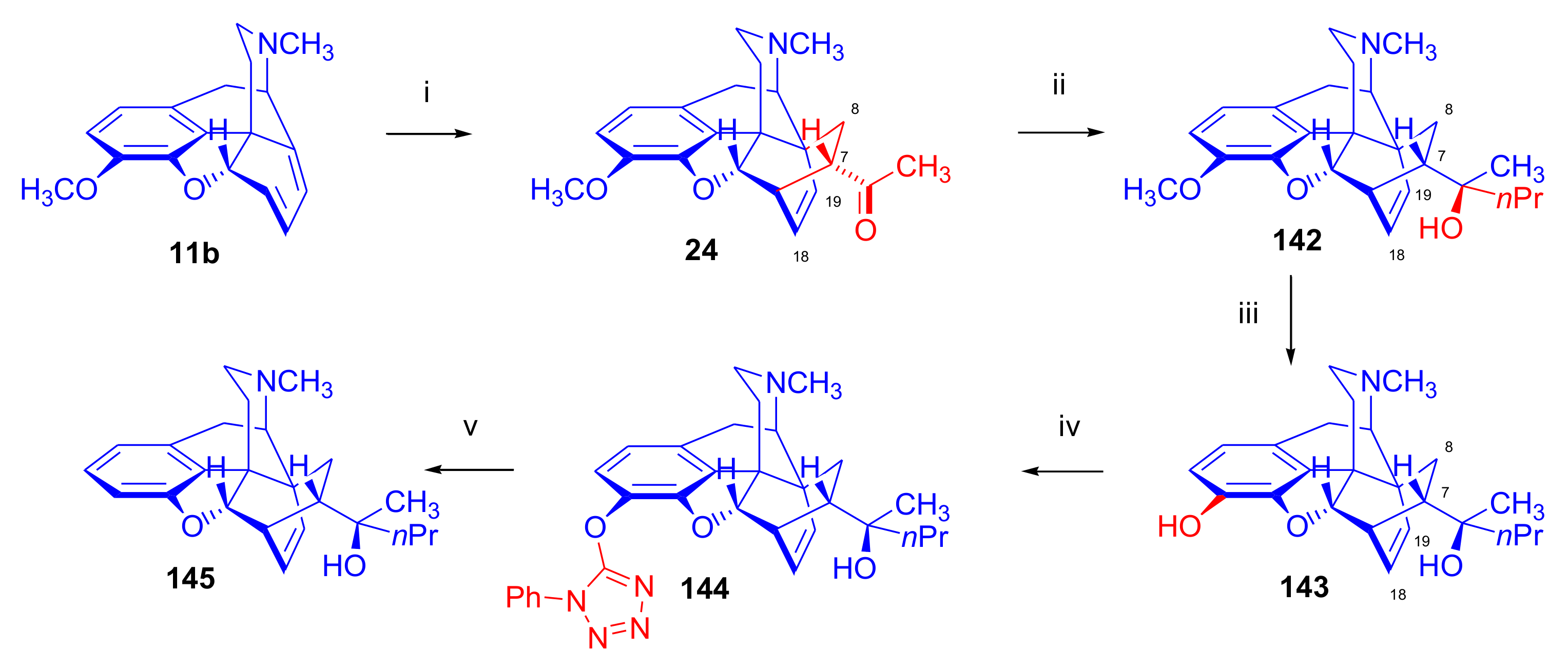{kind=link}
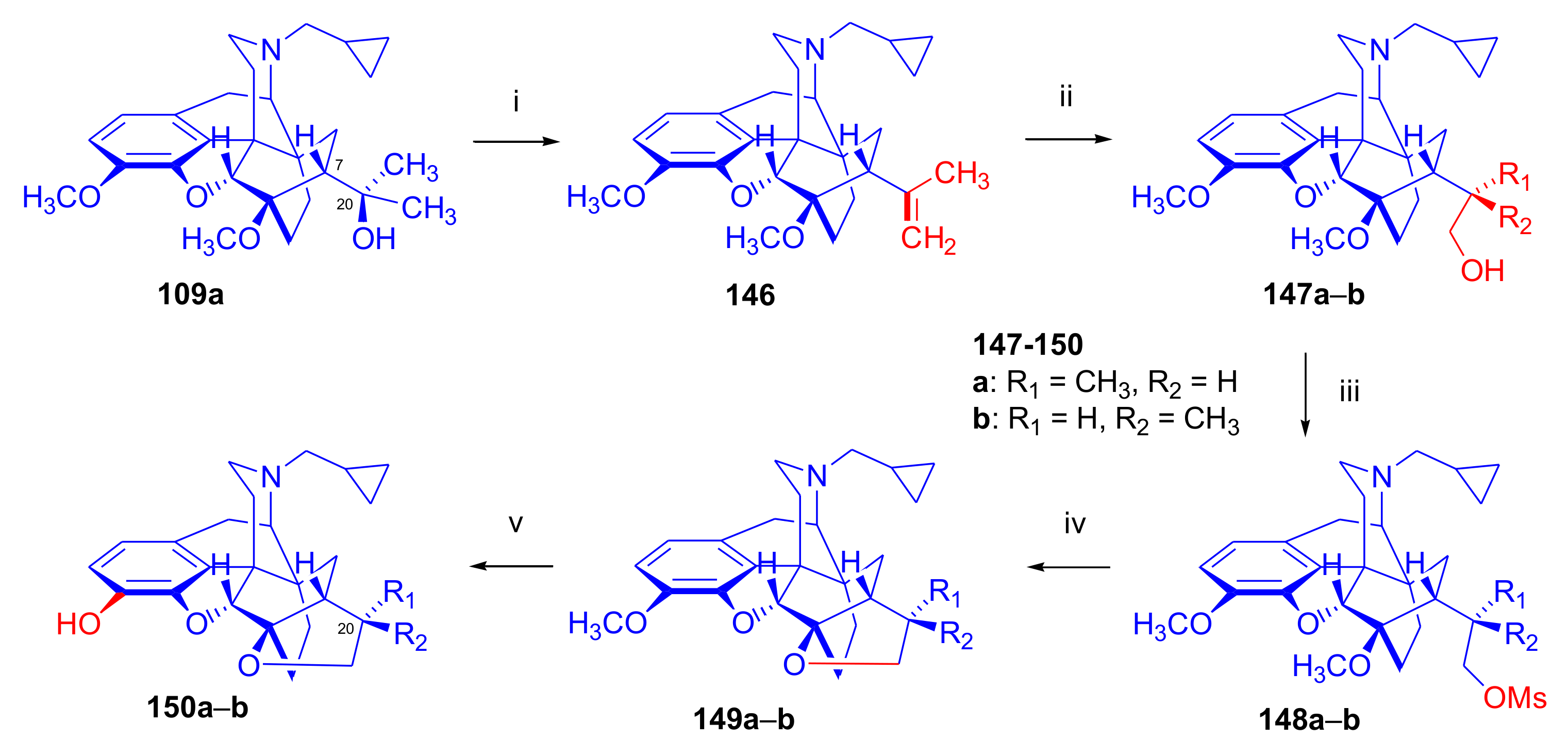{kind=link}
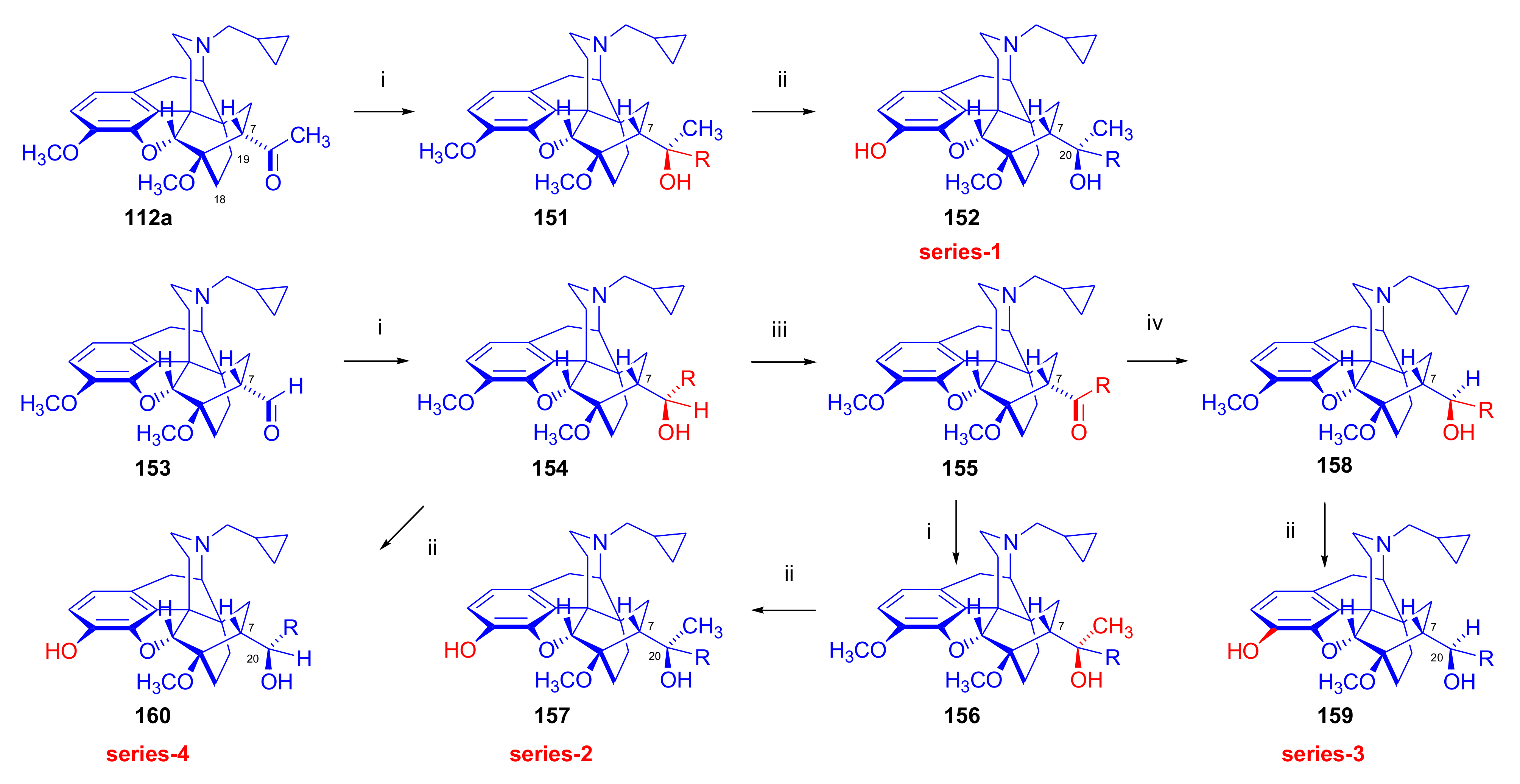{kind=link}
{kind=link}
{kind=link}
{kind=link}
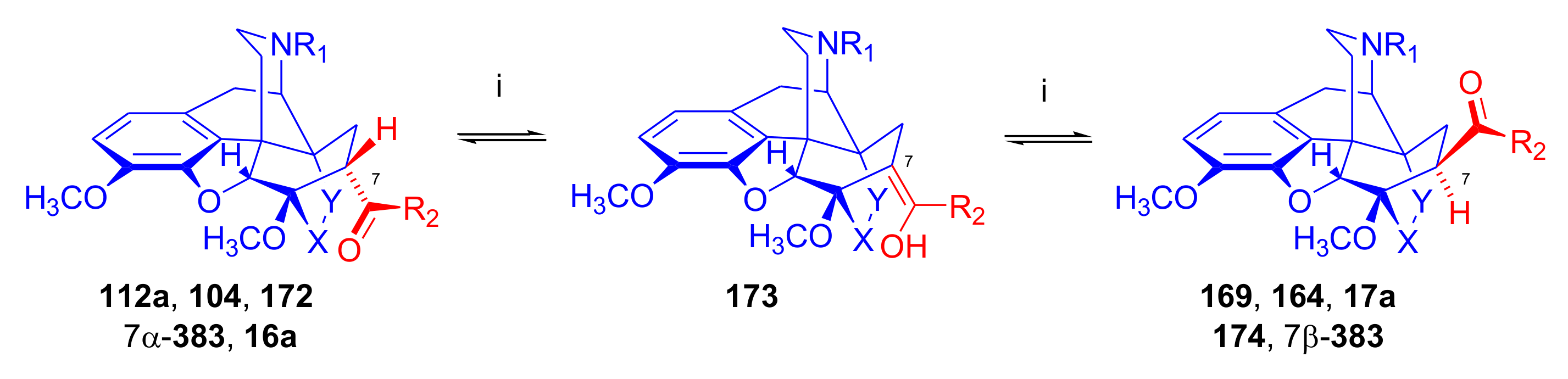{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
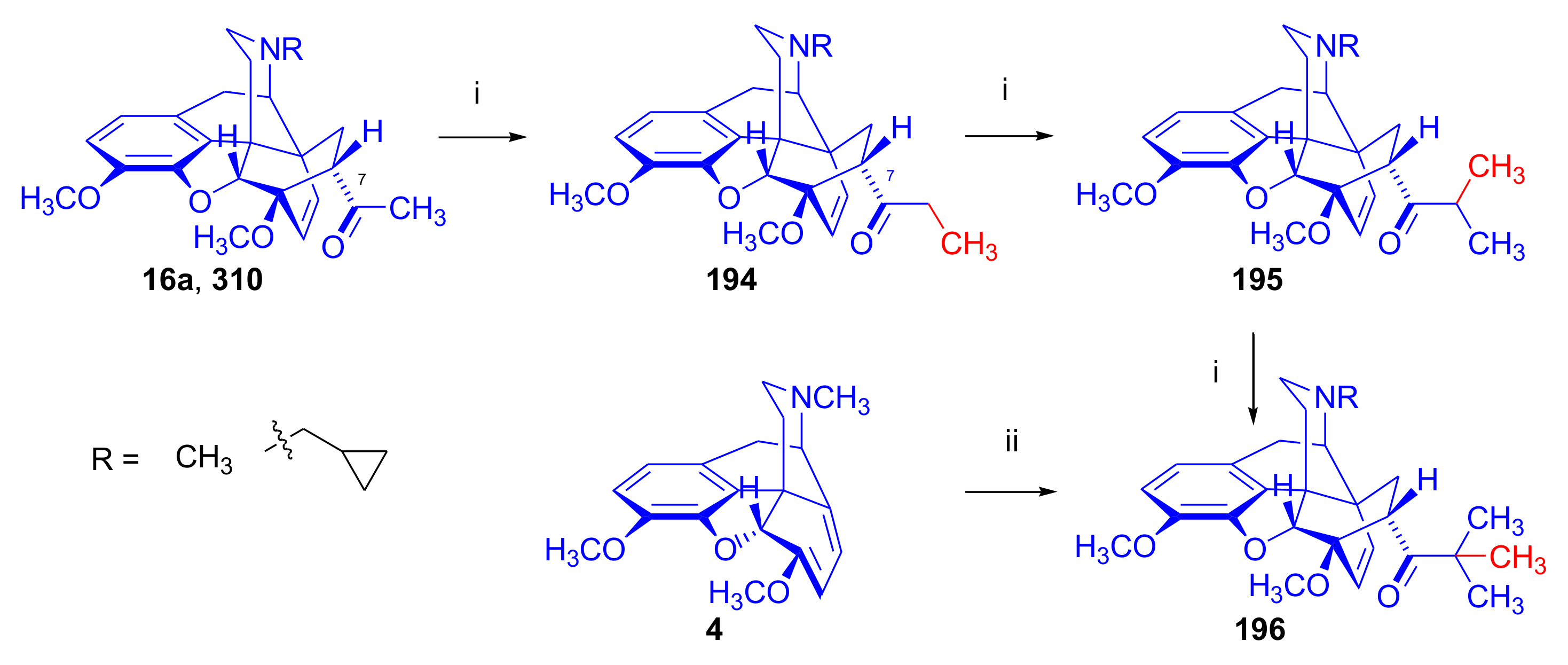{kind=link}
{kind=link}
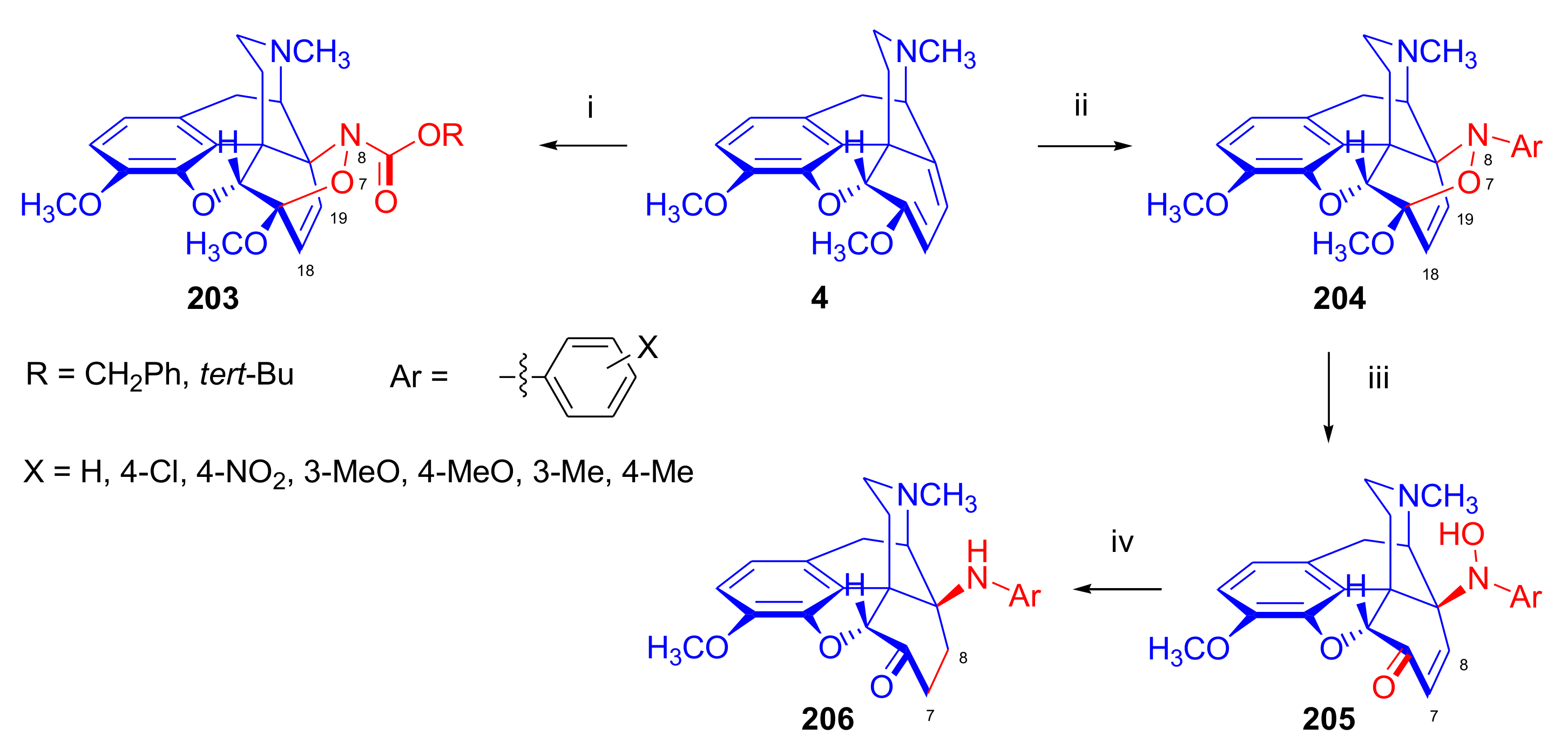{kind=link}
{kind=link}
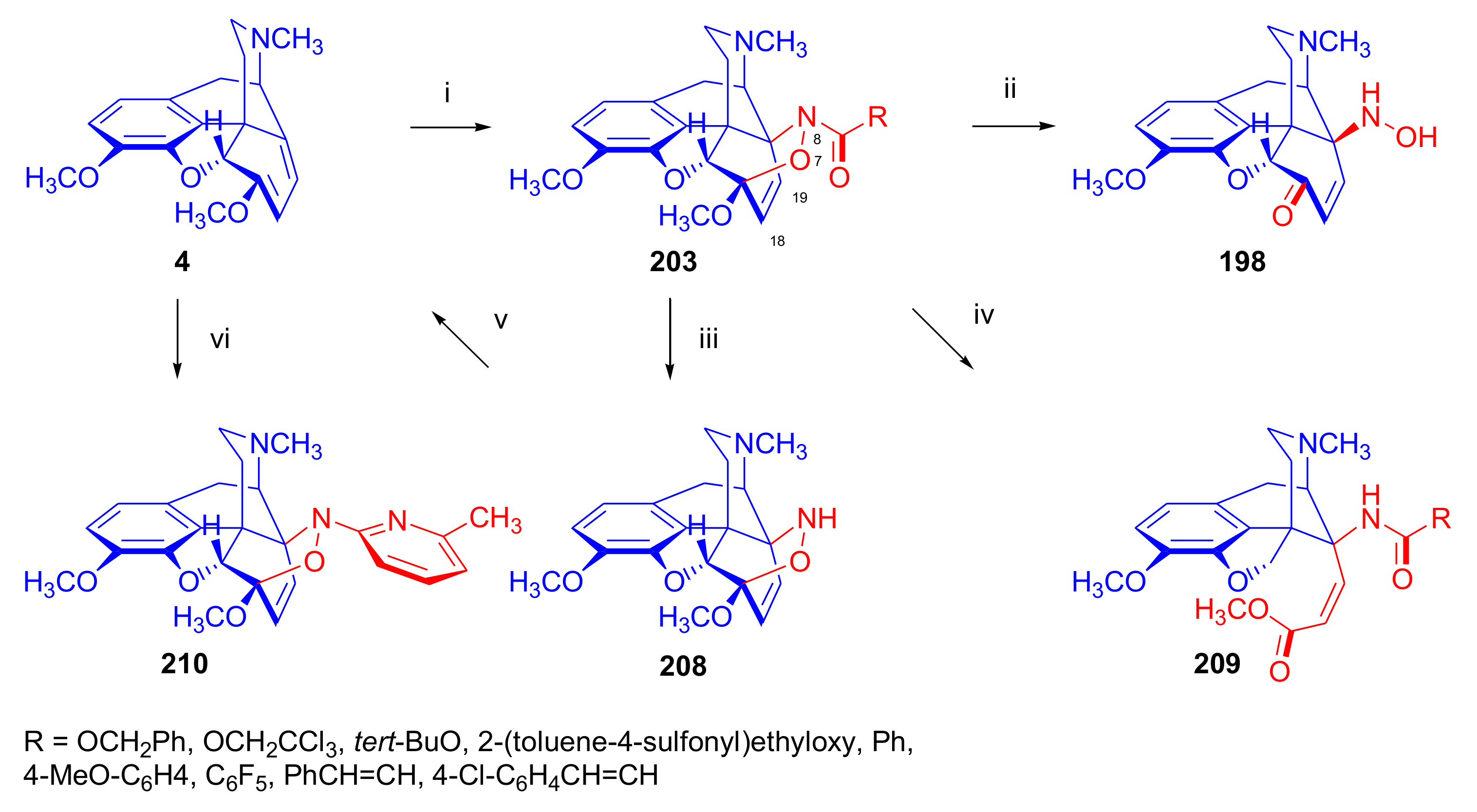{kind=link}
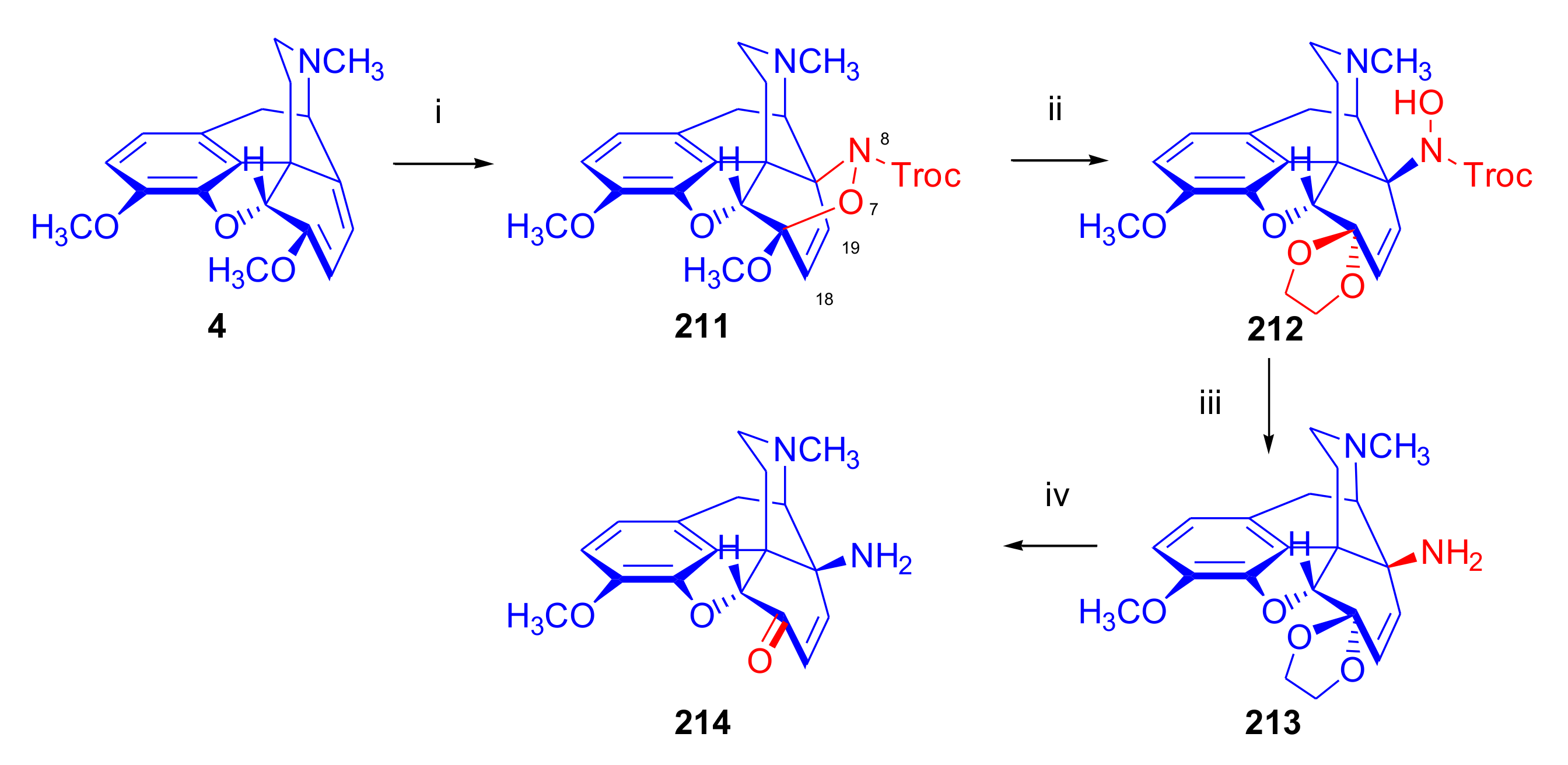{kind=link}
{kind=link}
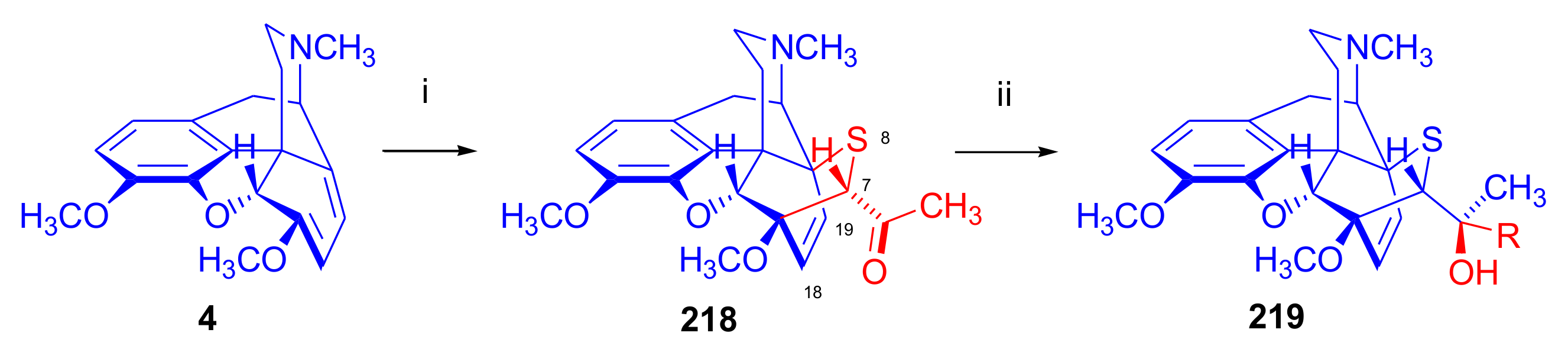{kind=link}
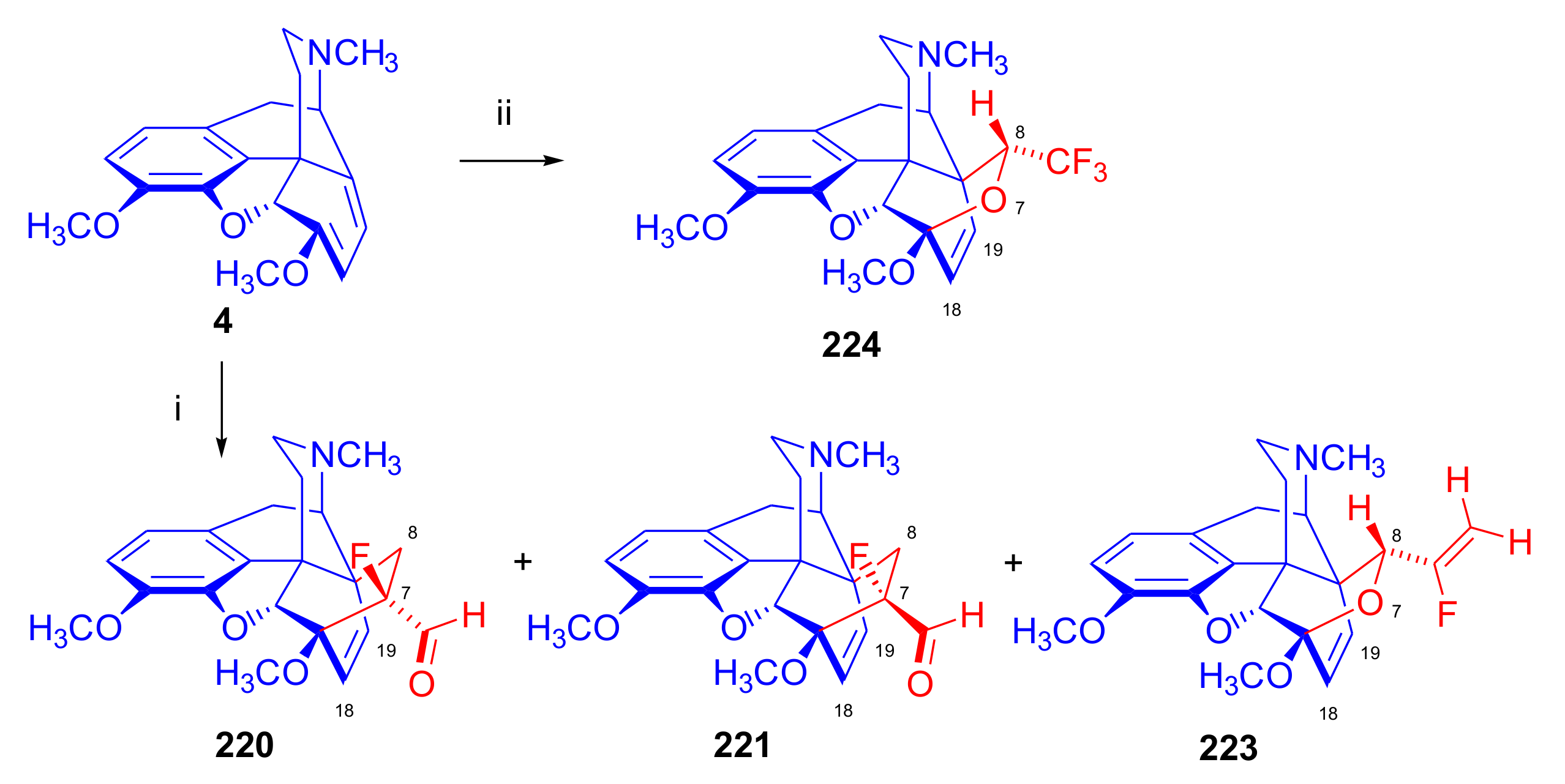{kind=link}
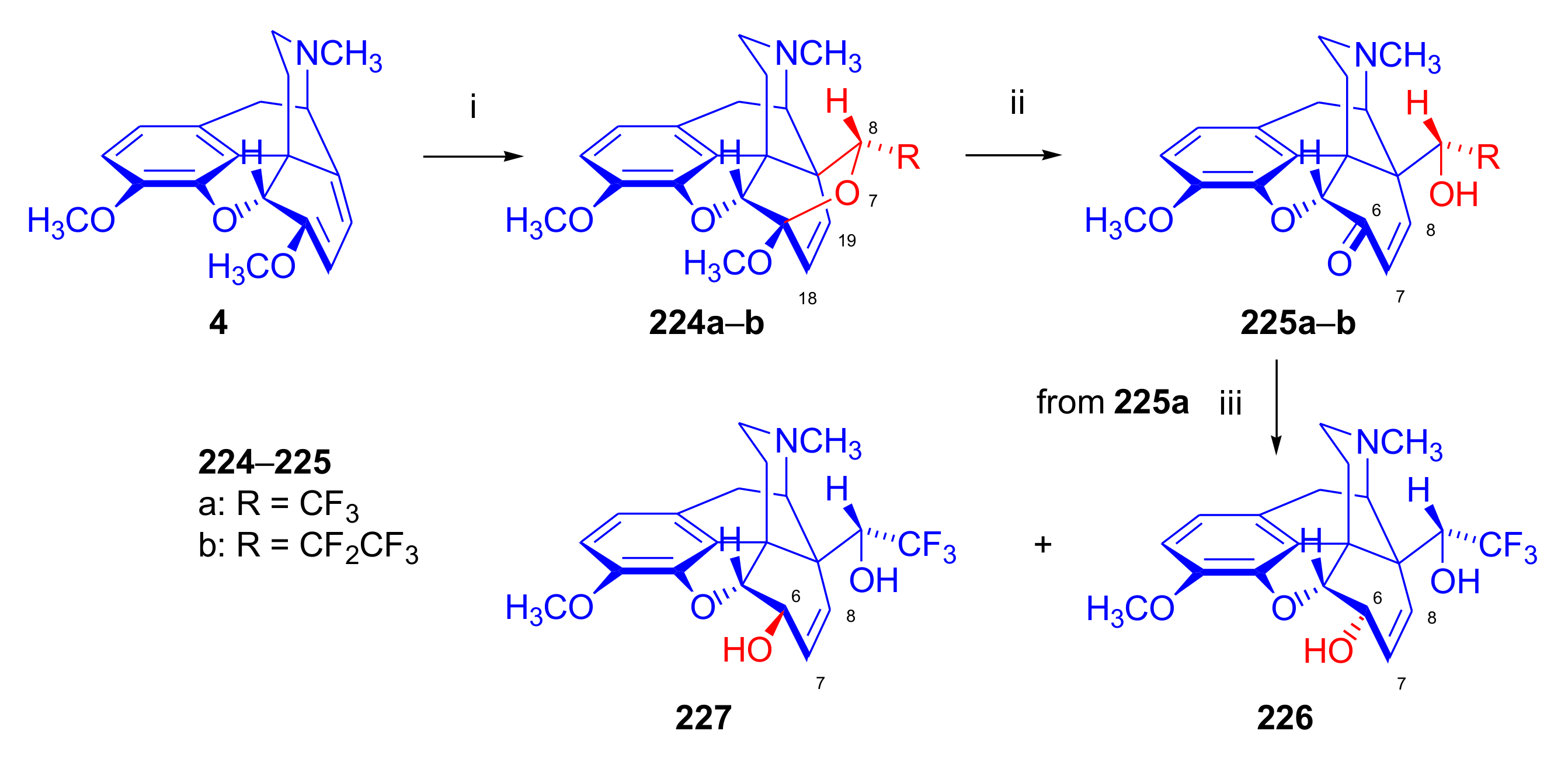{kind=link}
{kind=link}
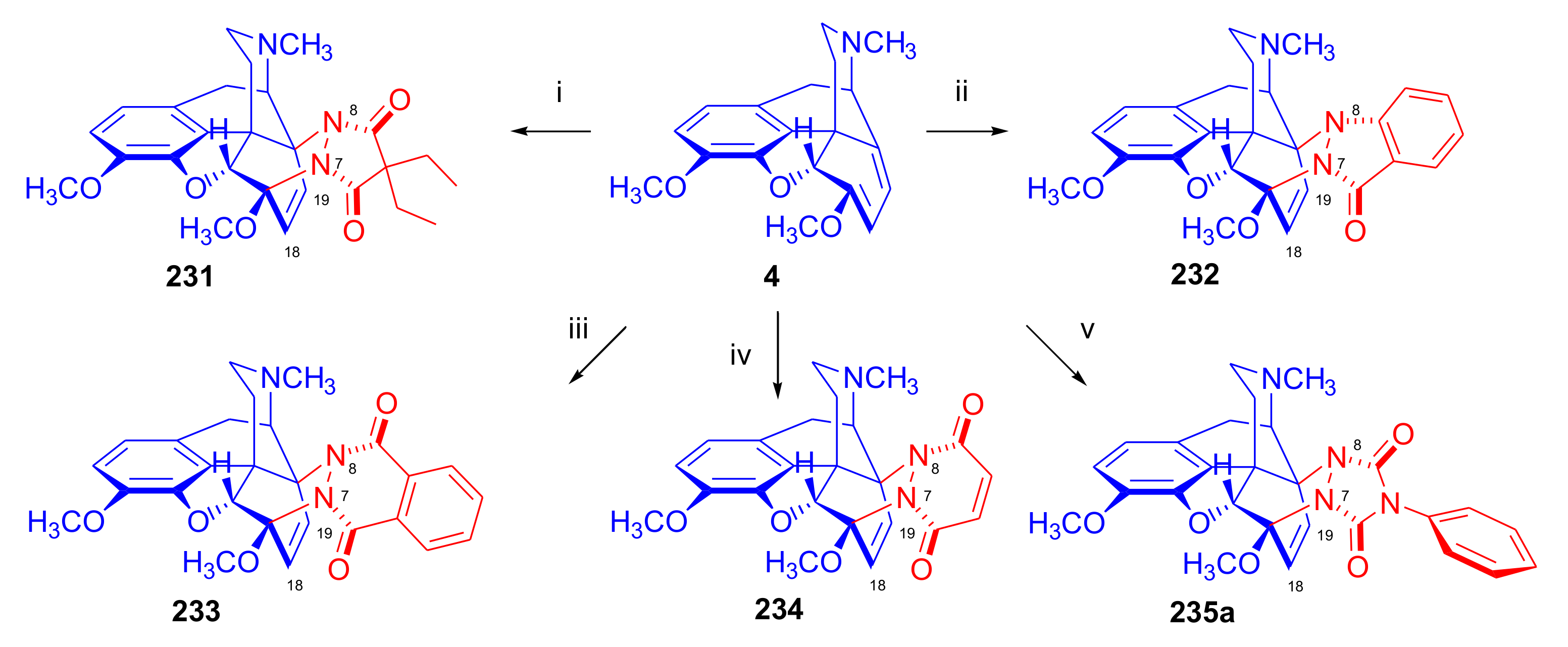{kind=link}
{kind=link}
{kind=link}
{kind=link}
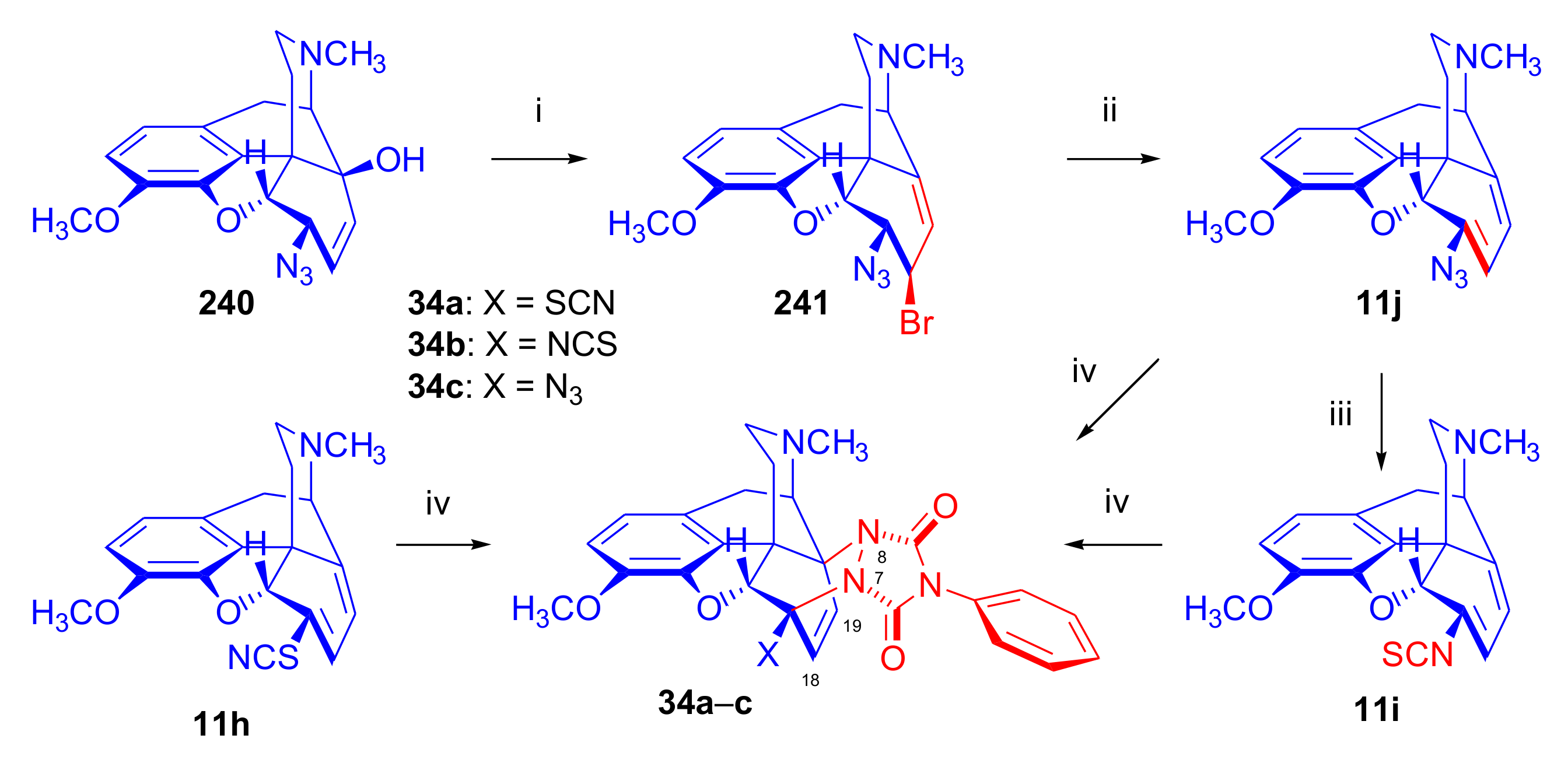{kind=link}
{kind=link}
{kind=link}
{kind=link}
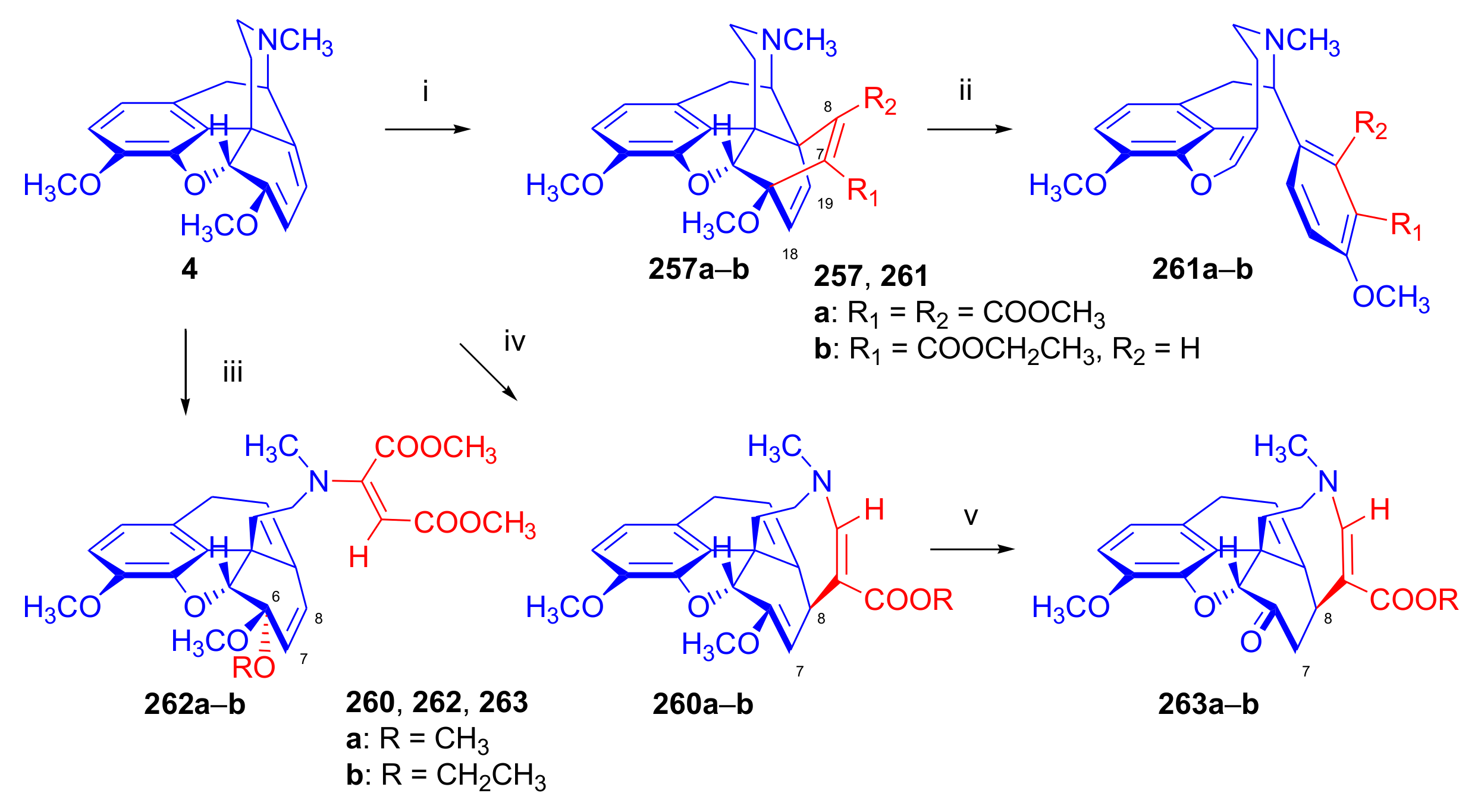{kind=link}
{kind=link}
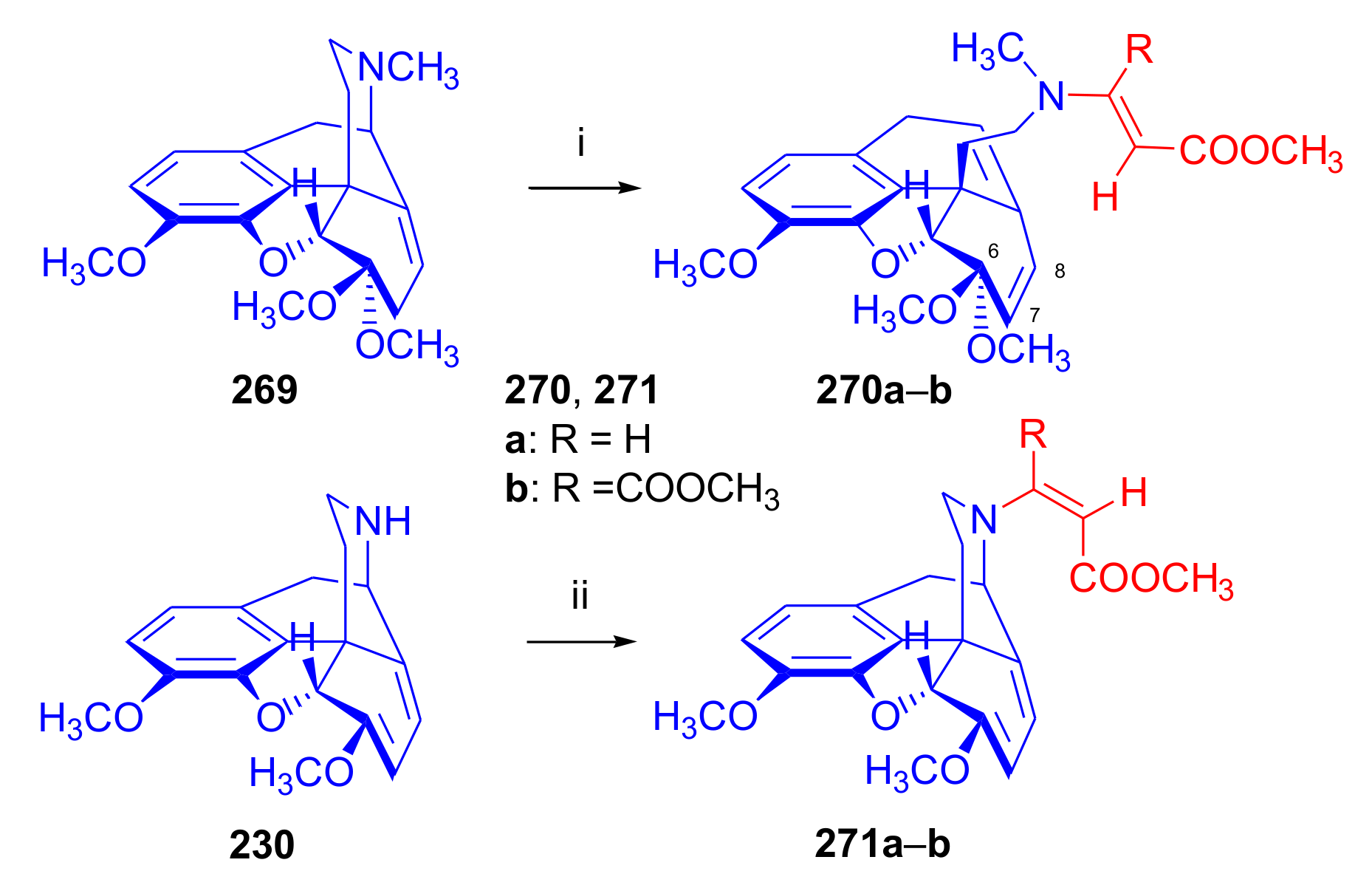{kind=link}
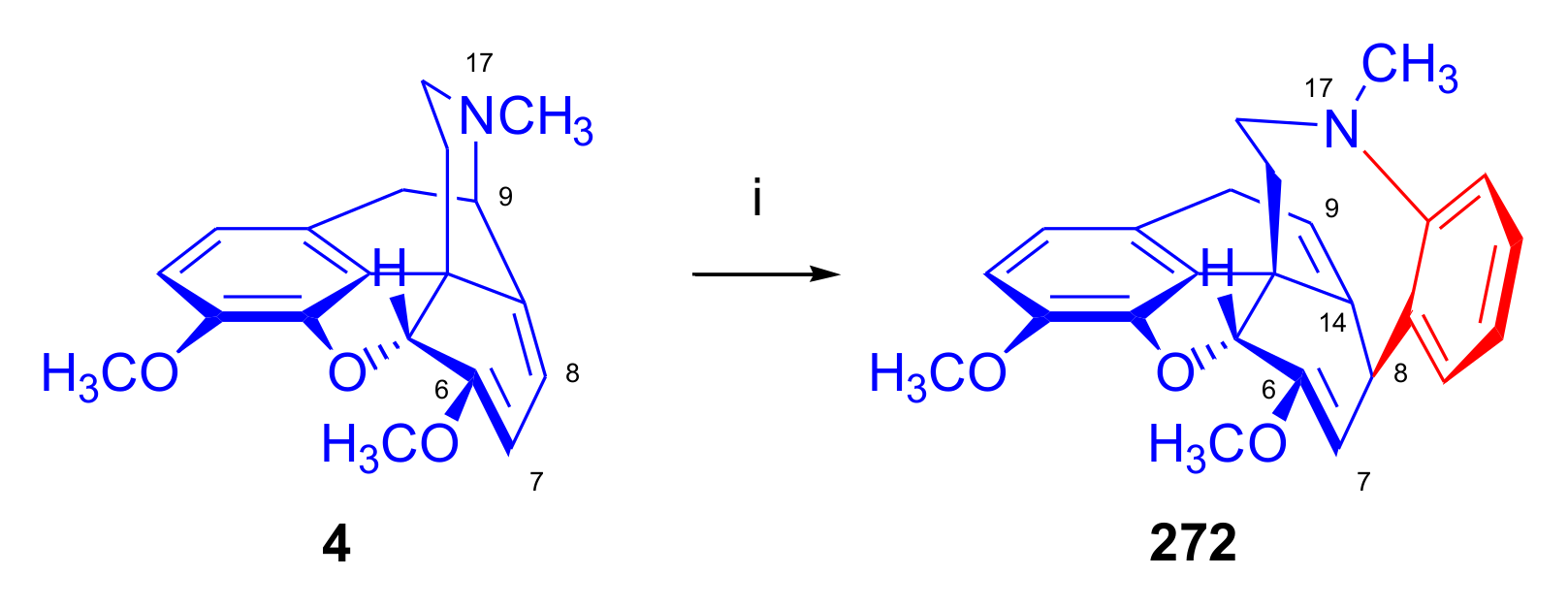{kind=link}
{kind=link}
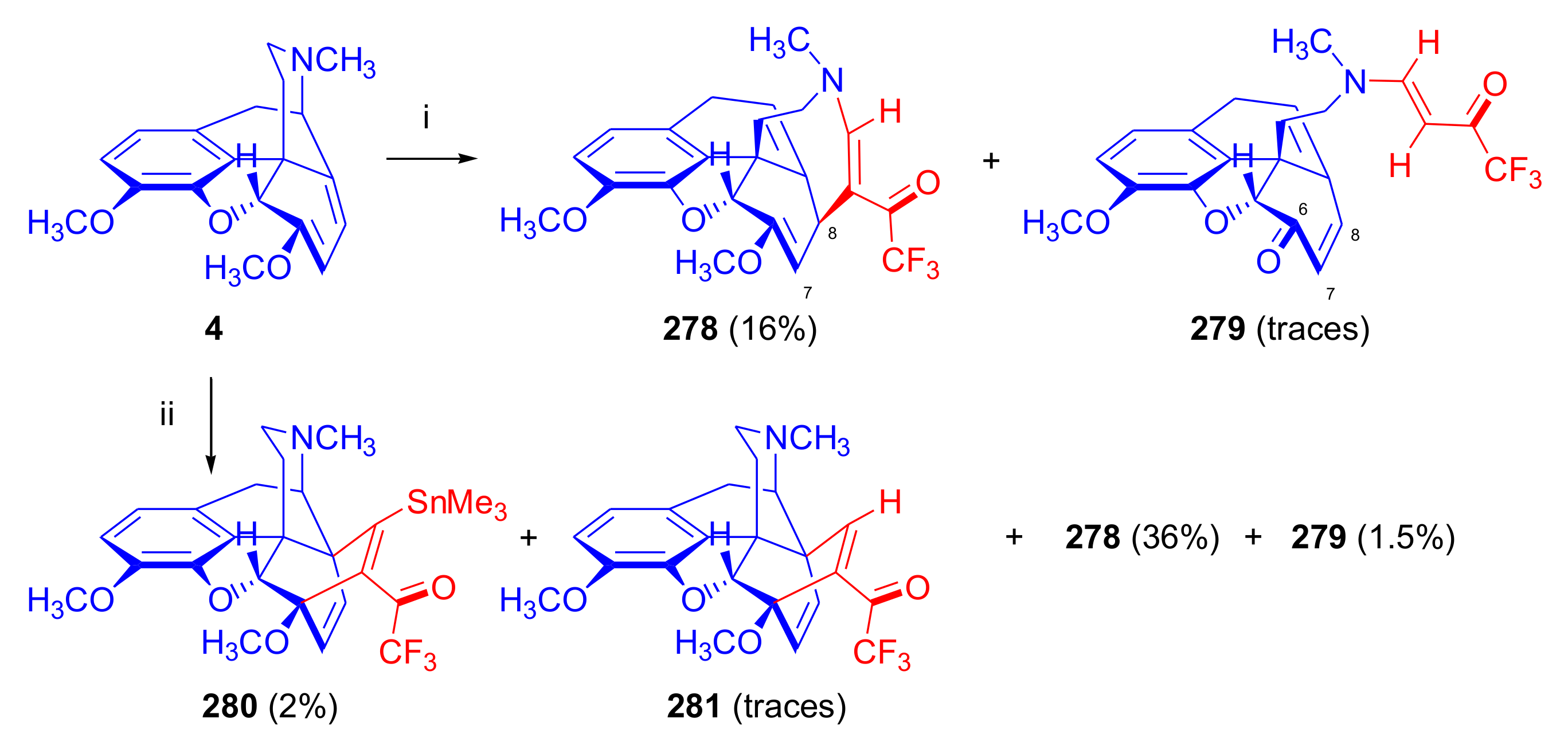{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
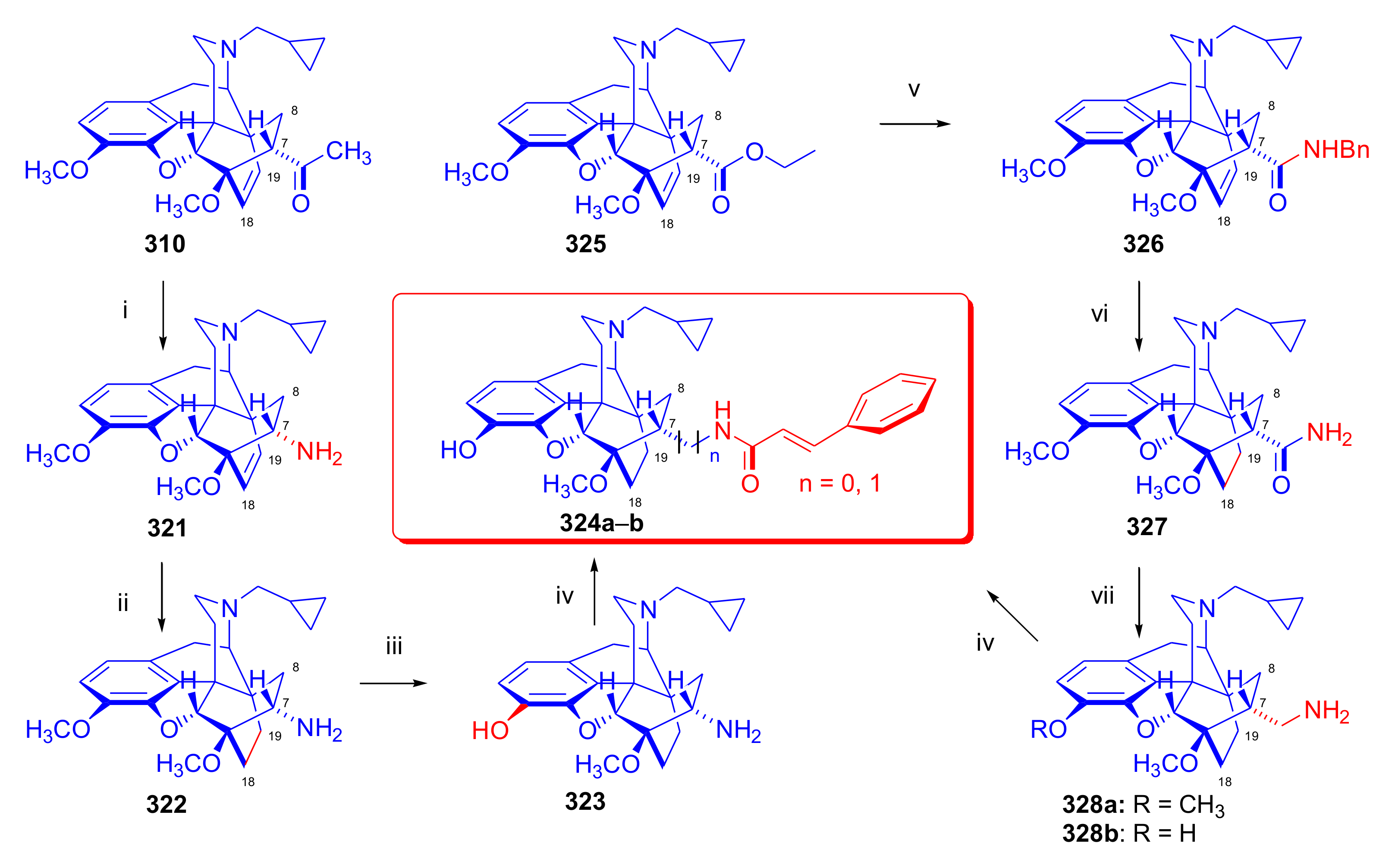{kind=link}
{kind=link}
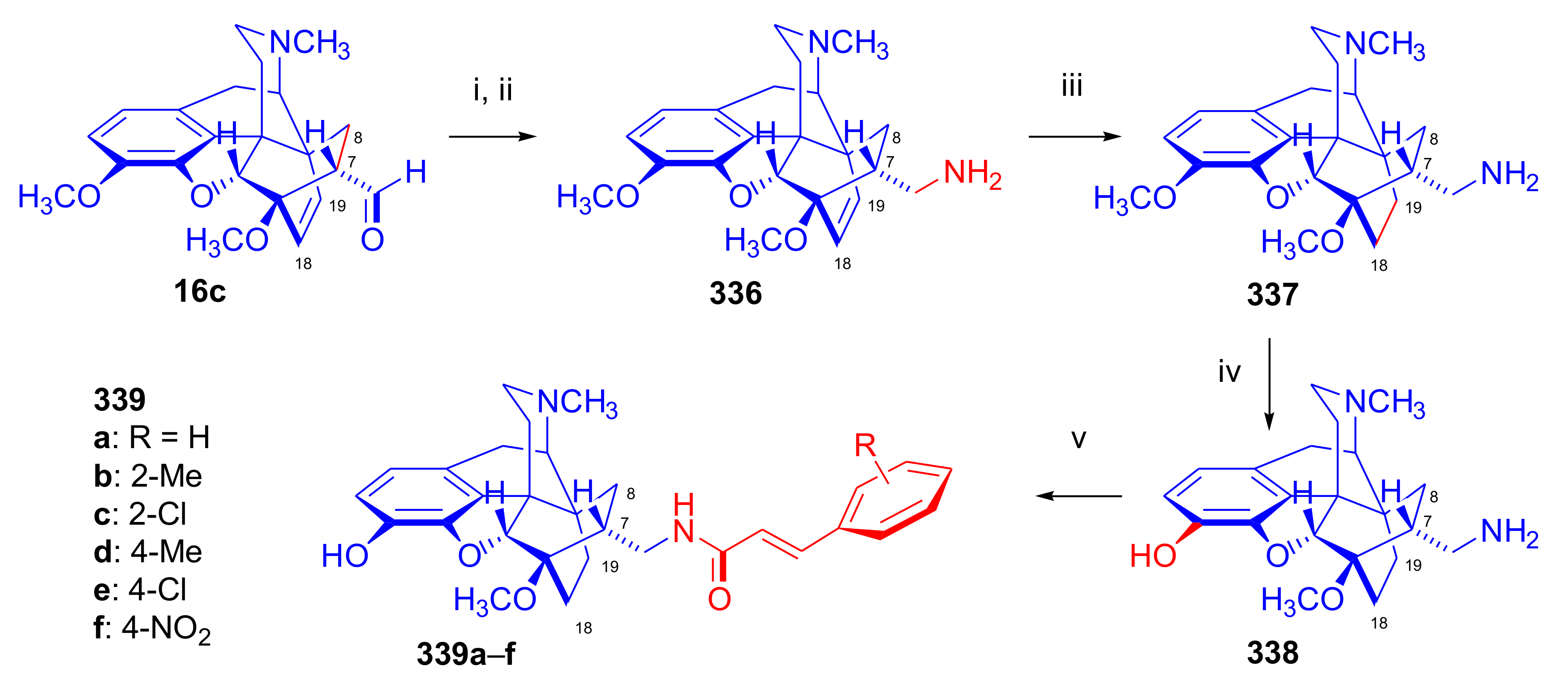{kind=link}
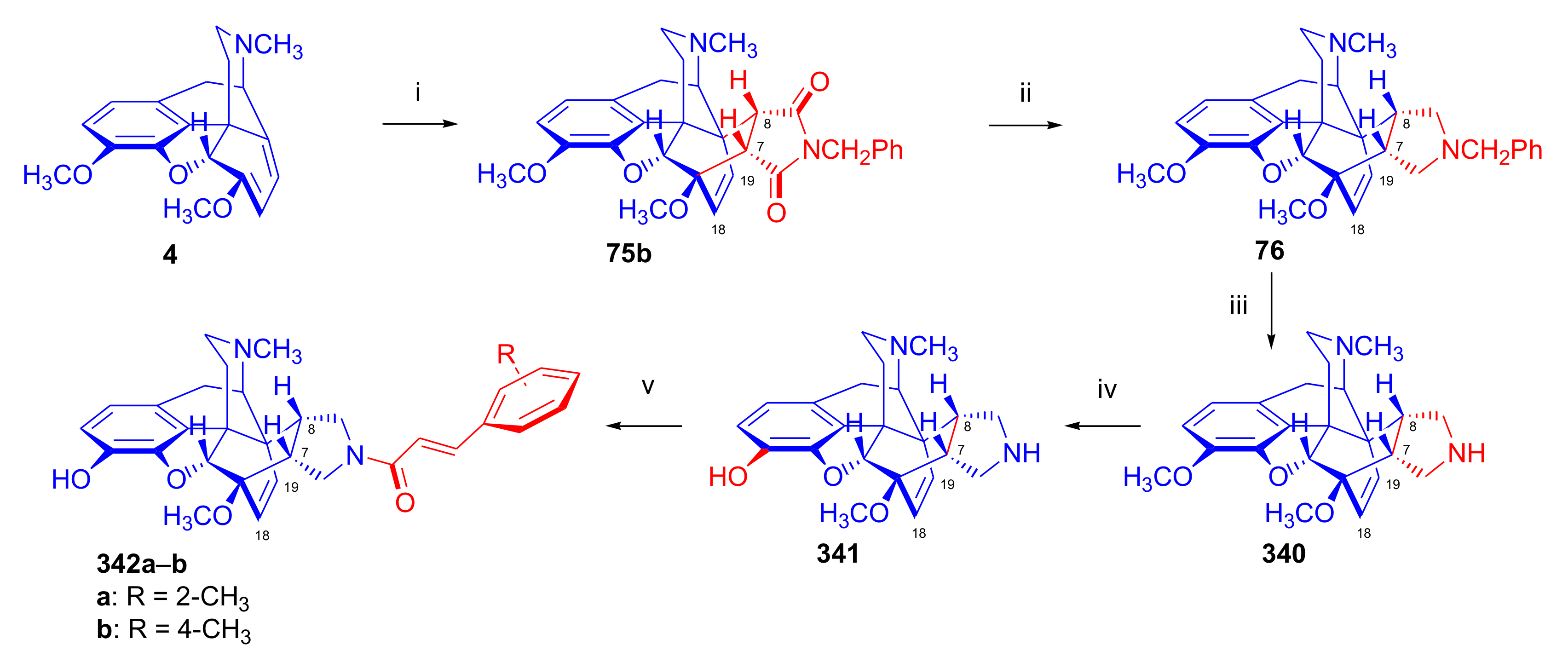{kind=link}
{kind=link}
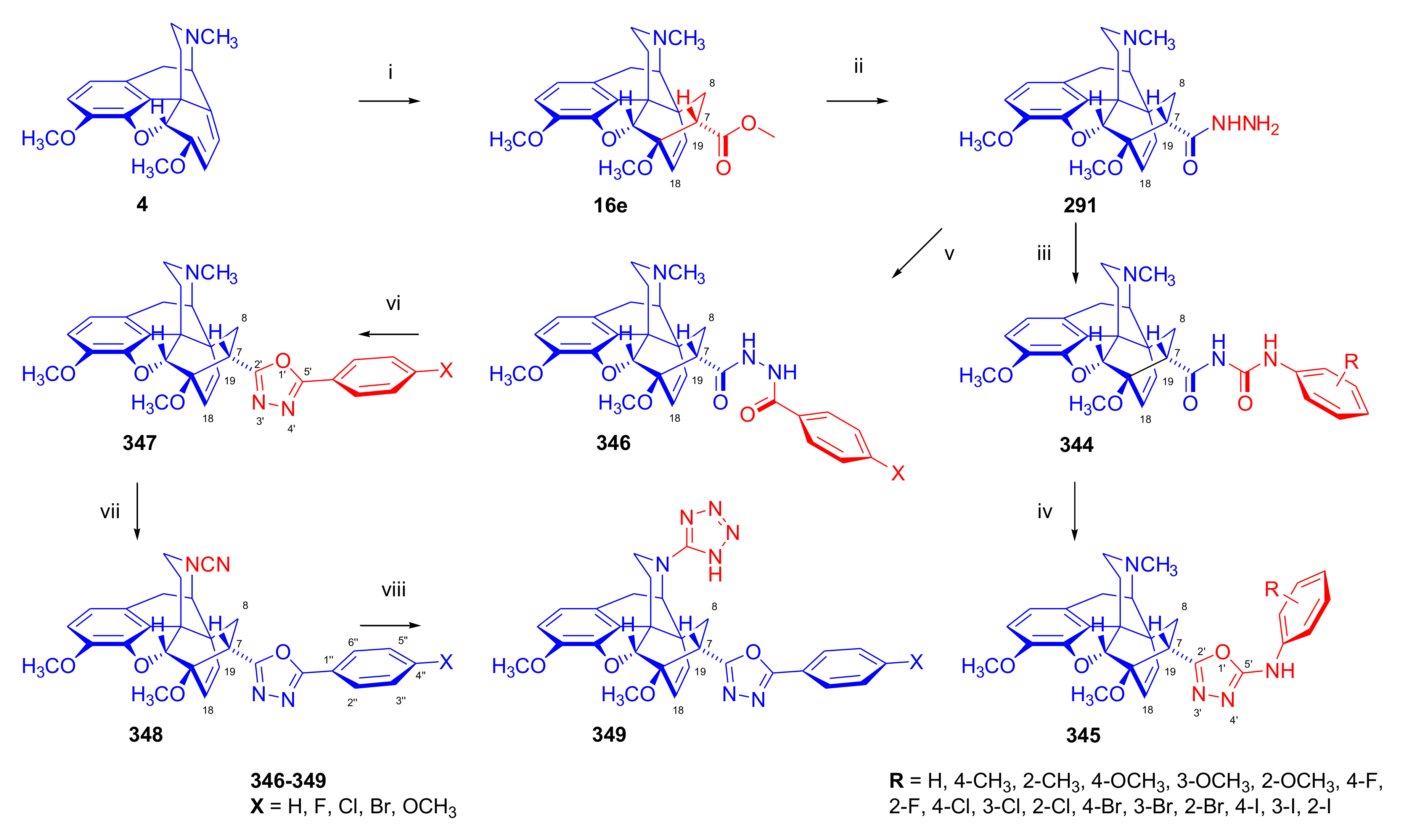{kind=link}
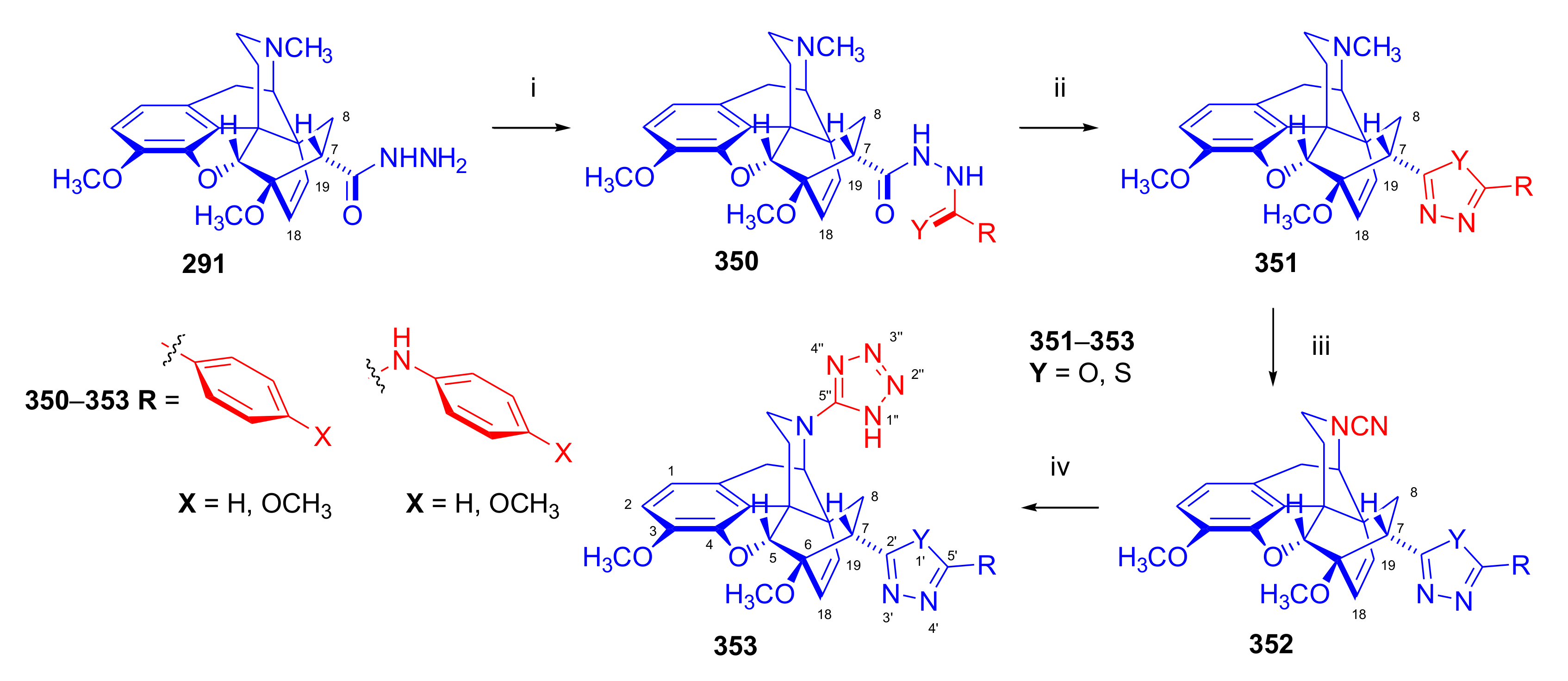{kind=link}
{kind=link}
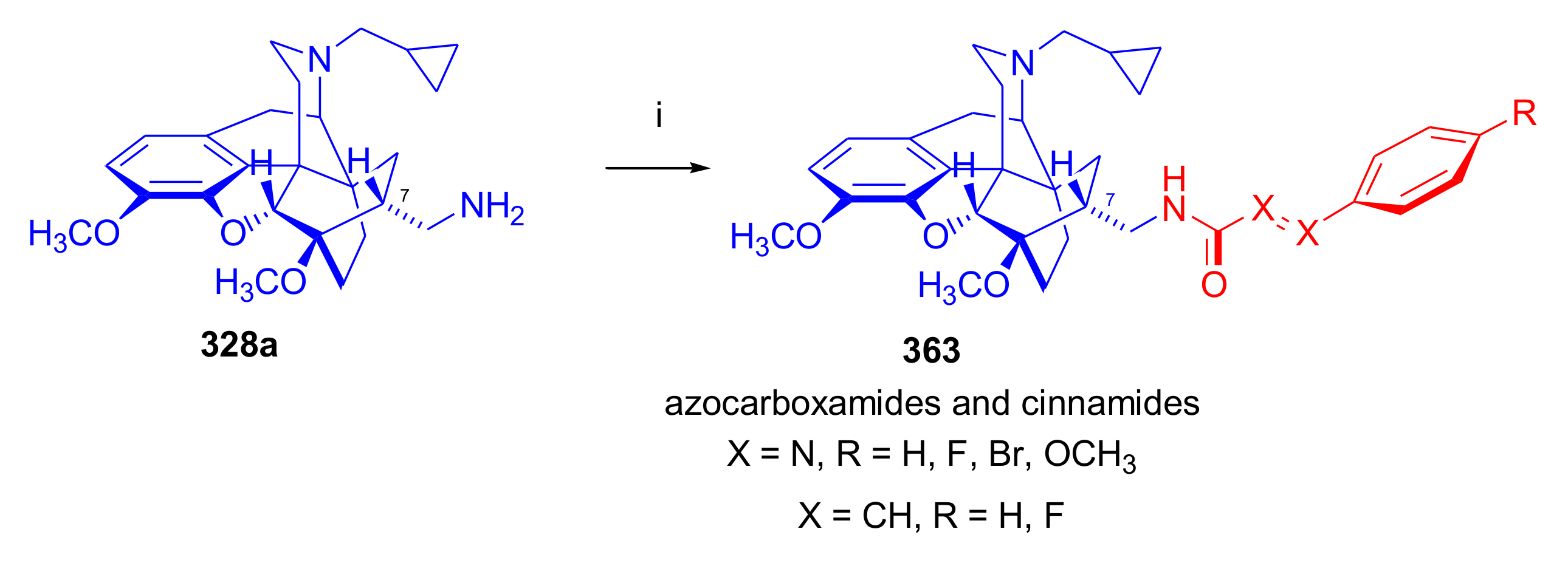{kind=link}
{kind=link}
{kind=link}
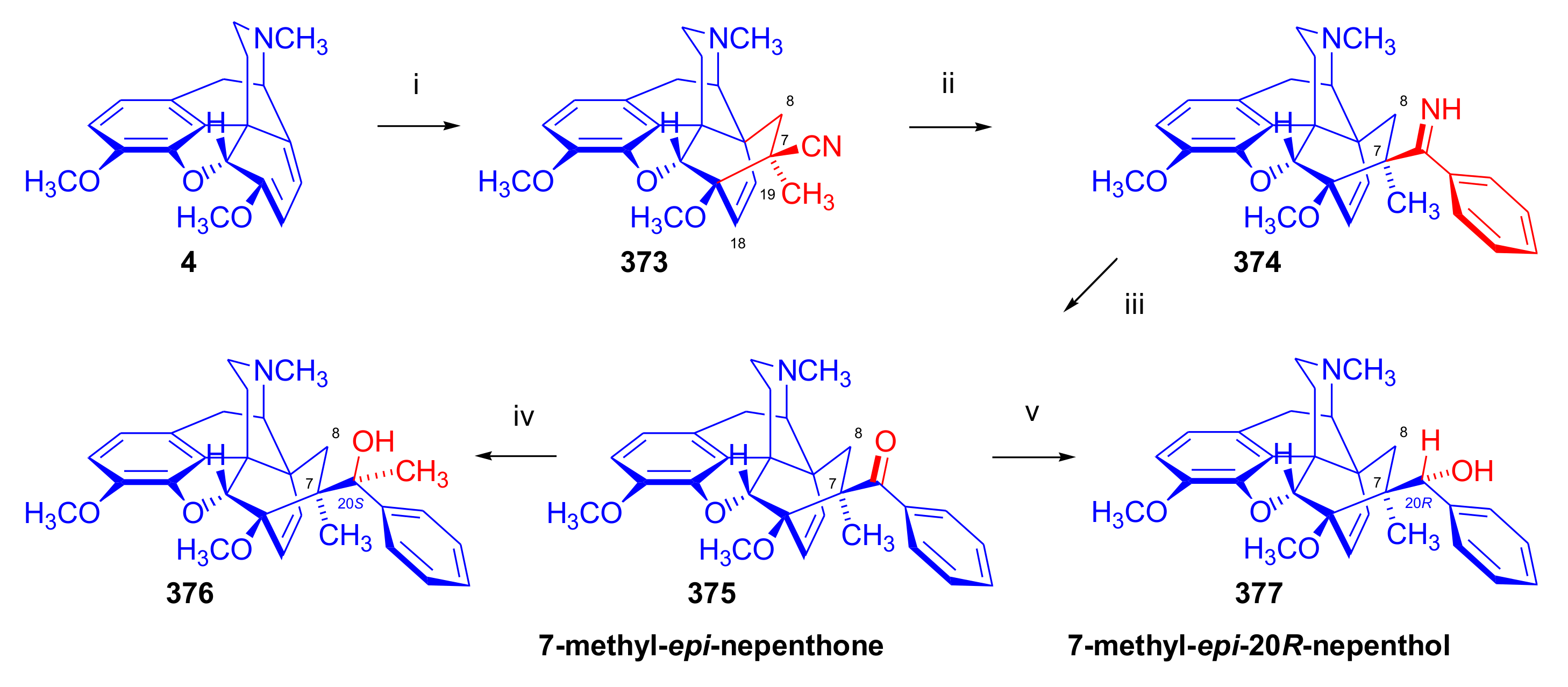{kind=link}
{kind=link}
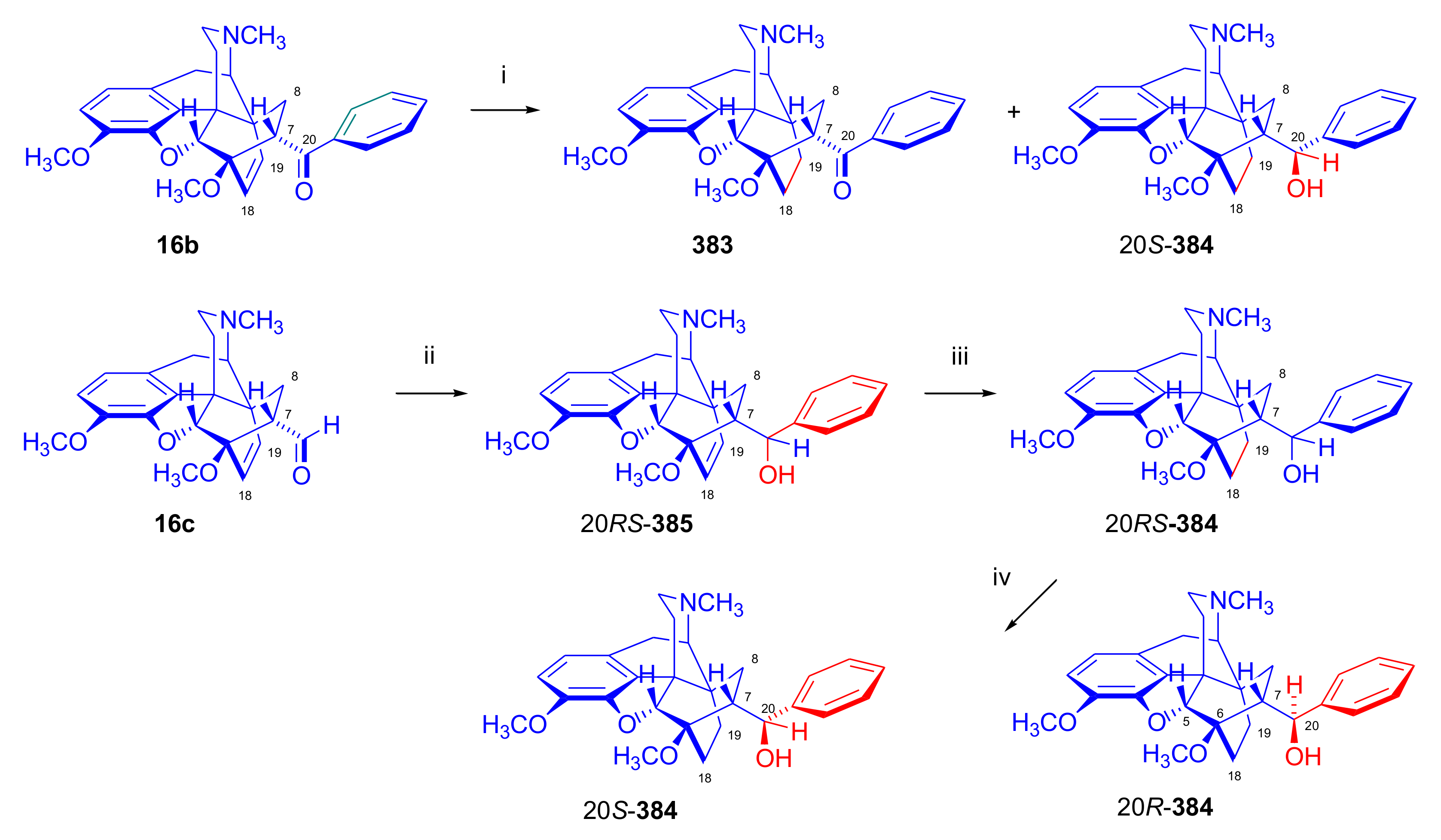{kind=link}
{kind=link}
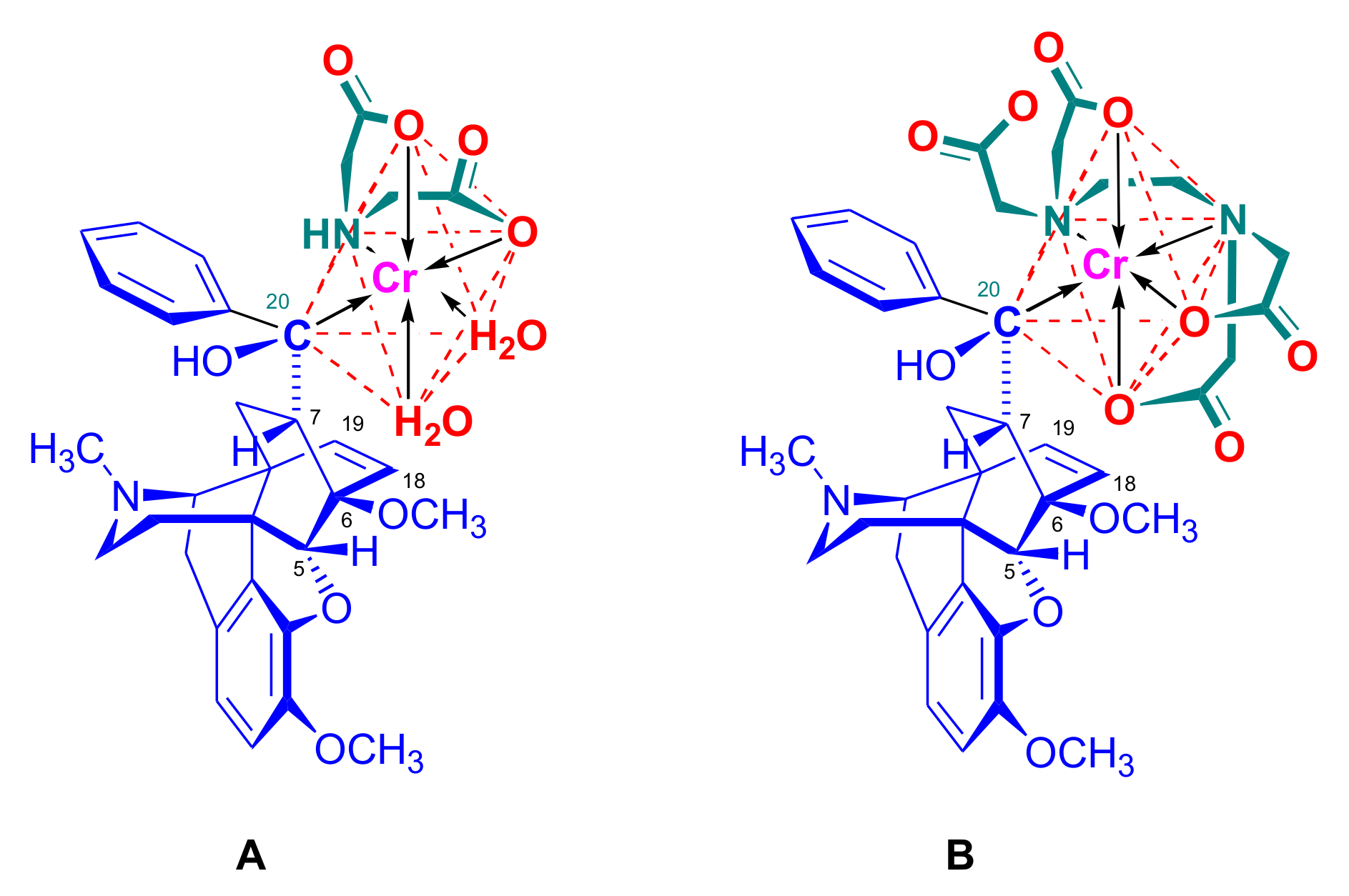{kind=link}
{kind=link}
{kind=link}
{kind=link}
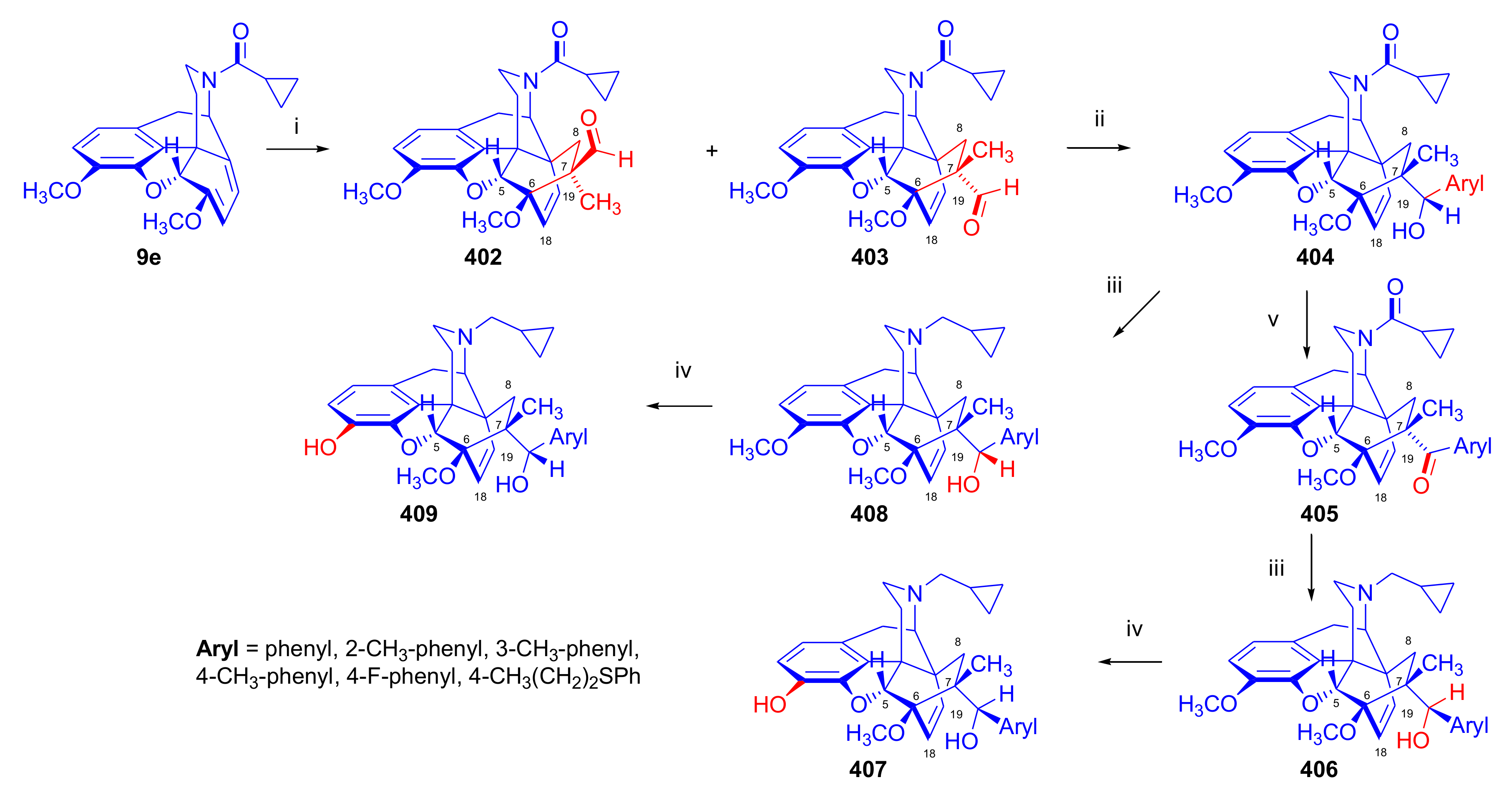{kind=link}
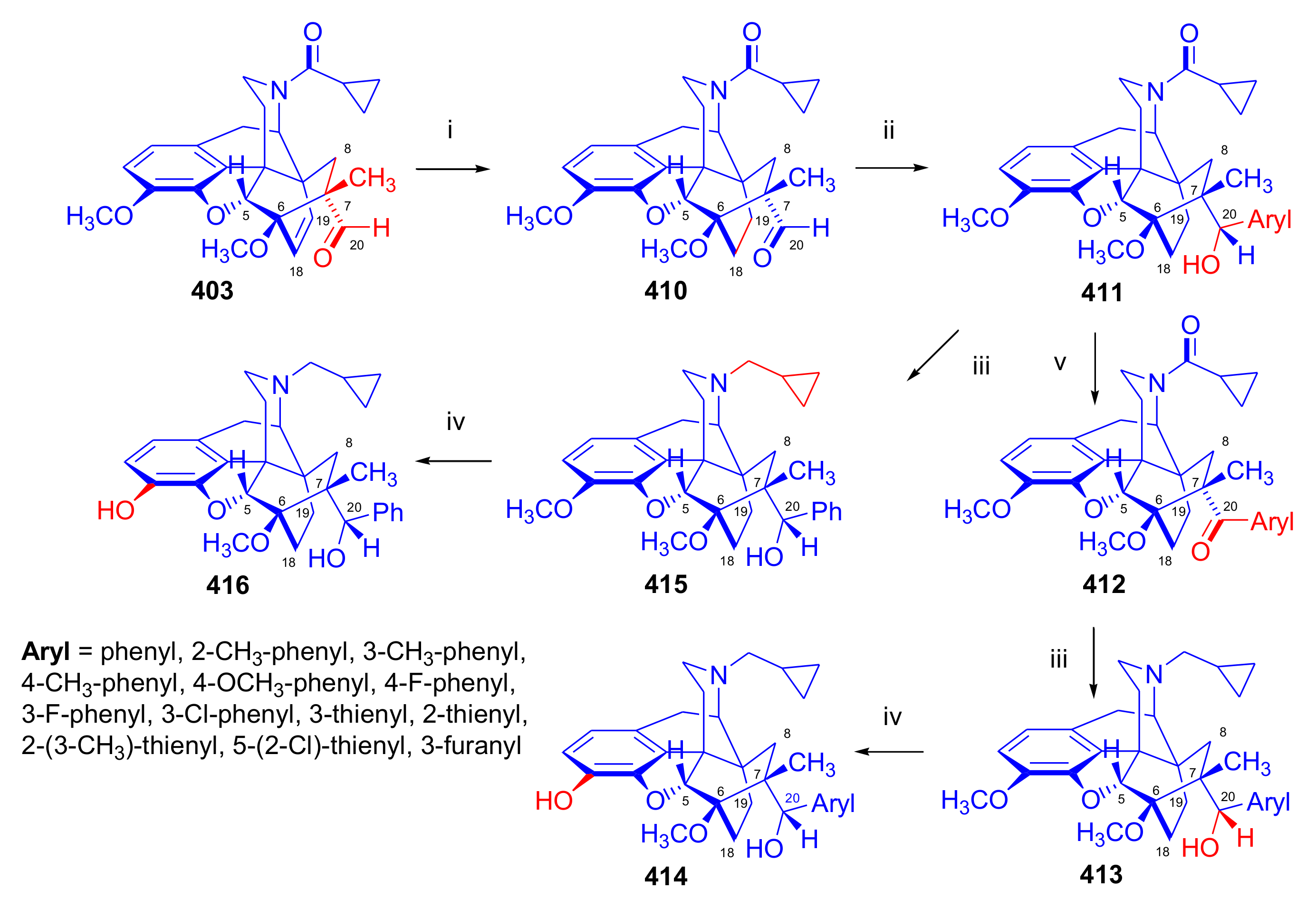{kind=link}
{kind=link}
{kind=link}
{kind=link}
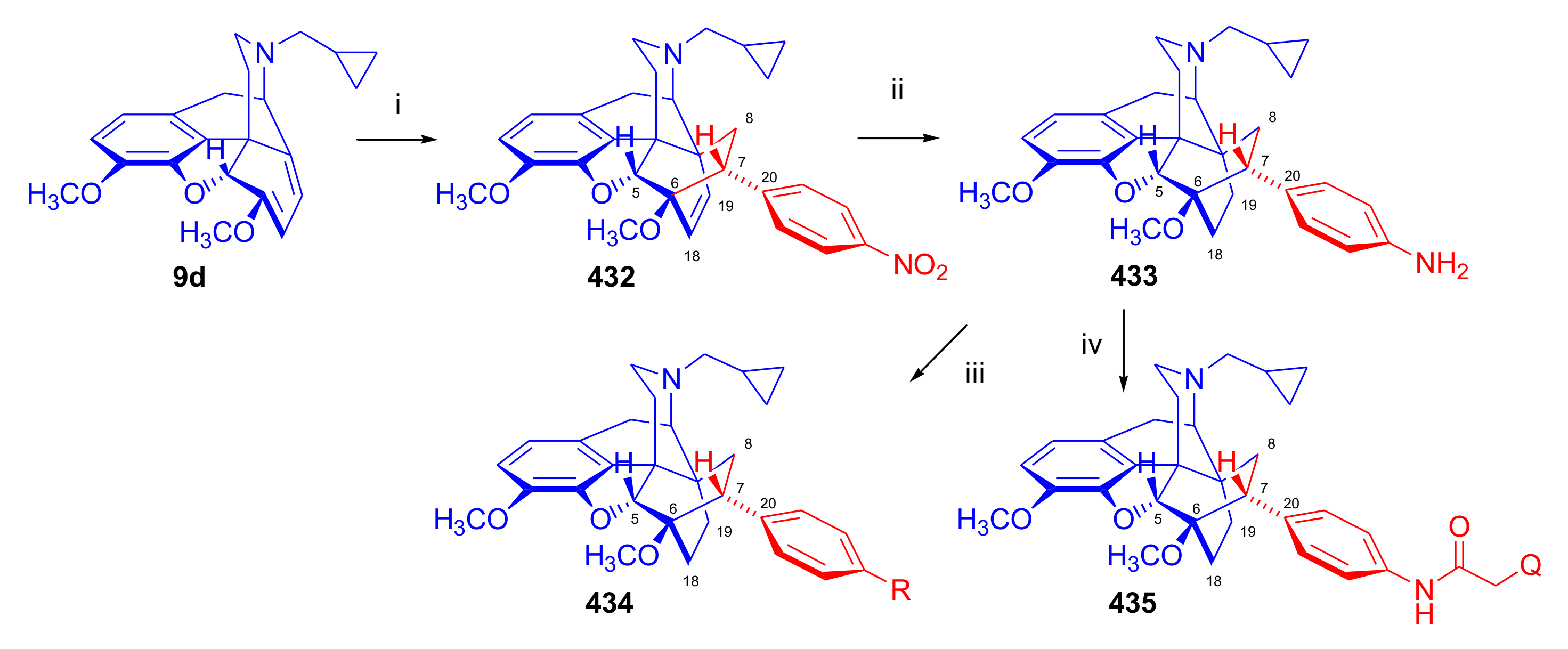{kind=link}
{kind=link}
{kind=link}
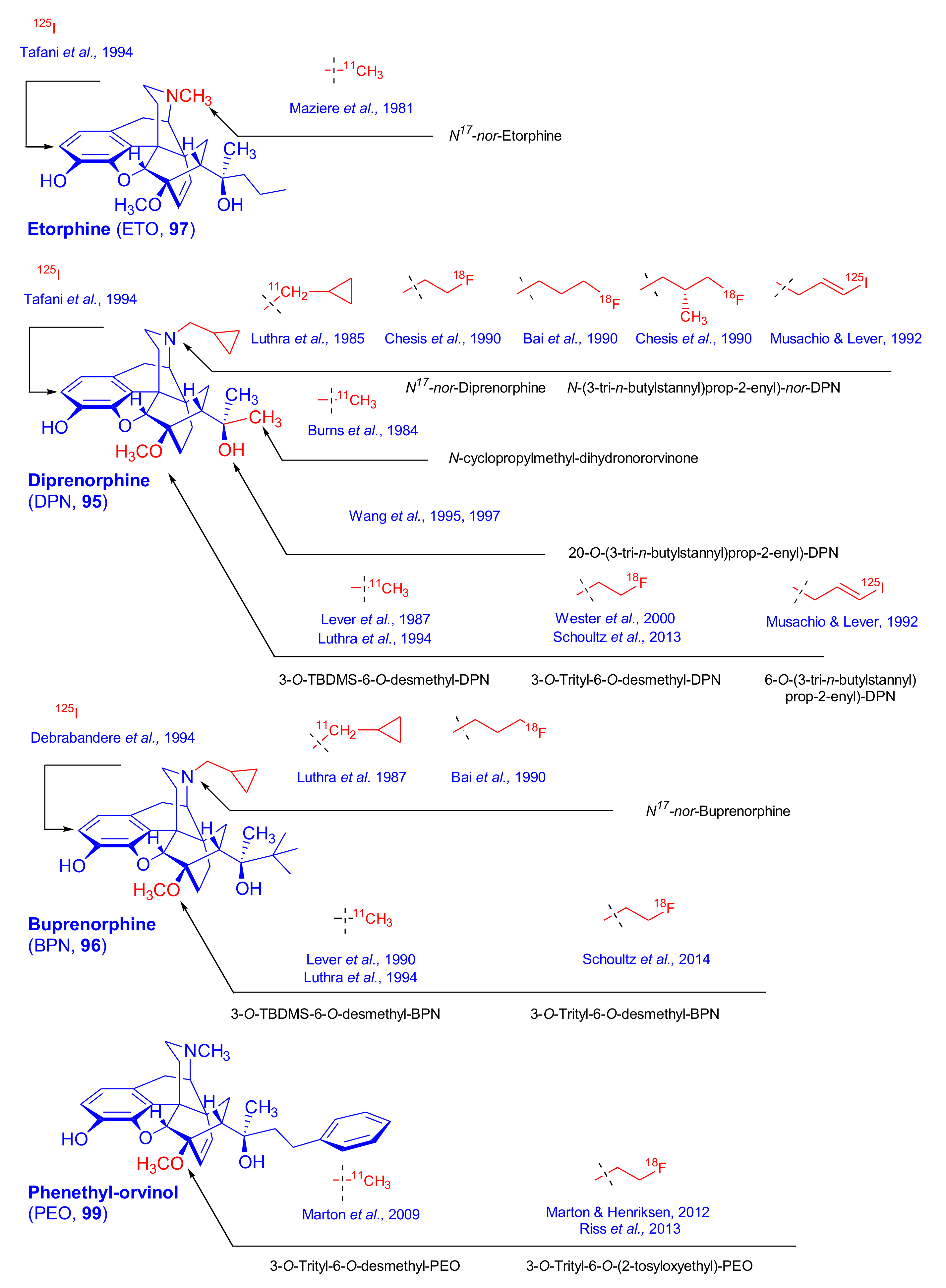{kind=link}
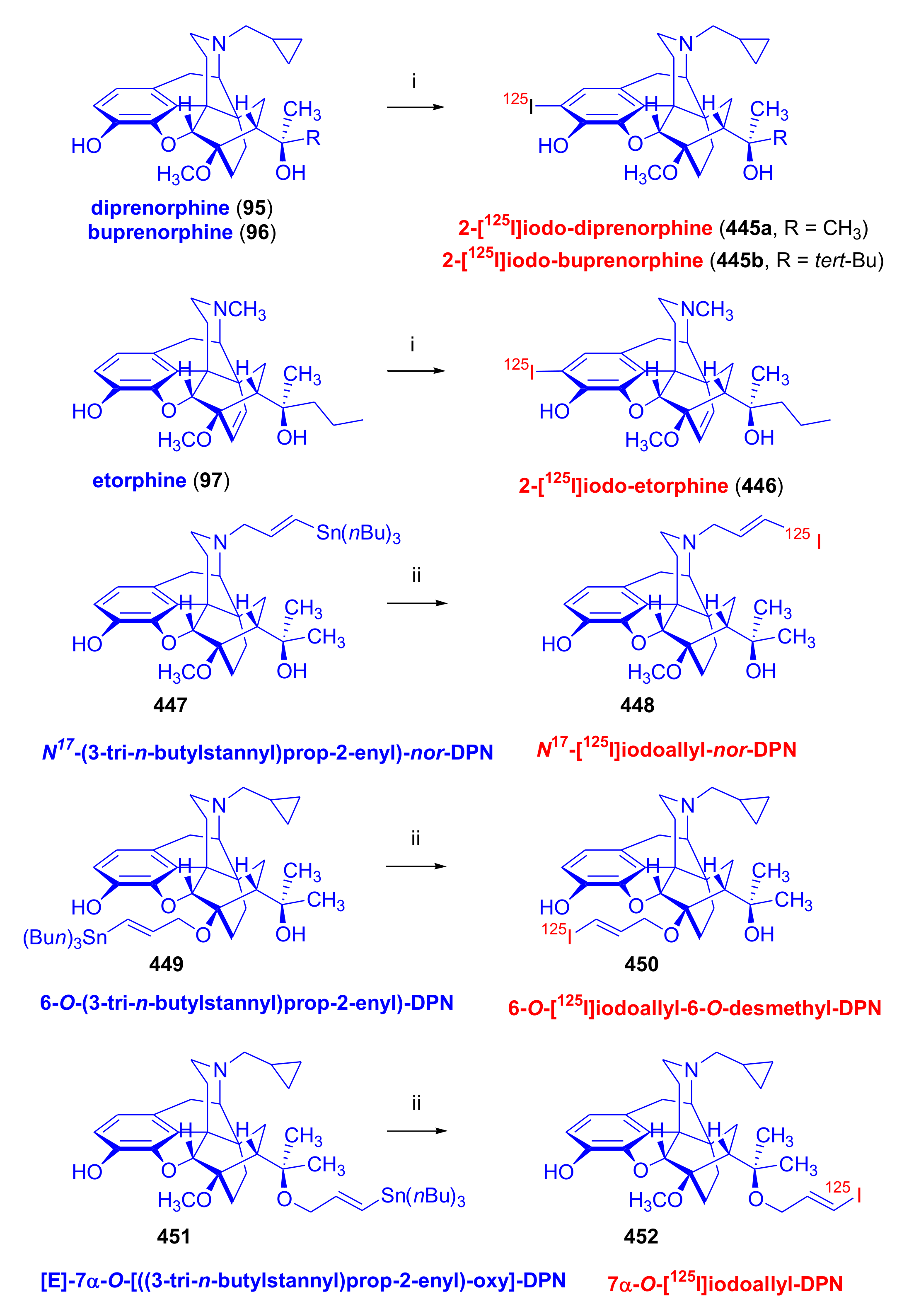{kind=link}
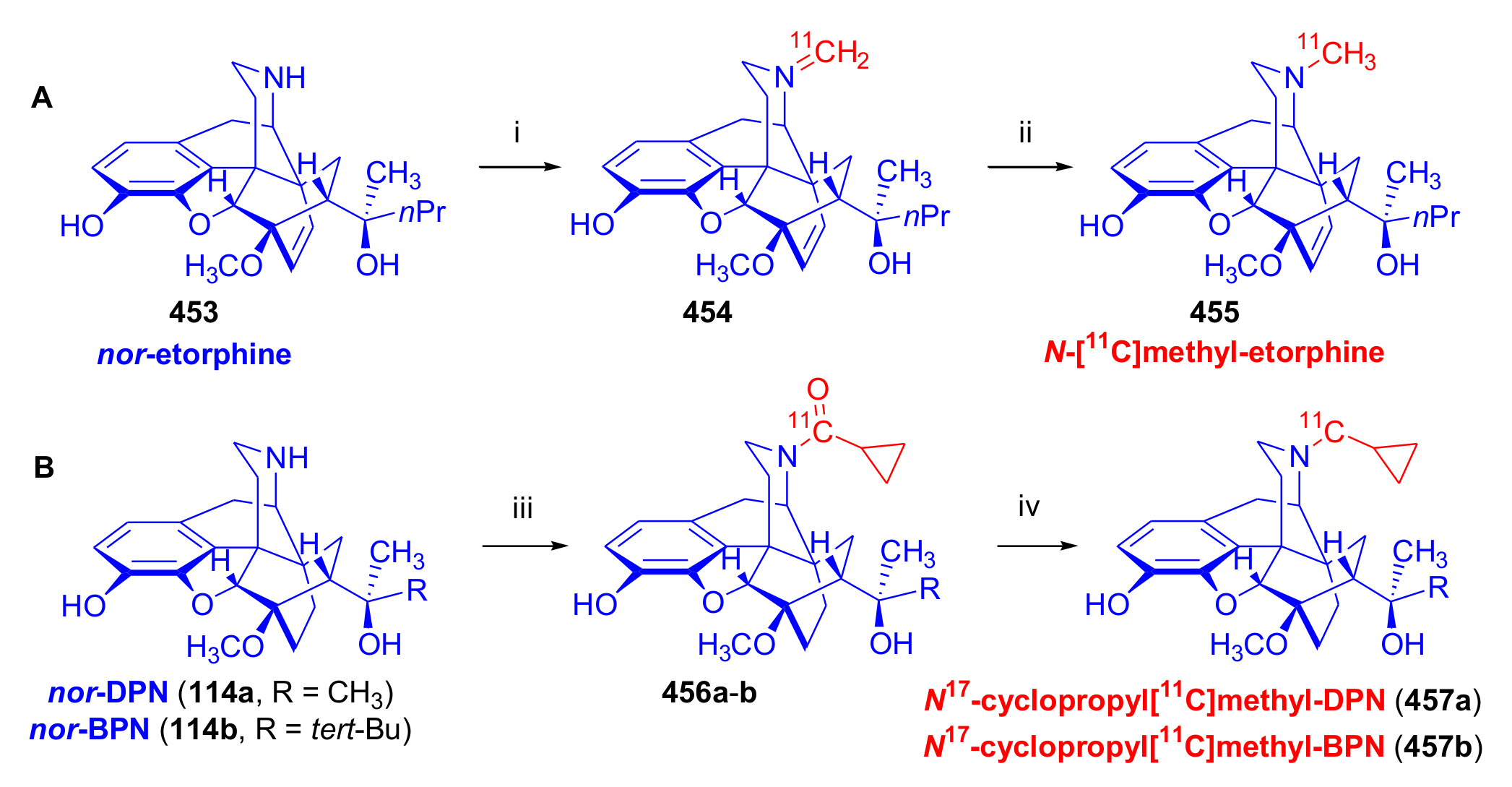{kind=link}
{kind=link}
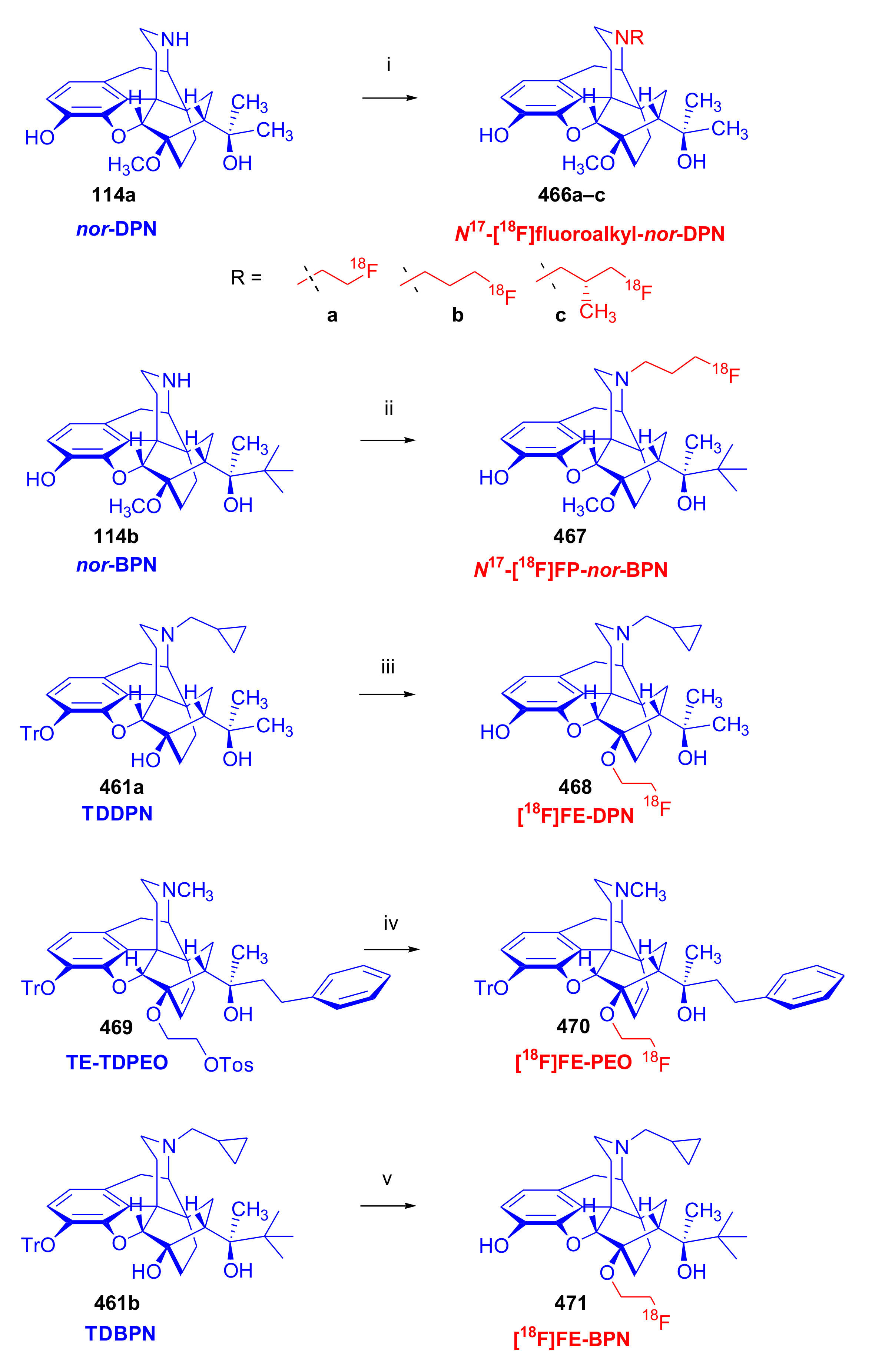{kind=link}
{kind=link}
| Comp. | Name | R1 | R2 | R3 | R4 | References |
|---|---|---|---|---|---|---|
| 4 | Thebaine | CH3 | - | - | - | Berényi [38] |
| 5 | Oripavine | H | - | - | - | Hosztafi [39] |
| 9a | N17-formyl-northebaine | CH3 | CHO | - | - | Maat [40] |
| 9b | N17-benzyl-northebaine | CH3 | Bn | - | - | Fleischhacker [41] |
| 9c | N17-benzoyl-northebaine | CH3 | Bz | - | - | Maurer [42] |
| 9d | N17-cyclopropylmethyl-northebaine | CH3 | CPM | - | - | Lewis [43] |
| 9e | N17-cyclopropylcarbonyl-northebaine | CH3 | CPCO | - | - | Barton [44] |
| 9f | N17-Boc-northebaine | CH3 | Boc | - | - | Sandulenko [45] |
| 10a | β-dihydrothebaine | H | CH3 | OCH3 | - | Bentley [46] |
| 10b | 4-O-acetyl-β-dihydrothebaine | Ac | CH3 | OCH3 | - | Hayakawa [47] |
| 10c | 4-O-phenyl-β-dihydrothebaine | Ph | CH3 | OCH3 | - | Ghosh [48] |
| 10d | 6-demethoxy-β-dihydrothebaine | H | CH3 | H | - | Linders [49] |
| 10e | 4-O-acetyl-N17-formyl-6-demethoxy-β-dihydrothebaine | Ac | CHO | H | - | Linders [50] |
| 11a | 6-deoxythebaine | CH3 | CH3 | CH3 | H | Knipmeyer [51] |
| 11b | 6-demethoxythebaine | CH3 | CH3 | H | H | Berényi [52] |
| 11c | 6-demethoxyoripavine | H | CH3 | H | H | Berényi [53] |
| 11d | N17-formyl-6-demethoxynorthebaine | CH3 | CHO | H | H | Linders [54] |
| 11e | 6-fluoro-6-demethoxythebaine | CH3 | CH3 | F | H | Berényi [55] |
| 11f | 6-chloro-6-demethoxythebaine | CH3 | CH3 | Cl | H | Berényi [56] |
| 11g | 6-bromo-6-demethoxythebaine | CH3 | CH3 | Br | H | Berényi [56] |
| 11h | 6-thiocyanato-6-demethoxythebaine | CH3 | CH3 | SCN | H | Berényi [57] |
| 11i | 6-isothiocyanato-6-demethoxythebaine | CH3 | CH3 | NCS | H | Csutorás [58] |
| 11j | 6-azido-6-demethoxythebaine | CH3 | CH3 | N3 | H | Csutorás [58] |
| 11k | 7-chloro-6-demethoxythebaine | CH3 | CH3 | H | Cl | Simon [59] |
| 11l | 7-bromo-6-demethoxythebaine | CH3 | CH3 | H | Br | Simon [59] |
| 11m | 7-thiocyanato-6-demethoxythebaine | CH3 | CH3 | H | SCN | Berényi [57] |
| 12a | 5β-methylthebaine | CH3 | CH3 | CH3 | H | Boden [60] |
| 12b | 5β-ethylthebaine | CH3 | CH3 | Et | H | Woudenberg [61] |
| 12c | 5β-butylthebaine | CH3 | CH3 | Bu | H | Woudenberg [61] |
| 12d | thebaine-5β-methanol | CH3 | CH3 | CH2OH | H | Woudenberg [62] |
| 12e | 10α-ethyl-5β-methylthebaine | CH3 | CH3 | CH3 | Et | Woudenberg [61] |
| 12f | 5β,10α-diethylthebaine | CH3 | CH3 | Et | Et | Woudenberg [61] |
| 12g | 5β-trimethylsilylthebaine | CH3 | CH3 | SiMe3 | H | Chen [63] |
| 13a | 5β-methyl-6-demethoxythebaine | CH3 | H | - | - | Woudenberg [64] |
| 13b | 7-chloro-5β-methyl-6-demethoxythebaine | CH3 | Cl | - | - | Woudenberg [65] |
| 13c | 7-methoxy-5β-methyl-6-demethoxythebaine | CH3 | OCH3 | - | - | Woudenberg [66] |
| 14a | 6-ethoxy-6-demethoxythebaine | CH3 | CH3 | EtO | - | Czakó [67] |
| 14b | 6-propoxy-6-demethoxythebaine | CH3 | CH3 | nPrO | - | Czakó [67] |
| 14c | 6-cyclopropylmethoxy-6-demethoxythebaine | CH3 | CH3 | CPMO | - | Czakó [67] |
| 14d | 6-phenyl-6-demethoxythebaine | CH3 | CH3 | Ph | - | Czakó [67] |
| 15a | 2′-aminothiazolo-[6,7:4′,5′]-morphinan-6,8-diene | NH2 | - | - | - | Sipos [68] |
| 15b | 2′-methylaminothiazolo-[6,7:4′,5′]-morphinan-6,8-diene | CH3 | - | - | - | Sipos [68] |
| 15c | 2′-phenylaminothiazolo-[6,7:4′,5′]-morphinan-6,8-diene | Ph | - | - | - | Sipos [68] |
| Starting 7-Acyl Compound | Pos.-7 | X-Y | R1 | R2 | T [°C] | τ [h] | Product Ratio | |
|---|---|---|---|---|---|---|---|---|
| 7α | 7β | |||||||
| 112a | 7α | CH2-CH2 | CPM a | CH3 | 70 | 16 | 67 | 33 |
| 104 | 7α | CH2-CH2 | CH3 | CH3 | 90 | 16 | 58 | 42 |
| 172 | 7α | CH2-CH2 | CH3 | tert-Bu b | 90 | 18 | 50 | 50 |
| 383 | 7α | CH2-CH2 | CH3 | Ph c | 70 | 4 | 56 | 44 |
| 16a | 7α | CH=CH | CH3 | CH3 | 70 | 4.5 | 67 | 17 |
| 17a | 7β | CH=CH | CH3 | CH3 | 70 | 4 | 83 | 17 |
| Ligand | Comp. | Ki [nM] | Action | References | |||
|---|---|---|---|---|---|---|---|
| μ-OR | δ-OR | κ-OR | NOP | ||||
| morphine | 1 | 2.06 | >10,000 | 134 | >10,000 | agonist μ-OR | Valenzano, 2004 [271] |
| DPN | 95 | 0.07 | 0.23 | 0.02 | ND | antagonist | Raynor, 1994 [272] |
| BPN | 96 | 1.5 | 6.1 | 2.5 | 77.4 | partial μOR agonist, κOR antagonist, partial NOP agonist | Cami-Cobeci, 2011 [189] |
| 5β-Me-BPN [a] | - | - | - | - | ND | inactive [a] | Aceto, 1984 [273] |
| PEO | 99 | 0.18 | 5.1 | 0.12 | ND | full agonist | Marton, 2009 [98] |
| FE-DPN | 468 | 0.24 | 8.0 | 0.20 | ND | antagonist | Schoultz, 2014 [274] |
| FE-BPN | 471 | 0.24 | 2.1 | 0.12 | ND | mixed agonist antagonist | Schoultz, 2014 [274] |
| FE-PEO | 470 | 0.10 | 0.49 | 0.08 | ND | full agonist | Schoultz, 2014 [274] |
| ETO | 97 | 0.33 | 1.54 | 0.22 | 56.9 | full agonist | Khroyan, 2011 [130] |
| ETO [b] | 97 | 0.62 (4.26) | ND | agonist | Biyashev, 2002 [113] | ||
| 7β-ETO [b] | 167a | 0.47 (11.5) | ND | agonist | Biyashev, 2002 [113] | ||
| 5β-Me-ETO [c] | - | 1.1 | 7.9 | 45.5 (κ1) 2670 (κ2) 12.7 (κ3) | ND | agonist | Maat, 1999 [275] |
| DHE | 98 | 0.45 | 1.82 | 0.57 | ND | agonist | Katsumata, 1995 [276] |
| DHE | 98 | 0.10 | 1.5 | 0.74 | 120 | full agonist | Olofsen, 2019 [126] |
| DHE [b] | 98 | 0.78 (13.1) | ND | agonist | Biyashev, 2002 [113] | ||
| 7β-DHE [b] | 167b | 1.61 (13.8) | ND | agonist | Biyashev, 2002 [113] | ||
| CYPR [d] | - | 0.076 | 0.68 | 0.79 | ND | antagonist | Smith, 1987 [277] |
| 16-Me-CYPR [d] | - | 1.77 | 0.73 | 59.6 | ND | pure antagonist | Smith, 1987 [277] |
| thienorphine | 100 | 0.22 | 0.69 | 0.14 | ND | κOR agonist | Li, 2007 [278] |
| BU08028 | 101 | 2.14 | 1.59 | 5.63 | 8.46 | mixed mOR/NOP agonist | Khroyan, 2011 [130] |
| BU127 | 396g | 0.71 | 1.91 | 0.49 | 43.2 | μOR and κOR antagonist, NOP agonist | Lewis, 2013 [279] Kumar, 2014 [114] |
| BU128 | - | 0.08 | 0.48 | 0.08 | 97 | μOR and κOR antagonist, NOP agonist | Lewis, 2013 [278] |
| BU10112 | - | 0.17 | 0.40 | 0.04 | 79 | μOR and κOR antagonist, NOP agonist | Lewis, 2013 [243] |
| BU10119 | 102 | 0.10 | 0.25 | 0.04 | 80 | μOR and κOR antagonist, NOP agonist | Lewis, 2013 [279] |
| BU10120 | 103 | 0.16 | 0.47 | 0.05 | 34 | μOR and κOR antagonist, NOP agonist | Lewis, 2013 [279] |
| SLL-020ACP [e] | 390a | 129.5 | 773.7 | 0.40 | ND | selective κOR agonist | Li, 2017 [164] |
| “SMH-15a” | 414 | 0.10 | 0.25 | 0.04 | 80 | κOR antagonist | Cueva, 2015 [131] |
| SLL-039 | 434 | 321.3 | 133.7 | 0.47 | ND | κOR agonist | Xiao, 2019 [266] |
| “LS-4j” | 435 | 114 | 293 | 1.8 | ND | agonist | Liu, 2021 [267] |
| Radioligand | No. | Decay-Corrected RCY [%] | Preparation Time [min.] | RCP [%] | Molar Activity (EOS) [GBq/μmol] | Reference |
|---|---|---|---|---|---|---|
| [N-methyl-11C]etorphine | 455 | 12–16 * | n.d. | n.d. | 0.03 | [299] (1981) |
| [20-methyl-11C]thevinol | - | 8 * | n.d. | n.d. | 7.4 | [300] (1984) |
| [N-cyclopropylmethyl-11C]DPN | 457a | 5–20 | 53 | 100 | >1.7 | [301] (1985) |
| [N-cyclopropylmethyl-11C]BPN | 457b | 20 | 57 | 100 | 6 | [302] (1987) |
| [6-O-methyl-11C]DPN | 460a | 28 | 30 | n.d. | 64.4 | [183] (1987) |
| [6-O-methyl-11C]DPN | 460a | 60–88 | 45 | >99 | 15.5–23.8 | [185] (1994) |
| [6-O-methyl-11C]DPN | 460a | 32 | 48 | 97 | 242.1 | [306] (2014) |
| [6-O-methyl-11C]DPN | 460a | 22.3 a | n.d. | 93.5 | 370 | [306] (2014) |
| [6-O-methyl-11C]DPN | 460a | n.d. | 40–50 | >96 | 4–36 | [308] (2000) |
| [6-O-methyl-11C]BPN | 460b | 23 | 24 | 97 | 41 | [184] (1990) |
| [6-O-methyl-11C]BPN | 460b | 69–93 | 50 | >99 | 12.8–21.2 | [185] (1994) |
| [6-O-methyl-11C]BPN | 460b | 60 | 40 | >97 | 7.6–20.9 | [304] (1996) |
| [6-O-methyl-11C]PEO | 465 | 41–73 | n.d. | >99 | 60 | [98] (2009) |
| Radioligand | No. | Decay-Corrected RCY [%] | Preparation Time [min.] | RCP [%] | Molar Activity (EOS) [GBq/μmol] | Reference |
|---|---|---|---|---|---|---|
| N17-[18F]FE-nor-DPN | 466a | 0.3–0.6 * | n.d. | n.d. | 4–10 | [309] (1990) |
| N17-[18F]FP-nor-DPN | 466b | 4–6 * | n.d. | n.d. | 31–67 | [309] (1990) |
| N17-[18F]FP-nor-DPN | 466b | 15 | 100 | n.d. | n.d. | [310] (1990) |
| N17-((S)-3-[18F]fluoro-2-methylpropyl)-nor-DPN | 466c | 4–6 * | n.d. | n.d. | 31–67 | [309] (1990) |
| [18F]FE-DPN | 468 | 22 | 100 | 98 | 37 | [308] (2000) |
| [18F]FE-DPN | 468 | 18–34 | 100 | n.d. | 50–300 | [274] (2014) |
| [18F]FE-PEO | 470 | 35 | 42 | >99 | 55–128 | [177] (2012) |
| [18F]FE-PEO | 470 | 28 | 90 | >97 | 52–224 | [314] (2013) |
| [18F]FE-PEO | 470 | 18–34 | 100 | n.d. | 50–300 | [274] (2014) |
| [18F]FE-BPN | 471 | 18–34 | 100 | n.d. | 50–300 | [274] (2014) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marton, J.; Fekete, A.; Cumming, P.; Hosztafi, S.; Mikecz, P.; Henriksen, G. Diels–Alder Adducts of Morphinan-6,8-Dienes and Their Transformations. Molecules 2022, 27, 2863. https://doi.org/10.3390/molecules27092863
Marton J, Fekete A, Cumming P, Hosztafi S, Mikecz P, Henriksen G. Diels–Alder Adducts of Morphinan-6,8-Dienes and Their Transformations. Molecules. 2022; 27(9):2863. https://doi.org/10.3390/molecules27092863
Chicago/Turabian StyleMarton, János, Anikó Fekete, Paul Cumming, Sándor Hosztafi, Pál Mikecz, and Gjermund Henriksen. 2022. "Diels–Alder Adducts of Morphinan-6,8-Dienes and Their Transformations" Molecules 27, no. 9: 2863. https://doi.org/10.3390/molecules27092863