
Covalently Immobilizable Tris(Pyridino)-Crown Ether for Separation of Amines Based on Their Degree of Substitution

 ,
, 
Abstract
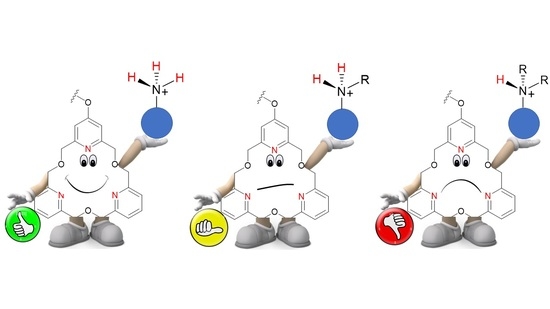
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Design of the New Selector Molecule
2.2. Synthesis
2.3. Molecular Recognition Studies
3. Conclusions
4. Experimental
4.1. General
4.2. Synthesis
4.2.1. Preparation of {6-[(Triphenylmethoxy)methyl]pyridin-2-yl}methyl 4-methylbenzene-1-sulfonate (5)
4.2.2. Preparation of {6-[(Benzyloxy)methyl]pyridin-2-yl}methanol (6)
4.2.3. Preparation of 2-[(Triphenylmethoxy)methyl]-6-[({6-[(triphenylmethoxy)methyl]pyridin-2-yl}methoxy)methyl]pyridine (7)
4.2.4. Preparation of 2-[(Benzyloxy)methyl]-6-[({6-[(triphenylmethoxy)methyl]pyridin-2-yl}methoxy)methyl]pyridine (8)
4.2.5. General Procedure for Removing Trityl Protective Group
4.2.6. Preparation of {6-[({6-[(Benzyloxy)methyl]pyridin-2-yl}methoxy)methyl]pyridin-2-yl}methanol (9)
4.2.7. Preparation of (6-{[(6-{[(4-Methylbenzenesulfonyl)oxy]methyl}pyridin-2-yl)methoxy]methyl}pyridin-2-yl)methyl 4-methylbenzene-1-sulfonate (11)
4.2.8. Preparation of 23-(Benzyloxy)-3,11,19-trioxa-25,26,27-triazatetracyclo[19.3.1.15,9.113,17]heptacosa-1(25),5,7,9(27),13(26),14,16,21,23-nonaene (1)
4.2.9. Preparation of 3,11,19-Trioxa-25,26,27-triazatetracyclo[19.3.1.15,9.113,17]heptacosa-1(24),5,7,9(27),13,15,17(26),21(25),22-nonaen-7-ol (14)
4.3. Molecular Recognition Studies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Yang, S.; Wu, F.; Yu, F.; Gu, L.; Wang, H.; Liu, Y.; Chu, Y.; Wang, F.; Fang, X.; Ding, C.-F. Distinction of chiral penicillamine using metal-ion coupled cyclodextrin complex as chiral selector by trapped ion mobility-mass spectrometry and a structure investigation of the complexes. Anal. Chim. Acta 2021, 1184, 339017. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Y.; Liu, Y.; Mao, Y.; Han, J.; Li, W.; Wang, Y. Recyclable aptamer-derived aqueous two-phase flotation for high-efficiency separation of mercury(II) ions modulated by aggregation states. Sep. Purif. Technol. 2021, 274, 118917. [Google Scholar] [CrossRef]
- Steed, J.W.; Atwood, J.L. Supramolecular Chemistry, 3rd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2022; pp. 1–20. ISBN 978-1-119-58251-9. [Google Scholar]
- Buschmann, H.-J.; Mutihac, L.; Mutihac, R. Physicochemical Parameters of the Transport of Amines and Amino Acids through Liquid Membranes by Macrocyclic Ligands. Sep. Sci. Technol. 1999, 34, 331–341. [Google Scholar] [CrossRef]
- Dörwald, F.Z. Lead Optimization for Medicinal Chemists: Pharmacokinetic Properties of Functional Groups and Organic Compounds, 1st ed.; Wiley-VHC: Weinheim, Germany, 2012; pp. 107–135. ISBN 978-3-527-33226-7. [Google Scholar]
- Ouellette, R.J.; Rawn, J.D. Organic Chemistry: Structure, Mechanism, Synthesis, 2nd ed.; Academic Press, Elsevier Inc.: Cambridge, USA, 2018; pp. 803–842. ISBN 978-0-12-812838-1. [Google Scholar]
- Shriner, R.L.; Hermann, C.K.; Morrill, T.C.; Curtin, D.Y.; Fuson, R.C. The Systematic Identification of Organic Compounds, 5th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2003; pp. 247–296. ISBN 0-471-21503-1. [Google Scholar]
- Buschmann, H.-J.; Schollmeyer, E.; Mutihac, L. The complexation of the ammonium ion by 18-crown-6 in different solvents and by noncyclic ligands, crown ethers and cryptands in methanol. Supramol. Sci. 1998, 5, 139–142. [Google Scholar] [CrossRef]
- Buschmann, H.-J.; Schollmeyer, E.; Mutihac, L. The Complexation of Amino Acids by Crown Ethers and Cryptands in Methanol. J. Incl. Phenom. Macrocycl. Chem. 1998, 30, 21–28. [Google Scholar] [CrossRef]
- Newcomb, M.; Gokel, G.W.; Cram, D.J. Pyridyl unit in host compounds. J. Am. Chem. Soc. 1974, 96, 6810–6811. [Google Scholar] [CrossRef]
- Newcomb, M.; Timko, J.M.; Walba, D.M.; Cram, D.J. Host-guest complexation. 3. Organization of pyridyl binding sites. J. Am. Chem. Soc. 1977, 99, 6392–6398. [Google Scholar] [CrossRef]
- Tsukube, H. Highly selective transport of biogenetic amines and drugs by using functionalized “acyclic crown ether”. Tetrahedron Lett. 1982, 23, 2109–2112. [Google Scholar] [CrossRef]
- Kupai, J.; Rojik, E.; Huszthy, P.; Szekely, G. Role of Chirality and Macroring in Imprinted Polymers with Enantiodiscriminative Power. ACS Appl. Mater. Interfaces 2015, 7, 9516–9525. [Google Scholar] [CrossRef]
- Kovács, E.; Deme, J.; Turczel, G.; Nagy, T.; Farkas, V.; Trif, L.; Kéki, S.; Huszthy, P.; Tuba, R. Synthesis and supramolecular assembly of fluorinated biogenic amine recognition host polymers. Polym. Chem. 2019, 10, 5626–5634. [Google Scholar] [CrossRef] [Green Version]
- Pál, D.; Móczár, I.; Szemenyei, B.; Marczona, D.; Kocsis, I.; Prikler, G.; Vezse, P.; Baranyai, P.; Huszthy, P. Pyridino-18-crown-6 ether type chemosensors containing a benzothiazole fluorophore unit: Synthesis and enantiomeric recognition studies. Tetrahedron 2019, 75, 2900–2909. [Google Scholar] [CrossRef]
- Horváth, G.; Huszthy, P.; Szarvas, S.; Szókán, G.; Redd, J.T.; Bradshaw, J.S.; Izatt, R.M. Preparation of a New Chiral Pyridino-Crown Ether-Based Stationary Phase for Enantioseparation of Racemic Primary Organic Ammonium Salts. Ind. Eng. Chem. Res. 2000, 39, 3576–3581. [Google Scholar] [CrossRef]
- Kupai, J.; Lévai, S.; Antal, K.; Balogh, G.T.; Tóth, T.; Huszthy, P. Preparation of pyridino-crown ether-based new chiral stationary phases and preliminary studies on their enantiomer separating ability for chiral protonated primary aralkylamines. Tetrahedron Asymmetry 2012, 23, 415–427. [Google Scholar] [CrossRef]
- Lévai, S.; Németh, T.; Fődi, T.; Kupai, J.; Tóth, T.; Huszthy, P.; Balogh, G.T. Studies of a pyridino-crown ether-based chiral stationary phase on the enantioseparation of biogenic chiral aralkylamines and α-amino acid esters by high-performance liquid chromatography. J. Pharm. Biomed. Anal. 2015, 115, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Newkome, G.R.; Lee, H.; Fronczek, F.R. A New Route to Macrocycles Possessing the 2,6-Pyridino Subunitvia Bis-1,3-Dithianes. Isr. J. Chem. 1986, 27, 87–95. [Google Scholar] [CrossRef]
- Leygue, N.; Picard, C.; Faure, P.; Bourrier, E.; Lamarque, L.; Zwier, J.M.; Galaup, C. Design of novel tripyridinophane-based Eu(iii) complexes as efficient luminescent labels for bioassay applications. Org. Biomol. Chem. 2021, 20, 182–195. [Google Scholar] [CrossRef]
- Behera, H.; Ramkumar, V.; Madhavan, N. Triamide macrocyclic chloride receptors via a one-pot tandem reduction–condensation–cyclization reaction. Org. Biomol. Chem. 2017, 15, 4937–4940. [Google Scholar] [CrossRef]
- Nolan, C.; Gunnlaugsson, T. Improved synthesis of a C3-symmetrical pyridinophane. Tetrahedron Lett. 2008, 49, 1993–1996. [Google Scholar] [CrossRef]
- Zubenko, A.D.; Fedorova, O.A. Aromatic and heteroaromatic azacrown compounds: Advantages and disadvantages of rigid macrocyclic ligands. Russ. Chem. Rev. 2020, 89, 750–786. [Google Scholar] [CrossRef]
- He, Y.B.; Cai, H.B.; Meng, L.Z.; Wu, C.T. Studies on the syntheses of azacrown compounds containing pyridine ring and central functional group. Chem. J. Chin. Univ. Chin. Ed. 1997, 18, 1973–1977. [Google Scholar]
- Weber, E.; Josel, H.P.; Puff, H.; Franken, S. Solid-state inclusion compounds of new host macrocycles with uncharged organic molecules. Host synthesis, inclusion properties, and x-ray crystal structure of an inclusion compound with n-propanol. J. Org. Chem. 1985, 50, 3125–3132. [Google Scholar] [CrossRef]
- Voegtle, F.; Oepen, G.; Rasshofer, W. Complexes between neutral molecules. V. Urea, thiourea and malonodinitrile complexes of noncyclic crown-type polyethers with pyridine N-oxide subunits. Liebigs Ann. Chem. 1979, 1, 1577–1584. [Google Scholar] [CrossRef]
- Cram, D.J. Multiheteromacrocycles. Federal Republic of Germany DE2539324, 1976. [Google Scholar]
- Diebold, A.; Fischer, J.; Weiss, R. Synthesis and spectroscopic and structural characterization of potentially hetero dinucleating ligands containing a meso-diphenylporphyrin. New J. Chem. 1996, 20, 959–970. [Google Scholar]
- Tsukube, H.; Shinoda, S.; Uenishi, J.; Hiraoka, T.; Imakoga, T.; Yonemitsu, O. Ag+-Specific Pyridine Podands: Effects of Ligand Geometry and Stereochemically Controlled Substitution on Cation Complexation and Transport Functions. J. Org. Chem. 1998, 63, 3884–3894. [Google Scholar] [CrossRef]
- Howáth, G.; Rusa, C.; Köntös, Z.; Gerencsér, J.; Huszthy, P. A new Efficient Method for the Preparation of 2,6-Pyridinedihiethyl Ditosylates from Dimethyl 2,60-Pyridinedicarboxylates. Synth. Commun. 1999, 29, 3719–3731. [Google Scholar] [CrossRef]
- Golcs, Á.; Ádám, B.Á.; Horváth, V.; Tóth, T.; Huszthy, P. Synthesis, Molecular Recognition Study and Liquid Membrane-Based Applications of Highly Lipophilic Enantiopure Acridino-Crown Ethers. Molecules 2020, 25, 2571. [Google Scholar] [CrossRef]
- Golcs, Á.; Kovács, K.; Vezse, P.; Tóth, T.; Huszthy, P. Acridino-Diaza-20-Crown-6 Ethers: New Macrocyclic Hosts for Optochemical Metal Ion Sensing. Molecules 2021, 26, 4043. [Google Scholar] [CrossRef]
- Wang, T.; Bradshaw, J.S.; Huszthy, P.; Izatt, R.M. Various aspects of enantiomeric recognition of (S,S)-dimethylpyridino-18-crown-6 by several organic ammonium salts. Supramol. Chem. 1996, 6, 251–255. [Google Scholar] [CrossRef]
- Izatt, R.M.; Wang, T.; Hathaway, J.K.; Zhang, X.X.; Curtis, J.C.; Bradshaw, J.S.; Zhu, C.Y.; Huszthy, P. Factors influencing enantiomeric recognition of primary alkylammonium salts by pyridino-18-crown-6 type ligands. J. Incl. Phenom. Mol. Recognit. Chem. 1994, 17, 157–175. [Google Scholar] [CrossRef]
- Szemenyei, B.; Móczár, I.; Pál, D.; Kocsis, I.; Baranyai, P.; Huszthy, P. Synthesis and Enantiomeric Recognition Studies of Optically Active Pyridino-Crown Ethers Containing an Anthracene Fluorophore Unit. Chirality 2016, 28, 562–568. [Google Scholar] [CrossRef]
- Riddick, J.A.; Bunger, W.B.; Sakano, T.K. Organic Solvents: Physical Properties and Methods of Purification. In Techniques of Chemistry, 4th ed.; Wiley: New York, NY, USA, 1986; ISBN 978-0-471-08467-9. [Google Scholar]
- Thordarson, P. Determining association constants from titration experiments in supramolecular chemistry. Chem. Soc. Rev. 2011, 40, 1305–1323. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vezse, P.; Benda, B.; Fekete, A.; Golcs, Á.; Tóth, T.; Huszthy, P. Covalently Immobilizable Tris(Pyridino)-Crown Ether for Separation of Amines Based on Their Degree of Substitution. Molecules 2022, 27, 2838. https://doi.org/10.3390/molecules27092838
Vezse P, Benda B, Fekete A, Golcs Á, Tóth T, Huszthy P. Covalently Immobilizable Tris(Pyridino)-Crown Ether for Separation of Amines Based on Their Degree of Substitution. Molecules. 2022; 27(9):2838. https://doi.org/10.3390/molecules27092838
Chicago/Turabian StyleVezse, Panna, Bianka Benda, András Fekete, Ádám Golcs, Tünde Tóth, and Péter Huszthy. 2022. "Covalently Immobilizable Tris(Pyridino)-Crown Ether for Separation of Amines Based on Their Degree of Substitution" Molecules 27, no. 9: 2838. https://doi.org/10.3390/molecules27092838