
Human Estrogen Receptor Alpha Antagonists, Part 3: 3-D Pharmacophore and 3-D QSAR Guided Brefeldin A Hit-to-Lead Optimization toward New Breast Cancer Suppressants

 , , , ,
, , , ,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Datasets Compilation

2.2. 3-D Pharmacophore and 3-D QSAR Modeling and Models’ Interpretation
2.3. Predictive Ability Assessment of the 3-D PhypI/3-D QSAR Model Ensemble
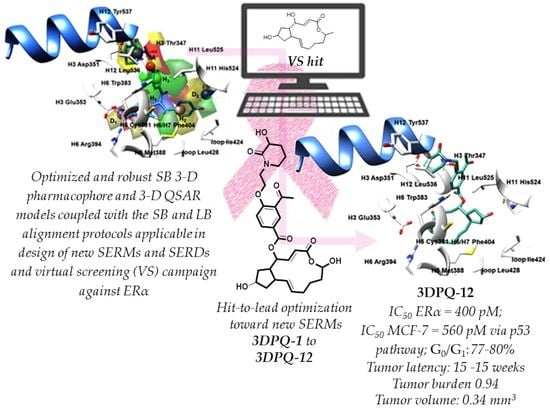
2.4. Virtual Screening, Anticancer Potency, and Binding Mode Analysis of Brefeldin A as a Hit for Hit-to-Lead Optimization towards Innovative SERMs
2.5. Rules for the Rational Design of Novel Brefeldin A Derivatives as SERMs
- The BFA’s C15-CH3 group was converted to C15-OH as a mixed HBA/HBD functional group to increase the compounds’ capacity for establishing hydrogen bonds with either H3 Glu353 and H6 Arg394 (or H11 His524) and hopefully the solubility (data not shown).
- The BFA’s C4-OH was substituted with 3-acetyl-4-hydroxybenzoic acid to provide interactions with H6 Trp383 and H3 Thr347, as well as to stabilize the H3 Thr347-Leu525-H12 Leu536 hydrophobic network, and consequent H12 dislocation. Choosing 3-acetyl-4-hydroxybenzoic acid as a BFA’s C4-OH substituent was an experimentally-guided decision since the tentative attempts to synthetically incorporate (see further text) the 1-(1,4-dihydroxynaphthalen-2-yl)ethenone as a fragment, perhaps more suitable to target H6 Trp383 by means of steric interactions, failed.
- The 3-acetyl-4-hydroxybenzoic acid’s p-OH was further substituted with either ethanolamine-based moieties, bearing primary and secondary amines, or various N-, O-, and N, O-heterocycles or 2-hydroxyethanesulfonic acid functions, capable of inducing the AF-2 function dislocation. The primary amine, secondary amine, and 2-hydroxyethanesulfonic acid were chosen as the AF-2 function invaders to reduce the steric pressure on H12, at the same time with the eligibility to establish HBs with H3 Asp351. On the other hand, as the 3-D PhypI/3-D QSAR model ensemble was not explicit on whether to keep the steric pressure on H12 or to reduce it completely, the various N-, O-, and N, O-heterocycles were chosen as bioisosteres of heterocycles found within the ERα binders (Table 1 and Table 2) in a way that their HBD functional groups could primarily engage H3 Asp351, thus influencing, alongside the steric pressure, the H12′s induced fitting, whereas the existing HBA functional groups could produce additional favorable interactions with the surrounding residues.
- The 12 designed compounds, belonging to the 3-D PhypI/3-D QSAR-based series, viz., 3DPQ, were then subjected to the SB/LB alignment (Supplementary Materials Figures S23 and S24) and the pIC50 prediction procedures against ERα (Table 6). This way, the designed compounds composed the ultimate prediction set [109,110] for the 3-D PhypI/3-D QSAR model ensemble, in which the SB and LB models’ associated q2pred and AAEP values were 0.858/0.045 and 0.732/0.1, respectively. Indeed, even eight compounds, namely 3DPQ-12, 3DPQ-3, 3DPQ-9, 3DPQ-4, 3DPQ-2, 3DPQ-1, 3DPQ-7, and 3DPQ-11 were predicted as more potent than 1ERR [13] (the most potent TR compound; see further text).
2.6. Synthesis of Brefeldin A Derivatives 3DPQ-1 to 3DPQ-12
2.7. Synthesized Compounds Antagonistic Potency and Relative Binding Affinities against ERα and ERβ
2.8. Synthesized Compounds Antiproliferative Activity against ERα(+)- and ERα(-)-Dependent Breast Cancer Cell Lines as Well as against ERα(+)-Dependent Endometrial Cancer Cell Lines
2.9. The Impact of Targeted ERα Antagonists on the MCF-7 Cells Signaling
2.10. Effects of Synthesized Compounds on Cytotoxicity and Cell Cycle Distribution of MCF-7 Cell Lines
2.11. Prediction of ADMETox Properties for the Compounds
2.12. In Vivo Anticancer Screening
3. Materials and Methods
3.1. ERα LBD-Partial Agonists/Antagonists Complexes Structures Preparation
3.2. 3-D Pharmacophore Hypotheses and 3-D QSAR Models Generation
3.3. SB Alignment Assessment
3.4. LB Alignment Assessment
3.5. The SB/LB Alignment Accuracy
3.6. Generation of Modeled and Designed Compounds
3.7. Test Sets and Designed Compounds Alignment
3.8. Virtual Screening
3.9. 3-D Pharmacophore Hypotheses and 3-D QSAR Models External Validation and Prediction Ability
3.10. Synthesis of Compounds 3DPQ-1 to 3DPQ-12
3.11. ADMETox Predictions for Compounds 3DPQ-1 to 3DPQ-12
3.12. Biochemical Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Bafna, D.; Ban, F.; Rennie, P.S.; Singh, K.; Cherkasov, A. Computer-Aided Ligand Discovery for Estrogen Receptor Alpha. Int. J. Mol. Sci. 2020, 21, 4193. [Google Scholar] [CrossRef] [PubMed]
- Shiau, A.K.; Barstad, D.; Radek, J.T.; Meyers, M.; Nettles, K.W.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A.; Agard, D.A.; Greene, G.L. Structural Characterization of a Subtype-Selective Ligand Reveals a Novel Mode of Estrogen Receptor Antagonism. Nat. Genet. 2002, 9, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.W.; Perkins, R.; Tong, W.; Hong, H. Versatility or Promiscuity: The Estrogen Receptors, Control of Ligand Selectivity and an Update on Subtype Selective Ligands. Int. J. Environ. Res. Public Health 2014, 11, 8709–8742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzo, P.; Ascenzi, P. Estrogen Signaling Multiple Pathways to Impact Gene Transcription. Curr. Genom. 2006, 7, 497–508. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.; Coombes, R.C. Estrogen Receptor Alpha in Human Breast Cancer: Occurrence and Significance. J. Mammary Gland Biol. Neoplasia 2000, 5, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Marc, R.; Monique, G.; Wurtz, J.M.; Moras, D. Estrogen Receptor Transcription and Transactivation: Structure-Function Relationship in DNA- and Ligand-Binding Domains of Estrogen Receptors. Breast Cancer Res. 2000, 2, 353–359. [Google Scholar]
- Farooq, A. Structural and Functional Diversity of Estrogen Receptor Ligands. Curr. Top. Med. Chem. 2015, 15, 1372–1384. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Zakharov, M.N.; Khan, S.H.; Miki, R.; Jang, H.; Toraldo, G.; Singh, R.; Bhasin, S.; Jasuja, R. The Dynamic Structure of the Estrogen Receptor. J. Amino Acids 2011, 2011, 812540. [Google Scholar] [CrossRef] [Green Version]
- Bentrem, D.; Dardes, R.; Liu, H.; MacGregor-Schafer, J.; Zapf, J.; Jordan, V. Molecular Mechanism of Action at Estrogen Receptor Alpha of a New Clinically Relevant Antiestrogen (GW7604) Related to Tamoxifen. Endocrinology 2001, 142, 838–846. [Google Scholar] [CrossRef]
- Safe, S.; Kim, K. Non-Classical Genomic Estrogen Receptor (ER)/Specificity Protein and ER/Activating Protein-1 Signaling Pathways. J. Mol. Endocrinol. 2008, 41, 263–275. [Google Scholar] [CrossRef]
- Heldring, N.; Pike, A.; Andersson, S.; Matthews, J.; Cheng, G.; Hartman, J.; Tujague, M.; Ström, A.; Treuter, E.; Warner, M.; et al. Estrogen Receptors: How Do They Signal and What Are Their Targets. Physiol. Rev. 2007, 87, 905–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwabe, J.; Chapman, L.; Finch, J.T.; Rhodes, D. The Crystal Structure of the Estrogen Receptor DNA-Binding Domain Bound to DNA: How Receptors Discriminate between their Response Elements. Cell 1993, 75, 567–578. [Google Scholar] [CrossRef]
- Brzozowski, A.M.; Pike, A.C.W.; Dauter, Z.; Hubbard, R.E.; Bonn, T.; Engström, O.; Öhman, L.; Greene, G.L.; Gustafsson, J.A.; Carlquist, M. Molecular Basis of Agonism and Antagonism in the Oestrogen Receptor. Nature 1997, 389, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Wardell, S.E.; Ellis, M.J.; Alley, H.M.; Eisele, K.; VanArsdale, T.; Dann, S.G.; Arndt, K.T.; Primeau, T.; Griffin, E.; Shao, J.; et al. Efficacy of SERD/SERM Hybrid-CDK4/6 Inhibitor Combinations in Models of Endocrine Therapy-Resistant Breast Cancer. Clin. Cancer Res. 2015, 21, 5121–5130. [Google Scholar] [CrossRef] [Green Version]
- Patel, H.K.; Bihani, T. Selective Estrogen Receptor Modulators (SERMs) and Selective Estrogen Receptor Degraders (SERDs) in Cancer Treatment. Pharmacol. Ther. 2018, 186, 1–24. [Google Scholar] [CrossRef]
- Maximov, P.Y.; Lee, T.M.; Jordan, V.C. The Discovery and Development of Selective Estrogen Receptor Modulators (SERMs) for Clinical Practice. Curr. Clin. Pharmacol. 2013, 8, 135–155. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Liu, W. Selective Estrogen Receptor Degraders (SERDs): A Promising Strategy for Estrogen Receptor Positive Endocrine-Resistant Breast Cancer. J. Med. Chem. 2020, 63, 15094–15114. [Google Scholar] [CrossRef]
- Bai, Z.; Gust, R. Breast Cancer, Estrogen Receptor and Ligands. Arch. Pharm. 2009, 342, 133–149. [Google Scholar] [CrossRef]
- Dadiboyena, S. Recent Advances in the Synthesis of Raloxifene: A Selective Estrogen Receptor Modulator. Eur. J. Med. Chem. 2012, 51, 17–34. [Google Scholar] [CrossRef]
- Begam, A.J.; Jubie, S.; Nanjan, M. Estrogen Receptor Agonists/Antagonists in Breast Cancer Therapy: A Critical Review. Bioorganic Chem. 2017, 71, 257–274. [Google Scholar] [CrossRef]
- Fanning, S.W.; Mayne, C.G.; Dharmarajan, V.; Carlson, K.E.; Martin, T.A.; Novick, S.J.; Toy, W.; Green, B.; Panchamukhi, S.; Katzenellenbogen, B.S.; et al. Estrogen Receptor Alpha Somatic Mutations Y537S and D538G Confer Breast Cancer Endocrine Resistance by Stabilizing the Activating Function-2 Binding Conformation. eLife 2016, 5, e12792. [Google Scholar] [CrossRef] [PubMed]
- Anstead, G.M.; Carlson, K.E.; Katzenellenbogen, J.A. The Estradiol Pharmacophore: Ligand Structure-Estrogen Receptor Binding Affinity Relationships and a Model for The Receptor Binding Site. Steroids 1997, 62, 268–303. [Google Scholar] [CrossRef]
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Yusharyahya, S.N.; Bramono, K.; Ascobat, P.; Hestiantoro, A.; Sutanto, N.R.; Fadilah, F. In silico Molecular Docking and Pharmacophore Modelling Studies of Trigonella foenum-graceum (fenugreek) Interactions with Estrogen Receptors α and β. J. Pharm. Sci. Res. 2019, 11, 3705–3711. [Google Scholar]
- McGregor, M.J.; Muskal, S.M. Pharmacophore Fingerprinting. 1. Application to QSAR and Focused Library Design. J. Chem. Inf. Comput. Sci. 1999, 39, 569–574. [Google Scholar] [CrossRef]
- Discovery Studio; Accelrys Software Inc.: San Diego, CA, USA, 2007.
- Catalyst Software Package; Accelrys Software Inc.: San Diego, CA, USA, 2007.
- Mukherjee, S.; Nagar, S.; Mullick, S.; Mukherjee, A.; Saha, A. Pharmacophore Mapping of Selective Binding Affinity of Estrogen Modulators through Classical and Space Modeling Approaches: Exploration of Bridged-Cyclic Compounds with Diarylethylene Linkage. J. Chem. Inf. Model. 2007, 47, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Nagar, S.; Mullick, S.; Mukherjee, A.; Saha, A. Pharmacophore Mapping of Arylbenzothiophene Derivatives for MCF Cell Inhibition Using Classical and 3D Space Modeling Approaches. J. Mol. Graph. Model. 2008, 26, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.A.; Nagar, S.; Das, S.; Mukherjee, A.; Saha, A. Molecular Design Based on Receptor-Independent Pharmacophore: Application to Estrogen Receptor Ligands. Biol. Pharm. Bull. 2008, 31, 1453–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brogi, S.; Kladi, M.; Vagias, C.; Papazafiri, P.; Roussis, V.; Tafi, A. Pharmacophore Modeling for Qualitative Prediction of Antiestrogenic Activity. J. Chem. Inf. Model. 2009, 49, 2489–2497. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Shen, J.; Cheng, F.; Xu, Z.; Liu, G.; Tang, Y. Computational Insights into Ligand Selectivity of Estrogen Receptors from Pharmacophore Modeling. Mol. Inf. 2011, 30, 539–549. [Google Scholar] [CrossRef]
- Brogi, S.; Papazafiri, P.; Roussis, V.; Tafi, A. 3D-QSAR using pharmacophore-based alignment and Virtual Screening for discovery of Novel MCF-7 Cell Line Inhibitors. Eur. J. Med. Chem. 2013, 67, 344–351. [Google Scholar] [CrossRef]
- Muchtaridi, M.; Yusuf, M.; Diantini, A.; Choi, S.B.; Al-Najjar, B.O.; Manurung, J.V.; Subarnas, A.; Achmad, T.H.; Wardhani, S.R.; Wahab, H.A. Potential Activity of Fevicordin-A from Phaleria macrocarpa (Scheff) Boerl. Seeds as Estrogen Receptor Antagonist Based on Cytotoxicity and Molecular Modelling Studies. Int. J. Mol. Sci. 2014, 15, 7225–7249. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Wei, W.; Yang, Y.; Zhang, T.; Shen, Z. Discovery of Novel Selective ERα/ERβ Ligands by Multi-pharmacophore Modeling and Virtual Screening. Chem. Pharm. Bull. 2015, 63, 780–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niinivehmas, S.P.; Manivannan, E.; Rauhamäki, S.; Huuskonen, J.; Pentikäinen, O.T. Identification of Estrogen Receptor Ligands with Virtual Screening Techniques. J. Mol. Graph. Model. 2016, 64, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Md Islam, A.; Patel, D.A.; Rathod, S.G.; Chunarkar, P.; Pillay, T.S. Identification of Structural Requirement of Estrogen Receptor Modulators using Pharmacoinformatics Techniques for Application to Estrogen Therapy. Med. Chem. Res. 2016, 25, 407–421. [Google Scholar] [CrossRef] [Green Version]
- Chu, Z.; Li, Y. Designing Modified Polybrominated Diphenyl Ether BDE-47, BDE-99, BDE-100, BDE-183, and BDE-209 Molecules with Decreased Estrogenic Activities using 3D-QSAR, Pharmacophore Models Coupled with Resolution V of the 210-3 Fractional Factorial Design and Molecular Docking. J. Hazard. Mater. 2019, 364, 151–162. [Google Scholar] [CrossRef]
- Yu, E.; Xu, Y.; Shi, Y.; Yu, Q.; Liu, J.; Xu, L. Discovery of Novel Natural Compound Inhibitors Targeting Estrogen Receptor α by an Integrated Virtual Screening Strategy. J. Mol. Model. 2019, 25, 278–288. [Google Scholar] [CrossRef]
- Scott, J.S.; Bailey, A.; Davies, R.D.; Degorce, S.L.; MacFaul, P.A.; Gingell, H.; Moss, T.; Norman, R.A.; Pink, J.H.; Rabow, A.A.; et al. Tetrahydroisoquinoline Phenols: Selective Estrogen Receptor Downregulator Antagonists with Oral Bioavailability in Rat. ACS Med. Chem. Lett. 2015, 7, 94–99. [Google Scholar] [CrossRef] [Green Version]
- Tria, G.S.; Abrams, T.; Baird, J.; Burks, H.E.; Firestone, B.; Gaither, L.A.; Hamann, L.G.; He, G.; Kirby, C.A.; Kim, S.; et al. Discovery of LSZ102, a Potent, Orally Bioavailable Selective Estrogen Receptor Degrader (SERD) for the Treatment of Estrogen Receptor Positive Breast Cancer. J. Med. Chem. 2018, 61, 2837–2864. [Google Scholar] [CrossRef]
- Mardianingrum, R.; Yusuf, M.; Hariono, M.; Gazzali, M.A.; Muchtaridi, M. α-Mangostin and its Derivatives against Estrogen Receptor Alpha. J. Biomol. Struct. Dyn. 2020, 40, 2621–2634. [Google Scholar] [CrossRef] [PubMed]
- Hariyanti, H.; Kusmardi, K.; Yanuar, A.; Hayun, H. Ligand Based Pharmacophore Modeling, Virtual Screening, and Molecular Docking Studies of Asymmetrical Hexahydro-2H-Indazole Analogs of Curcumin (AIACs) to Discover Novel Estrogen Receptors Alpha (ERα) Inhibitor. Indones. J. Chem. 2021, 21, 137–147. [Google Scholar] [CrossRef]
- Jereva, D.; Fratev, F.; Tsakovska, I.; Alov, P.; Pencheva, T.; Pajeva, I. Molecular Dynamics Simulation of the Human Estrogen Receptor Alpha: Contribution to the Pharmacophore of the Agonists. Math. Comput. Simul. 2017, 133, 124–134. [Google Scholar] [CrossRef]
- Gangloff, M.; Ruff, M.; Eiler, S.; Duclaud, S.; Wurtz, J.M.; Moras, D. Crystal Structure of a Mutant hERalpha Ligand-Binding Domain Reveals Key Structural Features for the Mechanism of Partial Agonism. J. Biol. Chem. 2001, 276, 15059–15065. [Google Scholar] [CrossRef] [Green Version]
- Nettles, K.W.; Bruning, J.B.; Gil, G.; O’Neill, E.E.; Nowak, J.; Guo, Y.; Kim, Y.; DeSombre, E.R.; Dilis, R.; Hanson, R.N.; et al. Structural Plasticity in the Oestrogen Receptor Ligand-Binding Domain. EMBO Rep. 2007, 8, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE), 2019.01; Chemical Computing Group ULC: Montreal, QC, Canada, 2017.
- Munir, A.; Azam, S.; Mehmood, A.; Khan, Z.; Mehmood, A.; Fazal, S. Structure-Based Pharmacophore Modeling, Virtual Screening and Molecular docking for the Treatment of ESR1 Mutations in Breast Cancer. Drug Des. 2016, 5, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Heldring, N.; Pawson, T.; McDonnell, D.; Treuter, E.; Gustafsson, J.A.; Pike, A.C. Structural Insights into Corepressor Recognition by Antagonist-Bound Estrogen Receptors. J. Biol. Chem. 2007, 282, 10449–10455. [Google Scholar] [CrossRef] [Green Version]
- Fanning, S.W.; Jeselsohn, R.; Dharmarajan, V.; Mayne, C.G.; Karimi, M.; Buchwalter, G.; Houtman, R.; Toy, W.; Fowler, C.E.; Han, R.; et al. The SERM/SERD Bazedoxifene Disrupts ESR1 Helix 12 to Overcome Acquired Hormone Resistance in Breast Cancer Cells. Elife 2018, 7, e37161. [Google Scholar] [CrossRef]
- Muchtaridi, M.; Syahidah, H.N.; Subarnas, A.; Yusuf, M.; Bryant, S.D.; Langer, T. Molecular Docking and 3D-Pharmacophore Modeling to Study the Interactions of Chalcone Derivatives with Estrogen Receptor Alpha. Pharmaceuticals 2017, 10, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muchtaridi, M.; Yusuf, M.; Syahidah, H.N.; Subarnas, A.; Zamri, A.; Bryant, S.D.; Langer, T. Cytotoxicity of Chalcone of Eugenia aquea Burm F. Leaves Against T47D Breast Cancer Cell Lines and Its Prediction as An Estrogen Receptor Antagonist Based on Pharmacophore-Molecular Dynamics Simulation. Adv Appl. Bioinform. Chem. 2019, 12, 33–43. [Google Scholar] [CrossRef] [Green Version]
- Sahayarayan, J.J.; Rajan, K.S.; Vidhyavathi, R.; Nachiappan, M.; Prabhu, D.; Alfarraj, S.; Arokiyaraj, S.; Daniel, A.N. In-silico Protein-Ligand Docking Studies against the Estrogen Protein of Breast Cancer Using Pharmacophore Based Virtual Screening Approaches. Saudi J. Biol. Sci. 2021, 28, 400–407. [Google Scholar] [CrossRef]
- Shylaja, R.; Loganathan, C.; Kabilan, S.; Vijayakumar, T.; Meganathan, C. Synthesis and Evaluation of the Antagonistic Activity of 3-acetyl-2H-benzo[g]chromen-2-one against Mutant Y537S Estrogen Receptor Alpha via E-Pharmacophore Modeling, Molecular Docking, Molecular Dynamics, and in-vitro Cytotoxicity Studies. J. Mol. Struct. 2021, 224, 129289. [Google Scholar] [CrossRef]
- Nwachukwu, J.C.; Srinivasan, S.; Zheng, Y.; Wang, S.; Min, J.; Dong, C.; Liao, Z.; Nowak, J.; Wright, N.J.; Houtman, R.; et al. Predictive Features of Ligand-Specific Signaling through the Estrogen Receptor. Mol. Syst. Biol. 2016, 12, 864. [Google Scholar] [CrossRef] [PubMed]
- Nwachukwu, J.C.; Srinivasan, S.; Bruno, N.E.; Nowak, J.; Wright, N.J.; Minutolo, F.; Rangarajan, E.S.; Izard, T.; Yao, X.Q.; Grant, B.J.; et al. Systems Structural Biology Analysis of Ligand Effects on ERα Predicts Cellular Response to Environmental Estrogens and Anti-hormone Therapies. Cell Chem. Biol. 2017, 24, 35–45. [Google Scholar] [CrossRef]
- Srinivasan, S.; Nwachukwu, J.C.; Bruno, N.E.; Dharmarajan, V.; Goswami, D.; Kastrati, I.; Novick, S.; Nowak, J.; Cavett, V.; Zhou, H.B.; et al. Full Antagonism of the Estrogen Receptor Without a Prototypical Ligand Side Chain. Nat. Chem. Biol. 2017, 13, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Speltz, T.E.; Fanning, S.W.; Mayne, C.G.; Fowler, C.; Tajkhorshid, E.; Greene, G.L.; Moore, T.W. Stapled Peptides with γ-Methylated Hydrocarbon Chains for the Estrogen Receptor/Coactivator Interaction. Angew. Chem. Int. Ed. Engl. 2016, 55, 4252–4555. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.-L.; Yang, X.; Ren, Z.; McDonnell, D.P.; Norris, J.; Willson, T.M.; Greene, G.L. Structural Basis for an Unexpected Mode of SERM-Mediated ER Antagonism. Mol. Cell 2005, 18, 413–424. [Google Scholar] [CrossRef]
- Wärnmark, A.; Treuter, E.; Gustafsson, J.A.; Hubbard, R.E.; Brzozowski, A.M.; Pike, A.C. Interaction of Transcriptional Intermediary Factor 2 Nuclear Receptor Box Peptides with the Coactivator Binding Site of Estrogen Receptor Alpha. J. Biol. Chem. 2002, 277, 21862–21868. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Wu, J.Y.; Birzin, E.T.; Frisch, K.; Chan, W.; Pai, L.Y.; Yang, Y.T.; Mosley, R.T.; Fitzgerald, P.M.D.; Sharma, N.; et al. Estrogen Receptor Ligands. II. Discovery of Benzoxathiins as Potent, Selective Estrogen Receptor α Modulators. J. Med. Chem. 2004, 47, 2171–2175. [Google Scholar] [CrossRef]
- Manas, E.S.; Unwalla, R.J.; Xu, Z.B.; Malamas, M.S.; Miller, C.P.; Harris, H.A.; Hsiao, C.; Akopian, T.; Hum, W.-T.; Malakian, K.; et al. Structure-Based Design of Estrogen Receptor-β Selective Ligands. J. Am. Chem. Soc. 2004, 126, 15106–15119. [Google Scholar] [CrossRef]
- Manas, E.S.; Xu, Z.B.; Unwalla, R.J.; Somers, W.S. Understanding the Selectivity of Genistein for Human Estrogen Receptor-Beta Using X-Ray Crystallography and Computational Methods. Structure 2004, 12, 2197–2207. [Google Scholar] [CrossRef] [Green Version]
- Blizzard, T.A.; DiNinno, F.; Morgan, J.D.; Chen, H.Y.; Wu, J.Y.; Kim, S.; Chan, W.; Birzin, E.T.; Yang, Y.T.; Pai, L.-Y.; et al. Estrogen Receptor Ligands. Part 9: Dihydrobenzoxathiin SERAMs with Alkyl Substituted Pyrrolidine Side Chains and Linkers. Bioorg. Med. Chem. Lett. 2005, 15, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Renaud, J.; Bischoff, S.F.; Buhl, T.; Floersheim, P.; Fournier, B.; Geiser, M.; Halleux, C.; Kallen, J.; Keller, H.; Ramage, P. Selective Estrogen Receptor Modulators with Conformationally Restricted Side Chains. Synthesis and Structure-Activity Relationship of ERα-Selective Tetrahydroisoquinoline Ligands. J. Med. Chem. 2005, 48, 364–379. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Blizzard, T.A.; Morgan, J.D.; Birzin, E.T.; Chan, W.; Yang, Y.T.; Pai, L.-Y.; Hayes, E.C.; DaSilva, C.A.; Warrier, S.; et al. Estrogen receptor ligands. Part 10: Chromanes: Old Scaffolds for New SERAMs. Bioorg. Med. Chem. Lett. 2005, 15, 1675–1681. [Google Scholar] [CrossRef] [PubMed]
- Kong, E.H.; Heldring, N.; Gustafsson, J.; Treuter, E.; Hubbard, R.E.; Pike, A.C.W. Delineation of A Unique Protein–Protein Interaction Site on the Surface of the Estrogen Receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 3593–3598. [Google Scholar] [CrossRef] [Green Version]
- Dykstra, K.D.; Guo, L.; Birzin, E.T.; Chan, W.; Yang, Y.T.; Hayes, E.C.; DaSilva, C.A.; Pai, L.Y.; Mosley, R.T.; Kraker, B.; et al. Estrogen Receptor Ligands. Part 16: 2-Aryl Indoles as Highly Subtype Selective Ligands for ERα. Bioorg. Med. Chem. Lett. 2007, 17, 2322–2328. [Google Scholar] [CrossRef]
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The Structural Basis of Estrogen Receptor/Coactivator Recognition and the Antagonism of This Interaction by Tamoxifen. Cell 1998, 95, 927–937. [Google Scholar] [CrossRef] [Green Version]
- Renaud, J.; Bischoff, S.F.; Buhl, T.; Floersheim, P.; Fournier, B.; Halleux, C.; Kallen, J.; Keller, H.; Schlaeppi, J.M.; Stark, W. Estrogen Receptor Modulators: Identification and Structure-Activity Relationships of Potent ERalpha-Selective Tetrahydroisoquinoline Ligands. J. Med. Chem. 2003, 46, 2945–2957. [Google Scholar] [CrossRef]
- Hsieh, R.W.; Rajan, S.S.; Sharma, S.K.; Greene, G.L. Molecular Characterization of a B-ring Unsaturated Estrogen: Implications for Conjugated Equine Estrogen Components of Premarin. Steroids 2008, 73, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Nettles, K.W.; Bruning, J.B.; Gil, G.; Nowak, J.; Sharma, S.K.; Hahm, J.B.; Kulp, K.; Hochberg, R.B.; Zhou, H.; Katzenellenbogen, J.A.; et al. NFκappaB Selectivity of Estrogen Receptor Ligands Revealed by Comparative Crystallographic Analyses. Nat. Chem. Biol. 2008, 4, 241–247. [Google Scholar] [CrossRef]
- Dai, S.Y.; Chalmers, M.J.; Bruning, J.; Bramlett, K.S.; Osborne, H.E.; Montrose-Rafizadeh, C.; Barr, R.J.; Wang, Y.; Wang, M.; Burris, T.P.; et al. Prediction of the Tissue-Specificity of Selective Estrogen Receptor Modulators by Using a Single Biochemical Method. Proc. Natl. Acad. Sci. USA 2008, 105, 7171–7176. [Google Scholar] [CrossRef] [Green Version]
- Degorce, S.L.; Bailey, A.; Callis, R.; De Savi, C.; Ducray, R.; Lamont, G.; MacFaul, P.; Maudet, M.; Martin, S.; Morgentin, R.; et al. Investigation of (E)-3-[4-(2-Oxo-3-aryl-chromen-4-yl)oxyphenyl]acrylic Acids as Oral Selective Estrogen Receptor Down-Regulators. J. Med. Chem. 2015, 58, 3522–3533. [Google Scholar] [CrossRef] [PubMed]
- Delfosse, V.; Grimaldi, M.; Pons, J.-L.; Boulahtouf, A.; le Maire, A.; Cavailles, V.; Labesse, G.; Bourguet, W.; Balaguer, P. Structural and mechanistic insights into bisphenols action provide guidelines for risk assessment and discovery of bisphenol A substitutes. Proc. Natl. Acad. Sci. USA 2012, 109, 14930–14935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osz, J.; Brélivet, Y.; Peluso-Iltis, C.; Cura, V.; Eiler, S.; Ruff, M.; Bourguet, W.; Rochel, N.; Moras, D. Structural basis for a molecular allosteric control mechanism of cofactor binding to nuclear receptors. Proc. Natl. Acad. Sci. USA 2012, 109, E588–E594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delfosse, V.; Grimaldi, M.; Cavaillès, V.; Balaguer, P.; Bourguet, W. Structural and Functional Profiling of Environmental Ligands for Estrogen Receptors. Environ. Health Perspect. 2014, 122, 1306–1313. [Google Scholar] [CrossRef] [Green Version]
- Delfosse, V.; le Maire, A.; Balaguer, P.; Bourguet, W. A Structural Perspective on Nuclear Receptors as Targets of Environmental Compounds. Acta Pharmacol. Sin. 2014, 36, 88–101. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Zhu, M.; Srinivasan, S.; Nwachukwu, J.; Cavett, V.; Min, J.; Carlson, K.E.; Wang, P.; Dong, C.; Katzenellenbogen, J.A.; et al. Development of Selective Estrogen Receptor Modulator (SERM)-Like Activity Through an Indirect Mechanism of Estrogen Receptor Antagonism: Defining the Binding Mode of 7-Oxabicyclo [2.2.1]hept-5-ene Scaffold Core Ligands. ChemMedChem 2012, 7, 1094–1100. [Google Scholar] [CrossRef] [Green Version]
- Mihović, N.; Tomašević, N.; Matić, S.; Mitrović, M.M.; Kostić, D.A.; Sabatino, M.; Antonini, L.; Ragno, R.; Mladenović, M. Human Estrogen Receptor α Antagonists. Part 1: 3-D QSAR-Driven Rational Design of Innovative Coumarin-Related Antiestrogens as Breast Cancer Suppressants through Structure-Based and Ligand-Based Studies. J. Chem. Inf. Model. 2021, 61, 5028–5053. [Google Scholar] [CrossRef]
- Kurtanović, N.; Tomašević, N.; Matić, S.; Mitrović, M.M.; Kostić, D.A.; Sabatino, M.; Antonini, L.; Ragno, R.; Mladenović, M. Human Estrogen Receptor α Antagonists, part 2: Synthesis Driven by Rational Design, in vitro Antiproliferative, and in vivo Anticancer Evaluation of Innovative Coumarin-Related Antiestrogens as Breast Cancer Suppressants. Eur. J. Med. Chem. 2022, 227, 113869. [Google Scholar] [CrossRef]
- Tanenbaum, D.M.; Wang, Y.; Williams, S.P.; Sigler, P.B. Crystallographic Comparison of the Estrogen and Progesterone Receptor’s Ligand Binding Domains. Proc. Natl. Acad. Sci. USA 1998, 95, 5998–6003. [Google Scholar] [CrossRef] [Green Version]
- Eiler, S.; Gangloff, M.; Duclaud, S.; Moras, D.; Ruff, M. Overexpression, Purification, and Crystal Structure of Native ER Alpha LBD. Protein Expr. Purif. 2001, 22, 165–173. [Google Scholar] [CrossRef]
- Fang, J.; Akwabi-Ameyaw, A.; Britton, J.E.; Katamreddy, S.R.; Navas, F.; Miller, A.B.; Williams, S.P.; Gray, D.W.; Orband-Miller, L.A.; Shearin, J.; et al. Synthesis of 3-alkyl Naphthalenes as Novel Estrogen Receptor Ligands. Bioorg. Med. Chem. Lett. 2008, 18, 5075–5077. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Nwachukwu, J.C.; Parent, A.A.; Cavett, V.; Nowak, J.; Hughes, T.S.; Kojetin, D.J.; Katzenellenbogen, J.A.; Nettles, K.W. Ligand-Binding Dynamics Rewire Cellular Signaling via Estrogen Receptor-α. Nat. Chem. Biol. 2013, 9, 326–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nwachukwu, J.C.; Srinivasan, S.; Bruno, N.E.; Parent, A.A.; Hughes, T.S.; Pollock, J.A.; Gjyshi, O.; Cavett, V.; Nowak, J.; Garcia-Ordonez, R.D.; et al. Resveratrol Modulates the Inflammatory Response via an Estrogen Receptor-Signal Integration Network. Elife 2014, 3, e02057. [Google Scholar] [CrossRef] [PubMed]
- De Savi, C.; Bradbury, R.H.; Rabow, A.A.; Norman, R.A.; de Almeida, C.; Andrews, D.M.; Ballard, P.; Buttar, D.; Callis, R.J.; Currie, G.S.; et al. Optimization of a Novel Binding Motif to (E)-3-(3,5-difluoro-4-((1R,3R)-2-(2-fluoro-2-methylpropyl)-3-methyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indol-1-yl)phenyl)acrylic Acid (AZD9496), a Potent and Orally Bioavailable Selective Estrogen Receptor Downregulator and Antagonist. J. Med. Chem. 2015, 58, 8128–8140. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef]
- Schrödinger Release 2015-2: Canvas; Schrödinger, LLC: New York, NY, USA, 2015.
- Ballante, F.; Caroli, A.; Wickersham, R.B., 3rd; Ragno, R. Hsp90 Inhibitors, Part 1: Definition of 3-D QSAutogrid/R Models as a Tool for Virtual Screening. J. Chem. Inf. Model. 2014, 54, 956–969. [Google Scholar] [CrossRef]
- Caroli, A.; Ballante, F.; Wickersham, R.B., 3rd; Corelli, F.; Ragno, R. Hsp90 Inhibitors, Part 2: Combining Ligand-Based and Structure-Based Approaches for Virtual Screening Application. J. Chem. Inf. Model. 2014, 54, 970–977. [Google Scholar] [CrossRef]
- Mladenović, M.; Patsilinakos, A.; Pirolli, A.; Sabatino, M.; Ragno, R. Understanding the Molecular Determinant of Reversible Human Monoamine Oxidase B Inhibitors Containing 2H-Chromen-2-One Core: Structure-Based and Ligand-Based Derived Three-Dimensional Quantitative Structure–Activity Relationships Predictive Models. J. Chem. Inf. Model. 2017, 57, 787–814. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Fanning, S.W.; Hodges-Gallagher, L.; Myles, D.C.; Sun, R.; Fowler, C.E.; Plant, I.N.; Green, B.D.; Harmon, C.L.; Greene, G.L.; Kushner, P.J. Specific Stereochemistry of OP-1074 Disrupts Estrogen Receptor Alpha Helix 12 and Confers Pure Antiestrogenic Activity. Nat. Commun. 2018, 9, 2368–2379. [Google Scholar] [CrossRef]
- Brozic, P.; Kocbek, P.; Sova, M.; Kristl, J.; Martens, S.; Adamski, J.; Gobec, S.; Lanisnik, R.T. Flavonoids and Cinnamic Acid Derivatives as Inhibitors of 17β-Hydroxysteroid Dehydrogenase Type 1. Mol. Cell. Endocrinol. 2009, 301, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Rathelot, P.; Azas, N.; El-Kashef, H.; Delmas, F.; Di Giorgio, C.; Timon-David, P.; Maldonado, J.; Vanelle, P. 1,3-Diphenylpyrazoles: Synthesis and Antiparasitic Activities of Azomethine Derivatives. Eur. J. Med. Chem. 2002, 37, 671–679. [Google Scholar] [CrossRef]
- Sun, J.; Huang, Y.R.; Harrington, W.R.; Sheng, S.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Antagonists Selective for Estrogen Receptor Alpha. Endocrinology 2002, 143, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.L.; Tamrazi, A.; Collins, M.L.; Katzenellenbogen, J.A. Design, Synthesis, and in Vitro Biological Evaluation of Small Molecule Inhibitors of Estrogen Receptor Alpha Coactivator Binding. J. Med. Chem. 2004, 47, 600–611. [Google Scholar] [CrossRef] [PubMed]
- Küçükoğlu, K.; Seçinti, H.; Özgür, A.; Seçen, H.; Tutar, Y. Synthesis, Molecular Docking, and Antitumoral Activity of Alnustone-Likecompounds Against Estrogen Receptor Alpha-Positive Human Breast Cancer. Turk. J. Chem. 2015, 39, 179–193. [Google Scholar] [CrossRef]
- Yang, W.; Yong, W.; AiQian, Z.; HongXia, Y.; LianSheng, W. Three-Dimensional Quantitative Structure-Activity Relationships of Flavonoids and Estrogen Receptors Based on Docking. Chin. Sci. Bull. 2010, 55, 1488–1494. [Google Scholar] [CrossRef]
- Stauffer, S.R.; Huang, Y.R.; Aron, Z.D.; Coletta, C.J.; Sun, J.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A. Triarylpyrazoles with Basic Side Chains: Development of Pyrazole-Based Estrogen Receptor Antagonists. Bioorg. Med. Chem. 2001, 9, 151–161. [Google Scholar] [CrossRef]
- Brian, E.F.; Deborah, S.M.; Shaun, R.S.; Zachary, D.A.; John, A.K. Novel Structural Templates for Estrogen-Receptor Ligands and Prospects for Combinatorial Synthesis of Estrogens. Chem. Biol. 1999, 6, 205–219. [Google Scholar] [CrossRef] [Green Version]
- Marvin Beans 15.4.27.0, 2015, ChemAxon. Available online: http://www.chemaxon.com (accessed on 1 January 2015).
- Herynk, M.H.; Fuqua, S.A.W. Estrogen Receptor Mutations in Human Disease. Endocr. Rev. 2004, 25, 869–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Tropsha, A. Best Practices for QSAR Model Development, Validation, and Exploitation. Mol. Inform. 2010, 29, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Ragno, R. www.3d-qsar.com: A Web Portal that Brings 3-D QSAR to all Electronic Devices-The Py-Comfa Web Application as Tool to Build Models from Pre-Aligned Datasets. J. Comput. Aided. Mol. Des. 2019, 33, 855–864. [Google Scholar] [CrossRef]
- Ragno, R.; Esposito, V.; Di Mario, M.; Masiello, S.; Viscovo, M.; Cramer, R.D. Teaching and Learning Computational Drug Design: Student Investigations of 3D Quantitative Structure–Activity Relationships through Web Applications. J. Chem. Educ. 2020, 97, 1922–1930. [Google Scholar] [CrossRef] [PubMed]
- Paek, S.-M. Recent Synthesis and Discovery of Brefeldin A Analogs. Mar. Drugs 2018, 16, 133. [Google Scholar] [CrossRef] [Green Version]
- Lippincott-Schwartz, J.; Yuan, L.C.; Bonifacino, J.S.; Klausner, R.D. Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: Evidence for membrane cycling from Golgi to ER. Cell 1989, 56, 801–813. [Google Scholar] [CrossRef]
- Dinter, A.; Berger, E.G. Golgi-disturbing agents. Histochem. Cell Biol. 1998, 109, 571–590. [Google Scholar] [CrossRef] [PubMed]
- Seehafer, K.; Rominger, F.; Helmchen, G.; Langhans, M.; Robinson, D.G.; Özata, B.; Brügger, B.; Strating, J.R.P.M.; Van Kuppeveld, F.J.M.; Klein, C.D. Synthesis and Biological Properties of Novel Brefeldin A Analogues. J. Med. Chem. 2013, 56, 5872–5884. [Google Scholar] [CrossRef] [PubMed]
- Anadu, N.O.; Davisson, V.J.; Cushman, M. Synthesis and Anticancer Activity of Brefeldin A Ester Derivatives. J. Med. Chem. 2006, 49, 3897–3905. [Google Scholar] [CrossRef]
- Argade, A.B.; Haugwitz, R.D.; Devraj, R.; Kozlowski, J.; Fanwick, A.P.E.; Cushman, M. Highly Efficient Diastereoselective Michael Addition of Various Thiols to (+)-Brefeldin A. J. Org. Chem. 1998, 63, 273–278. [Google Scholar] [CrossRef]
- Kozikowski, A.P.; Shum, P.W.; Basu, A.; Lazo, J.S. Synthesis of Structural Analogues of Lyngbyatoxin A and Their Evaluation as Activators of Protein Kinase C. J. Med. Chem. 1991, 34, 2420–2430. [Google Scholar] [CrossRef]
- Maki, B.E.; Scheidt, K.A. N-Heterocyclic Carbene-Catalyzed Oxidation of Unactivated Aldehydes to Esters. Org. Lett. 2008, 10, 4331–4334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blakemore, P.R.; Kocienski, P.J.; Marzcak, S.; Wicha, J. The Modified Julia Olefination in Vitamin D2 Synthesis. Synthesis 1999, 1999, 1209–1215. [Google Scholar] [CrossRef]
- Förster, S.; Persch, E.; Tverskoy, O.; Rominger, F.; Helmchen, G.; Klein, C.; Gönen, B.; Brügger, B. Syntheses and Biological Properties of Brefeldin Analogues. Eur. J. Org. Chem. 2010, 2011, 878–891. [Google Scholar] [CrossRef]
- Haynes, R.K.; Lam, W.W.-L.; Yeung, L.-L.; Williams, I.D.; Ridley, A.C.; Starling, S.M.; Vonwiller, S.C.; Hambley, T.W.; Lelandais, P. Highly Diastereoselective Conjugate Addition of Lithiated γ-Crotonolactone (But-2-en-4-olide) to Cyclic Enones to Give Syn-Adducts: Application to a Brefeldin Synthesis. J. Org. Chem. 1997, 62, 4552–4553. [Google Scholar] [CrossRef]
- Trost, B.M.; Crawley, M.L. A “Chiral Aldehyde” Equivalent as a Building Block Towards Biologically Active Targets. Chem. Eur. J. 2004, 10, 2237–2252. [Google Scholar] [CrossRef]
- Inanaga, J.; Hirata, K.; Saeki, H.; Katsuki, T.; Yamaguchi, M. A Rapid Esterification by Means of Mixed Anhydride and Its Application to Large-ring Lactonization. Bull. Chem. Soc. Jpn. 1979, 52, 1989–1993. [Google Scholar] [CrossRef] [Green Version]
- Williams, D.R.; Jass, P.A.; Tse, H.L.A.; Gaston, R.D. Total synthesis of (+)-breynolide. J. Am. Chem. Soc. 1990, 112, 4552–4554. [Google Scholar] [CrossRef]
- He, B.; Wang, Y.; Zheng, Y.; Chen, W.; Zhu, Q. Synthesis and Cytotoxic Evaluation of Acylated Brefeldin A Derivatives as Potential Anticancer Agents. Chem. Biol. Drug Des. 2013, 82, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Affini, A.; Hagenow, S.; Zivkovic, A.; Marco-Contelles, J.; Stark, H. Novel Indanone Derivatives as MAO B/H3R Dual-Targeting Ligands for Treatment of Parkinson’s Disease. Eur. J. Med. Chem. 2018, 148, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Hu, Z.; Luo, J.; Tang, C.; Zhang, S.; Ning, W.; Dong, C.; Huang, J.; Liu, X.; Zhou, H.-B. Dual Functional Small Molecule Fluorescent Probes for Image-Guided Estrogen Receptor-Specific Targeting Coupled Potent Antiproliferative Potency For Breast Cancer Therapy. Bioorganic Med. Chem. 2017, 25, 3531–3539. [Google Scholar] [CrossRef]
- Li, X.; Wu, C.; Lin, X.; Cai, X.; Liu, L.; Luo, G.; You, Q.; Xiang, H. Synthesis and Biological Evaluation of 3-Aryl-quinolin Derivatives as Anti-Breast Cancer Agents Targeting ERα and VEGFR-2. Eur. J. Med. Chem. 2018, 161, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.-B.; Sheng, S.; Compton, D.R.; Kim, Y.; Joachimiak, A.; Sharma, S.; Carlson, K.E.; Katzenellenbogen, B.S.; Nettles, K.W.; Greene, G.L.; et al. Structure-Guided Optimization of Estrogen Receptor Binding Affinity and Antagonist Potency of Pyrazolopyrimidines with Basic Side Chains. J. Med. Chem. 2006, 50, 399–403. [Google Scholar] [CrossRef]
- Luo, G.; Li, X.; Zhang, G.; Wu, C.; Tang, Z.; Liu, L.; You, Q.; Xiang, H. Novel SERMs Based On 3-Aryl-4-Aryloxy-2H-Chromen-2-One Skeleton—A Possible Way to Dual ERa/VEGFR-2 Ligands for Treatment of Breast Cancer. Eur. J. Med. Chem. 2017, 140, 252–273. [Google Scholar] [CrossRef]
- Callis, R.; Rabow, A.; Tonge, M.; Bradbury, R.; Challinor, M.; Roberts, K.; Jones, K.; Walker, G. A Screening Assay Cascade to Identify and Characterize Novel Selective Estrogen Receptor Downregulators (SERDs). SLAS Discov. Adv. Sci. Drug Discov. 2015, 20, 748–759. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.C.; Chuang, Y.C.; Chang, Y.S.; Lai, M.D.; Teng, Y.N.; Su, I.J.; Wang, C.C.; Lee, K.H.; Hung, J.H. Endoplasmic Reticulum Stress Stimulates p53 Expression Through NF-κB Activation. PLoS ONE 2012, 7, e39120. [Google Scholar] [CrossRef] [Green Version]
- Konduri, S.D.; Medisetty, R.; Liu, W.; Kaipparettu, B.A.; Srivastava, P.; Brauch, H.; Fritz, P.; Swetzig, W.M.; Gardner, A.E.; Khan, S.A.; et al. Mechanisms of Estrogen Receptor Antagonism toward p53 and its Implications in Breast Cancer Therapeutic Response and Stem Cell Regulation. Proc. Natl. Acad. Sci. USA 2010, 107, 15081–15086. [Google Scholar] [CrossRef] [Green Version]
- Perissi, V.; Jepsen, K.; Glass, C.K.; Rosenfeld, M.G. Deconstructing repression: Evolving models of co-repressor action. Nat. Rev. Genet. 2010, 11, 109–123. [Google Scholar] [CrossRef]
- Yang, X.J.; Seto, E. The Rpd3/Hda1 Family of Lysine Deacetylases: From Bacteria and Yeast to Mice and Men. Nat. Rev. Mol. Cell Biol. 2008, 9, 206–218. [Google Scholar] [CrossRef] [Green Version]
- Dalvai, M.; Bystricky, K. Cell Cycle and Anti-Estrogen Effects Synergize to Regulate Cell Proliferation and ER Target Gene Expression. PLoS ONE 2010, 5, e11011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elekofehinti, O.O.; Iwaloye, O.; Josiah, S.S.; Lawal, A.O.; Akinjiyan, M.O.; Ariyo, E.O. Molecular Docking Studies, Molecular Dynamics and ADME/Tox Reveal Therapeutic Potentials of STOCK1N-69160 against Papain-Like Protease of SARS-CoV-2. Mol. Divers. 2021, 25, 1761–1773. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Q.; Wilkinson, B. Drug Discovery Beyond the ‘Rule-Of Five’. Curr. Opin. Biotechnol. 2007, 18, 478–488. [Google Scholar] [CrossRef]
- Sun, H.; Li, Y.; Shen, M.; Tian, S.; Xu, L.; Pan, P.; Guan, Y.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 5. Improved docking performance using high solute dielectric constant MM/GBSA and MM/PBSA rescoring. Phys. Chem. Chem. Phys. 2014, 16, 22035–22045. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A Rule of Three for Fragment-Based Lead Discovery? Drug Discov. Today 2003, 8, 876–877. [Google Scholar] [CrossRef]
- Köster, H.; Craan, T.; Brass, S.; Herhaus, C.; Zentgraf, M.; Neumann, L.; Heine, A.; Klebe, G. A Small Nonrule of 3 Compatible Fragment Library Provides High Hit Rate of Endothiapepsin Crystal Structures with Various Fragment Chemotypes. J. Med. Chem. 2011, 54, 7784–7796. [Google Scholar] [CrossRef]
- Schrödinger Release 2015-2: QikProp; Schrödinger, LLC: New York, NY, USA, 2015.
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. AdmetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2018, 35, 1067–1069. [Google Scholar] [CrossRef]
- Mokale, S.N.; Begum, A.; Sakle, N.; Shelke, V.R.; Bhavale, S.A. Design, synthesis and anticancer screening of 3-(3-(substituted phenyl) acryloyl)-2H-chromen-2ones as selective anti-breast cancer agent. Biomed. Pharmacother. 2017, 89, 966–972. [Google Scholar] [CrossRef]
- Quick, A.J.; Stanley-Brown, M.; Bancroft, F.W. A Study of the Coagulation Defect in Hemophilia and in Jaundice. Am. J. Med. Sci. 1935, 190, 501. [Google Scholar] [CrossRef]
- Bergmeyer, H.U.; Bowers, G.N.; Hørder, M.; Moss, D.W. Provisional Recommendations on IFCC Methods for the Measurement of Catalytic Concentrations of Enzymes. Part 2. IFCC Method for Aspartat Aminotransferase. Clin. Chim. Acta 1976, 70, 19–42. [Google Scholar] [CrossRef]
- Bergmeyer, H.U.; Hørder, M. IFCC Methods for Measurement of Catalityc Concentrations of Enzymes. Clin. Chim. Acta 1980, 105, 147–172. [Google Scholar] [CrossRef]
- Walters, M.I.; Gerarde, H.W. An Ultramicromethod for the Determination of Conjugated and Total Bilirubin in Serum or Plasma. Microchem. J. 1970, 15, 231–243. [Google Scholar] [CrossRef]
- Jendrassik, L.; Gróf, P. Vereinfachte Photometrische Methoden zur Bestimmung des Blutbilirubins. Biochem. Z. 1938, 297, 82–89. [Google Scholar]
- Ellman, G.L. Tissue Sulfhydryl Groups. Arch. Biochem. Biophys. 1959, 82, 70–77. [Google Scholar] [CrossRef]
- Góth, L.A. Simple Method for Determination of Serum Catalase Activity and Revision of Reference Range. Clin. Chim. Acta 1991, 196, 143–152. [Google Scholar] [CrossRef]
- Ohkawa, H.; Ohishi, N.; Yagi, K. Assay for Lipid Peroxides in Animal Tissues by Thiobarbituric Acid Reaction. Anal. Biochem. 1979, 95, 351–358. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.L.; Farr, A.L.; Randall, R.I. Protein Measurement with Folin Phenol Reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Green, C.E.; Swezey, R.; Bakke, J.; Shinn, W.; Furimsky, A.; Bejugam, N.; Shankar, G.N.; Jong, L.; Kapetanovic, I.M. Improved oral bioavailability in rats of SR13668, a novel anti-cancer agent. Cancer Chemother. Pharmacol. 2010, 67, 995–1006. [Google Scholar] [CrossRef] [Green Version]
- Buttar, D.; Colclough, N.; Gerhardt, S.; MacFaul, P.A.; Phillips, S.D.; Plowright, A.; Whittamore, P.; Tam, K.; Maskos, K.; Steinbacher, S.; et al. A Combined Spectroscopic and Crystallographic Approach to probing Drug–Human Serum Albumin Interactions. Bioorg. Med. Chem. 2010, 18, 7486–7496. [Google Scholar] [CrossRef] [PubMed]
- Soars, M.G.; Grime, K.; Sproston, J.L.; Webborn, P.J.H.; Riley, R.J. Use of Hepatocytes to Assess the Contribution of Hepatic Uptake to Clearance in Vivo. Drug Metab. Dispos. 2007, 35, 859–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanković, N.; Mladenović, M.; Matić, S.; Stanić, S.; Mihailović, M.; Mihailović, V.; Katanić, J.; Boroja, T.; Vuković, N.; Sukdolak, S. Serum Albumin Binding Analysis and Toxicological Screening of Novel Chroman-2,4-Diones as Oral Anticoagulants. Chem. Interact. 2015, 227, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, H.D.; A McClain, S.; Lee, H.-M.; Zimmerman, T.; Deng, J.; Johnson, F.; Gu, Y.; Golub, L.M. The Maximum-Tolerated Dose and Pharmacokinetics of a Novel Chemically Modified Curcumin in Rats. J. Exp. Pharmacol. 2022, 14, 73–85. [Google Scholar] [CrossRef]
- Case, D.A.; Darden, T.A.; Cheatham, T.E., III; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M.; et al. AMBER 12; University of California: San Francisco, CA, USA, 2012. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB | Ligand Structure | pIC50 | Ref. | PDB | Ligand Structure | pIC50 | Ref. |
|---|---|---|---|---|---|---|---|
| 1ERE * PA a H12: CC b |  | 9.24 | [13] | 1XP9 * SERM H12: OC |  | 8.80 | [64] |
| 1ERR * SERM c H12: OC d |  | 9.52 | [13] | 1XPC * SERM H12: OC |  | 8.70 | [64] |
| 1GWQ ** PA H12: CC |  | 5.85 | [60] | 1XQC ** SERM H12: OC |  | 7.20 | [65] |
| 1R5K * SERD e H12: OC |  | 7.40 | [59] | 1YIM * SERM H12: OC |  | 8.80 | [66] |
| 1SJ0 * SERM H12: OC |  | 9.09 | [61] | 1YIN * SERM H12: OC |  | 8.80 | [66] |
| 1X7E ** PA H12: CC |  | 5.90 | [62] | 2BJ4 * SERM H12: OC |  | 8.60 | [67] |
| 1X7R * PA H12: CC |  | 8.01 | [63] | 2IOG * SERM H12: OC |  | 8.09 | [68] |
| 1XP1 * SERM H12: OC |  | 9.30 | [64] | 2IOK * SERM H12: OC |  | 9.00 | [68] |
| 1XP6 * SERM H12: OC |  | 9.30 | [64] | 3ERD * PA H12: CC |  | 9.48 | [69] |
| PDB | Ligand Structure | pIC50 | Ref. | PDB | Ligand Structure | pIC50 | Ref. |
|---|---|---|---|---|---|---|---|
| 1L2I * PA a H12: CC b |  | 8.50 | [2] | 2R6W * SERM H12: OC |  | 8.60 | [73] |
| 1UOM * SERM c H12: OC d |  | 7.70 | [70] | 2R6Y * SERM H12: OC |  | 8.90 | [73] |
| 2B1Z ** PA H12: CC |  | 7.10 | [71] | 2QA8 * PA H12: CC |  | 8.01 | [72] |
| 2QA6 ** PA H12: CC |  | 7.30 | [72] | 5AK2 * SERD e H12: OC |  | 8.40 | [74] |
| PDB | Ligand Structure | pKi | Ref. | PDB | Ligand Structure | pKi | Ref. |
|---|---|---|---|---|---|---|---|
| 3ERT (WT) PA a H12: CC b |  | 9.60 | [69] | 4MG9 (MUT) PA H12: CC |  | 6.00 | [77] |
| 3UU7 (MUT) PA H12: CC |  | 8.79 | [75] | 4MGA (MUT) PA H12: CC |  | 6.00 | [77] |
| 3UUA (MUT) PA H12: CC |  | 8.79 | [75] | 4MGC (MUT) PA H12: CC |  | 7.00 | [77] |
| 3UUC (WT) PA H12: CC |  | 5.70 | [75] | 4MGD (MUT) PA H12: CC |  | 6.00 | [77] |
| 4DMA (WT) PA H12: CC |  | 5.60 | [76] | 4TUZ (MUT) PA H12: CC |  | 10.00 | [78] |
| 4MG6 (MUT) PA H12: CC |  | 6.00 | [77] | 4ZN9 (MUT) PA H12: CC |  | 9.60 | [79] |
| 4MG8 (MUT) PA H12: CC |  | 10.00 | [77] |
| ADDRRRP.11 | ADDHHHP.13 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
 |  | |||||||||
| HID a | S b | S-I c | P-H d | S e | V f | VOL g | SE h | M i | A j | I k |
| ADDRRRP.11 | 3.741 | 0.967 | 6.429 | 0.81 | 0.991 | 0.426 | 2.678 | 17 | 9.52 | 1.751 |
| ADDHHHP.13 | 3.743 | 0.963 | 6.432 | 0.83 | 0.993 | 0.431 | 2.674 | 17 | 9.30 | 1.755 |
| PLSF l | r2 m | SD n | Fo | Pp | Stability q | q2LOOr | q2LSOs | q2YS LOOt | q2YS LSOu | |
| ADDRRRP.11 | 5 | 0.949 | 0.264 | 61.3 | 4.38e−15 | 0.971 | 0.825 | 0.627 | −0.234 | −0.247 |
| ADDHHHP.13 | 5 | 0.951 | 0.257 | 61.4 | 4.41e−15 | 0.977 | 0.826 | 0.659 | −0.241 | −0.258 |
| Entry | pKi | EC Pred. pKi a | AAEP d | SB Pred. pKi a | AAEP d | LB Pred. pKi a | AAEP d | |||
|---|---|---|---|---|---|---|---|---|---|---|
| LOO b | LSO c | LOO b | LSO c | LOO b | LSO c | |||||
| 3ERT | 9.60 | 8.76 | 8.64 | 0.90 | 8.36 | 8.34 | 1.25 | 7.99 | 8.12 | 1.55 |
| 3UU7 | 8.79 | 8.14 | 6.91 | 1.27 | 8.09 | 7.22 | 1.14 | 7.85 | 7.14 | 1.30 |
| 3UUA | 8.79 | 8.15 | 7.54 | 0.94 | 7.05 | 7.12 | 1.71 | 8.07 | 7.37 | 1.07 |
| 3UUC | 5.70 | 4.36 | 4.39 | 1.33 | 4.45 | 4.06 | 1.45 | 5.67 | 6.77 | 0.55 |
| 4DMA | 5.60 | 6.54 | 7.69 | 1.52 | 7.91 | 7.59 | 2.15 | 8.86 | 7.7 | 2.68 |
| 4MG6 | 6.00 | 4.76 | 4.77 | 1.24 | 4.17 | 3.03 | 2.40 | 4.16 | 4.82 | 1.51 |
| 4MG8 | 10.00 | 8.86 | 8.87 | 1.14 | 9.16 | 7.76 | 1.54 | 8.99 | 8.85 | 1.08 |
| 4MG9 | 6.00 | 7.12 | 6.52 | 0.82 | 6.19 | 4.10 | 1.05 | 4.51 | 5.96 | 0.77 |
| 4MGA | 6.00 | 8.13 | 6.99 | 1.56 | 7.13 | 6.89 | 1.01 | 7.41 | 4.98 | 1.22 |
| 4MGC | 7.00 | 8.66 | 6.7 | 0.98 | 6.36 | 6.54 | 0.55 | 7.58 | 5.85 | 0.87 |
| 4MGD | 6.00 | 7.66 | 9.04 | 2.35 | 8.46 | 7.13 | 1.80 | 9.19 | 9.48 | 3.34 |
| 4TUZ | 10.00 | 8.64 | 8.88 | 1.24 | 9.17 | 7.52 | 1.66 | 9.06 | 8.7 | 1.12 |
| 4ZN9 | 9.60 | 8.96 | 8.92 | 0.66 | 8.74 | 7.06 | 1.70 | 8.78 | 8.49 | 0.97 |
| # | Ligand Structure | 3DPhypI/3-D QSAR pred. pIC50 | # | Ligand Structure | 3DPhypI/3-D QSAR pred. pIC50 b | ||
|---|---|---|---|---|---|---|---|
| SB a | LB b | SB a | LB b | ||||
| 3DPQ-1 |  | 9.20 | 9.17 | 3DPQ-7 |  | 9.26 | 9.11 |
| 3DPQ-2 |  | 9.21 | 9.12 | 3DPQ-8 |  | 9.04 | 8.95 |
| 3DPQ-3 |  | 9.37 | 9.29 | 3DPQ-9 |  | 9.31 | 9.26 |
| 3DPQ-4 |  | 9.26 | 9.22 | 3DPQ-10 |  | 9.18 | 9.05 |
| 3DPQ-5 |  | 9.05 | 8.92 | 3DPQ-11 |  | 9.12 | 9.28 |
| 3DPQ-6 |  | 9.01 | 8.91 | 3DPQ-12 |  | 9.42 | 9.35 |
| Comp. | ERα a | ERβ b | logRBA c | logRBA d | Ka Erα e | Ka Erβ f |
|---|---|---|---|---|---|---|
| (IC50 nM) | (IC50 nM) | ERα | ERβ | (nM) | (nM) | |
| 3DPQ-1 | 0.57 ± 0.54 g,†,‡,§ | 74.33 ± 0.46 †,‡,§ | 2.19 ‡,§ | 0.08 †,‡,§ | 0.13 †,‡ | 41.76 †,‡,§ |
| 3DPQ-2 | 0.54 ± 0.31 †,‡,§ | 77.24 ± 0.42 †,‡,§ | 2.22 †,‡,§ | 0.06 †,‡,§ | 0.12 †,‡ | 43.39 †,‡,§ |
| 3DPQ-3 | 0.44 ± 0.31 †,‡,§ | 74.86 ± 0.14 †,‡,§ | 2.31 †,‡,§ | 0.08 †,‡,§ | 0.10 †,‡ | 42.06 †,‡,§ |
| 3DPQ-4 | 0.47 ± 0.12 †,‡,§ | 82.45 ± 0.54 †,‡,§ | 2.28 †,‡,§ | 0.03 †,‡,§ | 0.11 †,‡ | 46.32 †,‡,§ |
| 3DPQ-5 | 0.81 ± 0.43 †,‡,§ | 74.41 ± 0.46 †,‡,§ | 2.04 ‡ | 0.08 †,‡,§ | 0.18 †,‡ | 41.80 †,‡,§ |
| 3DPQ-6 | 0.84 ± 0.11 †,‡,§ | 86.56 ± 0.33 †,‡,§ | 2.03 ‡ | 0.01 †,‡,§ | 0.19 ‡ | 48.63 †,‡,§ |
| 3DPQ-7 | 0.64 ± 0.13 †,‡,§ | 72.34 ± 0.17 †,‡,§ | 2.14 †,‡ | 0.09 †,‡,§ | 0.14 †,‡ | 40.64 †,‡,§ |
| 3DPQ-8 | 0.81 ± 0.14 †,‡,§ | 72.35 ± 0.78 †,‡,§ | 2.04 ‡ | 0.09 †,‡,§ | 0.18 †,‡ | 40.65 †,‡,§ |
| 3DPQ-9 | 0.45 ± 0.14 †,‡,§ | 83.56 ± 0.46 †,‡,§ | 2.30 †,‡,§ | 0.03 †,‡,§ | 0.10 †,‡ | 46.94 †,‡,§ |
| 3DPQ-10 | 0.77 ± 0.14 †,‡,§ | 79.54 ± 0.76 †,‡,§ | 2.06 ‡ | 0.05 †,‡,§ | 0.17 †,‡ | 44.69 †,‡,§ |
| 3DPQ-11 | 0.70 ± 0.33 †,‡,§ | 76.52 ± 0.48 †,‡,§ | 2.10 ‡ | 0.07 †,‡,§ | 0.16 †,‡ | 42.99 †,‡,§ |
| 3DPQ-12 | 0.40 ± 0.43 †,‡,§ | 89.45 ± 0.31 †,‡,§ | 2.35 †,‡,§ | 0.00 †,‡,§ | 0.09 †,‡,§ | 50.25 †,‡,§ |
| E2 h | 0.88 ± 0.24 ‡,§ | 0.88 ± 0.32 ‡,§ | 2.00 | 2.00 ‡,§ | 0.20 ‡,§ | 0.49 ‡,§ |
| 4-OHT. i | 1.13 ± 0.24 †,§ | 3.62 ± 0.43 †,§ | 1.90 § | 1.39 † | 0.25 †,§ | 2.03 †,§ |
| Ral. j | 0.73 ± 0.35 †,‡ | 3.39 ± 0.16 †,‡ | 2.09 ‡ | 1.42 † | 0.16 †,‡ | 1.90 †,‡ |
| Control k | NA l | NA | NA | NA | NA | NA |
| Comp. | MCF-7 a | MDA-MB-231 b | SI c | MRC-5 d | MCF-7 DR e | PR MCF-7 f | Ishikawa g |
|---|---|---|---|---|---|---|---|
| (IC50 nM) | (IC50 nM) | (IC50 nM) | (IC50 nM) | (IC50 nM) | (IC50 nM) | ||
| 3DPQ-1 | 0.76 ± 0.24 h,‡,§ | 72.44 ± 0.32 ‡,§ | 95.31 ‡,§ | >100 | >100 | >100 | 0.94 ± 0.36 g,‡,§ |
| 3DPQ-2 | 0.73 ± 0.42 ‡,§ | 72.42 ± 0.47 ‡,§ | 99.20 ‡,§ | >100 | >100 | >100 | 0.99 ± 0.35 ‡ |
| 3DPQ-3 | 0.61 ± 0.56 ‡,§ | 86.63 ± 0.68 ‡,§ | 142.02 ‡,§ | >100 | >100 | >100 | 0.84 ± 0.74 ‡,§ |
| 3DPQ-4 | 0.64 ± 0.15 ‡,§ | 67.31 ± 0.34 ‡,§ | 105.17 ‡,§ | >100 | >100 | >100 | 0.92 ± 0.43 ‡,§ |
| 3DPQ-5 | 1.02 ± 0.64 ‡,§ | 52.64 ± 0.69 ‡,§ | 51.61 ‡,§ | >100 | >100 | >100 | 1.42 ± 0.32 ‡,§ |
| 3DPQ-6 | 1.14 ± 0.49 ‡,§ | 52.31 ± 0.46 ‡,§ | 45.89 ‡,§ | >100 | >100 | >100 | 1.46 ± 0.43 ‡,§ |
| 3DPQ-7 | 0.78 ± 0.52 ‡,§ | 51.96 ± 0.68 ‡,§ | 66.61 ‡,§ | >100 | >100 | >100 | 1.74 ± 0.43 ‡,§ |
| 3DPQ-8 | 1.06 ± 0.45 ‡,§ | 42.56 ± 0.35 ‡,§ | 40.15 ‡,§ | >100 | >100 | >100 | 1.98 ± 0.32 ‡,§ |
| 3DPQ-9 | 0.62 ± 0.15 ‡,§ | 81.63 ± 0.42 ‡,§ | 131.66 ‡,§ | >100 | >100 | >100 | 0.89 ± 0.24 ‡,§ |
| 3DPQ-10 | 0.97 ± 0.34 ‡,§ | 41.97 ± 0.32 ‡,§ | 42.27 ‡,§ | >100 | >100 | >100 | 1.55 ± 0.42 ‡,§ |
| 3DPQ-11 | 0.81 ± 0.22 ‡,§ | 67.12 ± 0.54 ‡,§ | 82.86 ‡,§ | >100 | >100 | >100 | 1.37 ± 0.47 ‡,§ |
| 3DPQ-12 | 0.56 ± 0.11 ‡,§ | 82.84 ± 0.61 ‡,§ | 147.93 ‡,§ | >100 | >100 | >100 | 0.77 ± 0.43 ‡,§ |
| E2 i | N m | NA | NA | NA | NA | NA | NA |
| 4-OHT. j | 1.19 ± 0.57 § | 37.10 ± 0.45 § | 31.18 § | >10 | >100 | >100 | 1.29 ± 0.43 § |
| Ral. k | 0.90 ± 0.19 ‡ | 93.41 ± 0.48 ‡ | 103.97 ‡ | >10 | >100 | >100 | 0.97 ± 0.35 ‡ |
| Control l | NA | NA | NA | NA | NA | NA | NA |
| Comp. | Cell Cycle (%) | |||||
|---|---|---|---|---|---|---|
| Stage | G0/G1 a,b | S c | G2/M d,e | |||
| Conc. (nM) | 0.1 (1) f | 1 (10) | 0.1 (1) f | 1 (10) | 0.1 (1) f | 1 (10) |
| 3DPQ-1 | 72.62 ± 2.47 *,†,‡,§ | 75.08 ± 2.13 *,†,‡,§ | 9.98 ± 1.65 *,†,‡,§ | 10.69 ± 1.42 *,†,‡,§ | 17.40 ± 3.63 *,†,‡,§ | 14.24 ± 2.54 *,†,‡,§ |
| 3DPQ-2 | 73.64 ± 5.32 *,†,‡,§ | 76.10 ± 1.43 *,†,‡,§ | 11.88 ± 0.87 *,†,‡,§ | 12.59 ± 1.57 *,†,‡,§ | 14.48 ± 2.54 *,†,‡,§ | 11.32 ± 3.25 *,†,‡,§ |
| 3DPQ-3 | 72.99 ± 1.32 *,†,‡,§ | 75.45 ± 1.53 *,†,‡,§ | 8.98 ± 1.64 *,†,‡,§ | 9.69 ± 0.94 *,†,‡,§ | 18.03 ± 1.65 *,†,‡,§ | 14.87 ± 2.43 *,†,‡,§ |
| 3DPQ-4 | 77.78 ± 3.54 *,†,‡,§ | 80.24 ± 2.53 *,†,‡,§ | 7.20 ± 2.88 *,†,‡,§ | 7.91 ± 0.1.54 *,†,‡,§ | 15.02 ± 4.23 *,†,‡,§ | 11.86 ± 3.43 *,†,‡,§ |
| 3DPQ-5 | 71.78 ± 0.67 *,†,‡,§ | 74.24 ± 2.15 *,†,‡,§ | 9.21 ± 1.95 *,†,‡,§ | 9.92 ± 0.76 *,†,‡,§ | 19.01 ± 3.55 *,†,‡,§ | 15.85 ± 4.43 *,†,‡,§ |
| 3DPQ-6 | 70.52 ± 1.53 *,†,‡,§ | 71.98 ± 2.44 *,†,‡,§ | 13.27 ± 2.64 *,†,‡,§ | 13.98 ± 1.33 *,†,‡,§ | 16.21 ± 3.25 *,†,‡,§ | 14.05 ± 2.43 *,†,‡,§ |
| 3DPQ-7 | 73.25 ± 2.54 *,†,‡,§ | 75.71 ± 1.43 *,†,‡,§ | 14.06 ± 1.58 *,†,‡,§ | 14.77 ± 1.46 *,†,‡,§ | 12.69 ± 2.64 *,†,‡,§ | 9.53 ± 3.54 *,†,‡,§ |
| 3DPQ-8 | 72.39 ± 1.43 *,†,‡,§ | 74.85 ± 2.54 *,†,‡,§ | 12.50 ± 1.22 *,†,‡,§ | 13.21 ± 2.15 *,†,‡,§ | 15.11 ± 2.56 *,†,‡,§ | 11.95 ± 2.45 *,†,‡,§ |
| 3DPQ-9 | 71.47 ± 0.99 *,†,‡,§ | 75.93 ± 152 *,†,‡,§ | 12.97 ± 1.65 *,†,‡,§ | 13.68 ± 1.74 *,†,‡,§ | 15.56 ± 2.65 *,†,‡,§ | 10.40 ± 3.54 *,†,‡,§ |
| 3DPQ-10 | 71.96 ± 1.43 *,†,‡,§ | 74.42 ± 2.12 *,†,‡,§ | 11.96 ± 2.41 *,†,‡,§ | 12.67 ± 2.46 *,†,‡,§ | 16.08 ± 1.56 *,†,‡,§ | 12.92 ± 4.32 *,†,‡,§ |
| 3DPQ-11 | 72.53 ± 0.47 *,†,‡,§ | 74.99 ± 2.54 *,†,‡,§ | 13.31 ± 1.66 *,†,‡,§ | 14.02 ± 1.43 *,†,‡,§ | 14.16 ± 2.13 *,†,‡,§ | 11.00 ± 3.43 *,†,‡,§ |
| 3DPQ-12 | 77.83 ± 0.92 *,†,‡,§ | 80.29 ± 1.24 *,†,‡,§ | 16.96 ± 1.23 *,†,‡,§ | 17.67 ± 1.32 *,†,‡,§ | 5.21 ± 2.54 *,†,‡,§ | 2.05 ± 1.43 *,†,‡,§ |
| E2 g | 17.34 ± 0.35 *,‡,§ | 25.34 ± 0.36 *,‡,§ | 28.15 ± 0.52 *,‡,§ | 29.52 ± 0.46 *,‡,§ | 54.51 ± 0.57 *,‡,§ | 45.14 ± 0.33 *,‡,§ |
| 4-OTH. h | 57.22 ± 0.37 *,†,§ | 63.26 ± 0.41 *,†,§ | 18.76 ± 0.41 *,†,§ | 21.14 ± 0.25 *,†,§ | 24.02 ± 0.53 *,,†§ | 15.60 ± 0.15 *,†,§ |
| Ral. i | 59.14 ± 0.54 *,†,‡ | 66.52 ± 0.56 *,†,‡ | 15.83 ± 0.53 *,†,‡ | 16.37 ± 0.46 *,†,‡ | 25.03 ± 0.35 *,†,‡ | 17.11 ± 0.46 *,†,‡ |
| Control j | 32.21 ± 0.45 | 34.97 ± 0.53 | 32.82 ± 0.35 | |||
| Comp. | mol_MWT a | donorHB b | acceptHB c | QPlogPo/w d | PSA e | R05 f | QPlogKshsa g | QPlogHERG h | QPPCaco i |
| 3DPQ-1 | 501.243 | 3 | 9 | 2.11 | 133.084 | 2 | −0.571 | −5.759 | 26.396 |
| 3DPQ-2 | 515.254 | 2 | 9 | 2.49 | 124.532 | 1 | −0.529 | −5.242 | 27.138 |
| 3DPQ-3 | 597.263 | 3 | 11 | 2.29 | 131.324 | 3 | −0.539 | −5.354 | 31.352 |
| 3DPQ-4 | 552.175 | 3 | 11 | 1.43 | 136.387 | 3 | −0.645 | −5.367 | 25.872 |
| 3DPQ-5 | 569.234 | 2 | 11 | 1.45 | 160.686 | 3 | −0.934 | −4.029 | 26.464 |
| 3DPQ-6 | 590.261 | 2 | 10 | 3.01 | 154.432 | 2 | 0.005 | −4.903 | 22.432 |
| 3DPQ-7 | 601.272 | 2 | 9 | 4.33 | 122.038 | 1 | 0.198 | −5.836 | 34.075 |
| 3DPQ-8 | 610.336 | 3 | 10 | 3.51 | 133.649 | 2 | 0.191 | −4.976 | 165.259 |
| 3DPQ-9 | 611.243 | 3 | 12 | 1.39 | 140.653 | 3 | −0.562 | −5.321 | 27.621 |
| 3DPQ-10 | 555.286 | 2 | 9 | 3.32 | 143.543 | 2 | 0.135 | −4.324 | 132.594 |
| 3DPQ-11 | 541.276 | 2 | 9 | 2.81 | 143.653 | 2 | 0..162 | −4.321 | 135.594 |
| 3DPQ-12 | 585.243 | 3 | 11 | 1.58 | 140.795 | 3 | −0.900 | −5.239 | 26.295 |
| E2 s | 278.434 | 2 | 3 | 2.487 | 47.727 | 0 | 0.214 | −1.994 | 1322.153 |
| 4-OTH. t | 407.679 | 1 | 5 | 4.201 | 36.102 | 0 | 0.669 | −3.909 | 669.539 |
| Ral u | 495.759 | 3 | 9 | 2.381 | 73.257 | 0 | 0.173 | −3.648 | 130.539 |
| QPPMDCK j | QPlogBB k | A l | B m | C n | D o | E p | F q | G r | |
| 3DPQ-1 | 26.435 | −1.964 | − | − | − | − | − | − | − |
| 3DPQ-2 | 31.095 | −1.892 | − | − | − | − | − | − | − |
| 3DPQ-3 | 34.542 | −2.963 | − | − | − | − | − | − | − |
| 3DPQ-4 | 31.921 | −2.735 | − | − | − | − | − | − | − |
| 3DPQ-5 | 32.351 | −2.029 | − | − | − | − | − | − | + |
| 3DPQ-6 | 23.658 | −2.432 | − | − | − | − | + | − | + |
| 3DPQ-7 | 14.190 | −3.977 | − | − | + | + | + | − | + |
| 3DPQ-8 | 70.677 | −3.237 | − | − | − | − | + | − | + |
| 3DPQ-9 | 36.284 | −2.876 | − | − | − | − | − | − | + |
| 3DPQ-10 | 16.325 | −3.321 | − | − | + | − | − | − | − |
| 3DPQ-11 | 18.362 | −3.431 | − | − | + | − | − | − | − |
| 3DPQ-12 | 32.285 | −2.682 | − | − | − | − | − | − | + |
| E2 s | 669.023 | −0.209 | − | − | − | − | − | − | − |
| 4-OTH. t | 354.743 | −0.136 | − | − | − | − | − | − | − |
| Ral u | 88.081 | −0.582 | − | − | − | − | − | − | − |
| Comp. | Dose | log D7.4 a | Tumor Latency | Tumor Burden | Tumor Volume | Rat PPB b | Rat CL c | BIO d | MFD e (5 days) | WL after MFD f (day 1, mg) g |
|---|---|---|---|---|---|---|---|---|---|---|
| (mg/kg) | (week) | (week) | (mm3) | (%free) | in vivo | (mg/kg) | (day 5, mg) h | |||
| 3DPQ-1 | 5 | 1.94 ‡,‖ | 9 * | 3.38 ± 0.31 i,*,†,‖ | 1.09 ± 0.23 *,†,‡,‖ | 1.33 ‡,‖ | 60 ‡,‖ | 91 | 1000 | 310.34 ± 0.34 i |
| 50 | 12 *,† | 2.04 ± 0.35 *,†,§,┴ | 0.68 ± 0.35 *,†,§,┴ | 1.22 §,┴ | 69 §,┴ | 94 | 300.23 ± 0.62 | |||
| 3DPQ-2 | 5 | 1.99 ‡,‖ | 9 * | 3.34 ± 0.57 *,†,‖ | 0.96 ± 0.41 *,†,‡,‖ | 1.15 ‡,‖ | 59 ‡,‖ | 92 | 1000 | 305.03 ± 0.66 |
| 50 | 12 *,† | 1.98 ± 0.45 *,†,§,┴ | 0.69 ± 0.23 *,†,§,┴ | 1.24 §,┴ | 64 §,┴ | 94 | 300.43 ± 0.65 | |||
| 3DPQ-3 | 5 | 2.07 ‡,‖ | 12 *,†,‡ | 2.18 ± 0.69 *,†,‡,‖ | 0.78 ± 0.43 *,†,‡,‖ | 1.34 ‡,‖ | 66 ‡,‖ | 90 | 1000 | 320,45 ± 0.62 |
| 50 | 15 *,†,§ | 1.16 ± 0.64 *,†,§,┴ | 0.66 ± 0.21 *,†,§,┴ | 1.47 §,┴ | 71 §,┴ | 93 | 300.31 ± 0.52 | |||
| 3DPQ-4 | 5 | 1.88 ‡,‖ | 10 *,† | 2.39 ± 0.56 *,†,‡,‖ | 0.98 ± 0.31 *,†,‡,‖ | 1.23 ‡,‖ | 64 ‡,‖ | 90 | 1000 | 320.73 ± 0.36 |
| 50 | 14 *,† | 1.33 ± 0.15 *,†,§,┴ | 0.41 ± 0.23 *,†,§,┴ | 1.51 §,┴ | 76 §,┴ | 93 | 305.56 ± 0.68 | |||
| 3DPQ-9 | 5 | 2.02 ‡,‖ | 12 *,†,‡ | 2.28 ± 0.47 *,†,‡,‖ | 0.77 ± 0.32 *,†,‡,‖ | 1.28 ‡,‖ | 62 ‡,‖ | 94 ‡ | 1000 | 315.54 ± 0.65 |
| 50 | 15 *,†,§ | 1.14 ± 0.65 *,†,§,┴ | 0.40 ± 0.43 *,†,§,┴ | 1.31 §,┴ | 78 §,┴ | 97 | 310.33 ± 0.95 | |||
| 3DPQ-12 | 5 | 2.06 ‡,‖ | 12 *,†,‡ | 2.24 ± 0.54 *,†,‡,‖ | 0.67 ± 0.22 *,†,‡,‖ | 1.24 ‡,‖ | 63 ‡,‖ | 93 ‡ | 1000 | 305.06 ± 0.94 |
| 50 | 15 *,†,§ | 0.94 ± 0.35 *,†,§,┴ | 0.34 ± 0.11 *,†,§,┴ | 1.31 §,┴ | 71 §,┴ | 96 | 299.56 ± 0.45 | |||
| 4-OTH. j | 5 | 3.64 ‖ | 7 * | 3.36 ± 0.38 *,†,‖ | 1.88 ± 0.35 *,†,‖ | 1.85 | 35 | 88 ‖ | 1000 | 305.84 ± 0.59 |
| 50 | 10 *,† | 3.22 ± 0.21 *,†,┴ | 1.35 ± 0.63 *,†,┴ | 2.52 ┴ | 42 | 94 | 297.65 ± 0.39 | |||
| Ral. k | 5 | 2.39 ‡ | 8 * | 3.11 ± 0.47 *,†,‡ | 1.67 ± 0.31 *,†,‡ | 1.85 | 36 | 93 ‡ | 1000 | 310.54 ± 0.45 |
| 50 | 13 *,† | 2.91 ± 0.22 *,†,§ | 1.41 ± 0.54 *,†§ | 1.90 § | 42 | 96 | 300.54 ± 0.48 | |||
| MNU l | 50 | NA o | 5 *,†,‡,§,‖,┴ | 4.55 ± 0.15 *,‡,§,‖,┴ | 4.48 ± 0.54 | NA | NA | NA | 100 | 305.44 ± 0.62 |
| C m | NA | 0 †,‡,§,‖,┴ | 0 †,‡,§,‖,┴ | 0 †,‡,§,‖,┴ | NA | NA | NA | NA | 210.54 ± 0.29 | |
| Placebo n | NA | NA | NA | NA | NA | NA | NA | NA | 300.54 ± 0.63 | |
| NA | NA | NA | NA | NA | NA | NA | NA | 325.43 ± 0.29 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurtanović, N.; Tomašević, N.; Matić, S.; Proia, E.; Sabatino, M.; Antonini, L.; Mladenović, M.; Ragno, R. Human Estrogen Receptor Alpha Antagonists, Part 3: 3-D Pharmacophore and 3-D QSAR Guided Brefeldin A Hit-to-Lead Optimization toward New Breast Cancer Suppressants. Molecules 2022, 27, 2823. https://doi.org/10.3390/molecules27092823
Kurtanović N, Tomašević N, Matić S, Proia E, Sabatino M, Antonini L, Mladenović M, Ragno R. Human Estrogen Receptor Alpha Antagonists, Part 3: 3-D Pharmacophore and 3-D QSAR Guided Brefeldin A Hit-to-Lead Optimization toward New Breast Cancer Suppressants. Molecules. 2022; 27(9):2823. https://doi.org/10.3390/molecules27092823
Chicago/Turabian StyleKurtanović, Nezrina, Nevena Tomašević, Sanja Matić, Elenora Proia, Manuela Sabatino, Lorenzo Antonini, Milan Mladenović, and Rino Ragno. 2022. "Human Estrogen Receptor Alpha Antagonists, Part 3: 3-D Pharmacophore and 3-D QSAR Guided Brefeldin A Hit-to-Lead Optimization toward New Breast Cancer Suppressants" Molecules 27, no. 9: 2823. https://doi.org/10.3390/molecules27092823