
Enantioselective Organocatalyzed Michael Addition of Isobutyraldehyde to Maleimides in Aqueous Media


Abstract
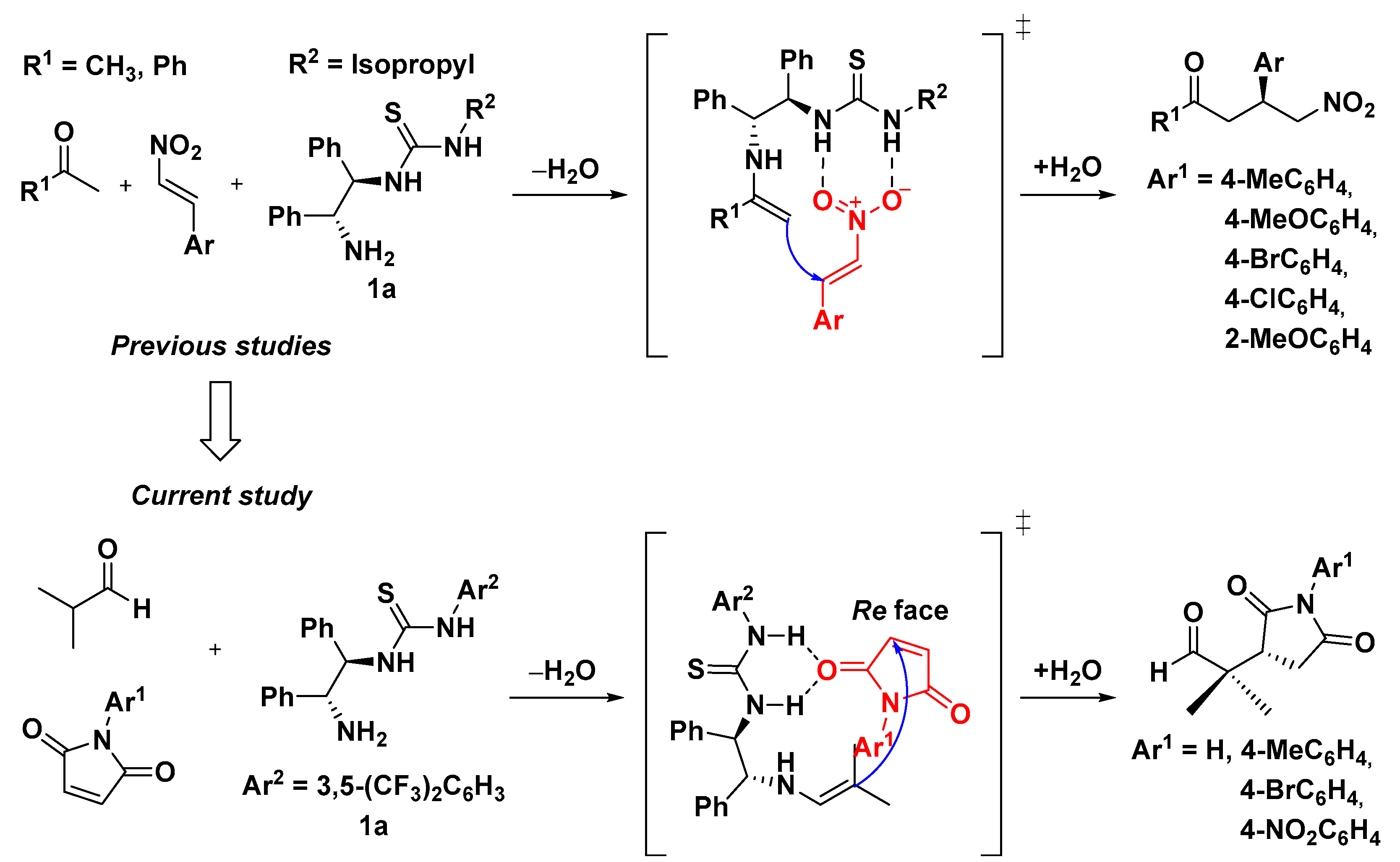
:1. Introduction
2. Results and Discussion
2.1. Asymmetric Michael Reaction of Maleimide and Aldehydes Using a Thiourea Catalyst
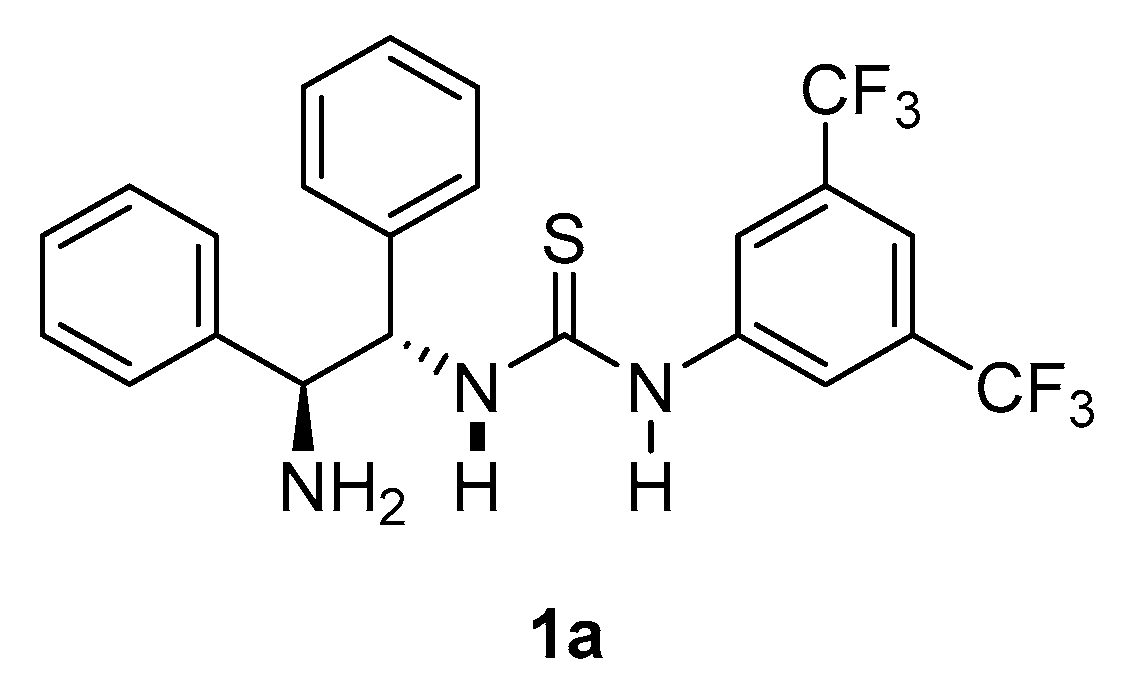
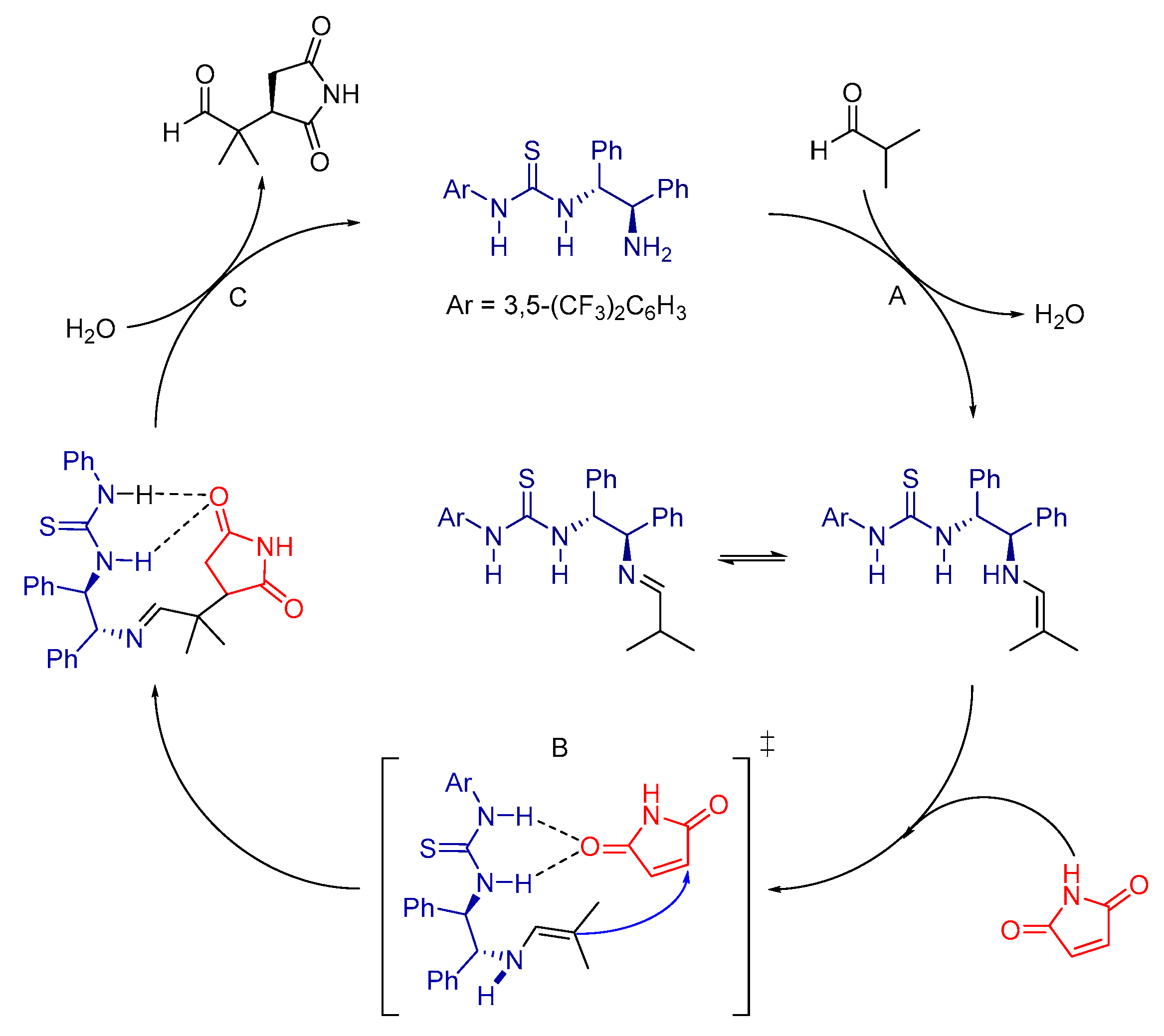
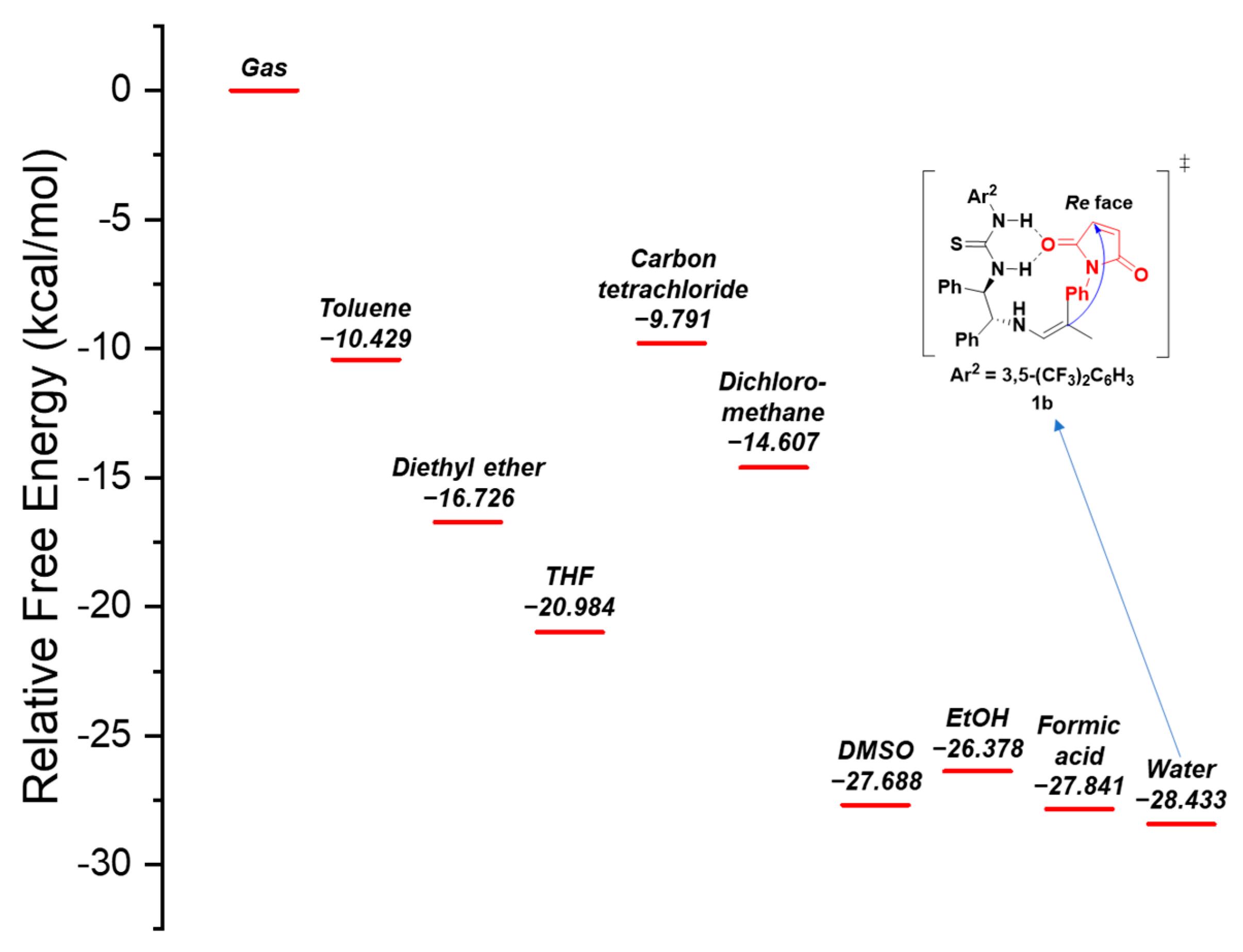
2.2. Reaction Mechanism Inferred through Expected Transition States
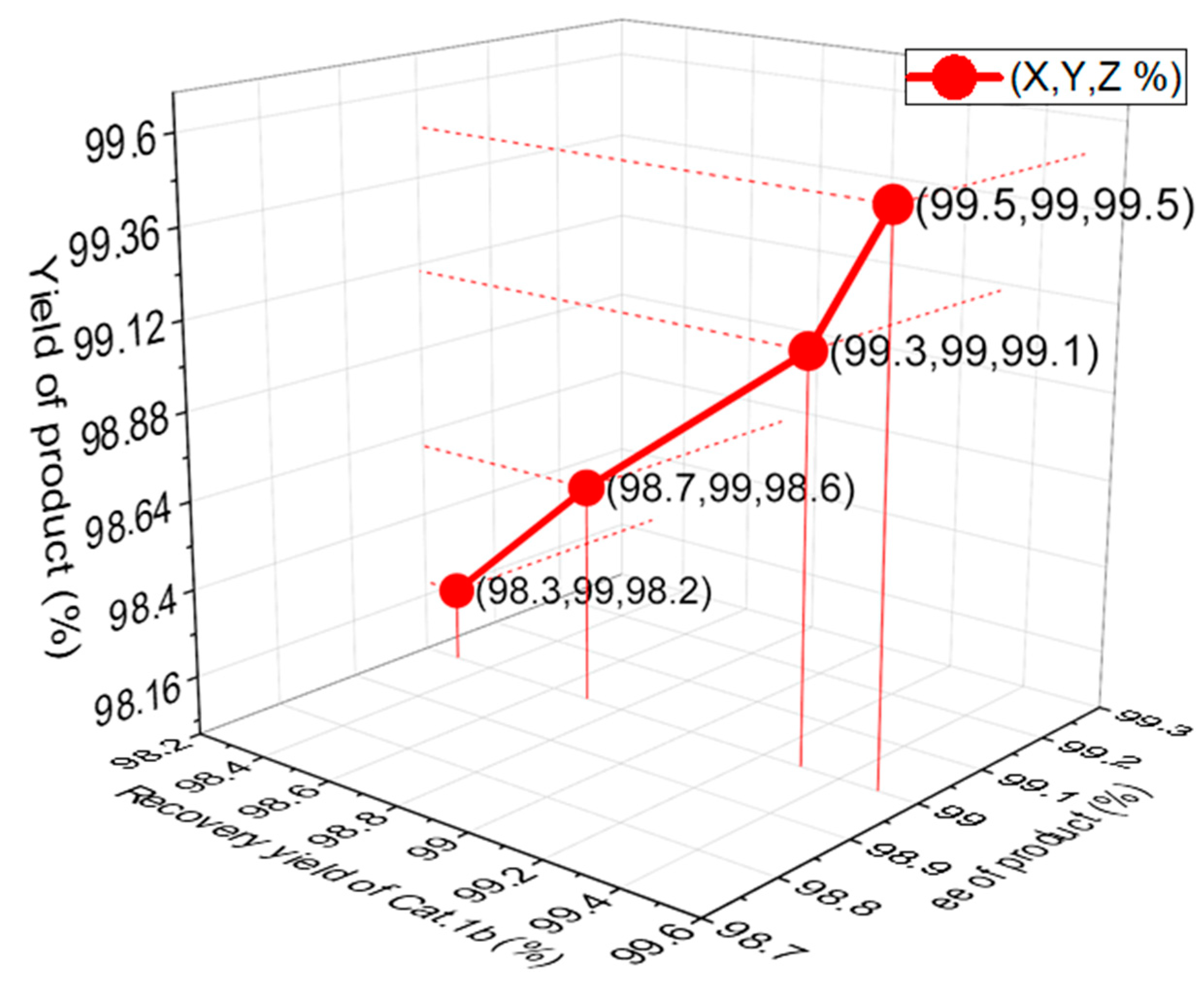
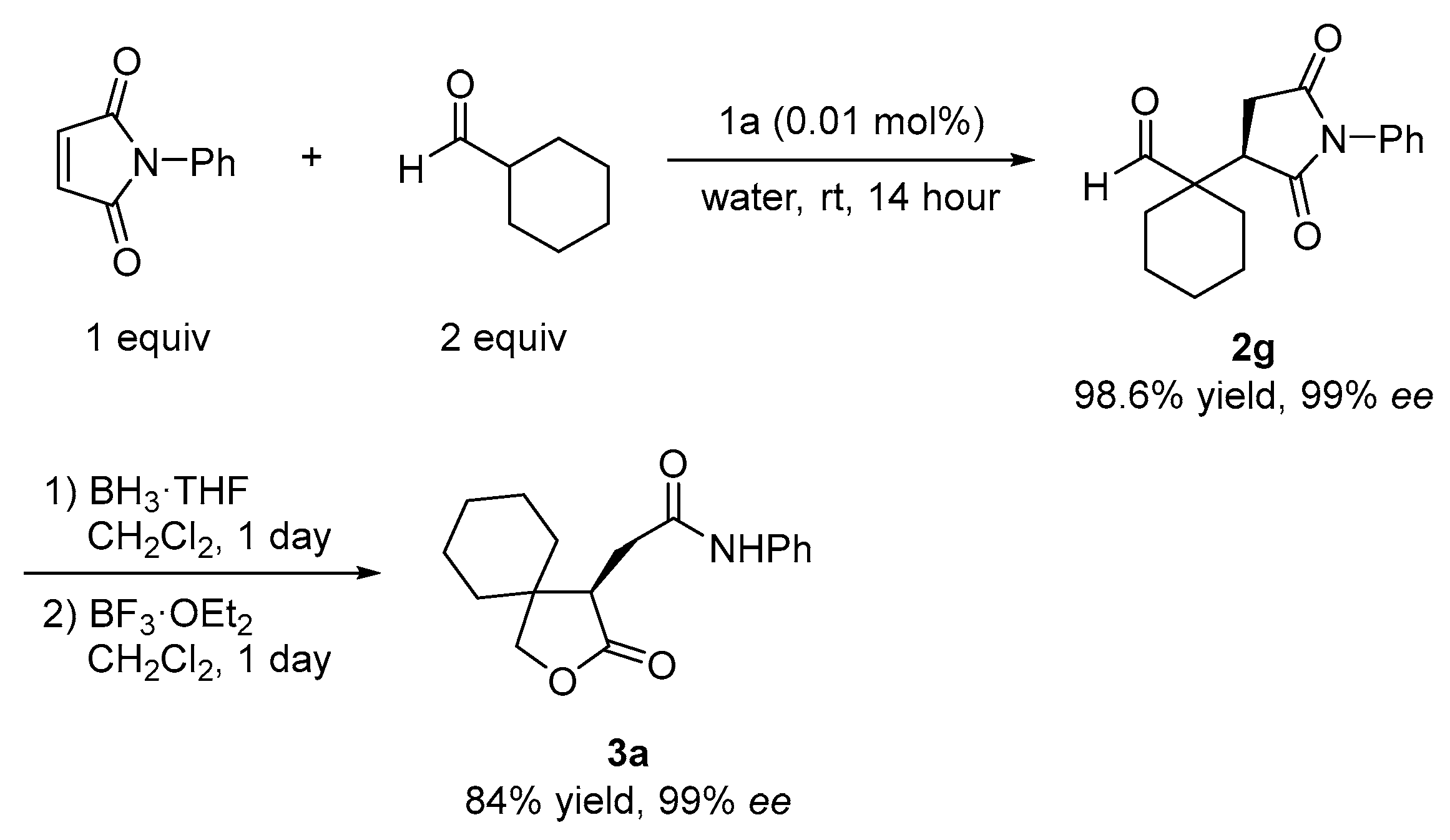
2.3. Pharmaceutical Applications Using Gram-Scale Asymmetric Reactions
3. Materials and Methods
3.1. Instruments and Reagents
3.2. Experimental Method
3.2.1. Synthesis of N-Mono-Thiourea Catalyst
3.2.2. Asymmetric Michael Reaction of Aldehyde and Maleimide Using a Chiral Thiourea Catalyst
3.2.3. Gram-Scale Asymmetric Michael Reaction of Aldehyde and Maleimide Using a Chiral Thiourea Catalyst
3.2.4. General Procedure of the Racemic Michael Addition
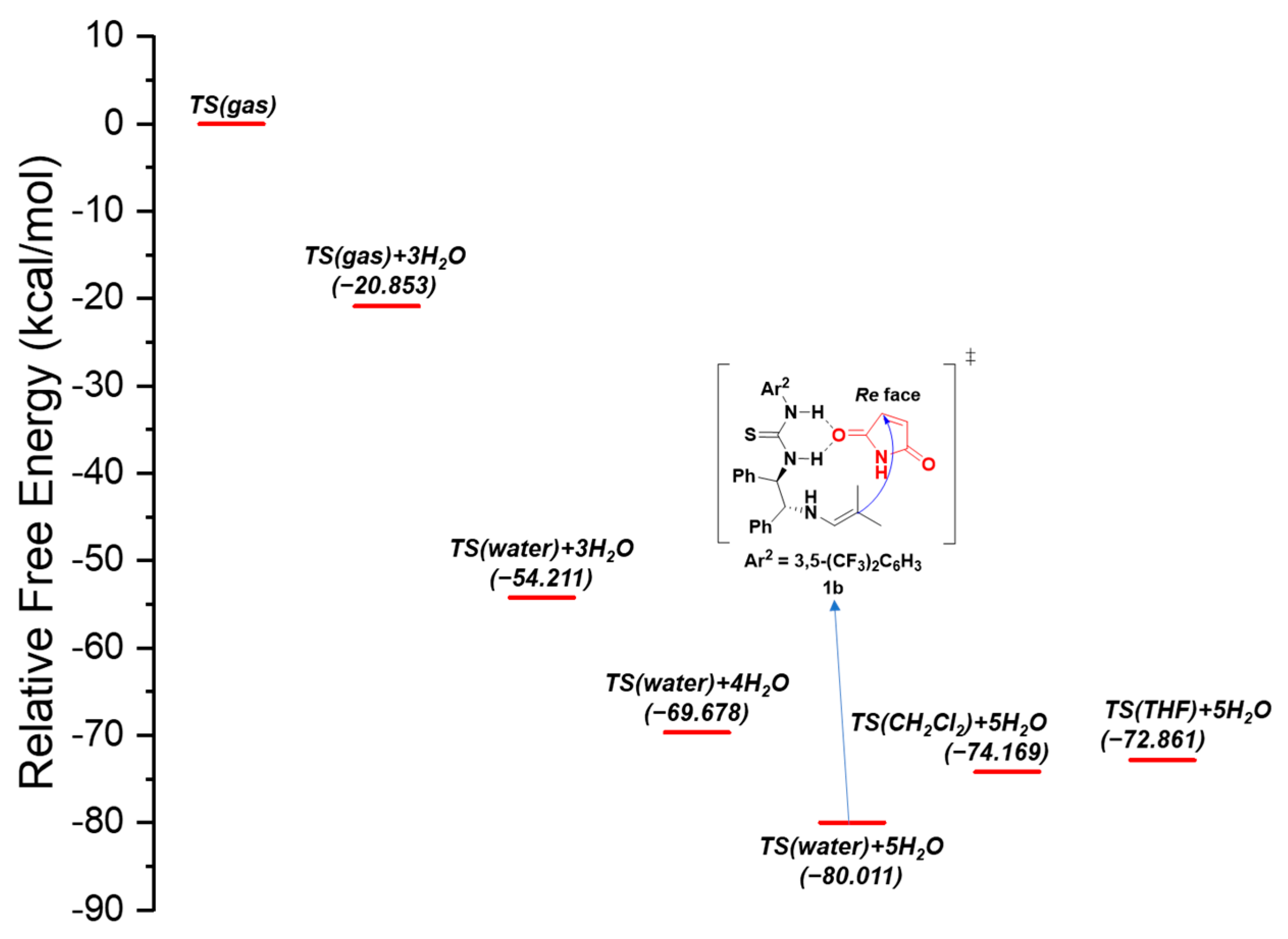
3.3. Results of Density Functional Theory Calculations and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- MacMillan, D.W.C. The advent and development of organocatalysis. Nature 2008, 455, 304. [Google Scholar] [CrossRef] [PubMed]
- Dalko, P.I.; Moisan, L. In the golden age of organocatalysis. Angew. Chem. Int. Ed. Engl. 2004, 43, 5138. [Google Scholar] [CrossRef]
- Doyle, A.G.; Jacobsen, E.N. Small-molecule H-bond donors in asymmetric catalysis. Chem. Rev. 2007, 107, 5713. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Yang, J.W.; Hoffmann, S.; List, B. Asymmetric enamine catalysis. Chem. Rev. 2007, 107, 5471. [Google Scholar] [CrossRef] [PubMed]
- CDavie, E.A.C.; Mennen, S.M.; Xu, Y.; Miller, S.J. Asymmetric catalysis mediated by synthetic peptides. Chem. Rev. 2007, 107, 5759. [Google Scholar] [CrossRef]
- Ballini, R.; Bosica, G.; Cioci, G.; Fiorini, D.; Petrini, M. Conjugate addition of nitroalkanes to N-substituted maleimides. Synthesis of 3-alkylsuccinimides and pyrrolidines. Tetrahedron 2003, 59, 3603. [Google Scholar] [CrossRef]
- Du, Z.-H.; Qin, W.-J.; Tao, B.-X.; Yuan, M.; Da, C.-S. N-primary-amine tetrapeptide-catalyzed highly asymmetric Michael addition of aliphatic aldehydes to maleimides. Org. Biomol. Chem. 2020, 18, 6899. [Google Scholar] [CrossRef] [PubMed]
- Mase, N.; Tanaka, F.; Barbas, C.F., III. Synthesis of β-hydroxyaldehydes with stereogenic quaternary carbon centers by direct organocatalytic asymmetric aldol reactions. Angew. Chem., Int. Ed. Engl. 2004, 43, 2420. [Google Scholar] [CrossRef]
- Chowdari, N.S.; Suri, J.T.; Barbas, C.F., III. Asymmetric synthesis of quaternary α- and β-amino acids and β-lactams via proline-catalyzed Mannich reactions with branched aldehyde donors. Org. Lett. 2004, 6, 2507. [Google Scholar] [CrossRef]
- Mase, N.; Thayumanavan, R.; Tanaka, F.; Barbas, C.F., III. Organocatalytic direct Michael reaction of ketones and aldehydes with β-nitrostyrene in brine. Org. Lett. 2004, 6, 2527. [Google Scholar] [CrossRef]
- Chowdari, N.S.; Barbas, C.F., III. Total synthesis of LFA-1 antagonist BIRT-377 via organocatalytic asymmetric construction of a quaternary stereocenter. Org. Lett. 2005, 7, 867. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.R.; Kuo, W.-H.; Jacobsen, E.N. Enantioselective catalytic α-alkylation of aldehydes via an SN1 pathway. J. Am. Chem. Soc. 2010, 132, 9286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, J.H.; Nam, S.H.; Kim, B.S.; Ha, D.C. Organocatalytic asymmetric Michael addition of ketones to α, β-unsaturated nitro compounds. Catalysts 2020, 10, 618. [Google Scholar] [CrossRef]
- Shim, J.H.; Kim, M.J.; Lee, J.Y.; Kim, K.H.; Ha, D.C. Organocatalytic asymmetric aldol reaction using protonated chiral 1,2-diamines. Tetrahedron Lett. 2020, 61, 152295. [Google Scholar] [CrossRef]
- Shim, J.H.; Lee, M.J.; Lee, M.H.; Kim, B.S.; Ha, D.C. Enantioselective organocatalytic Michael reactions using chiral (R,R)-1,2-diphenylethylenediamine-derived thioureas. RSC Adv. 2020, 10, 31808. [Google Scholar] [CrossRef]
- Shim, J.H.; Park, S.J.; Ahn, B.K.; Lee, J.Y.; Kim, B.S.; Ha, D.C. Enantioselective thiolysis and aminolysis of cyclic anhydrides using a chiral diamine-derived thiourea catalyst. ACS Omega 2021, 6, 34501. [Google Scholar] [CrossRef]
- Shim, J.H.; Ahn, B.K.; Lee, J.Y.; Kim, H.S.; Ha, D.C. Organocatalysis for the asymmetric Michael addition of cycloketones and α,β-unsaturated nitroalkenes. Catalysts 2021, 11, 1004. [Google Scholar] [CrossRef]
- Shim, J.H.; Hong, Y.; Kim, J.H.; Kim, H.S.; Ha, D.-C. Organocatalytic asymmetric Michael addition in aqueous media by a hydrogen-bonding catalyst and application for inhibitors of GABAB receptor. Catalysts 2021, 11, 1134. [Google Scholar] [CrossRef]
- Shim, J.H.; Cheun, S.H.; Kim, H.S.; Ha, D.-C. Organocatalysis for the asymmetric Michael addition of aldehydes and α,β-unsaturated nitroalkenes. Catalysts 2022, 12, 121. [Google Scholar] [CrossRef]
- Shim, J.H.; Lee, J.Y.; Kim, H.S.; Ha, D.-C. Protonated Chiral 1,2-Diamine Organocatalysts for N-Selective Nitroso Aldol Reaction. Catalysts 2022, 12, 435. [Google Scholar] [CrossRef]
- Xue, F.; Liu, L.; Zhang, S.; Duan, W.; Wang, W. A simple primary amine thiourea catalyzed highly enantioselective conjugate addition of α,α-disubstituted aldehydes to maleimides. Chem. Eur. J. 2010, 16, 7979–7982. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.-w.; Liu, Y.-x.; Li, P.l.; Ren, H.; Zhu, Y.; Tao, J.C. A highly efficient large-scale asymmetric Michael addition of isobutyraldehyde to maleimides promoted by a novel multifunctional thiourea. Tetrahedron Asymmetry 2011, 22, 1740–1748. [Google Scholar] [CrossRef]
- Orlandi, S.; Pozzi, G.; Ghisetti, M.; Benaglia, M. Synthesis and catalytic activity of fluorous chiral primary amine-thioureas. New J. Chem. 2013, 37, 4140. [Google Scholar] [CrossRef]
- Kokotos, C.G. An Asymmetric Michael Addition of α,α-Disubstituted Aldehydes to Maleimides Leading to a One-Pot Enantioselective Synthesis of Lactones Catalyzed by Amino Acids. Org. Lett. 2013, 15, 2406–2409. [Google Scholar] [CrossRef]
- Ferrandiz, J.F.; Bengoa, E.G.; Chinchilla, R. Solvent-induced reversal of enantioselectivity in the synthesis of succinimides by the addition of aldehydes to maleimides catalysed by carbamate-monoprotected 1,2-diamines. Eur. J. Org. Chem. 2015, 6, 1218. [Google Scholar] [CrossRef]
- Qiaoa, Y.; Headley, A.D. A simple and highly effective water-compatible organocatalytic system for asymmetric direct Michael reactions of linear aldehydes to maleimides. Green Chem. 2013, 15, 2690–2694. [Google Scholar] [CrossRef]
- Kolcsára, V.J.; Szőllősi, G. Chitosan as a chiral ligand and organocatalyst: Preparation conditions–property–catalytic performance relationships. Catal. Sci. Technol. 2021, 11, 7652–7666. [Google Scholar] [CrossRef]
- Du, Z.-H.; Qin, W.-J.; Tao, B.-X.; Yuan, M.; Da, C.-S. Silver-catalyzed direct C–H oxidative carbamoylation of quinolines with oxamic acids. Org. Biomol. Chem. 2020, 18, 6899. [Google Scholar] [CrossRef]
- Landeros, J.M.; Cruz-Hernandez, C.; Juaristi, E. α-amino acids and α,β-dipeptides intercalated into hydrotalcite: Efficient catalysts in the asymmetric Michael addition reaction of aldehydes to N-substituted maleimides. Eur. J. Org. Chem. 2021, 37, 5117. [Google Scholar] [CrossRef]
- Avila, A.; Chinchilla, R.; Najera, C. Enantioselective Michael addition of α,α-disubstituted aldehydes to maleimides organocatalyzed by chiral primary amine-guanidines. Tetrahedron Asymmetry 2012, 23, 1625. [Google Scholar] [CrossRef]
- Nugent, T.C.; Sadiq, A.; Bibi, A.; Heine, T.; Zeonjuk, L.L.; Vankova, N.; Bassil, B. Noncovalent bifunctional organocatalysts: Powerful tools for contiguous quaternary-tertiary stereogenic carbon formation, scope, and origin of enantioselectivity. Chem. Eur. J. 2012, 18, 4088. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Chavez, J.; Luna-Morales, S.; Cruz-Aguilar, D.-A.; Díaz-Salazar, H.; Narváez, W.E.V.; Silva-Gutiérrez, R.S.; Hernández-Ortega, S.; Rocha-Rinza, T.; Hernández-Rodríguez, M. The effect of chiral N-substituents with methyl or trifluoromethyl groups on the catalytic performance of mono- and bifunctional thioureas. Org. Biomol. Chem. 2019, 17, 10045. [Google Scholar] [CrossRef] [PubMed]
- de Simone, N.A.D.; Meninno, S.; Talotta, C.; Gaeta, C.; Neri, P.; Lattanzi, A. Solvent-free enantioselective Michael reactions catalyzed by a calixarene-based primary amine thiourea. J. Org. Chem. 2018, 83, 10318. [Google Scholar] [CrossRef]
- Torregrosa-Chinillach, A.; Moragues, A.; Pérez-Furundarena, H.; Chinchilla, R.; Gómez-Bengoa, E. Enantioselective Michael addition of aldehydes to maleimides organocatalyzed by a chiral primary amine-salicylamide. Molecules 2018, 23, 3299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiza, A.; Spiliopoulou, N.; Shahu, A.; Kokotos, C.G. Combining organocatalysis with photoorganocatalysis: Photocatalytic hydroacylation of asymmetric organocatalytic Michael addition products. New J. Chem. 2018, 42, 18844. [Google Scholar] [CrossRef]
- Muramulla, S.; Ma, J.-A.; Zhao, J.C.-G. Michael addition of ketones and aldehydes to maleimides catalyzed by modularly designed organocatalysts. Adv. Synth. Catal. 2013, 355, 1260. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. Mol. Phys. 1970, 19, 55341. [Google Scholar] [CrossRef]
- Miura, T.; Masuda, A.; Ina, M.; Nakashima, K.; Nishida, S.; Tada, N.; Itoh, A. Asymmetric Michael reactions of α, α-disubstituted aldehydes with maleimides using a primary amine thiourea organocatalyst. Tetrahedron Asymmetry 2011, 22, 1605–1609. [Google Scholar] [CrossRef]
- Bai, J.F.; Peng, L.; Wang, L.L.; Wang, L.X.; Xu, X.Y. Chiral primary amine thiourea promoted highly enantioselective Michael reactions of isobutylaldehyde with maleimides. Tetrahedron 2010, 66, 8928–8932. [Google Scholar] [CrossRef]
- Yu, F.; Jin, Z.; Huang, H.; Ye, T.; Liang, X.; Ye, J. A highly efficient asymmetric Michael addition of α, α-disubstituted aldehydes to maleimides catalyzed by primary amine thiourea salt. Org. Biomol. Chem. 2010, 8, 4767–4774. [Google Scholar] [CrossRef]
- Ma, Z.W.; Liu, X.F.; Liu, J.T.; Liu, Z.J.; Tao, J.C. Highly enantioselective Michael addition of α, α-disubstituted aldehydes to maleimides catalyzed by new primary amine-squaramide bifunctional organocatalysts. Tetrahedron Lett. 2017, 58, 4487–4490. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
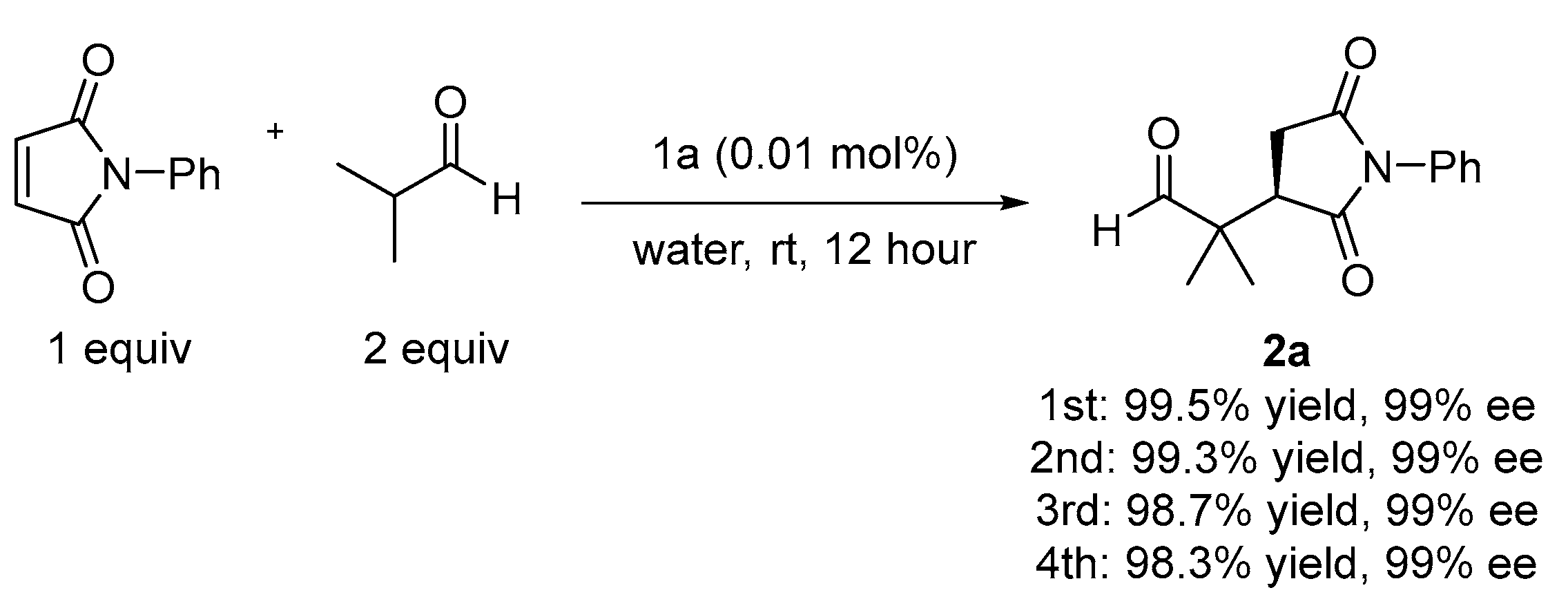{kind=link}
{kind=link}
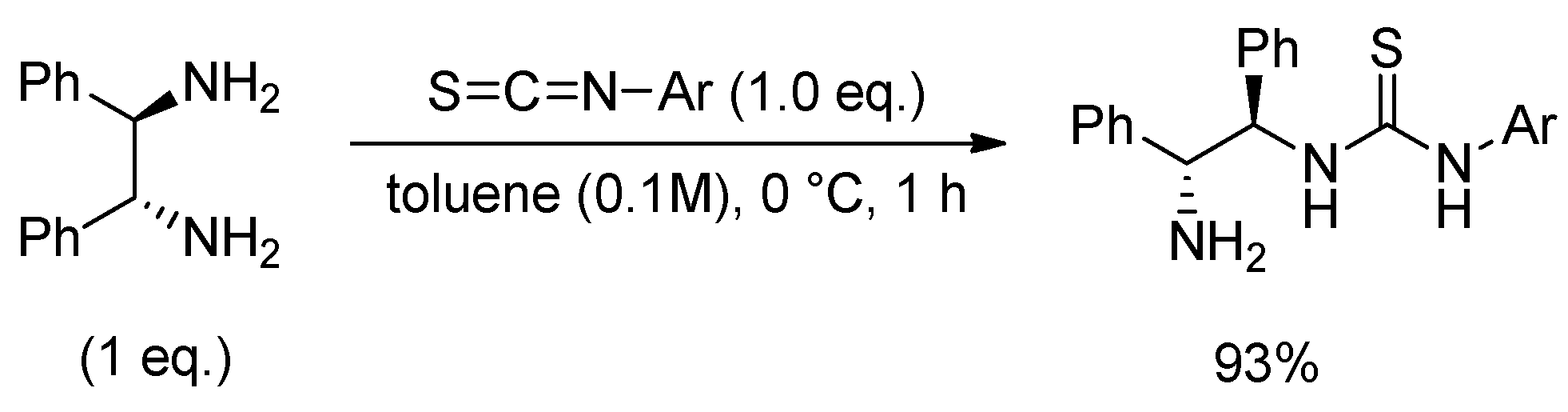{kind=link}

| Entry | Time (h) | Additive | Solvent | Yield (%) a | ee (%) b |
|---|---|---|---|---|---|
| 1 | 24 | Trifluoro-acetic acid | CH2Cl2 | 78 | 98 |
| 2 | 24 | Acetic acid | CH2Cl2 | 83 | 94 |
| 3 | 24 | Salicylic acid | CH2Cl2 | 82 | 94 |
| 4 | 24 | Benzoic acid | Toluene | 78 | 99 |
| 5 | 24 | Benzoic acid | THF | 98 | 99 |
| 6 c | 0.67 | - | Water | 99 | 99 |
| 7 d | 6 | - | Water | 99 | 99 |
| 8 e | 12 | - | Water | 97 | 99 |

| Entry | Ar | Time (h) | Yield (%) a | ee (%) b |
|---|---|---|---|---|
| 1 | H | 10 | 98 | 99 |
| 2 | 4-MeC6H4 | 12 | 99 | 99 |
| 3 | 4-BrC6H4 | 12 | 98 | 99 |
| 4 | 4-NO2C6H4 | 12 | 97 | 99 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shim, J.H.; Cheun, S.H.; Kim, H.S.; Ha, D.-C. Enantioselective Organocatalyzed Michael Addition of Isobutyraldehyde to Maleimides in Aqueous Media. Molecules 2022, 27, 2759. https://doi.org/10.3390/molecules27092759
Shim JH, Cheun SH, Kim HS, Ha D-C. Enantioselective Organocatalyzed Michael Addition of Isobutyraldehyde to Maleimides in Aqueous Media. Molecules. 2022; 27(9):2759. https://doi.org/10.3390/molecules27092759
Chicago/Turabian StyleShim, Jae Ho, Seok Hyun Cheun, Hyeon Soo Kim, and Deok-Chan Ha. 2022. "Enantioselective Organocatalyzed Michael Addition of Isobutyraldehyde to Maleimides in Aqueous Media" Molecules 27, no. 9: 2759. https://doi.org/10.3390/molecules27092759