
Impact of Different [Tc(N)PNP]-Scaffolds on the Biological Properties of the Small cRGDfK Peptide: Synthesis, In Vitro and In Vivo Evaluations

 , , , , , and
, , , , , and
Abstract
:
1. Introduction
2. Results
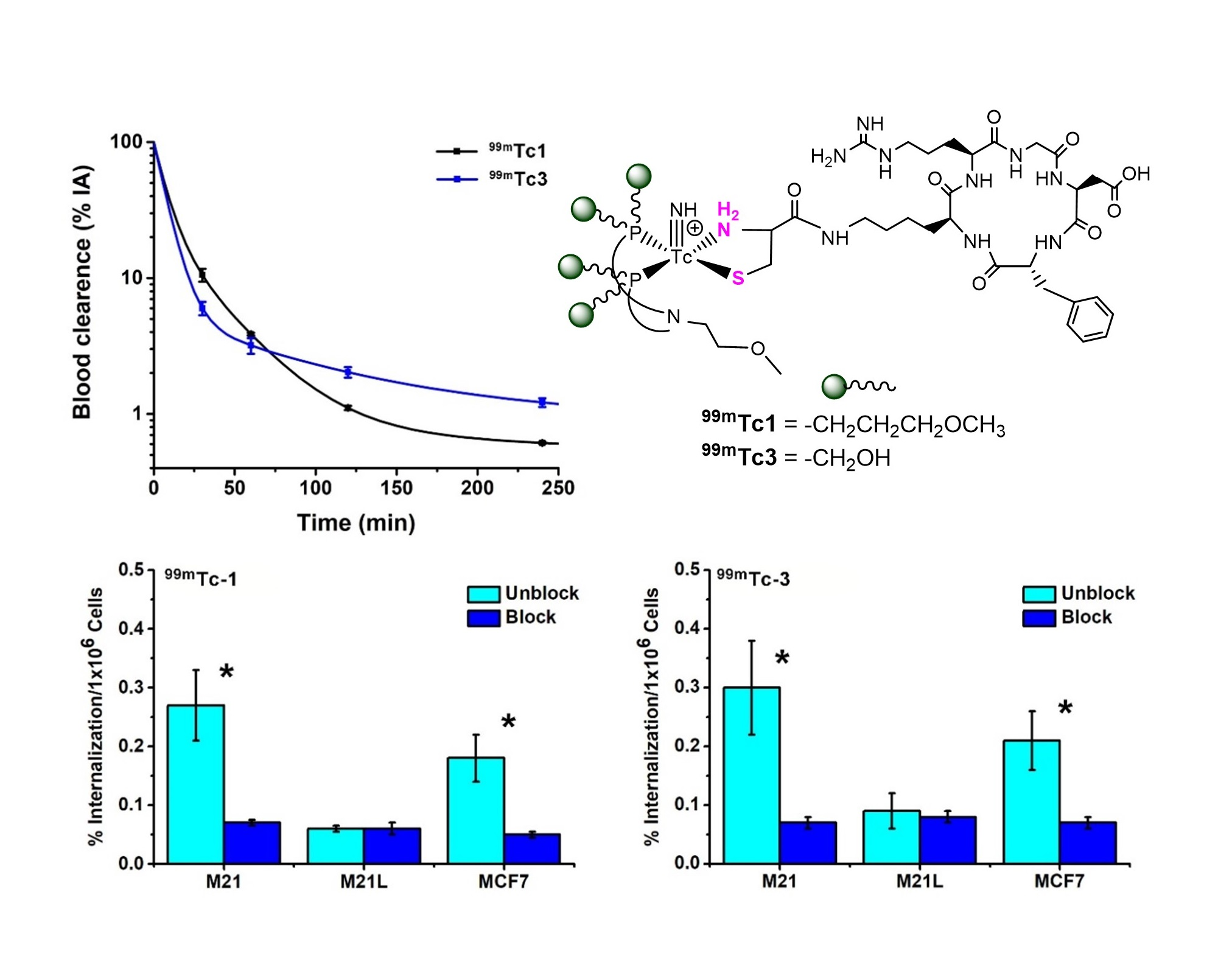
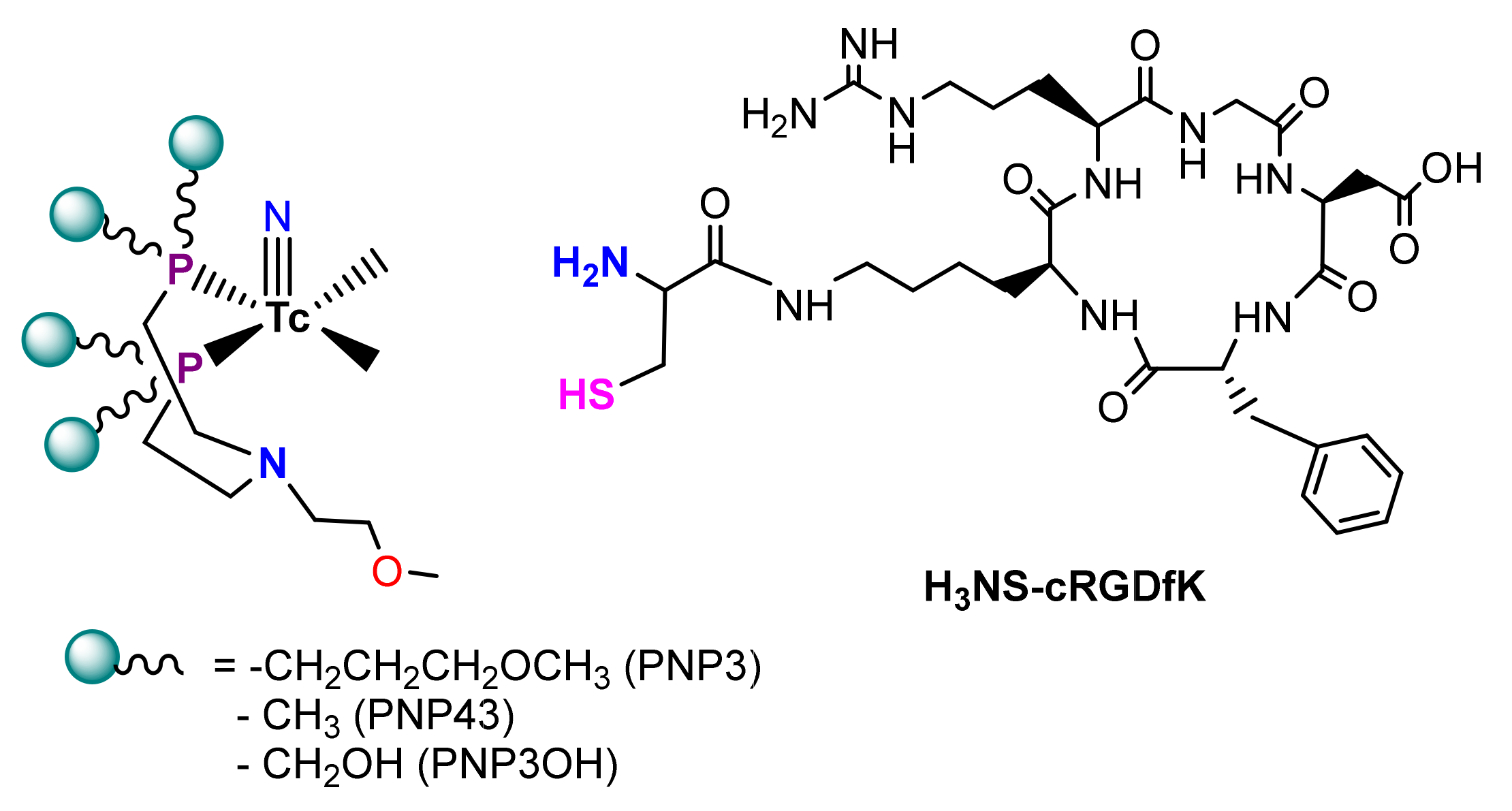
2.1. Peptide Synthesis
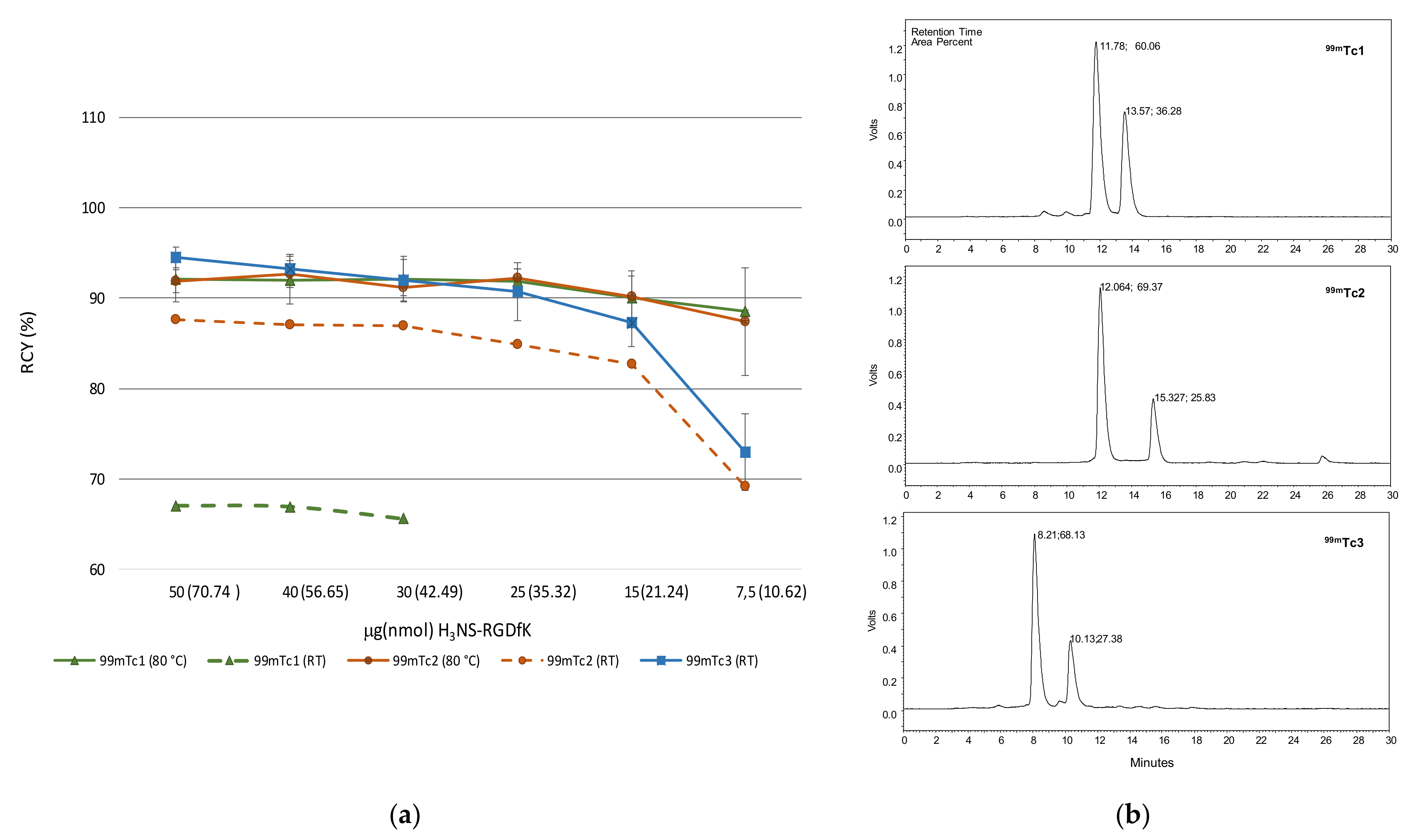
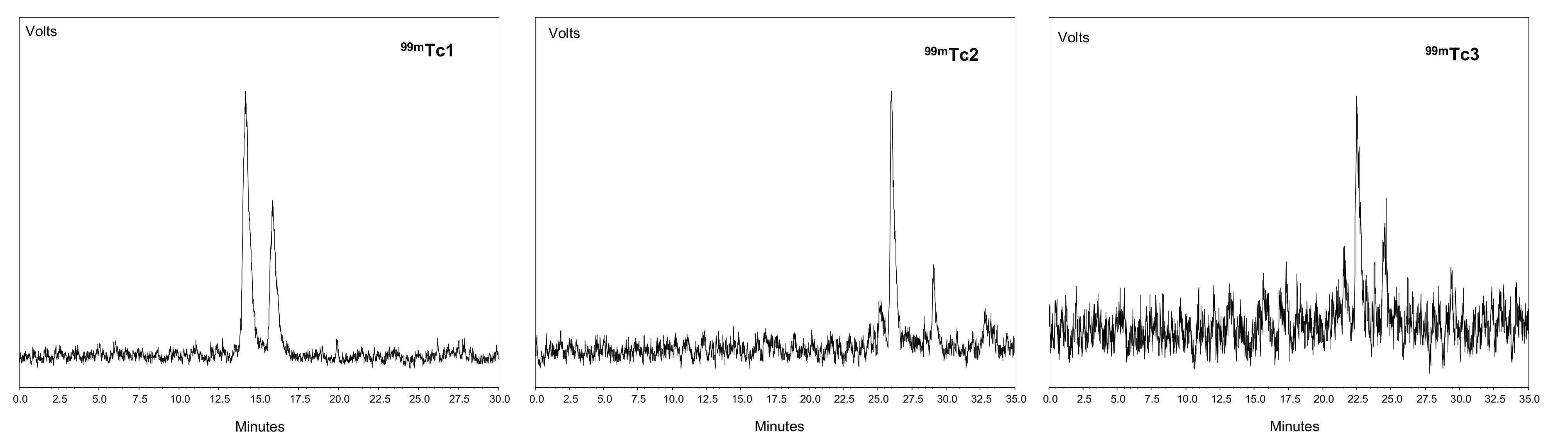
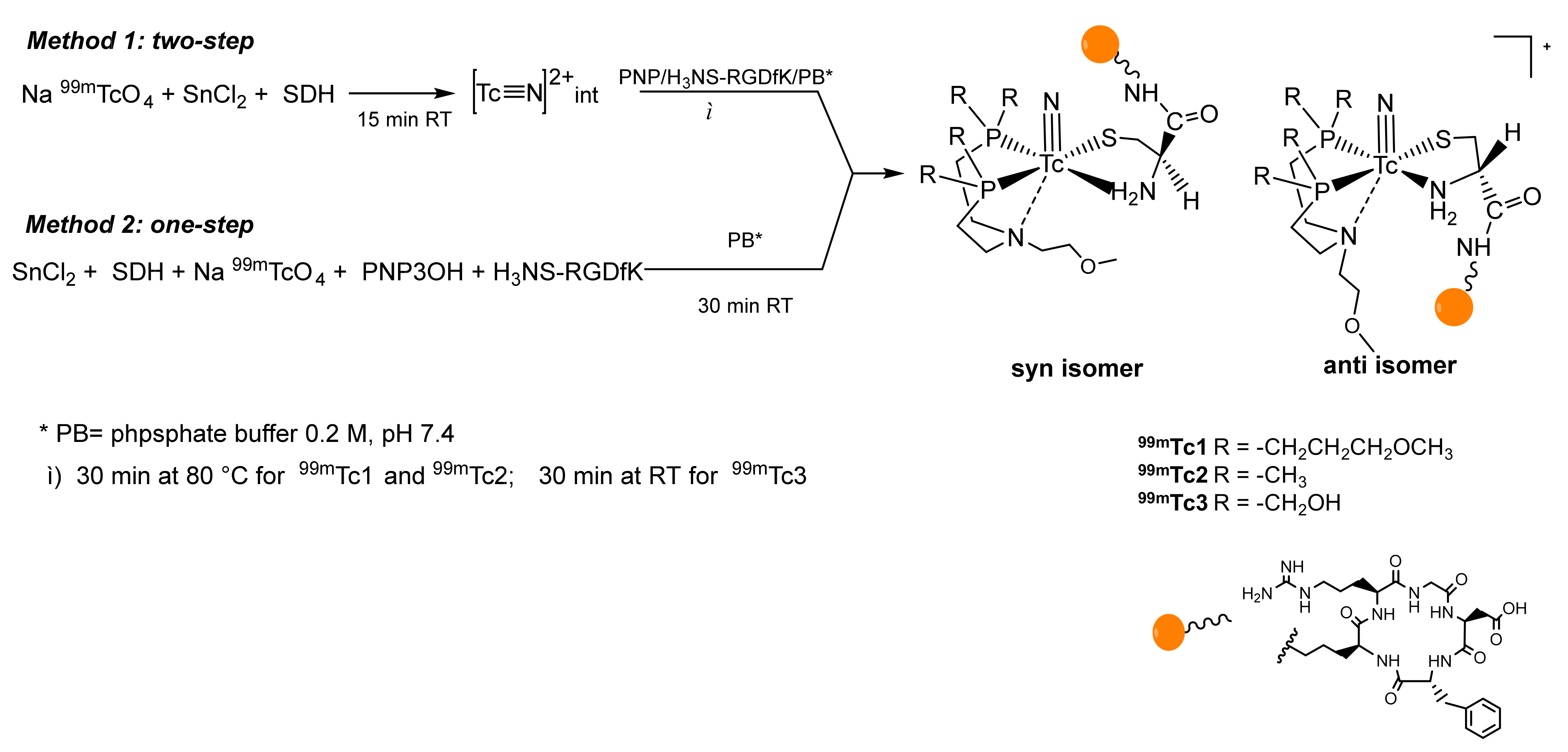
2.2. Radiosyntheses
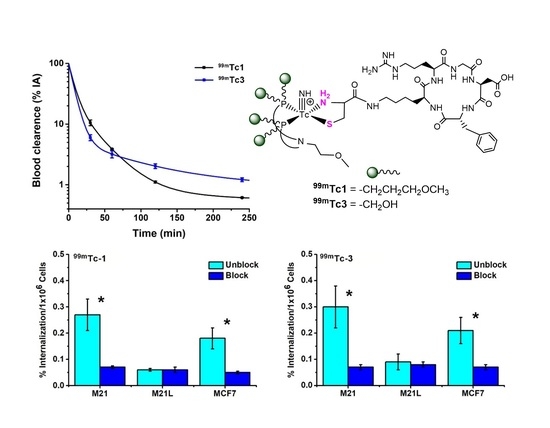
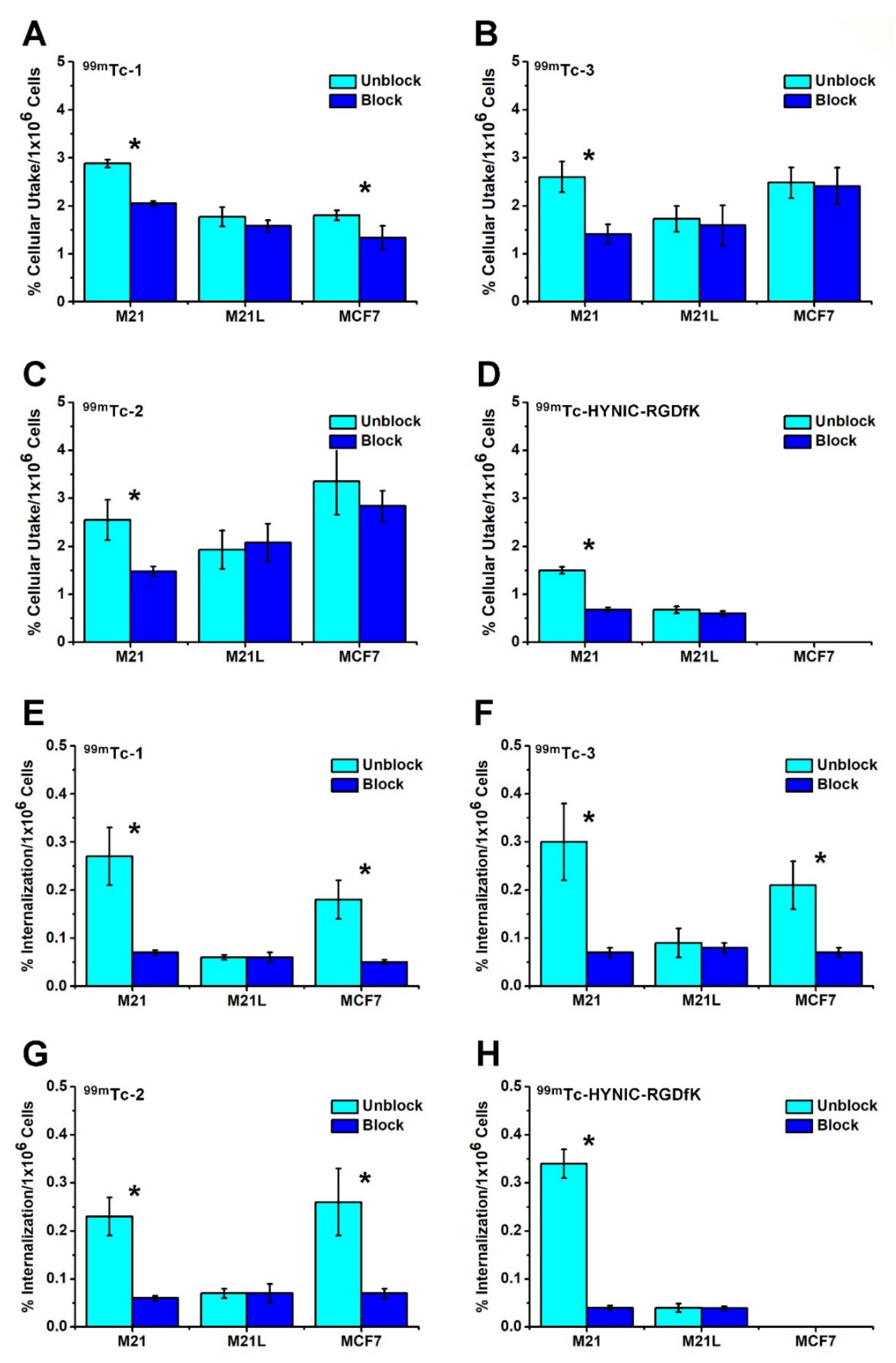
2.3. Cell Studies
2.4. Metabolism and In Vivo Studies
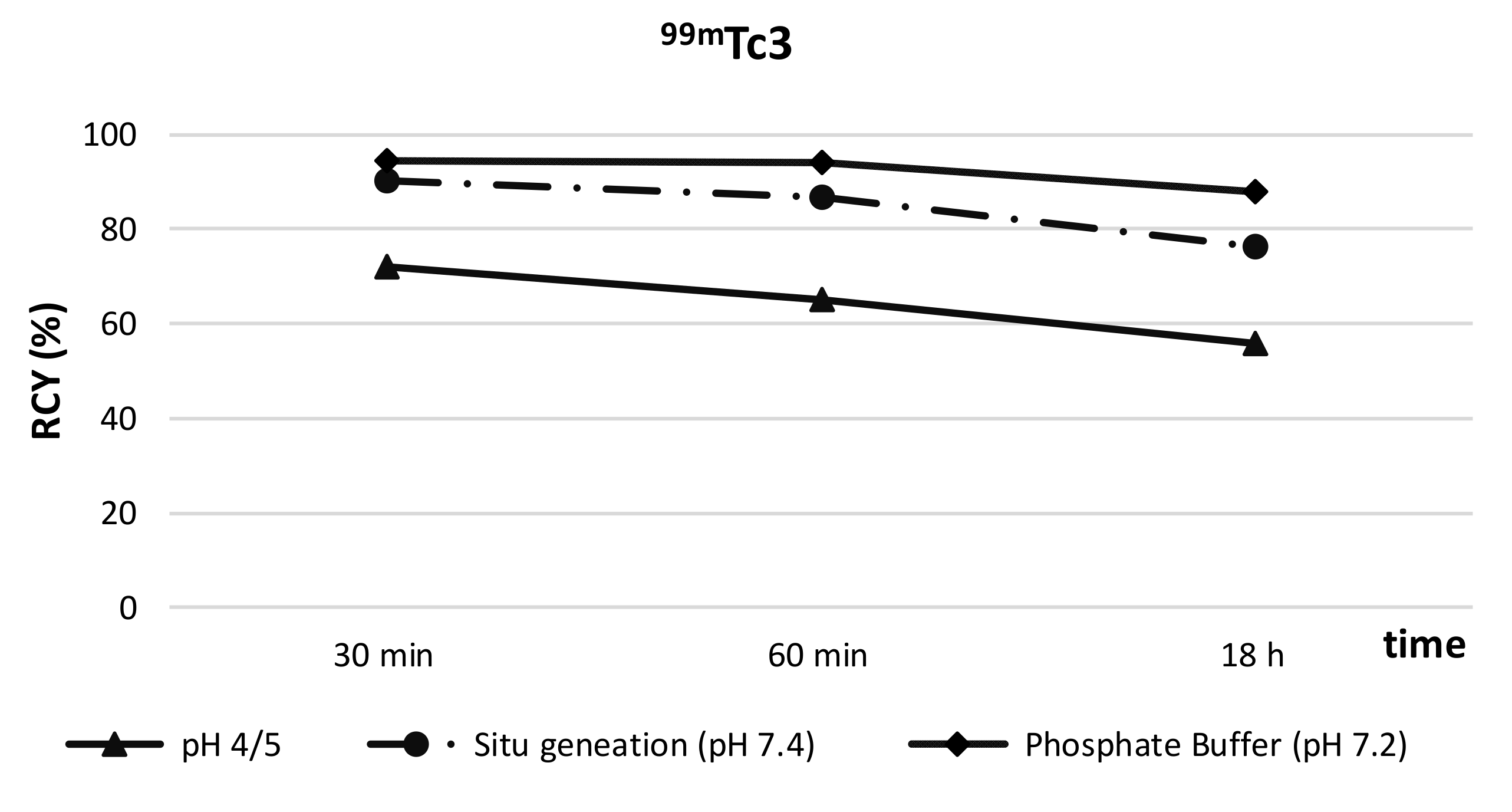
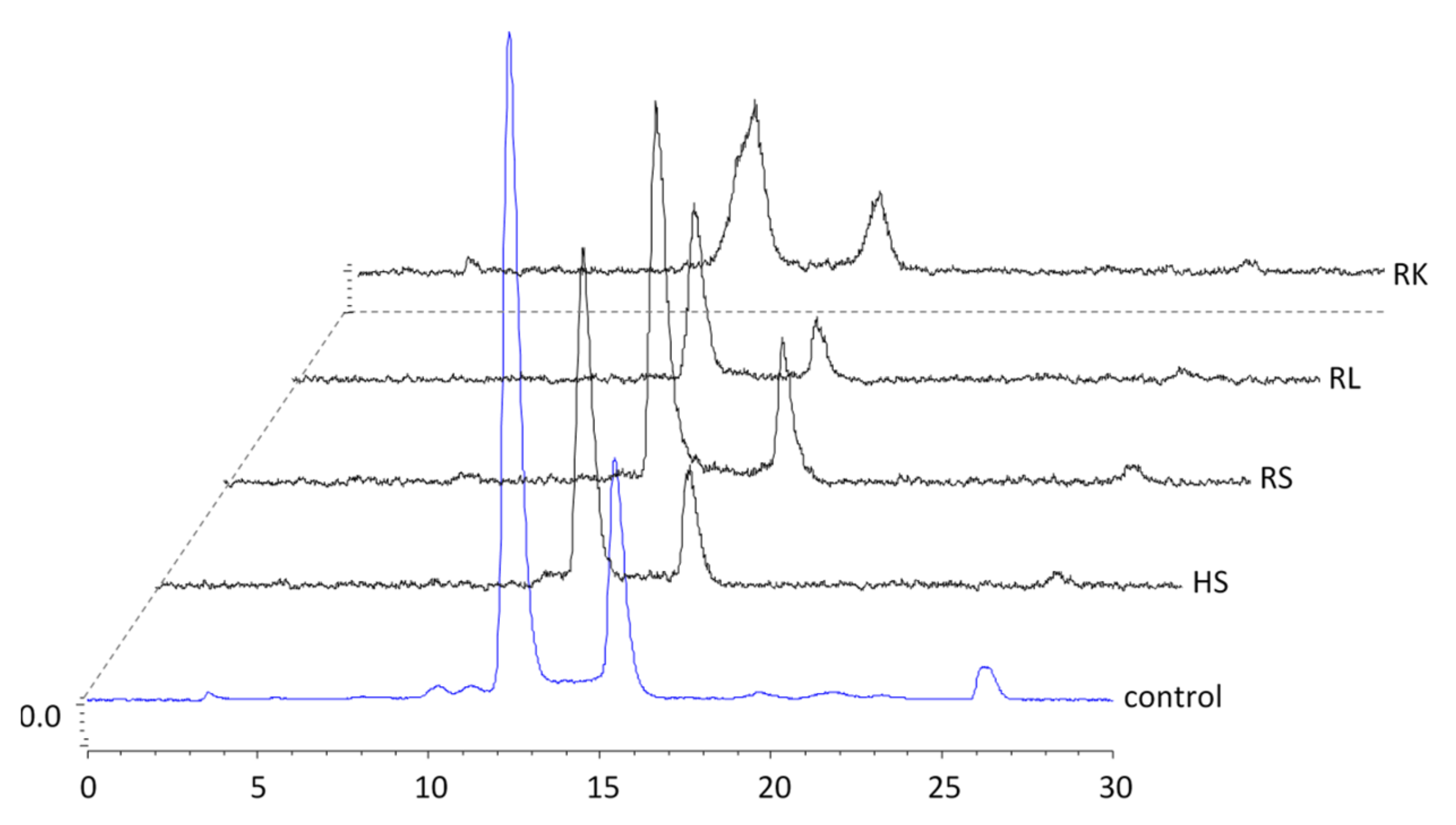
2.4.1. In Vitro Metabolism
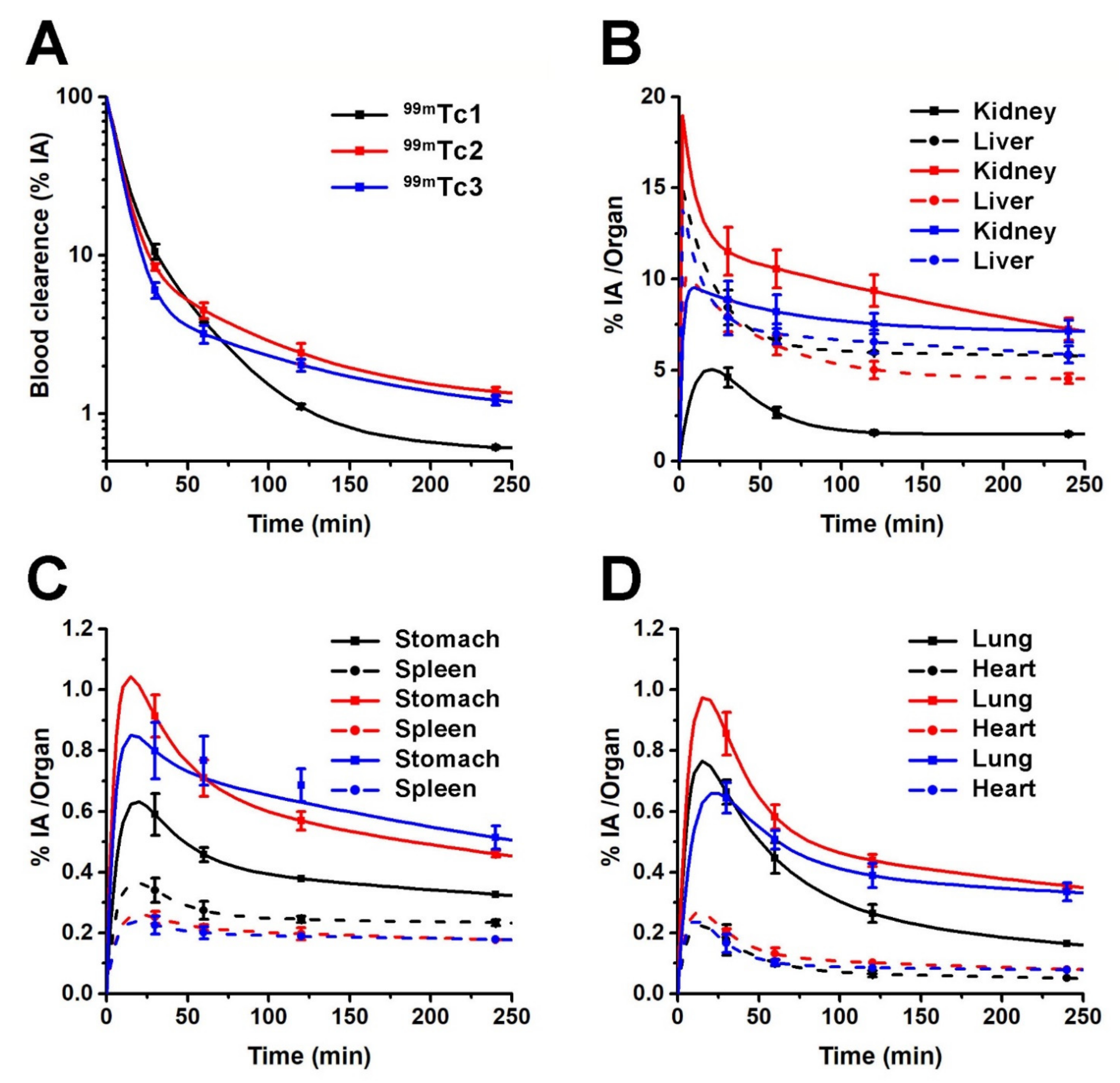
2.4.2. In Vivo Studies
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Bolzati, C.; Refosco, F.; Marchiani, A.; Ruzza, P. 99mTc-Radiolabelled Peptides for Tumour Imaging: Present and Future. Curr. Med. Chem. 2010, 17, 2656–2683. [Google Scholar] [CrossRef] [PubMed]
- Duatti, A. Review on 99mTc Radiopharmaceuticals with Emphasis on New Advancements. Nucl. Med. Biol. 2020, 92, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Bolzati, C.; Carta, D.; Salvarese, N.; Refosco, F. Chelating Systems for 99mTc/188Re in the Development of Radiolabeled Peptide Pharmaceuticals. Anti-Cancer Agents Med. Chem. 2012, 12, 428–464. [Google Scholar] [CrossRef] [PubMed]
- Alberto, R.; Schibli, R.; Egli, A.; Schubiger, A.P.; Abram, U.; Kaden, T.A. A Novel Organometallic Aqua Complex of Technetium for the Labeling of Biomolecules: Synthesis of [99mTc(OH2)3(CO)3]+ from [99mTcO4]− in Aqueous Solution and Its Reaction with a Bifunctional Ligand. J. Am. Chem. Soc. 1998, 120, 7987–7988. [Google Scholar] [CrossRef]
- Charron, C.L.; Hickey, J.L.; Nsiama, T.K.; Cruickshank, D.R.; Turnbull, W.L.; Luyt, L.G. Molecular Imaging Probes Derived from Natural Peptides. Nat. Prod. Rep. 2016, 33, 761–800. [Google Scholar] [CrossRef]
- Opalinska, M.; Hubalewska-Dydejczyk, A.; Sowa-Staszczak, A. Radiolabeled peptides: Current and new perspectives. Q. J. Nucl. Med. Mol. Imaging 2017, 61, 153–167. [Google Scholar] [CrossRef]
- Rezazadeh, F.; Sadeghzadeh, N. Tumor Targeting with 99mTc Radiolabeled Peptides: Clinical Application and Recent Development. Chem. Biol. Drug Des. 2019, 93, 205–221. [Google Scholar] [CrossRef]
- Tolmachev, V.; Orlova, A. Influence of Labelling Methods on Biodistribution and Imaging Properties of Radiolabelled Peptides for Visualisation of Molecular Therapeutic Targets. Curr. Med. Chem. 2010, 17, 2636–2655. [Google Scholar] [CrossRef]
- Boschi, A.; Bolzati, C.; Benini, E.; Malagò, E.; Uccelli, L.; Duatti, A.; Piffanelli, A.; Refosco, F.; Tisato, F. A Novel Approach to the High-Specific-Activity Labeling of Small Peptides with the Technetium-99m Fragment [99mTc(N)(PXP)]2+ (PXP = Diphosphine Ligand). Bioconjugate Chem. 2001, 12, 1035–1042. [Google Scholar] [CrossRef]
- Carta, D.; Salvarese, N.; Morellato, N.; Gao, F.; Sihver, W.; Pietzsch, H.J.; Biondi, B.; Ruzza, P.; Refosco, F.; Carpanese, D.; et al. Melanoma Targeting with [99mTc(N)(PNP3)]-Labeled α-Melanocyte Stimulating Hormone Peptide Analogs: Effects of Cyclization on the Radiopharmaceutical Properties. Nucl. Med. Biol. 2016, 43, 788–801. [Google Scholar] [CrossRef]
- Bolzati, C.; Salvarese, N.; Carpanese, D.; Seraglia, R.; Meléndez-Alafort, L.; Rosato, A.; Capasso, D.; Saviano, M.; Del Gatto, A.; Comegna, D.; et al. [99mTc][Tc(N)PNP43]-Labeled RGD Peptides As New Probes for a Selective Detection of Avβ3 Integrin: Synthesis, Structure–Activity and Pharmacokinetic Studies. J. Med. Chem. 2018, 61, 9596–9610. [Google Scholar] [CrossRef]
- Bolzati, C.; Dolmella, A. Nitrido Technetium-99m Core in Radiopharmaceutical Applications: Four Decades of Research. Inorganics 2020, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Boschi, A.; Uccelli, L.; Bolzati, C.; Duatti, A.; Sabba, N.; Moretti, E.; Domenico, G.D.; Zavattini, G.; Refosco, F.; Giganti, M. Synthesis and Biologic Evaluation of Monocationic Asymmetric 99mTc-Nitride Heterocomplexes Showing High Heart Uptake and Improved Imaging Properties. J. Nucl. Med. 2003, 44, 806–814. [Google Scholar]
- Bolzati, C.; Cavazza-Ceccato, M.; Agostini, S.; Refosco, F.; Yamamichi, Y.; Tokunaga, S.; Carta, D.; Salvarese, N.; Bernardini, D.; Bandoli, G. Biological in Vitro and in Vivo Studies of a Series of New Asymmetrical Cationic [99mTc(N)(DTC-Ln)(PNP)]+ Complex (DTC-Ln = Alicyclic Dithiocarbamate and PNP = Diphosphinoamine). Bioconjugate Chem. 2010, 21, 928–939. [Google Scholar] [CrossRef]
- Salvarese, N.; Carta, D.; Marzano, C.; Gerardi, G.; Melendez-Alafort, L.; Bolzati, C. [99mTc][Tc(N)(DASD)(PNPn)]+ (DASD = 1,4-Dioxa-8-Azaspiro[4,5]Decandithiocarbamate, PNPn = Bisphosphinoamine) for Myocardial Imaging: Synthesis, Pharmacological and Pharmacokinetic Studies. J. Med. Chem. 2018, 61, 11114–11126. [Google Scholar] [CrossRef]
- Bolzati, C.; Boschi, A.; Uccelli, L.; Tisato, F.; Refosco, F.; Cagnolini, A.; Duatti, A.; Prakash, S.; Bandoli, G.; Vittadini, A. Chemistry of the Strong Electrophilic Metal Fragment [99Tc(N)(PXP)]2+ (PXP = Diphosphine Ligand). A Novel Tool for the Selective Labeling of Small Molecules. J. Am. Chem. Soc. 2002, 124, 11468–11479. [Google Scholar] [CrossRef]
- Tisato, F.; Refosco, F.; Porchia, M.; Bolzati, C.; Bandoli, G.; Dolmella, A.; Duatti, A.; Boschi, A.; Jung, C.M.; Pietzsch, H.-J.; et al. The Crucial Role of the Diphosphine Heteroatom X in the Stereochemistry and Stabilization of the Substitution-Inert [M(N)(PXP)]2+ Metal Fragments (M = Tc, Re; PXP = Diphosphine Ligand). Inorg. Chem. 2004, 43, 8617–8625. [Google Scholar] [CrossRef]
- Bolzati, C.; Boschi, A.; Duatti, A.; Prakash, S.; Uccelli, L.; Refosco, F.; Tisato, F.; Bandoli, G. Geometrically Controlled Selective Formation of Nitrido Technetium(V) Asymmetrical Heterocomplexes with Bidentate Ligands. J. Am. Chem. Soc. 2000, 122, 4510–4511. [Google Scholar] [CrossRef]
- Bolzati, C.; Salvarese, N.; Spolaore, B.; Vittadini, A.; Forrer, D.; Brunello, S.; Ghiani, S.; Maiocchi, A. Water-Soluble [Tc(N)(PNP)] Moiety for Room-Temperature 99mTc Labeling of Sensitive Target Vectors. Mol. Pharm. 2022, 19, 876–894. [Google Scholar] [CrossRef]
- Alday-Parejo, B.; Stupp, R.; Rüegg, C. Are Integrins Still Practicable Targets for Anti-Cancer Therapy? Cancers 2019, 11, 978. [Google Scholar] [CrossRef] [Green Version]
- Debordeaux, F.; Chansel-Debordeaux, L.; Pinaquy, J.-B.; Fernandez, P.; Schulz, J. What about Avβ3 Integrins in Molecular Imaging in Oncology? Nucl. Med. Biol. 2018, 62–63, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Kapp, T.G.; Rechenmacher, F.; Neubauer, S.; Maltsev, O.V.; Cavalcanti-Adam, E.A.; Zarka, R.; Reuning, U.; Notni, J.; Wester, H.-J.; Mas-Moruno, C.; et al. A Comprehensive Evaluation of the Activity and Selectivity Profile of Ligands for RGD-Binding Integrins. Sci. Rep. 2017, 7, 39805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decristoforo, C.; Santos, I.; Pietzsch, H.J.; Kuenstler, J.U.; Duatti, A.; Smith, C.J.; Rey, A.; Alberto, R.; Guggenberg, E.V.; Haubner, R. Comparison of in Vitro and in Vivo Properties A of [99mTc]CRGD Peptides Labeled Using Different Novel Tc-Cores. Q. J. Nucl. Med. Mol. Imaging 2007, 51, 9. [Google Scholar]
- Kunstler, J.-U.; Seidel, G.; Bergmann, R.; Gniazdowska, E.; Walther, M.; Schiller, E.; Decristoforo, C.; Stephan, H.; Haubner, R.; Steinbach, J.; et al. Novel 99mTc ‘4 + 1’ Peptide Conjugates: Tuning the Biodistribution by Variation of Coligands. Eur. J. Med. Chem. 2010, 45, 3645–3655. [Google Scholar] [CrossRef]
- Damjanovich, L.; Albelda, S.M.; Mette, S.A.; Buck, C.A. Distribution of Integrin Cell Adhesion Receptors in Normal and Malignant Lung Tissue. Am. J. Respir. Cell Mol. Biol. 1992, 6, 197–206. [Google Scholar] [CrossRef]
- Monnier, Y.; Farmer, P.; Bieler, G.; Imaizumi, N.; Sengstag, T.; Alghisi, G.C.; Stehle, J.-C.; Ciarloni, L.; Andrejevic-Blant, S.; Moeckli, R.; et al. CYR61 and AVβ5 Integrin Cooperate to Promote Invasion and Metastasis of Tumors Growing in Preirradiated Stroma. Cancer Res. 2008, 68, 7323–7331. [Google Scholar] [CrossRef] [Green Version]
- Seguin, L.; Desgrosellier, J.S.; Weis, S.M.; Cheresh, D.A. Integrins and Cancer: Regulators of Cancer Stemness, Metastasis, and Drug Resistance. Trends Cell Biol. 2015, 25, 234–240. [Google Scholar] [CrossRef] [Green Version]
- Liu, S. Bifunctional Coupling Agents for Radiolabeling of Biomolecules and Target-Specific Delivery of Metallic Radionuclides. Adv. Drug Deliv. Rev. 2008, 60, 1347–1370. [Google Scholar] [CrossRef] [Green Version]
- Schottelius, M.; Laufer, B.; Kessler, H.; Wester, H.-J. Ligands for Mapping Avβ3-Integrin Expression in Vivo. Acc. Chem. Res. 2009, 42, 969–980. [Google Scholar] [CrossRef]
- Beer, A.J.; Schwaiger, M. Imaging of Integrin Avβ3 Expression. Cancer Metastasis Rev. 2008, 27, 631–644. [Google Scholar] [CrossRef]
- Edwards, D.; Jones, P.; Haramis, H.; Battle, M.; Lear, R.; Barnett, D.J.; Edwards, C.; Crawford, H.; Black, A.; Godden, V. 99mTc-NC100692—A Tracer for Imaging Vitronectin Receptors Associated with Angiogenesis: A Preclinical Investigation. Nucl. Med. Biol. 2008, 35, 365–375. [Google Scholar] [CrossRef]
- Meyer, A.; Auernheimer, J.; Modlinger, A.; Kessler, H. Targeting RGD Recognizing Integrins: Drug Development, Biomaterial Research, Tumor Imaging and Targeting. Curr. Pharm. Des. 2006, 12, 2723–2747. [Google Scholar] [CrossRef]
- Beer, A.J.; Kessler, H.; Wester, H.-J.; Schwaiger, M. PET Imaging of Integrin AVβ3 Expression. Theranostics 2011, 1, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Danhier, F.; Breton, A.L. RGD-Based Strategies To Target Alpha(v) Beta(3) Integrin in Cancer Therapy and Diagnosis. Mol. Pharm. 2012, 9, 2961–2973. [Google Scholar] [CrossRef]
- Meléndez-Alafort, L.; Rosato, A.; Ferro-Flores, G.; Penev, I.; Uzunov, N. Development of a Five-compartmental Model and Software for Pharmacokinetic Studies. Comptes Rendus De L’acad’emie Bulg. Des Sci. 2017, 70, 1649–1654. [Google Scholar]
- Grieco, P.; Gitu, P.m.; Hruby, V.j. Preparation of ‘Side-Chain-to-Side-Chain’ Cyclic Peptides by Allyl and Alloc Strategy: Potential for Library Synthesis. J. Pept. Res. 2001, 57, 250–256. [Google Scholar] [CrossRef]
- Blackburn, C.; Kates, S.A. [9] Solid-Phase Synthesis of Cyclic Homodetic Peptides. In Methods in Enzymology; Solid-Phase Peptide Synthesis; Academic Press: Cambridge, MA, USA, 1997; Volume 289, pp. 175–198. [Google Scholar]
- Bolzati, C.; Caporale, A.; Agostini, S.; Carta, D.; Cavazza-Ceccato, M.; Refosco, F.; Tisato, F.; Schievano, E.; Bandoli, G. Avidin-Biotin System: A Small Library of Cysteine Biotinylated Derivatives Designed for the [Tc-99m(N)(PNP)](2+) Metal Fragment. Nucl. Med. Biol. 2007, 34, 511–522. [Google Scholar] [CrossRef]
- Meyer, T.; Marshall, J.F.; Hart, I.R. Expression of Av Integrins and Vitronectin Receptor Identity in Breast Cancer Cells. Br. J. Cancer 1998, 77, 530. [Google Scholar] [CrossRef] [Green Version]
- Decristoforo, C.; Faintuch-Linkowski, B.; Rey, A.; von Guggenberg, E.; Rupprich, M.; Hernandez-Gonzales, I.; Rodrigo, T.; Haubner, R. [99mTc]HYNIC-RGD for Imaging Integrin αvβ3 Expression. Nucl. Med. Biol. 2006, 33, 945–952. [Google Scholar] [CrossRef]
- Bolzati, C.; Mahmood, A.; Malagò, E.; Uccelli, L.; Boschi, A.; Jones, A.G.; Refosco, F.; Duatti, A.; Tisato, F. The [99mTc(N)(PNP)]2+ Metal Fragment: A Technetium-Nitrido Synthon for Use with Biologically Active Molecules. The N-(2-Methoxyphenyl)Piperazyl-Cysteine Analogues as Examples. Bioconjugate Chem. 2003, 14, 1231–1242. [Google Scholar] [CrossRef]
- Boschi, A.; Uccelli, L.; Duatti, A.; Bolzati, C.; Refosco, F.; Tisato, F.; Romagnoli, R.; Baraldi, P.G.; Varani, K.; Borea, P.A. Asymmetrical Nitrido Tc-99m Heterocomplexes as Potential Imaging Agents for Benzodiazepine Receptors. Bioconjugate Chem. 2003, 14, 1279–1288. [Google Scholar] [CrossRef] [PubMed]
- Cazzola, E.; Benini, E.; Pasquali, M.; Mirtschink, P.; Walther, M.; Pietzsch, H.-J.; Uccelli, L.; Boschi, A.; Bolzati, C.; Duatti, A. Labeling of Fatty Acid Ligands with the Strong Electrophilic Metal Fragment [99mTc(N)(PNP)]2+ (PNP = Diphosphane Ligand). Bioconjugate Chem. 2008, 19, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Kothari, K.; Tóth, G.; Szemenyei, E.; Sarma, H.D.; Környei, J.; Venkatesh, M. 99mTc-Labeled Annexin V Fragments: A Potential SPECT Radiopharmaceutical for Imaging Cell Death. Nucl. Med. Biol. 2006, 33, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Agostini, S.; Bolzati, C.; Didonè, E.; Cavazza-Ceccato, M.; Refosco, F.; Aloj, L.; Arra, C.; Aurilio, M.; Tornesello, A.L.; Tesauro, D.; et al. The [Tc(N)(PNP)]2+ Metal Fragment Labeled Cholecystokinin-8 (CCK8) Peptide for CCK-2 Receptors Imaging: In Vitro and in Vivo Studies. J. Pept. Sci. 2007, 13, 211–219. [Google Scholar] [CrossRef]
- Haubner, R.; Weber, W.A.; Beer, A.J.; Vabuliene, E.; Reim, D.; Sarbia, M.; Becker, K.-F.; Goebel, M.; Hein, R.; Wester, H.-J.; et al. Noninvasive Visualization of the Activated αvβ3 Integrin in Cancer Patients by Positron Emission Tomography and [18F]Galacto-RGD. PLoS Med. 2005, 2, e70. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | HPLC Rt min | a/b | %RCY | % Protein Binding * | Log P | ||
|---|---|---|---|---|---|---|---|
| H3NS-cRGDfK | 13.00 1 | ||||||
| [Tc(N)(H2NS-cRGDfK)(PNP3)]+ | 99mTc1 | a | 14.09 1; 11.39 2 | 61.76/31.98 | 93.74 ± 0.05 | 14.95 ± 1.95 | −1.72 1 |
| b | 15.79 1; 13.23 2 | ||||||
| [Tc(N)(H2NS-cRGDfK)(PNP43)]+ | 99mTc2 | a | 12.78 2; 26.04 3 | 60.64/27.80 | 89.04 ± 0.09 | 13.63 ± 2.95 | −1.95 |
| b | 15.45 2; 29.08 3 | ||||||
| [Tc(N)(H2NS-cRGDfK)(PNP3OH)]+ | 99mTc3 | a | 8.21 2; 22.75 3 | 67.56/23.33 | 90.89 ± 0.11 | 7.91 ± 2.01 | −2.39 |
| b | 10.12 2; 24.86 3 | ||||||
| [Tc-HYNIC-cRGDfK] | 9.02 2 | 2.3 ± 0.11 | −3.52 |
| Cell Line (Tumor Type) | ΔMFI αvβ3 | αvβ3 | ΔMFI αvβ5 | αvβ5 |
|---|---|---|---|---|
| M21 (Human melanoma) | 25 | +++ | 1 | ---- |
| MCF7 (Human breast cancer) | 4 | ---- | 67 | ++++ |
| M21-L (Human melanoma) | 1 | ---- | 0 | ---- |
| Range for MFI values of αvβ3 | Range for MFI values of αvβ5 | |||
 |  | |||
| 99mTc1 | 99mTc2 | 99mTc3 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Organs | 30 min | 60 min | 120 min | 240 min | 30 min | 60 min | 120 min | 240 min | 30 min | 60 min | 120 min | 240 min |
| Blood | 0.81 ± 0.05 | 0.30 ± 0.04 | 0.09 ± 0.01 | 0.05 ± 0.01 | 0.65 ± 0.04 | 0.35 ± 0.07 | 0.19 ± 0.04 | 0.11 ± 0.02 | 0.46 ± 0.07 | 0.25 ± 0.03 | 0.16 ± 0.01 | 0.09 ± 0.01 |
| Heart | 0.26 ± 0.05 | 0.15 ± 0.01 | 0.10 ± 0.02 | 0.08 ± 0.01 | 0.30 ± 0.01 | 0.20 ± 0.04 | 0.15 ± 0.06 | 0.12 ± 0.01 | 0.25 ± 0.06 | 0.15 ± 0.00 | 0.13 ± 0.02 | 0.12 ± 0.03 |
| Lungs | 0.64 ± 0.09 | 0.43 ± 0.01 | 0.25 ± 0.03 | 0.16 ± 0.04 | 0.82 ± 0.09 | 0.56 ± 0.10 | 0.42 ± 0.02 | 0.34 ± 0.04 | 0.62 ± 0.08 | 0.49 ± 0.01 | 0.37 ± 0.06 | 0.32 ± 0.02 |
| Liver | 0.96 ± 0.10 | 0.75 ± 0.02 | 0.68 ± 0.06 | 0.66 ± 0.07 | 0.90 ± 0.13 | 0.73 ± 0.01 | 0.57 ± 0.03 | 0.52 ± 0.07 | 0.90 ± 0.09 | 0.80 ± 0.06 | 0.75 ± 0.09 | 0.67 ± 0.04 |
| Spleen | 0.65 ± 0.09 | 0.51 ± 0.04 | 0.47 ± 0.04 | 0.45 ± 0.00 | 0.47 ± 0.02 | 0.40 ± 0.05 | 0.38 ± 0.04 | 0.34 ± 0.07 | 0.43 ± 0.05 | 0.39 ± 0.04 | 0.36 ± 0.02 | 0.34 ± 0.04 |
| Kidneys | 2.71 ± 0.18 | 1.57 ± 0.13 | 0.92 ± 0.20 | 0.88 ± 0.09 | 6.77 ± 0.86 | 6.20 ± 0.68 | 5.51 ± 0.42 | 4.27 ± 0.31 | 5.22 ± 0.44 | 4.83 ± 0.35 | 4.44 ± 0.32 | 4.22 ± 0.45 |
| Stomach | 0.53 ± 0.07 | 0.41 ± 0.02 | 0.34 ± 0.04 | 0.29 ± 0.02 | 0.82 ± 0.07 | 0.64 ± 0.08 | 0.51 ± 0.08 | 0.41 ± 0.09 | 0.72 ± 0.04 | 0.64 ± 0.08 | 0.57 ± 0.03 | 0.46 ± 0.04 |
| Intestine | 0.65 ± 0.16 | 0.60 ± 0.12 | 0.40 ± 0.07 | 0.38 ± 0.01 | 1.19 ± 0.14 | 1.69 ± 0.17 | 1.94 ± 0.25 | 1.90 ± 0.19 | 1.29 ± 0.13 | 1.95 ± 0.17 | 3.76 ± 0.27 | 4.17 ± 0.59 |
| Muscle | 0.21 ± 0.09 | 0.13 ± 0.06 | 0.10 ± 0.06 | 0.06 ± 0.01 | 0.22 ± 0.03 | 0.14 ± 0.03 | 0.12 ± 0.01 | 0.09 ± 0.00 | 0.17 ± 0.01 | 0.13 ± 0.01 | 0.12 ± 0.00 | 0.08 ± 0.00 |
| Urine | 27.2 ± 11.7 | 33.1 ± 5.6 | 54.3 ± 1.9 | 55.9 ± 7.1 | 17.7 ± 3.6 | 28.5 ± 4.6 | 41.5 ± 4.5 | 43.5 ± 3.4 | 27.8 ± 5.6 | 31.2 ± 3.0 | 44.2 ± 10.9 | 30.2 ± 9.45 |
| Organ | MRT (min) | ||
|---|---|---|---|
| 99mTc1 | 99mTc2 | 99mTc3 | |
| Blood | 14.7 | 16.9 | 14.5 |
| Heart | 0.3 | 0.4 | 0.4 |
| Lung | 0.9 | 1.6 | 1.6 |
| Liver | 30.5 | 25.9 | 26.7 |
| Kidney | 9.2 | 30.7 | 36.7 |
| Spleen | 1.2 | 0.8 | 0.8 |
| Stomach | 1.5 | 2.1 | 2.1 |
| Organs | 99mTc1 | 99mTc2 | 99mTc3 | 99mTc_HYNIC [40] | |
|---|---|---|---|---|---|
| 1 h | 4 h | ||||
| Blood | 0.07 ± 0.02 | 0.12 ± 0.03 | 0.05 ± 0.01 | 0.98 ± 0.04 | 0.45 ± 0.02 |
| Heart | 0.47 ± 0.03 | 0.48 ± 0.07 | 0.44 ± 0.09 | 1.01 ± 0.05 | 0.64 ± 0.09 |
| Lungs | 1.25 ± 0.23 | 1.04 ± 0.08 | 1.32 ± 0.20 | ||
| Liver | 3.59 ± 0.31 | 2.43 ± 0.23 | 3.22 ± 0.24 | 2.63 ± 0.14 | 1.71 ± 0.28 |
| Spleen | 1.61 ± 0.01 | 1.26 ± 0.04 | 1.43 ± 0.19 | ||
| Kidneys | 4.10 ± 0.37 | 3.38 ± 0.36 | 3.34 ± 0.18 | 3.68 ± 0.13 | 2.81 ± 0.37 |
| Stomach * | 1.22 ± 0.15 | 1.02 ± 0.16 | 1.05 ± 0.14 | 2.50 ± 0.75 | 1.28 ± 0.22 |
| Intestine | 2.30 ± 0.10 | 2.45 ± 0.29 | 3.09 ± 0.62 | 2.04 ± 0.18 | 1.29 ± 0.25 |
| Muscle | 0.37 ± 0.04 | 0.31 ± 0.05 | 0.23 ± 0.03 | 0.76 ± 0.69 | 0.34 ± 0.05 |
| M21 | 3.17 ± 0.60 | 1.08 ± 0.24 | 3.92 ± 0.69 | 2.73 ± 0.26 | 2.06 ± 0.37 |
| M21L | 1.73 ± 0.19 | 0.42 ± 0.05 | 0.70 ± 0.16 | 0.85 ± 0.20 | |
| M21/M21L | 1.83 | 2.57 | 5.60 | 3.21 | |
| M21/blood | 29.96 | 11.26 | 73.42 | 2.79 | 4.58 |
| M21/muscle | 9.15 | 3.54 | 16.72 | 3.59 | 6.05 |
| M21/liver | 0.88 | 0.44 | 1.11 | 1.03 | 1.20 |
| M21/lung | 2.53 | 1.04 | 2.98 | ||
| M21/kidneys | 0.77 | 0.32 | 1.23 | 0.74 | 0.73 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salvarese, N.; Carpanese, D.; Meléndez-Alafort, L.; De Nardo, L.; Calderan, A.; Biondi, B.; Ruzza, P.; Rosato, A.; Bolzati, C. Impact of Different [Tc(N)PNP]-Scaffolds on the Biological Properties of the Small cRGDfK Peptide: Synthesis, In Vitro and In Vivo Evaluations. Molecules 2022, 27, 2548. https://doi.org/10.3390/molecules27082548
Salvarese N, Carpanese D, Meléndez-Alafort L, De Nardo L, Calderan A, Biondi B, Ruzza P, Rosato A, Bolzati C. Impact of Different [Tc(N)PNP]-Scaffolds on the Biological Properties of the Small cRGDfK Peptide: Synthesis, In Vitro and In Vivo Evaluations. Molecules. 2022; 27(8):2548. https://doi.org/10.3390/molecules27082548
Chicago/Turabian StyleSalvarese, Nicola, Debora Carpanese, Laura Meléndez-Alafort, Laura De Nardo, Andrea Calderan, Barbara Biondi, Paolo Ruzza, Antonio Rosato, and Cristina Bolzati. 2022. "Impact of Different [Tc(N)PNP]-Scaffolds on the Biological Properties of the Small cRGDfK Peptide: Synthesis, In Vitro and In Vivo Evaluations" Molecules 27, no. 8: 2548. https://doi.org/10.3390/molecules27082548