
Reactive Acrylamide-Modified DNA Traps for Accurate Cross-Linking with Cysteine Residues in DNA–Protein Complexes Using Mismatch Repair Protein MutS as a Model

 , , , , and
, , , , and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. The Design of Modified DNA Duplexes for Cross-Linking with the MutS Protein
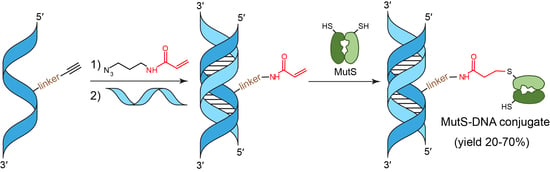
2.2. Synthesis of Oligonucleotides Containing a dU Residue Carrying an Acrylamide Group
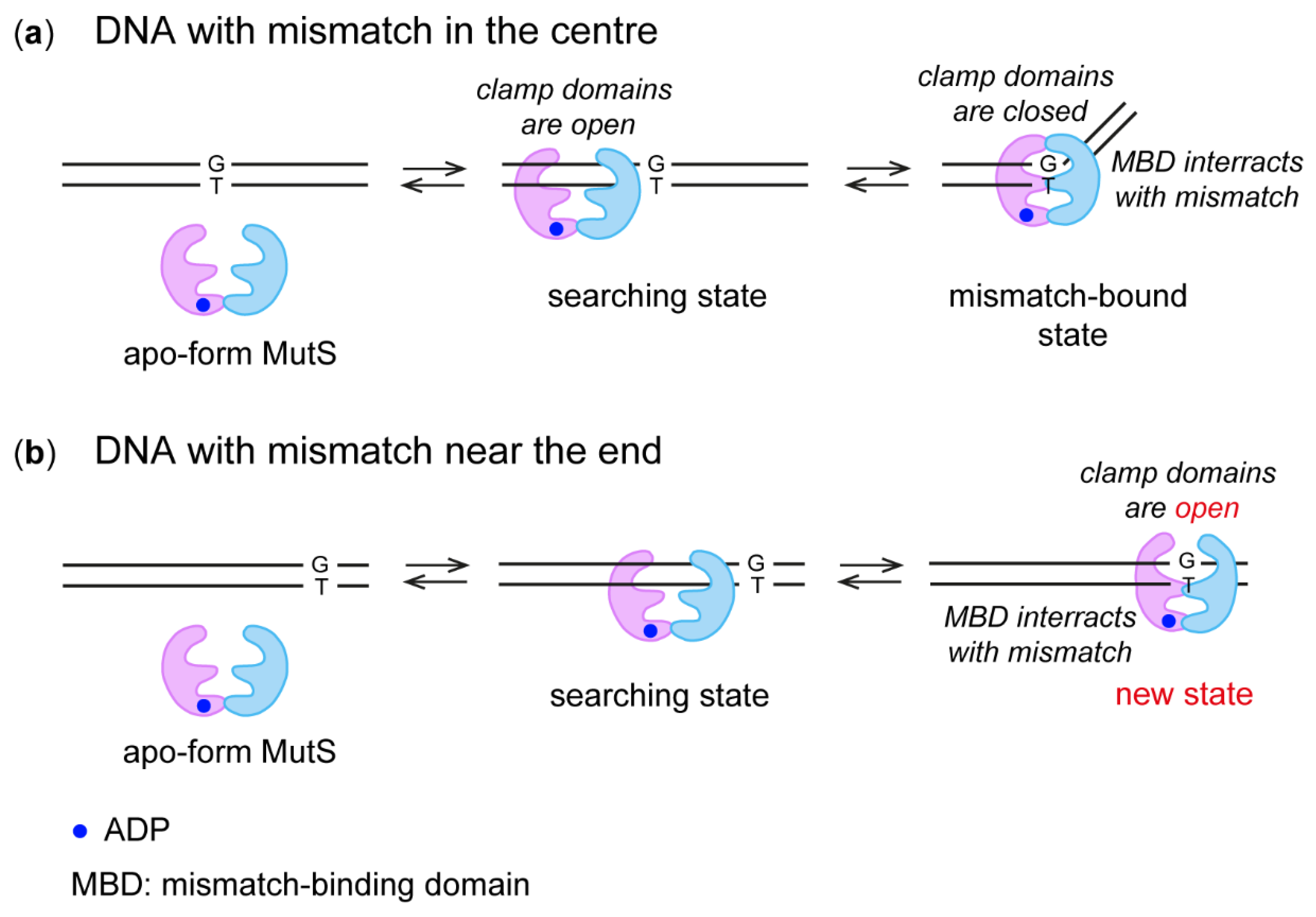
2.3. MutS Binding to DNA Duplexes Carrying the Ethynyl Group
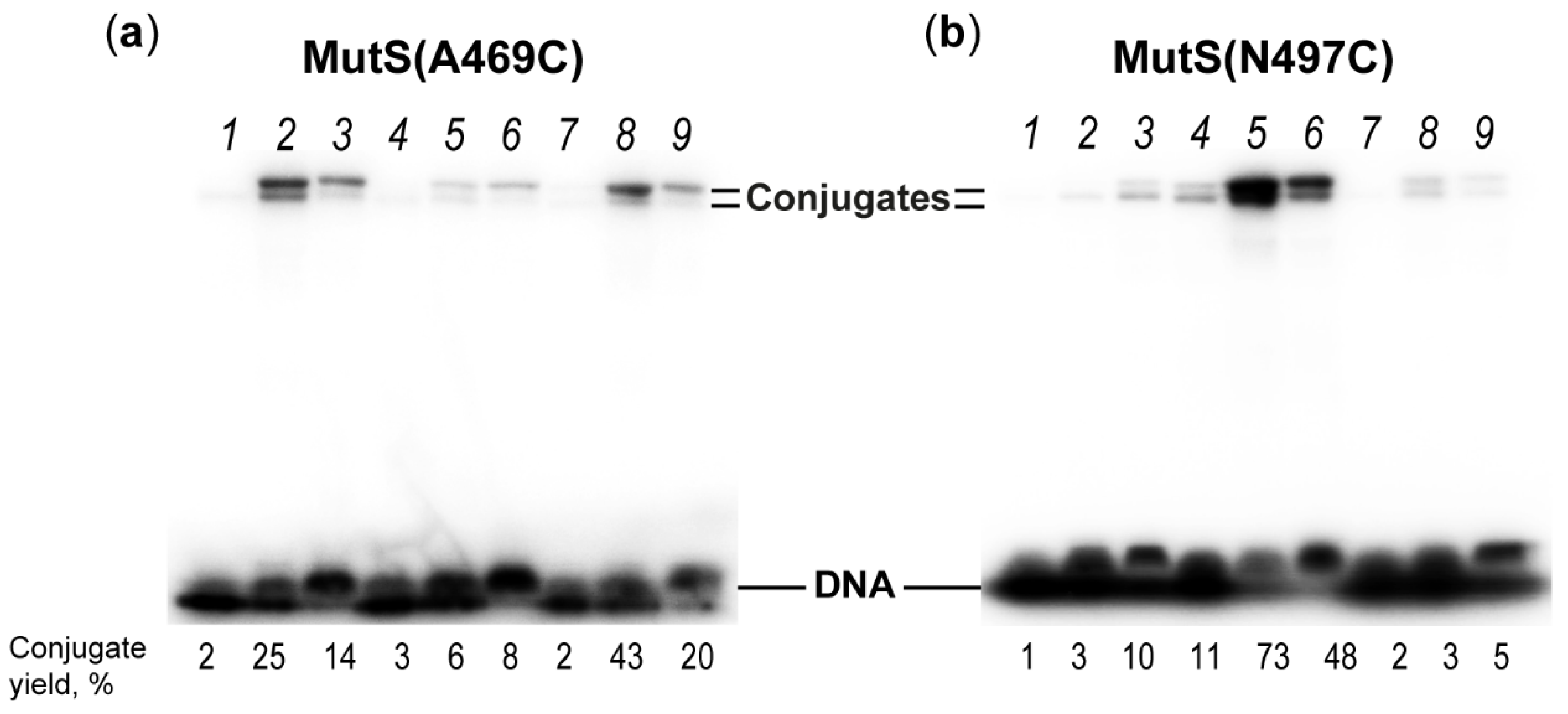
2.4. Interaction of MutS Variants with 17-mer DNA Duplexes Carrying the Acrylamide Group on a Linker of Various Lengths
3. Materials and Methods
3.1. Proteins
3.2. Modelling of Modified DNA and DNA–Protein Complexes
3.3. The Synthesis of Oligonucleotides
3.4. The Effect of Temperature on the Stability of DNA Duplexes Carrying the Ethynyl Group
3.5. 32P Labelling of the Oligonucleotides and Preparation of DNA Duplexes
3.6. Complex Formation between the MutS(A469C) Variant and Ethynyl-Containing DNAs
3.7. Cross-Linking of MutS Protein Variants with Reactive DNA Containing the Acrylamide Group
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lambert, M.; Jambon, S.; Depauw, S.; David-Cordonnier, M.-H. Targeting transcription factors for cancer treatment. Molecules 2018, 23, 1479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, M.; Jia, K.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; He, Y.; Zhou, C. Alterations of DNA damage repair in cancer: From mechanisms to applications. Ann. Transl. Med. 2020, 8, 1685. [Google Scholar] [CrossRef] [PubMed]
- Ilina, E.S.; Khodyreva, S.N.; Lavrik, O.I. Unusual interaction of human apurinic/apyrimidinic endonuclease 1 (APE1) with abasic sites via the Schiff-base-dependent mechanism. Biochimie 2018, 150, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Khodyreva, S.; Lavrik, O. Non-canonical interaction of DNA repair proteins with intact and cleaved AP sites. DNA Repair 2020, 90, 102847. [Google Scholar] [CrossRef]
- Hyjek-Składanowska, M.; Stasińska, A.R.; Napiórkowska-Gromadzka, A.; Bartłomiejczak, A.; Seth, P.P.; Chmielewski, M.K.; Nowotny, M. Disulfide bridge cross-linking between protein and the RNA backbone as a tool to study RNase H1. Bioorg. Med. Chem. 2020, 28, 115741. [Google Scholar] [CrossRef]
- Monakhova, M.; Ryazanova, A.; Hentschel, A.; Viryasov, M.; Oretskaya, T.; Friedhoff, P.; Kubareva, E. Chromatographic isolation of the functionally active MutS protein covalently linked to deoxyribonucleic acid. J. Chromatogr. A 2015, 1389, 19–27. [Google Scholar] [CrossRef]
- Abrosimova, L.A.; Samsonova, A.R.; Perevyazova, T.A.; Yunusova, A.K.; Artyukh, R.I.; Romanova, E.A.; Zheleznaya, L.A.; Oretskaya, T.S.; Kubareva, E.A. The role of cysteine residues in the interaction of nicking endonuclease BspD6I with DNA. Mol. Biol. 2020, 54, 599–610. [Google Scholar] [CrossRef]
- Koniev, O.; Wagner, A. Developments and recent advancements in the field of endogenous amino acid selective bond forming reactions for bioconjugation. Chem. Soc. Rev. 2015, 44, 5495–5551. [Google Scholar] [CrossRef] [Green Version]
- Seio, K.; Ohno, Y.; Ohno, K.; Takeshita, L.; Kanamori, T.; Masaki, Y.; Sekine, M. Photo-controlled binding of MutS to photo-caged DNA duplexes incorporating 4-O-(2-nitrobenzyl) or 4-O-[2-(2-nitrophenyl)propyl]thymidine. Bioorg. Med. Chem. Lett. 2016, 26, 4861–4863. [Google Scholar] [CrossRef]
- Zatsepin, T.S.; Abrosimova, L.A.; Monakhova, M.V.; Hien, L.T.; Pingoud, A.; Kubareva, E.A.; Oretskaya, T.S. Design of photocontrolled biomolecules based on azobenzene derivatives. Russ. Chem. Rev. 2013, 82, 942–963. [Google Scholar] [CrossRef]
- Lee, J.-B.; Cho, W.-K.; Park, J.; Jeon, Y.; Kim, D.; Lee, S.H.; Fishel, R. Single-molecule views of MutS on mismatched DNA. DNA Repair 2014, 20, 82–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fishel, R. Mismatch repair. J. Biol. Chem. 2015, 290, 26395–26403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, P. Molecular mechanisms of DNA mismatch repair. Mutat. Res. Repair 2001, 486, 71–87. [Google Scholar] [CrossRef]
- Schofield, M.J.; Hsieh, P. DNA mismatch repair: Molecular mechanisms and biological function. Annu. Rev. Microbiol. 2003, 57, 579–608. [Google Scholar] [CrossRef]
- Liu, J.; Hanne, J.; Britton, B.M.; Bennett, J.; Kim, D.; Lee, J.-B.; Fishel, R. Cascading MutS and MutL sliding clamps control DNA diffusion to activate mismatch repair. Nature 2016, 539, 583–587. [Google Scholar] [CrossRef] [Green Version]
- Natrajan, G. Structures of Escherichia coli DNA mismatch repair enzyme MutS in complex with different mismatches: A common recognition mode for diverse substrates. Nucleic Acids Res. 2003, 31, 4814–4821. [Google Scholar] [CrossRef] [Green Version]
- Lamers, M.H.; Georgijevic, D.; Lebbink, J.H.; Winterwerp, H.H.K.; Agianian, B.; de Wind, N.; Sixma, T.K. ATP increases the affinity between MutS ATPase domains. J. Biol. Chem. 2004, 279, 43879–43885. [Google Scholar] [CrossRef] [Green Version]
- Lebbink, J.H.G.; Georgijevic, D.; Natrajan, G.; Fish, A.; Winterwerp, H.H.K.; Sixma, T.K.; de Wind, N. Dual role of MutS glutamate 38 in DNA mismatch discrimination and in the authorization of repair. EMBO J. 2006, 25, 409–419. [Google Scholar] [CrossRef] [Green Version]
- Lamers, M.H.; Perrakis, A.; Enzlin, J.H.; Winterwerp, H.H.K.; de Wind, N.; Sixma, T.K. The crystal structure of DNA mismatch repair protein MutS binding to a G·T mismatch. Nature 2000, 407, 711–717. [Google Scholar] [CrossRef]
- Groothuizen, F.S.; Fish, A.; Petoukhov, M.V.; Reumer, A.; Manelyte, L.; Winterwerp, H.H.K.; Marinus, M.G.; Lebbink, J.H.G.; Svergun, D.I.; Friedhoff, P.; et al. Using stable MutS dimers and tetramers to quantitatively analyze DNA mismatch recognition and sliding clamp formation. Nucleic Acids Res. 2013, 41, 8166–8181. [Google Scholar] [CrossRef] [Green Version]
- Perry, S.A.; Kubareva, E.A.; Monakhova, M.V.; Trikin, R.M.; Kosaretskiy, E.M.; Romanova, E.A.; Metelev, V.G.; Friedhoff, P.; Oretskaya, T.S. DNA with a 2-pyridyldithio group at the C2’ atom: A promising tool for the crosslinking of the MutS protein preserving its functional activity. Russ. J. Bioorg. Chem. 2021, 47, 447–460. [Google Scholar] [CrossRef]
- Fernandez-Leiro, R.; Bhairosing-Kok, D.; Kunetsky, V.; Laffeber, C.; Winterwerp, H.H.; Groothuizen, F.; Fish, A.; Lebbink, J.H.G.; Friedhoff, P.; Sixma, T.K.; et al. The selection process of licensing a DNA mismatch for repair. Nat. Struct. Mol. Biol. 2021, 28, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Monakhova, M.; Ryazanova, A.; Kunetsky, V.; Li, P.; Shilkin, E.; Kisil, O.; Rao, D.N.; Oretskaya, T.; Friedhoff, P.; Kubareva, E. Probing the DNA-binding center of the MutL protein from the Escherichia coli mismatch repair system via crosslinking and Förster resonance energy transfer. Biochimie 2020, 171–172, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Dadová, J.; Orság, P.; Pohl, R.; Brázdová, M.; Fojta, M.; Hocek, M. Vinylsulfonamide and acrylamide modification of DNA for cross-linking with proteins. Angew. Chem. Int. Ed. 2013, 52, 10515–10518. [Google Scholar] [CrossRef]
- Heinze, R.J.; Sekerina, S.; Winkler, I.; Biertümpfel, C.; Oretskaya, T.S.; Kubareva, E.; Friedhoff, P. Covalently trapping MutS on DNA to study DNA mismatch recognition and signaling. Mol. Biosyst. 2012, 8, 1861. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.-J. 3DNA: A software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003, 31, 5108–5121. [Google Scholar] [CrossRef] [Green Version]
- Acharya, S.; Foster, P.L.; Brooks, P.; Fishel, R. The coordinated functions of the E. coli MutS and MutL proteins in mismatch repair. Mol. Cell 2003, 12, 233–246. [Google Scholar] [CrossRef]
- Sánchez, A.; Pedroso, E.; Grandas, A. Maleimide-dimethylfuran exo adducts: Effective maleimide protection in the synthesis of oligonucleotide conjugates. Org. Lett. 2011, 13, 4364–4367. [Google Scholar] [CrossRef]
- Allabush, F.; Mendes, P.M.; Tucker, J.H.R. Acrylamide-dT: A polymerisable nucleoside for DNA incorporation. RSC Adv. 2019, 9, 31511–31516. [Google Scholar] [CrossRef] [Green Version]
- Rehman, F.N.; Audeh, M.; Abrams, E.S.; Hammond, P.W.; Kenney, M.; Boles, T.C. Immobilization of acrylamide-modified oligonucleotides by co-polymerization. Nucleic Acids Res. 1999, 27, 649–655. [Google Scholar] [CrossRef]
- Graham, D.; Parkinson, J.A.; Brown, T. DNA duplexes stabilized by modified monomer residues: Synthesis and stability. J. Chem. Soc. Perkin Trans. 1998, 1, 1131–1138. [Google Scholar] [CrossRef]
- He, J. Propynyl groups in duplex DNA: Stability of base pairs incorporating 7-substituted 8-aza-7-deazapurines or 5-substituted pyrimidines. Nucleic Acids Res. 2002, 30, 5485–5496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branch, P.; Aquilina, G.; Bignami, M.; Karran, P. Defective mismatch binding and a mutator phenotype in cells tolerant to DNA damage. Nature 1993, 362, 652–654. [Google Scholar] [CrossRef] [PubMed]
- Ni, T.T.; Marsischky, G.T.; Kolodner, R.D. MSH2 and MSH6 are required for removal of adenine misincorporated opposite 8-oxo-guanine in S. cerevisiae. Mol. Cell 1999, 4, 439–444. [Google Scholar] [CrossRef]
- Mazurek, A.; Berardini, M.; Fishel, R. Activation of human MutS homologs by 8-oxo-guanine DNA damage. J. Biol. Chem. 2002, 277, 8260–8266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.M.; Wang, H.; Romano, L.J. Human MutSα specifically binds to DNA containing aminofluorene and acetylaminofluorene adducts. J. Biol. Chem. 1996, 271, 24084–24088. [Google Scholar] [CrossRef] [Green Version]
- Feng, W.Y.; Lee, E.H.; Hays, J.B. Recombinagenic processing of UV-light photoproducts in nonreplicating phage DNA by the Escherichia coli methyl-directed mismatch repair system. Genetics 1991, 129, 1007–1020. [Google Scholar] [CrossRef]
- Mu, D.; Tursun, M.; Duckett, D.R.; Drummond, J.T.; Modrich, P.; Sancar, A. Recognition and repair of compound DNA lesions (base damage and mismatch) by human mismatch repair and excision repair systems. Mol. Cell. Biol. 1997, 17, 760–769. [Google Scholar] [CrossRef] [Green Version]
- Obmolova, G.; Ban, C.; Hsieh, P.; Yang, W. Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature 2000, 407, 703–710. [Google Scholar] [CrossRef]
- Su, X. Surface plasmon resonance spectroscopy and quartz crystal microbalance study of muts binding with single thymine-guanine mismatched DNA. Front. Biosci. 2005, 10, 268. [Google Scholar] [CrossRef] [Green Version]
- Blackwell, L.J.; Bjornson, K.P.; Allen, D.J.; Modrich, P. Distinct MutS DNA-binding modes that are differentially modulated by ATP binding and hydrolysis. J. Biol. Chem. 2001, 276, 34339–34347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metelev, V.; Romanenkov, A.; Kubareva, E.; Zubin, E.; Polouchine, N.; Zatsepin, T.; Molochkov, N.; Oretskaya, T. Structure-based cross-linking of NF-κB p50 homodimer and decoy bearing a novel 2′-disulfide trapping site. IUBMB Life 2006, 58, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Nirwal, S.; Kulkarni, D.S.; Sharma, A.; Rao, D.N.; Nair, D.T. Mechanism of formation of a toroid around DNA by the mismatch sensor protein. Nucleic Acids Res. 2018, 46, 256–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhairosing-Kok, D.; Groothuizen, F.S.; Fish, A.; Dharadhar, S.; Winterwerp, H.H.K.; Sixma, T.K. Sharp kinking of a coiled-coil in MutS allows DNA binding and release. Nucleic Acids Res. 2019, 47, 8888–8898. [Google Scholar] [CrossRef]
- Borsellini, A.; Kunetsky, V.; Friedhoff, P.; Lamers, M.H. Cryogenic electron microscopy structures reveal how ATP and DNA binding in MutS coordinates sequential steps of DNA mismatch repair. Nat. Struct. Mol. Biol. 2022, 29, 59–66. [Google Scholar] [CrossRef]
- Feng, G.; Winkler, M.E. Single-step purifications of His6-MutH, His6-MutL and His6-MutS repair proteins of Escherichia coli K-12. Biotechniques 1995, 19, 956–965. [Google Scholar]
- Farzan, V.M.; Ulashchik, E.A.; Martynenko-Makaev, Y.V.; Kvach, M.V.; Aparin, I.O.; Brylev, V.A.; Prikazchikova, T.A.; Maklakova, S.Y.; Majouga, A.G.; Ustinov, A.V.; et al. Automated solid-phase click synthesis of oligonucleotide conjugates: From small molecules to diverse N-acetylgalactosamine clusters. Bioconjug. Chem. 2017, 28, 2599–2607. [Google Scholar] [CrossRef]
- Aralov, A.V.; Gubina, N.; Cabrero, C.; Tsvetkov, V.B.; Turaev, A.V.; Fedeles, B.I.; Croy, R.G.; Isaakova, E.A.; Melnik, D.; Dukova, S.; et al. 7,8-Dihydro-8-oxo-1,N6-ethenoadenine: An exclusively Hoogsteen-paired thymine mimic in DNA that induces A→T transversions in Escherichia coli. Nucleic Acids Res. 2022, 50, gkac148. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position of Cys Residue in MutS | Distance between Indicated Cys and DNA, Å | Conjugate MutS–DNA Yield, % Linker (Its Length, Å) | |||
|---|---|---|---|---|---|
| s (12.6 Å) | m (22.3 Å) | l (32.6 Å) | |||
| Modification at 5th position from mismatch | |||||
| 469 | Subunit A | 10 | 2 ± 1 | 27 ± 4 | 15 ± 1 |
| Subunit B | 13 | ||||
| 497 | Subunit A | 14 | 2 ± 1 | 4 ± 1 | 6 ± 1 |
| Subunit B | 20 | ||||
| Modification at 8th position from mismatch | |||||
| 469 | Subunit A | 19 | 1 | 2 ± 1 | 5 ± 1 |
| Subunit B | 12 | ||||
| 497 | Subunit A | 9 | 12 ± 1 | 76 ± 2 | 53 ± 6 |
| Subunit B | 25 | ||||
| Modification at 11th position from mismatch | |||||
| 469 | Subunit A | 31 | 2 ± 1 | 42 ± 2 | 17 ± 3 |
| Subunit B | 19 | ||||
| 497 | Subunit A | 19 | 2 ± 1 | 4 ± 2 | 3 ± 1 |
| Subunit B | 37 | ||||
| Duplex | Tm, °C |
|---|---|
| 17AT | 81.6 ± 0.3 |
| 17AT-dUsethynyl | 80.7 ± 0.3 |
| 17AT-dUmethynyl | 81.3 ± 0.3 |
| 17AT-dUlethynyl | 80.6 ± 0.5 |
| 17GT | 77.1 ± 0.3 |
| 17GT-dUsethynyl-5 | 77.5 ± 0.1 |
| 17GT-dUmethynyl-5 | 76.9 ± 0.3 |
| 17GT-dUlethynyl-5 | 76.6 ± 0.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monakhova, M.V.; Kubareva, E.A.; Kolesnikov, K.K.; Anashkin, V.A.; Kosaretskiy, E.M.; Zvereva, M.I.; Romanova, E.A.; Friedhoff, P.; Oretskaya, T.S.; Zatsepin, T.S. Reactive Acrylamide-Modified DNA Traps for Accurate Cross-Linking with Cysteine Residues in DNA–Protein Complexes Using Mismatch Repair Protein MutS as a Model. Molecules 2022, 27, 2438. https://doi.org/10.3390/molecules27082438
Monakhova MV, Kubareva EA, Kolesnikov KK, Anashkin VA, Kosaretskiy EM, Zvereva MI, Romanova EA, Friedhoff P, Oretskaya TS, Zatsepin TS. Reactive Acrylamide-Modified DNA Traps for Accurate Cross-Linking with Cysteine Residues in DNA–Protein Complexes Using Mismatch Repair Protein MutS as a Model. Molecules. 2022; 27(8):2438. https://doi.org/10.3390/molecules27082438
Chicago/Turabian StyleMonakhova, Mayya V., Elena A. Kubareva, Kirill K. Kolesnikov, Viktor A. Anashkin, Egor M. Kosaretskiy, Maria I. Zvereva, Elena A. Romanova, Peter Friedhoff, Tatiana S. Oretskaya, and Timofei S. Zatsepin. 2022. "Reactive Acrylamide-Modified DNA Traps for Accurate Cross-Linking with Cysteine Residues in DNA–Protein Complexes Using Mismatch Repair Protein MutS as a Model" Molecules 27, no. 8: 2438. https://doi.org/10.3390/molecules27082438