3.2. General Procedure for the Synthesis of α-Hydroxyphosphonates (1a–d, 3a)
A mixture of 11.0 mmol of substituted aldehyde (benzaldehyde, 1.2 g; 4-chlorobenzaldehyde, 1.5 g; 3-trifluoromethylbenzaldehyde, 1.5 mL; 3,4,5-trifluorobenzaldehyde, 1.2 mL), 11.0 mmol of dialkyl phosphite (dimethyl phosphite, 1.1 mL; diethyl phosphite, 1.4 mL) and 1.1 mmol (0.15 mL) of triethylamine in acetone (1.0 mL) was stirred at reflux for 30 min–6 h (See
Table 8). After adding pentane (6.0 mL), the reaction mixture was cooled to 5 °C whereupon the product crystallized from the mixture as a white solid. Filtration afforded products
1a,b,d, and
3a in a pure form with yields of 87–95%. In one case, the crude product was purified by column chromatography on silica gel applying dichloromethane–methanol (97:3) as the eluent to afford product (
1c) as an oil.
3.2.1. Diethyl α-Hydroxy-α-Phenyl-Methylphosphonate (1a)
31P NMR (CDCl
3) δ 21.6, δ
P (CDCl
3) 21.7 [
11,
38]; [M + H]
+ = 245.
3.2.2. Diethyl α-Hydroxy-α-(4-Chlorophenyl)-Methylphosphonate (1b)
31P NMR (CDCl
3) δ 20.8, δ
P (CDCl
3) 21.0 [
11,
38]; [M + H]
+ = 278.
3.2.3. Diethyl α-Hydroxy-α-(3-Trifluoromethylphenyl)-Methylphosphonate (1c)
31P NMR (CDCl3) δ 20.5; 13C NMR (CDCl3) δ 16.3 (d, J = 5.9 Hz, OCH2CH3), 63.2 (d, J = 7.6 Hz, OCH2CH3), 63.8 (d, J = 7.1 Hz, OCH2CH3), 70.1 (d, J = 160.2 Hz, PCH), 123.9 (dq, J = 5.6; 3.7 Hz, C2), 124.1 (q, 272.5 Hz, CF3), 124.6 (qd/dq, 3.6 Hz, C4), 128.5 (d, 2.5 Hz, C2′), 130.4 (dq, 5.4, 1.2 Hz, C5), 130.5 (qd, 32.1, 2.8 Hz, CCF3), 138.2 (d, 2.0 Hz, C1); 1H NMR (CDCl3) δ 1.23 (d, J = 7.1, 3H, OCH2CH3), 1.26 (d, J = 6.9, 3H, OCH2CH3), 4.00–4.13 (m, 2H, OCH2CH3), 5.1 (d, J = 10.9, 1H, PCH), 7.48 (t, J = 7.8, 1H, ArH), 7.57 (d, J = 7.9, 1H, ArH), 7.67 (d, J = 7.9, 1H, ArH), 7.75–7.78 (m, 1H, ArH); [M + H]+ = 313, [M + Na]+found = 335.0631, calculated: 335.0636, C12H16F3O4PNa.
3.2.4. Diethyl α-Hydroxy-α-(3,4,5-Trifluorophenyl)-Methylphosphonate (1d)
31P NMR (CDCl3) δ 19.8; 13C NMR (CDCl3) δ 16.13 (d, J = 3.4 Hz, OCH2CH3), 16.17 (d, J = 3.4 Hz, OCH2CH3), 63.3 (d, J = 7.5, OCH2CH3), 63.8 (d, J = 6.4, OCH2CH3), 69.1 (d, J = 163.3, PCO), 111.0 (dt, J = 17.2, C2), 133.8 (d, J = 8.3, C1), 139.03 (dt, J = 251.0, 15.5 Hz, C4), 148.66–152.84 (m, C3); 1H NMR (CDCl3) 1.28 (t, J = 5.9, 3H, OCH2CH3), 1.33 (t, J = 5.9, 3H, OCH2CH3), 4.02–4.27 (m, 2H, OCH2CH3), 4.98 (d, J = 11.2, 1H, PCH), 7.11–7.22 (m, 2H, ArH); [M + H]+ = 299, [M + Na]+found = 321.0475, calculated: 321.0480, C11H14F3O4PNa.
3.2.5. Dimethyl α-Hydroxy-α-Phenyl-Methylphosphonate (3a)
31P NMR (CDCl
3) δ 23.8, δ
P (CDCl
3) 23.8 [
11,
42]; [M + H]
+ = 217.
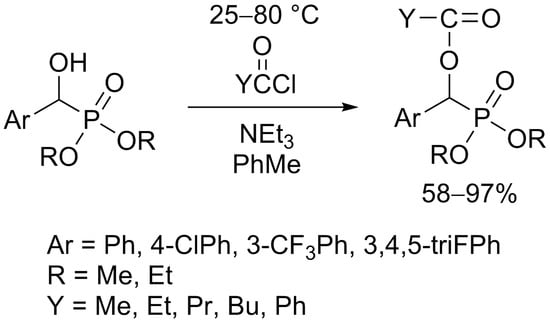
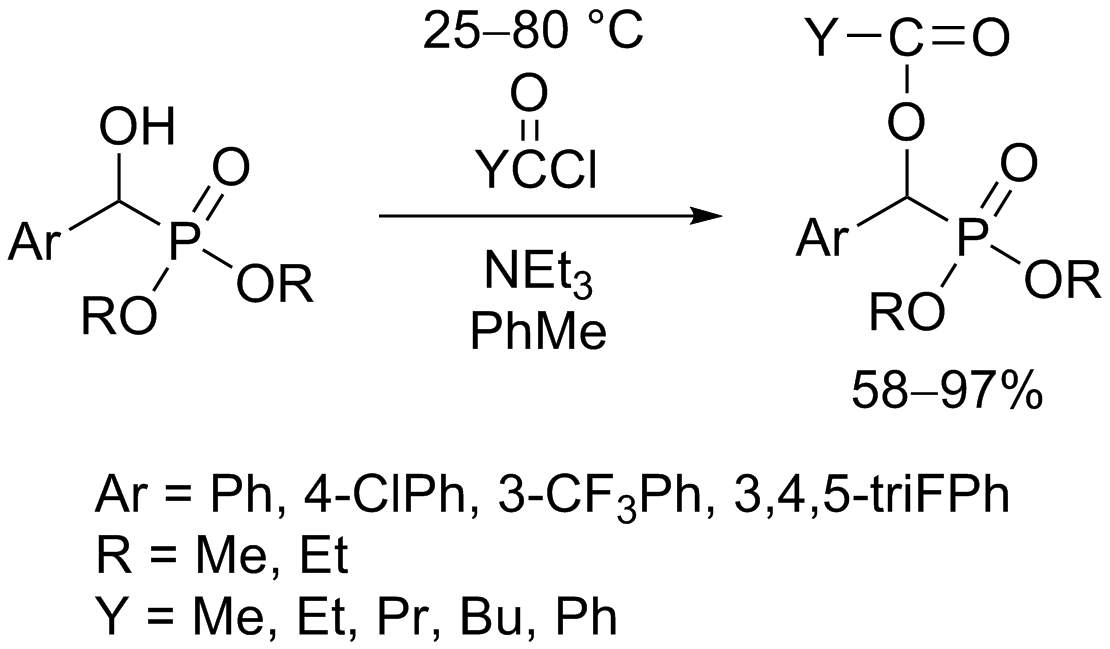
3.3. General Procedure for the Synthesis of Acylated Diethyl and Dimethyl α-Hydroxyphosphonates (2a–f, 4a–d)
To 1.2 mmol of α-hydroxyphosphonate (diethyl hydroxy-benzylphosphonate, 0.28 g; diethyl hydroxy-4-chlorobenzylphosphonate, 0.32 g; diethyl hydroxy-3-trifluoromethyl-benzylphosphonate, 0.36 g; diethyl hydroxy-3,4,5-trifluorobenzylphosphonate, 0.34 g), and 1.3 mmol (0.18 mL) of triethylamine in toluene (4.0 mL), 3.5 mmol (0.25 mL) of acetyl chloride, or 1.7 mmol of other acyl chlorides (butyryl chloride, 0.18 mL; benzoyl chloride, 0.20 mL) were added and the mixture was kept at 25–80 °C for 24 h (See
Table 2) in a sealed tube. The precipitated triethylamine hydrochloride was filtered off, and the volatile components were removed in vacuo. The crude product so obtained was purified by column chromatography on silica gel applying dichloromethane–methanol (97:3) as the eluent to give products
2a–
f in yields of 69–97% as oils.
To 1.2 mmol (0.25 g) of dimethyl α-hydroxy-benzylphosphonate and 1.2 mmol (0.18 mL) of triethylamine in toluene (4.0 mL) was added to 3.5 mmol (0.25 mL) of acetyl chloride or 1.7 mmol of other acyl chlorides (propionyl chloride, 0.18 mL; butyryl chloride, 0.18 mL; valeryl chloride, 0.18 mL; benzoyl chloride, 0.20 mL) and the mixture was stirred at 25–80 °C for 1–1.5 days (See
Table 3). A similar work-up as described above afforded products
4a–
d in yields of 45–87%.
The following compounds were thus prepared:
3.3.1. Diethyl α-Acetyloxy-α-Phenyl-Methylphosphonate (2a)
31P NMR (CDCl3) δ 17.7; 13C NMR (CDCl3) δ 16.23 (d, J = 5.6, OCH2CH3), 16.36 (d, J = 5.6, OCH2CH3), 20.8 (s, CCH3), 63.23 (d, J = 3.3, OCH2CH3), 63.28 (d, J = 4.1, OCH2CH3), 70.4 (d, J = 170.1, PCH), 127.9 (d, J = 5.9, C2*), 128.4 (d, J = 2.2, C3*), 128.7 (d, J = 2.9, C4), 133.5 (d, J = 2.2, C1), 169.2 (d, J = 8.8, C(O)), *may be reversed; 1H NMR (CDCl3) δ 1.21 (t, J = 7.1, 3H, OCH2CH3), 1.27 (t, J = 7.1, 3H, OCH2CH3), 2.2 (s, 3H, C(O)CH3), 3.89–4.15 (m, 2H, OCH2CH3), 6.1 (d, J = 13.6, 1H, PCH), 7.31–7.40 (m, 3H, ArH), 7.49 (dt, J = 8.0, 1.8, 2H); [M + H]+ = 287, [M + Na]+found = 309.0866, calculated: 309.0868, C13H19O5PNa.
3.3.2. Diethyl α-Acetyloxy-α-(4-Chlorophenyl)-Methylphosphonate (2b)
31P NMR (CDCl3) δ 14.4; 13C NMR (CDCl3) δ 16.4 (d, J = 5.7, OCH2CH3), 16.5 (d, J = 5.5, OCH2CH3), 20.9 (s, CCH3), 63.4 (d, J = 7.1, OCH2CH3), 63.5 (d, J = 6.4, OCH2CH3), 69.9 (d, J = 170.6, PCH), 128.8 (d, J = 2.3, C2*), 129.3 (d, J = 5.8, C3*), 132.2 (d, J = 2.4, C4), 134.7 (d, J = 3.7, C1), 169.2 (d, J = 8.9, C(O)), *may be reversed; 1H NMR (CDCl3) δ 1.23 (t, J = 7.0, 3H, OCH2CH3), 1.28 (t, J = 7.1, 3H, OCH2CH3), 2.2 (s, 3H, C(O)CH3), 3.87–4.17 (m, 2H, OCH2CH3), 6.1 (d, J = 13.7, 1H, PCH), 7.33–7.36 (m, 2H, ArH), 7.41–7.44 (m, 2H, ArH); [M + H]+ = 321, [M + Na]+found = 343.0475, calculated: 343.0478, C13H18ClO5PNa.
3.3.3. Diethyl α-Acetyloxy-α-(3-Trifluoromethylphenyl)-Methylphosphonate (2c)
31P NMR (CDCl3) δ 16.7; 13C NMR (CDCl3) δ 16.1 (d, J = 6.8 Hz, OCH2CH3), 16.2 (d, J = 6.8 Hz, OCH2CH3), 20.6 (s, CCH3), 63.35 (d, J = 7.1, OCH2CH3), 63.45 (d, J = 7.1, OCH2CH3), 69.8 (d, J = 169.8, PCH), 123.8 (q, J = 272.6, CF3), 124.4 (dq J = 3.9, C2), 125.3 (dq/qd, C4), 129.0 (d, J = 2.2, C2), 130.8 (dq, J = 32.6, 2.3, CCF3), 131.1 (dq, J = 1.2, C3), 134.8 (d, J = 2.2, C1), 169.0 (d, J = 8.7, C(O)); 1H NMR (CDCl3) δ 1.21 (d, J = 7.1, 3H, OCH2CH3), 1.26 (d, J = 7.1, 3H, OCH2CH3), 2.2 (s, 3H, C(O)CH3), 3.94–4.17 (m, 2H, OCH2CH3), 6.2 (d, J = 13.6, 1H, PCH), 7.48 (t, J = 8.0, 2H, ArH), 7.58 (d, J = 8.0, 1H, ArH), 7.66 (d, J= 7.9, 1H, ArH), 7.71 (s, 1H, ArH); [M + H]+ = 355, [M + Na]+found = 377.0739, calculated: 377.0742, C14H18F3O5PNa.
3.3.4. Diethyl α-Acetyloxy-α-(3,4,5-Trifluorophenyl)-Methylphosphonate (2d)
31P NMR (CDCl3) δ 16.1; 13C NMR (CDCl3) δ 16.26 (d, J = 6.1 Hz, OCH2CH3), 16.34 (d, J = 5.9 Hz, OCH2CH3), 20.6 (d, J = 2.3 Hz, CCH3), 63.5 (d, J = 6.7 Hz, OCH2CH3), 63.6 (d, J = 7.1 Hz, OCH2CH3), 68.9 (d, J = 171.2 Hz, PCH), 112.1 (dt, J = 17.1, 5.6 Hz, C2), 130.0 (d, J = 6.8, C1), 139.8 (d, J = 253.4, C4), 150.0 (d, J = 10.2, C3), 152.0 (d, J = 10.0, C3), 168.9 (d, J = 8.8, C(O)); 1H NMR (CDCl3) δ 1.28 (d, J = 7.0, 3H, OCH2CH3), 1.33 (d, J = 7.0, 3H, OCH2CH3), 2.2 (s, 3H, C(O)CH3), 3.96–4.26 (m, 2H, OCH2CH3), 6.0 (d, J = 14.1, 1H, PCH), 7.1 (ArH); [M + H]+ = 341, [M + Na]+found = 363.0585, calculated: 363.0585, C13H16F3O5PNa.
3.3.5. Diethyl α-Butyryloxy-α-Phenyl-Methylphosphonate (2e)
2e: 31P NMR (CDCl3) δ 17.9; 13C NMR (CDCl3) δ 13.7 (s, CH2CH3), 16.4 (d, J = 5.9, OCH2CH3), 16.5 (d, J = 5.8, OCH2CH3), 18.5 (s, CH2CH3), 36.1 (s, CH2CH2CH3), 63.36 (d, J = 3.6, OCH2CH3), 63.41 (d, J = 4.0, OCH2CH3), 70.2 (d, J = 170.0, PCH), 127.9 (d, J = 5.8, C2*), 128.5 (d, J = 2.2, C3*), 128.7 (d, J = 3.0, C4), 133.6 (d, J = 2.2, C1), 172.0 (d, J = 8.7, C(O), *may be reversed; 1H NMR (CDCl3) δ 1.0 (t, J = 7.4, 3H, CH2CH3), 1.21 (t, J = 7.1, 3H, OCH2CH3), 1.26 (t, J = 7.1, 3H, OCH2CH3), 2.30–2.36 (m, 2H, C(O)CH2), 2.48–2.53 (m, 2H, CH2CH3), 3.85–4.17 (m, 4H, OCH2CH3), 6.2 (d, J = 13.5, 1H, PCH), 7.32–7.43 (m, 3H, ArH), 7.46–7.50 (m, 2H, ArH); [M + H]+ = 315, [M + Na]+found = 337.1178, calculated: 337.1181, C15H23O5PNa.
3.3.6. Diethyl α-Benzoyloxy-α-Phenyl-Methylphosphonate (2f)
31P NMR (CDCl3) δ 18.6; 13C NMR (CDCl3) δ 16.3 (d, J = 5.7, OCH2CH3), 16.4 (d, J = 5.7, OCH2CH3), 63.5 (d, J = 6.7, OCH2CH3), 63.6 (d, J = 7.0, OCH2CH3), 70.9 (d, J = 170.6, PCH), 127.9 (d, J = 5.8, C2*), 128.5 (d, J = 3.9, C3*), 129.9 (d, J = 3.2, C4), 133.4 (d, J = 2.1, C1), 165.0 (d, J = 9.1, C(O)), *may be reversed; 1H NMR (CDCl3) δ 1.20 (t, J = 6.9, 3H, OCH2CH3), 1.24 (t, J = 7.0, 3H, OCH2CH3), 3.68–4.20 (m, 2H, OCH2CH3), 6.4 (d, J = 13.2, PCH), 7.30–7.60 (ArH); [M + H]+ = 313, [M + Na]+found = 371.1024, calculated: 371.1024, C18H21O5PNa.
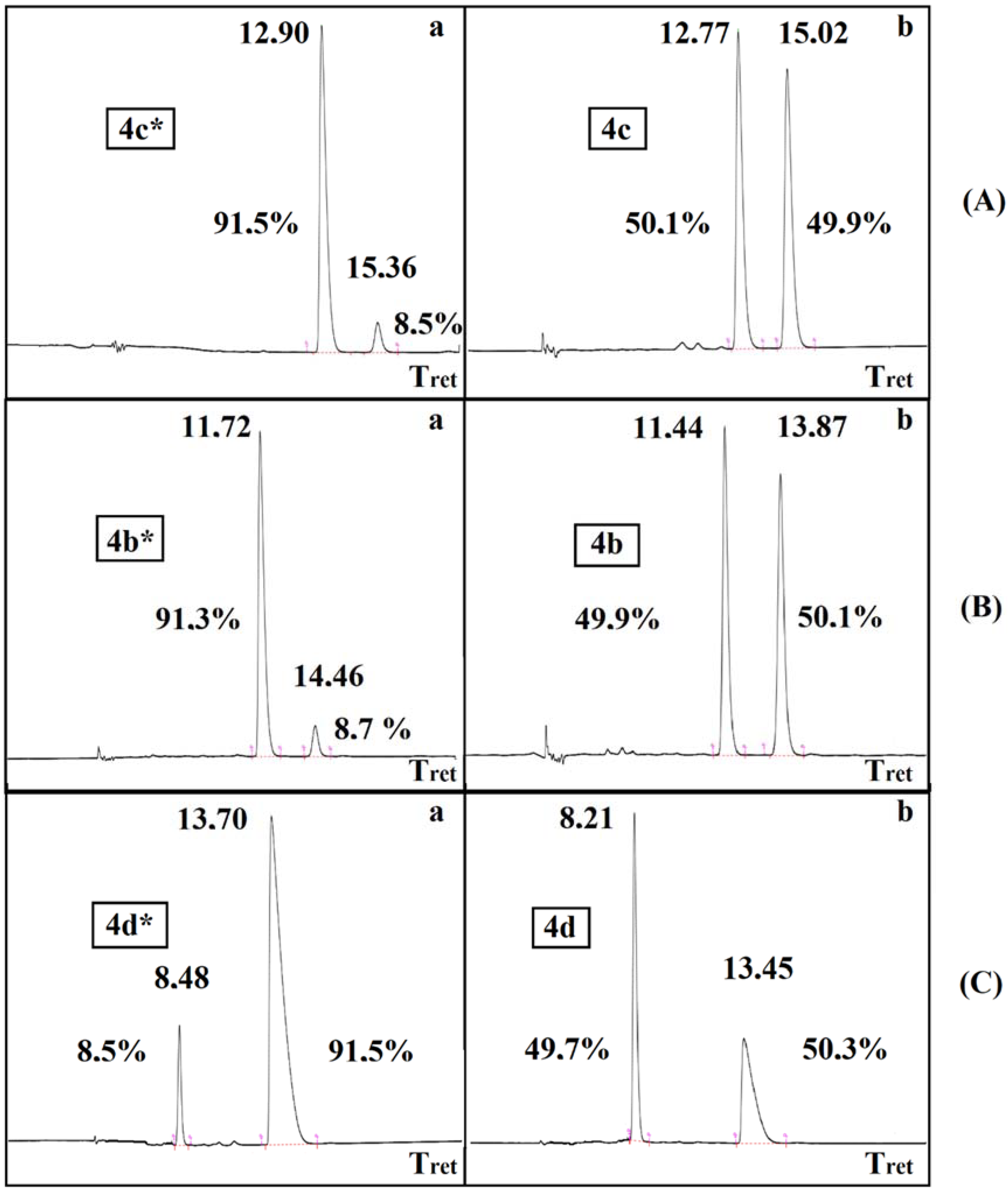
3.3.7. Dimethyl α-Valeryloxy-α-Phenyl-Methylphosphonate (4a)
31P NMR (CDCl3) δ 18.8; 13C NMR (CDCl3) δ 13.6 (CH2CH3), 22.1 (CH2CH3), 26.8 (CH2CH2CH3), 33.8 (CH2C(O)), 53.69 (d, J = 6.5, OCH3), 53.73 (d, J = 7.1, OCH3), 69.7 (d, J = 169.8, PCH), 127.7 (d, J = 5.8, C2*), 128.5 (d, J = 2.2, C3*), 128.7 (d, J = 2.8, C4), 133.2 (d, J = 2.1, C1), 171.9 (d, J = 8.4, C(O)), *may be reversed; 1H NMR (CDCl3) δ 0.91 (t, J = 7.3, 3H, CH2CH3), 1.27–1.41 (m, 2H, CH2CH3), 1.55–1.71 (m, 2H, CH2CH2CH3), 2.44 (td, J = 7.4, J = 1.6, 2H, CH2C(O)), 3.65 (d, J = 10.6, 3H, OCH3), 3.72 (d, J = 10.7, 3H, OCH3), 6.19 (d, J = 13.5, 1H, PCH), 7.30–7.42 (m, 3H, ArH), 7.43–7.52 (m, 2H, ArH).
3.3.8. Dimethyl α-Propionyloxy-α-Phenyl-Methylphosphonate (4b)
31P NMR (CDCl3) δ 20.19; 13C NMR (CDCl3) δ 9.0 (s, CH2CH3), 27.5 (s, CH2CH3), 53.76 (d, J = 6.0 Hz, OCH3), 53.84 (d, J = 6.0 Hz, OCH3), 69.9 (d, J = 170.0 Hz, PCH), 127.77 (d, J = 5.8 Hz, C2*), 128.6 (d, J = 2.2 Hz, C3*), 128.8 (d, J = 2.9 Hz, C4), 133.3 (d, J = 2.3 Hz, C1), 172.7 (dd, J = 8.7, Hz, C(O)), *may be reversed; 1H NMR (CDCl3) δ 1.16 (t, J = 7.5 Hz, 3H, CH2CH3), 2.53–2.38 (m, 2H, CH2CH3), 3.64 (d, J = 10.5 Hz, 3H, OCH3), 3.70 (d, J = 10.7 Hz, 3H, OCH3), 6.17 (d, J = 13.5 Hz, 1H, PCH), 7.30–7.39 (m, 3H, Ar), 7.45–7.49 (m, 2H, ArH).
3.3.9. Dimethyl α-Acetyloxy-α-Phenyl-Methylphosphonate (4c)
31P NMR (CDCl3) δ 20.1; 13C NMR (CDCl3) δ 20.7 (CH3), 53.69 (d, J = 6.8, OCH3), 53.74 (d, J = 6.8, OCH3), 69.9 (d, J = 170.0, PCH), 127.8 (d, J = 5.7, C2*), 128.5 (d, J = 2.6, C3*), 128.8 (d, J = 2.9, C4), 133.2 (d, J = 2.3, C1), 169.1 (d, J = 8.8, C(O); *may be reversed; 1H NMR (CDCl3) δ 2.2 (s, 3H, C(O)CH3), 3.65 (d, J = 10.6, 3H, OCH3), 3.73 (d, J = 10.7, 3H, OCH3), 6.2 (d, J = 13.5, 1H, PCH), 7.33–7.40 (m, 3H, ArH), 7.48–7.51 (m, 2H, ArH); [M + H]+ = 259. [M + Na]+found = 281.0555, calculated: 281.0555, C11H15O5PNa.
3.3.10. Dimethyl α-Benzoyloxy-α-Phenyl-Methylphosphonate (4d)
31P NMR (CDCl3) δ 20.26; 13C NMR (CDCl3) δ 54.0 (d, J = 6.5 Hz, OCH3), 54.2 (d, J = 7.0 Hz, OCH3), 70.5 (d, J = 170.5 Hz, PCH), 127.9 (d, J = 5.8 Hz, C2a), 128.6 (s, C2′b), 128.7 (d, J = 2.3 Hz, C3a), 128.96 (d, J = 2.9 Hz, C1), 129.9 (s, C3′b), 130.04 (d, J = 1.5 Hz, C4), 133.2 (d, J = 2.2 Hz, C1′), 133.6 (s, C4′), 164.90 (d, J = 8.8 Hz, C(O)), a,bmay be reversed; 1H NMR (CDCl3) δ 3.69 (d, J = 10.6 Hz, 3H, OCH3), 3.74 (d, J = 10.7 Hz, 3H, OCH3), 6.4 (d, J = 13.3 Hz, 1H, PCH), 7.31–7.41 (m, 2H, Ar), 7.44–7.50 (m, 3H, Ar), 7.56–7.63 (m, 3H, Ar), 8.1 (dd, J = 8.0, 1.4 Hz, 2H, ArH).
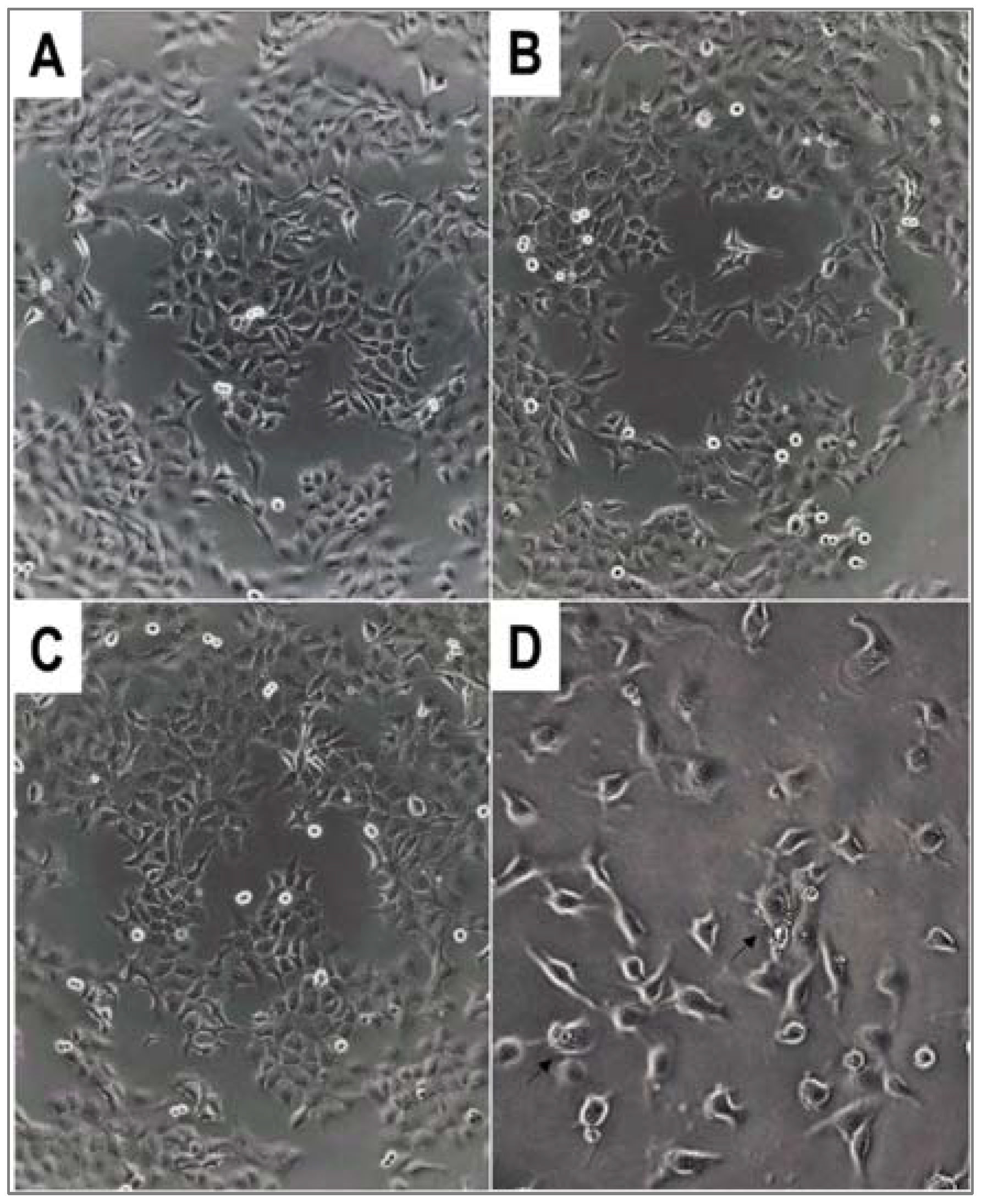
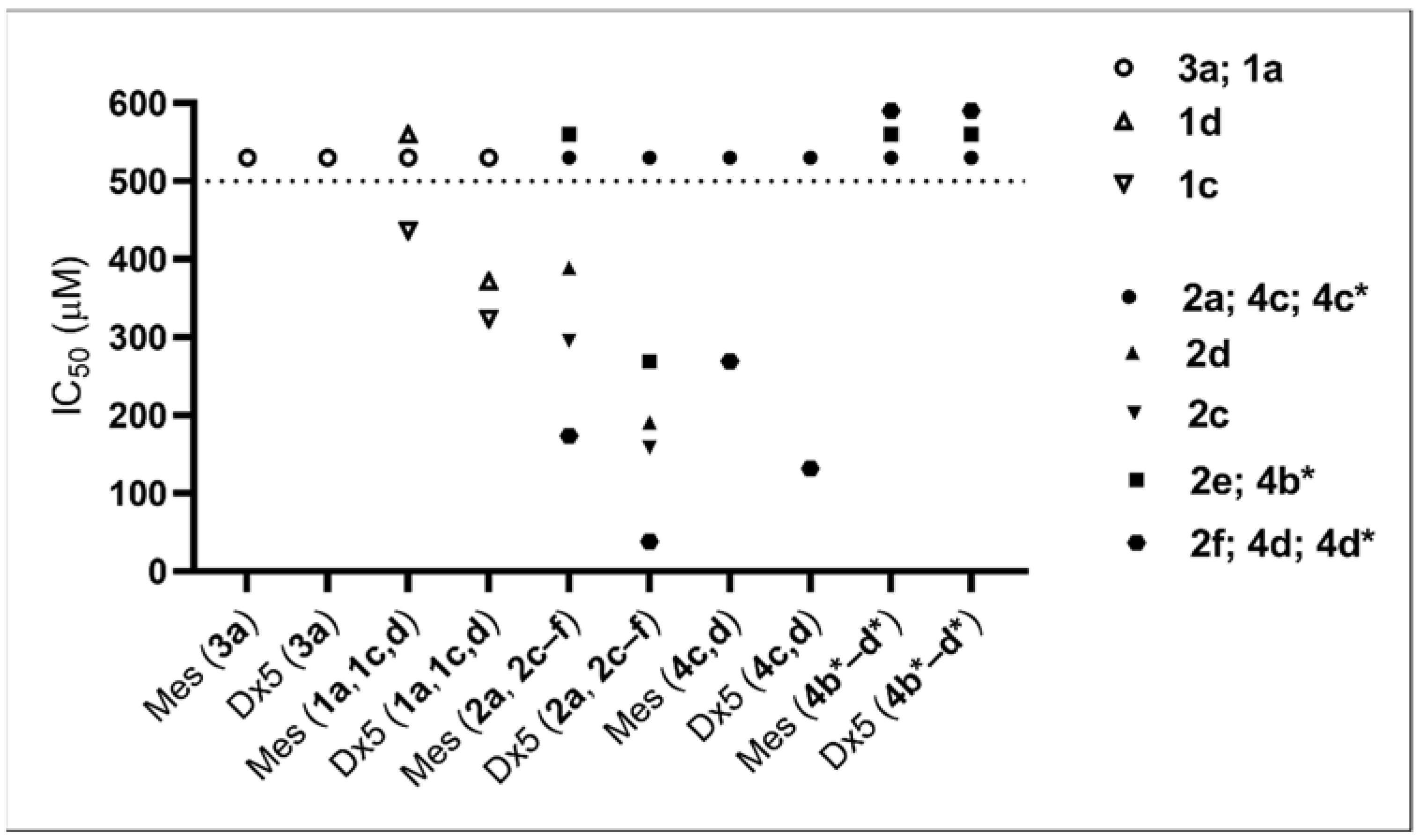
3.5. Cell Lines and Culture Conditions—In Vitro Cytostasis Assays
In vitro cytostatic effect of the compounds was studied on MDA-MB-231 human breast adenocarcinoma [
30], A431 human epidermoid carcinoma [
31], PC-3 human prostate adenocarcinoma [
32], Ebc-1 human lung squamous cell carcinoma [
33], MDA-MB-453 human metastatic epithelial breast carcinoma [
34], A2058 human melanoma [
35], HT-29 human colorectal carcinoma [
36], and Kaposi’s sarcoma (KS) [
37] cells. MDA-MB 435 cell line was a generous gift of Dr. Angels Fabra, Hospital of Duran e Reynalds, Barcelona, Spain, 1995 and obtained from Dr. Janet E. Price [
36]. The other cell lines were generous gifts of Dr. József Tóvári (Department of Experimental Pharmacology, National Institute of Oncology, Budapest, Hungary). MDA-MB-231, MDA-MB-453, PC-3, Ebc-1, and A431 cells were cultured in DMEM medium (Lonza, Basel, Switzerland) supplemented with 10% FBS (EuroClone, Pero, Italy), 2 mM L-glutamine (BioSera, Nuaille, France), penicillin-streptomycin antibiotics mixture (50 IU/mL and 50 μg/mL, respectively), 1 mM sodium pyruvate (both obtained from Lonza, Basel, Switzerland), and 1% nonessential amino acid mixture (BioSera, Nuaille, France). KS, A2058, and HT-29 cells were cultured in RPMI-1640 medium (Lonza, Basel, Switzerland) supplemented with 10% FBS (EuroClone, Pero, Italy), 2 mM L-glutamine (EuroClone, Pero, Italy), and penicillin-streptomycin antibiotics mixture (50 IU/mL and 50 μg/mL, respectively) (Lonza, Basel, Switzerland). The cultures were maintained at 37 °C in a humidified atmosphere with 5% CO
2. The cells were grown to confluency and then divided into 96-well tissue culture plates (Sarstedt, Nümbrecht, Germany) with the initial cell number of 5.0 × 10
3 cells/well. After 24 h incubation at 37 °C, the cells were treated with the compounds in 200 μL final volume containing 1.0
v/v% DMSO (Merck, Darmstadt, Germany) at 50 μM concentration overnight, whereas control cells were treated with serum-free medium only, or with DMSO (c = 1.0
v/v%) at the same conditions. After incubation, the cells were washed twice with serum-free medium. Subsequently, the cells were cultured for additional 72 h in 10% serum containing medium at 37 °C; then, the MTT (Merck, Darmstadt, Germany) solution (at c = 0.37 mg/mL final concentration) was added to each well. The respiratory chain [
45] and other electron transport systems [
46] reduce MTT, and thereby form non-water-soluble violet formazan crystals within the cell [
47]. The amount of these crystals may be determined by spectrophotometry and serves as an estimate for the number of mitochondria, and hence, the number of living cells in the well [
48]. After 3 h of incubation with MTT, the cells were centrifuged with 2000 rpm for 5 min and then the supernatant was removed. The obtained formazan crystals were dissolved in DMSO (100 µL) and the optical density (OD) of the samples was measured at λ = 540 nm and 620 nm, respectively, using ELISA Reader (iEMS Reader, Labsystems, Vantaa, Finland). OD
620 values were subtracted from OD
540 values. The percent of cytostasis was calculated with the following equation:
where values OD
treated and OD
control correspond to the optical densities of the treated and the control wells, respectively. In each case, two independent experiments were carried out with four parallel measurements. Statistical analysis of data was performed using Student’s t test at the 95% confidence level.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}




