
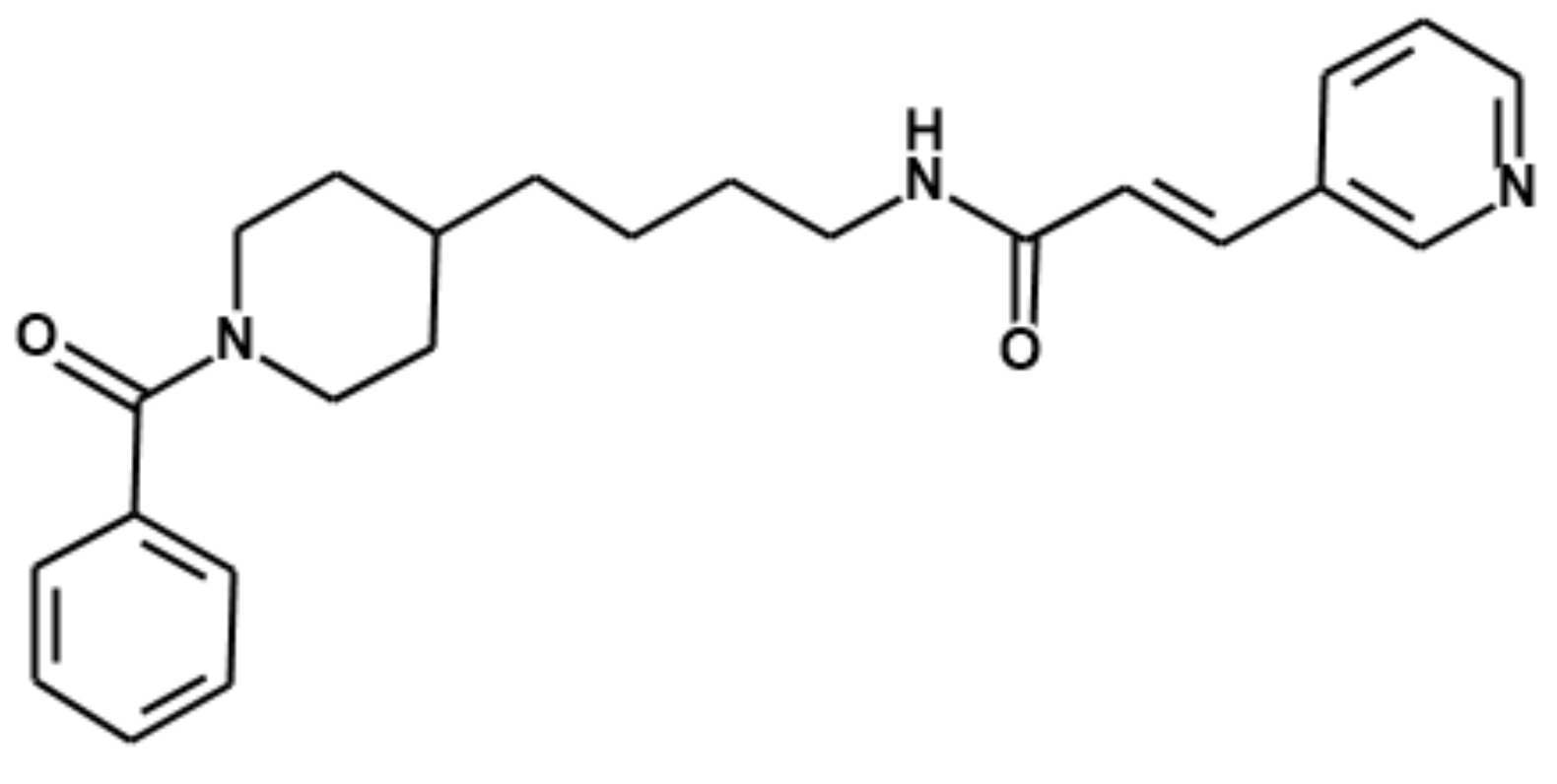
Quantitative Analysis of Daporinad (FK866) and Its In Vitro and In Vivo Metabolite Identification Using Liquid Chromatography-Quadrupole-Time-of-Flight Mass Spectrometry


Abstract
:1. Introduction
2. Results and Discussion
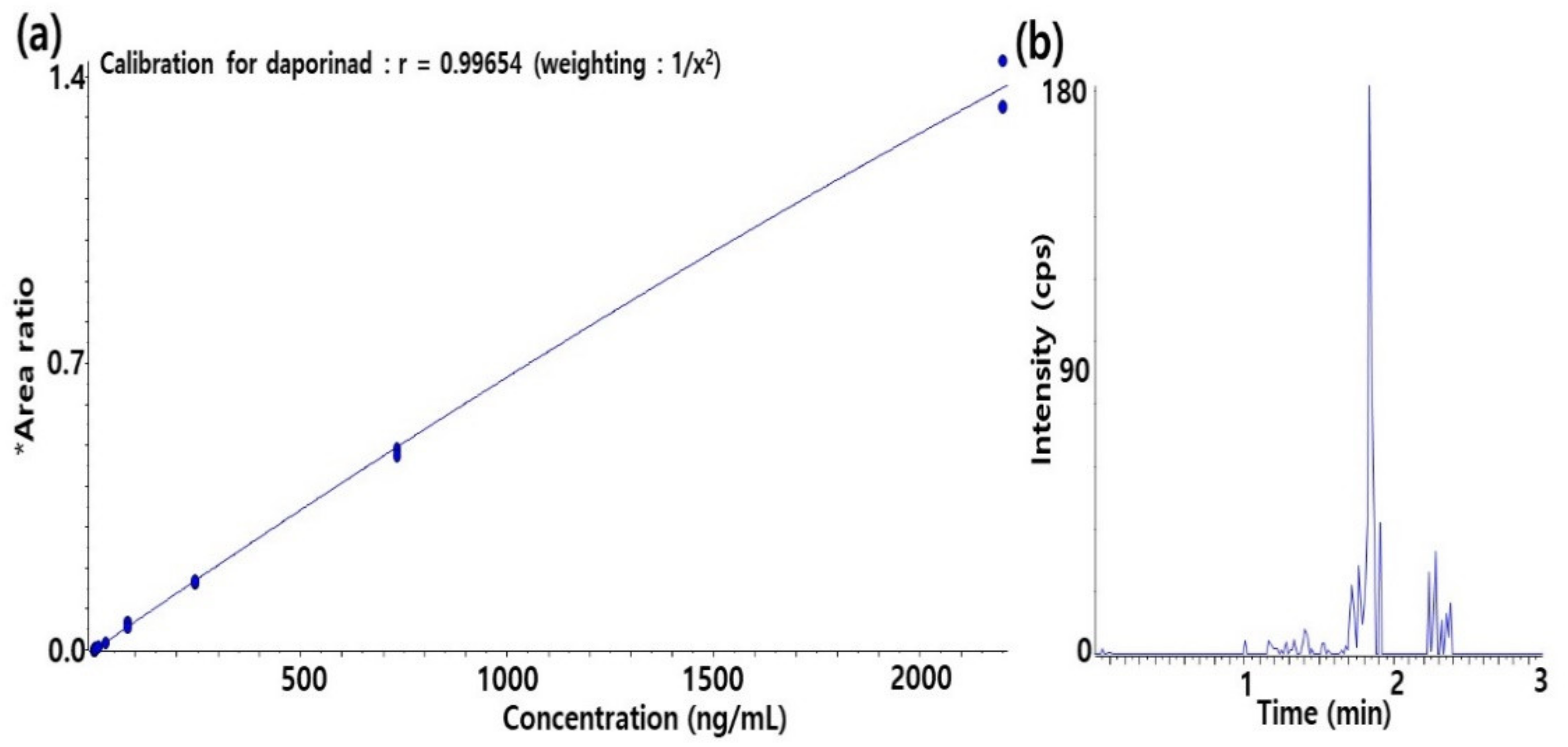
2.1. LC-qTOF-MS Method Development and Qualification for Daporinad in Mouse Plasma
2.1.1. Method Development for Quantification of Daporinad in Mouse Plasma
2.1.2. Preliminary Stability Tests for Daporinad in Mouse Plasma
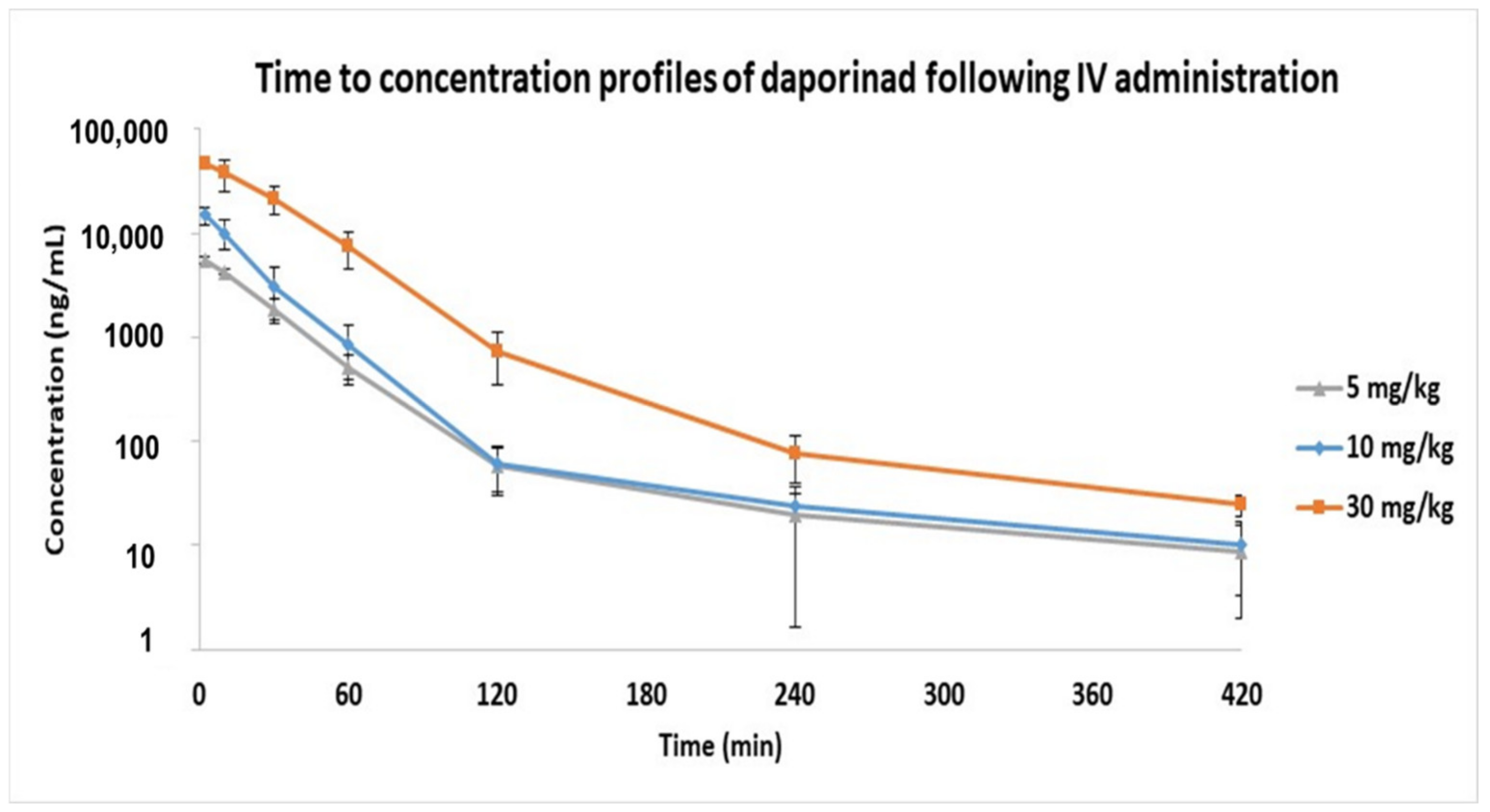
2.2. Application for Intravenous (IV) Pharmacokinetic (PK) Studies in Mouse
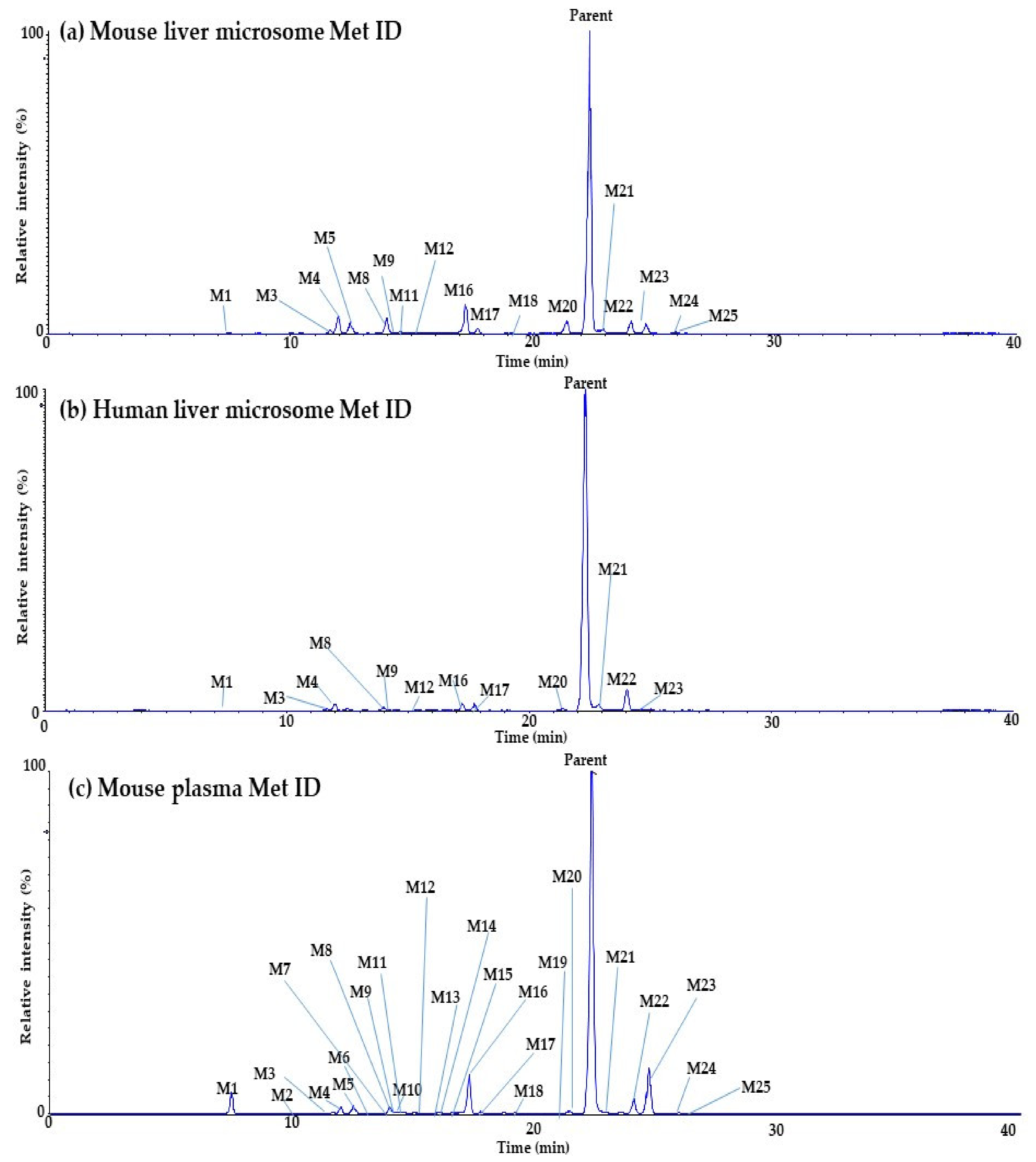
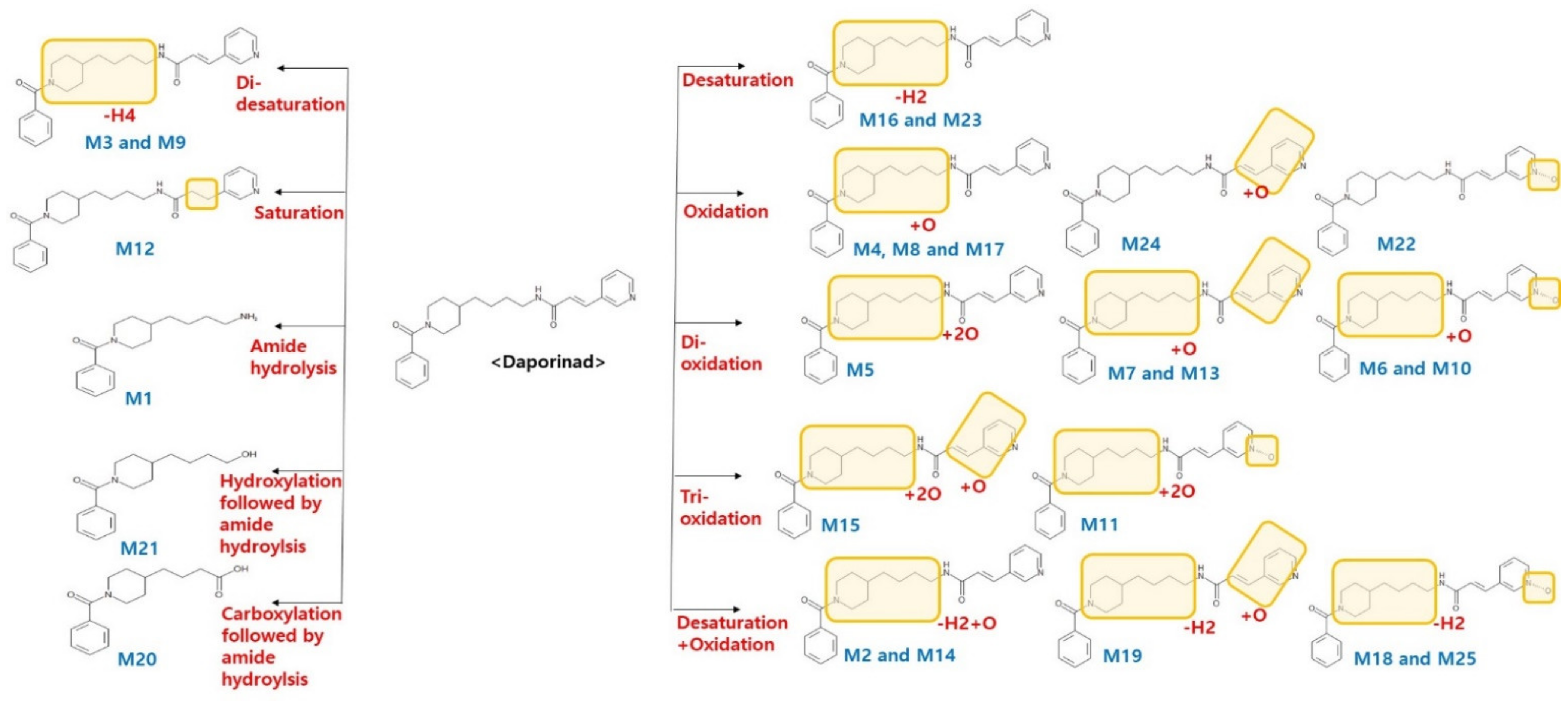
2.3. In Vitro and In Vivo Metabolite Identification (Met ID) for Daporinad
3. Materials and Methods
3.1. Reagents and Chemicals
3.2. Preparation of Stock, Standard (STD) Working and Quality Control (QC)
Working Solutions
3.3. Sample Preparation for Method Qualification and PK Analysis
3.4. Sample Preparation for In Vitro Met ID In Mouse and Human Liver Microsomes
3.5. Sample Preparation for In Vivo Met ID in Mouse PK Samples
3.6. Sample Preparation for N-Oxide Metabolites Identification in In Vitro and In Vivo Samples
3.7. LC-qTOF-MS Condition
3.8. Method Qualification
3.9. Application for Animal Study
3.10. Software
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kim, S.-Y. Cancer energy metabolism: Shutting power off cancer factory. Biomol. Ther. 2018, 26, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varghese, E.; Samuel, S.M.; Líšková, A.; Samec, M.; Kubatka, P.; Büsselberg, D. Targeting glucose metabolism to overcome resistance to anticancer chemotherapy in breast cancer. Cancers 2020, 12, 2252. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahra, I.B.; Laurent, K.; Giuliano, S.; Larbret, F.; Ponzio, G.; Gounon, P.; Le Marchand-Brustel, Y.; Giorgetti-Peraldi, S.; Cormont, M.; Bertolotto, C. Targeting cancer cell metabolism: The combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 2010, 70, 2465–2475. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Harris, A.L. Development of cancer metabolism as a therapeutic target: New pathways, patient studies, stratification and combination therapy. Br. J. Cancer 2020, 122, 1–3. [Google Scholar] [CrossRef]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial metabolism as a target for cancer therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef]
- Navas, L.E.; Carnero, A. NAD+ metabolism, stemness, the immune response, and cancer. Signal Transduct. Target. Ther. 2021, 6, 2. [Google Scholar] [CrossRef]
- Amjad, S.; Nisar, S.; Bhat, A.A.; Frenneaux, M.P.; Fakhro, K.; Haris, M.; Reddy, R.; Patay, Z.; Baur, J.; Bagga, P. Role of NAD+ in regulating cellular and metabolic signaling pathways. Mol. Metab. 2021, 49, 101195. [Google Scholar] [CrossRef]
- Yaku, K.; Okabe, K.; Hikosaka, K.; Nakagawa, T. NAD metabolism in cancer therapeutics. Front. Oncol. 2018, 8, 622. [Google Scholar] [CrossRef]
- Demarest, T.G.; Babbar, M.; Okur, M.N.; Dan, X.; Croteau, D.L.; Fakouri, N.B.; Mattson, M.P.; Bohr, V.A. NAD+ metabolism in aging and cancer. Annu. Rev. Cancer Biol. 2019, 3, 105–130. [Google Scholar] [CrossRef]
- Ghanem, M.S.; Monacelli, F.; Nencioni, A. Advances in NAD-Lowering Agents for Cancer Treatment. Nutrients 2021, 13, 1665. [Google Scholar] [CrossRef] [PubMed]
- Chowdhry, S.; Zanca, C.; Rajkumar, U.; Koga, T.; Diao, Y.; Raviram, R.; Liu, F.; Turner, K.; Yang, H.; Brunk, E. NAD metabolic dependency in cancer is shaped by gene amplification and enhancer remodelling. Nature 2019, 569, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Mottahedeh, J.; Haffner, M.C.; Grogan, T.R.; Hashimoto, T.; Crowell, P.D.; Beltran, H.; Sboner, A.; Bareja, R.; Esopi, D.; Isaacs, W.B. CD38 is methylated in prostate cancer and regulates extracellular NAD+. Cancer Metab. 2018, 6, 13. [Google Scholar] [CrossRef]
- Luengo, A.; Li, Z.; Gui, D.Y.; Sullivan, L.B.; Zagorulya, M.; Do, B.T.; Ferreira, R.; Naamati, A.; Ali, A.; Lewis, C.A. Increased demand for NAD+ relative to ATP drives aerobic glycolysis. Mol. Cell 2021, 81, 691–707.e696. [Google Scholar] [CrossRef] [PubMed]
- Kumaraswamy, G.K.; Guerra, T.; Qian, X.; Zhang, S.; Bryant, D.A.; Dismukes, G.C. Reprogramming the glycolytic pathway for increased hydrogen production in cyanobacteria: Metabolic engineering of NAD+-dependent GAPDH. Energy Environ. Sci. 2013, 6, 3722–3731. [Google Scholar] [CrossRef]
- Giddings, E.L.; Champagne, D.P.; Wu, M.-H.; Laffin, J.M.; Thornton, T.M.; Valenca-Pereira, F.; Culp-Hill, R.; Fortner, K.A.; Romero, N.; East, J. Mitochondrial ATP fuels ABC transporter-mediated drug efflux in cancer chemoresistance. Nat. Commun. 2021, 12, 13. [Google Scholar] [CrossRef] [PubMed]
- Galli, U.; Colombo, G.; Travelli, C.; Tron, G.C.; Genazzani, A.A.; Grolla, A.A. Recent advances in NAMPT inhibitors: A novel immunotherapic strategy. Front. Pharmacol. 2020, 11, 656. [Google Scholar] [CrossRef]
- Liu, H.-w.; Smith, C.B.; Schmidt, M.S.; Cambronne, X.A.; Cohen, M.S.; Migaud, M.E.; Brenner, C.; Goodman, R.H. Pharmacological bypass of NAD+ salvage pathway protects neurons from chemotherapy-induced degeneration. Proc. Natl. Acad. Sci. USA 2018, 115, 10654–10659. [Google Scholar] [CrossRef] [Green Version]
- Gibson, A.E.; Yeung, C.; Issaq, S.H.; Collins, V.J.; Gouzoulis, M.; Zhang, Y.; Ji, J.; Mendoza, A.; Heske, C.M. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) with OT-82 induces DNA damage, cell death, and suppression of tumor growth in preclinical models of Ewing sarcoma. Oncogenesis 2020, 9, 80. [Google Scholar] [CrossRef]
- Zhang, B.; Shi, D.; Zhang, X.; Liang, G.; Liu, W.; Qiao, S. FK866 inhibits the epithelial-mesenchymal transition of hepatocarcinoma MHCC97-H cells. Oncol. Lett. 2018, 16, 7231–7238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautam, P.; Karhinen, L.; Szwajda, A.; Jha, S.K.; Yadav, B.; Aittokallio, T.; Wennerberg, K. Identification of selective cytotoxic and synthetic lethal drug responses in triple negative breast cancer cells. Mol. Cancer 2016, 15, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, R.; Yuan, Z.; Yang, L.; Li, L.; Li, D.; Lv, C. Inhibition of NAMPT decreases cell growth and enhances susceptibility to oxidative stress. Oncol. Rep. 2017, 38, 1767–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, E.; Li, R.; Yang, J.; Zhang, J.; Li, A.; Yang, Y.; Liu, S.; Liu, A.; Jiang, X. FK866 attenuates acute hepatic failure through c-jun-N-terminal kinase (JNK)-dependent autophagy. Sci. Rep. 2017, 7, 2206. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-Q.; Lu, J.-T.; Jiang, W.-X.; Lu, Y.-B.; Wu, M.; Wei, E.-Q.; Zhang, W.-P.; Tang, C. NAMPT inhibitor and metabolite protect mouse brain from cryoinjury through distinct mechanisms. Neuroscience 2015, 291, 230–240. [Google Scholar] [CrossRef]
- Podsednik, A.; Jiang, J.; Jacob, A.; Li, L.Z.; Xu, H.N. Optical Redox Imaging of Treatment Responses to Nampt Inhibition and Combination Therapy in Triple-Negative Breast Cancer Cells. Int. J. Mol. Sci. 2021, 22, 15563. [Google Scholar] [CrossRef]
- Lucena-Cacace, A.; Otero-Albiol, D.; Jiménez-García, M.P.; Muñoz-Galvan, S.; Carnero, A. NAMPT is a potent oncogene in colon cancer progression that modulates cancer stem cell properties and resistance to therapy through Sirt1 and PARP. Clin. Cancer Res. 2018, 24, 1202–1215. [Google Scholar] [CrossRef] [Green Version]
- Sauer, H.; Kampmann, H.; Khosravi, F.; Sharifpanah, F.; Wartenberg, M. The nicotinamide phosphoribosyltransferase antagonist FK866 inhibits growth of prostate tumour spheroids and increases doxorubicin retention without changes in drug transporter and cancer stem cell protein expression. Clin. Exp. Pharmacol. Physiol. 2021, 48, 422–434. [Google Scholar] [CrossRef]
- Hasmann, M.; Schemainda, I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003, 63, 7436–7442. [Google Scholar]
- Holen, K.; Saltz, L.B.; Hollywood, E.; Burk, K.; Hanauske, A.-R. The pharmacokinetics, toxicities, and biologic effects of FK866, a nicotinamide adenine dinucleotide biosynthesis inhibitor. Investig. New Drugs 2008, 26, 45–51. [Google Scholar] [CrossRef]
- Franceschini, N.; Oosting, J.; Tamsma, M.; Niessen, B.; Bruijn, I.B.-d.; van den Akker, B.; Kruisselbrink, A.B.; Palubeckaitė, I.; Bovée, J.V.; Cleton-Jansen, A.-M. Targeting the NAD Salvage Synthesis Pathway as a Novel Therapeutic Strategy for Osteosarcomas with Low NAPRT Expression. Int. J. Mol. Sci. 2021, 22, 26273. [Google Scholar] [CrossRef] [PubMed]
- Karpov, A.S.; Abrams, T.; Clark, S.; Raikar, A.; D’Alessio, J.A.; Dillon, M.P.; Gesner, T.G.; Jones, D.; Lacaud, M.; Mallet, W. Nicotinamide phosphoribosyltransferase inhibitor as a novel payload for antibody–drug conjugates. ACS Med. Chem. Lett. 2018, 9, 838–842. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.D.; Liu, G.X. Daporinad in vitro metabolite profiling via rat, dog, monkey and human liver microsomes by liquid chromatography/quadrupole-orbitrap mass spectrometry. Rapid Commun. Mass Spectrom. 2021, 35, e9150. [Google Scholar] [CrossRef] [PubMed]
- Bateman, K.P.; Cohen, L.; Emary, B.; Pucci, V. Standardized workflows for increasing efficiency and productivity in discovery stage bioanalysis. Bioanalysis 2013, 5, 1783–1794. [Google Scholar] [CrossRef] [PubMed]
- Somei, M.; Kato, K.; Inoue, S. Titanium (III) chloride for the reduction of heteroaromatic and aromatic nitro compounds. Chem. Pharm. Bull. 1980, 28, 2515–2518. [Google Scholar] [CrossRef] [Green Version]
- Seaton, Q.F.; Lawley, C.W.; Akers, H.A. The reduction of aliphatic and aromatic N-oxides to the corresponding amines with titanium (III) chloride. Anal. Biochem. 1984, 138, 238–241. [Google Scholar] [CrossRef]
- Hop, C.E.; Wang, Z.; Chen, Q.; Kwei, G. Plasma-pooling methods to increase throughput for in vivo pharmacokinetic screening. J. Pharm. Sci. 1998, 87, 901–903. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run | Statistics | Low QC (15.0 ng/mL) | Medium QC (165 ng/mL) | High QC (1820 ng/mL) |
|---|---|---|---|---|
| Intra-run 1 | Mean accuracy (%) | 96.9 | 107 | 104 |
| Precision (CV %) | 10.3 | 6.00 | 10.1 | |
| Intra-run 2 | Mean accuracy (%) | 106 | 90.7 | 97.8 |
| Precision (CV %) | 12.7 | 4.16 | 1.68 | |
| Intra-run 3 | Mean accuracy (%) | 106 | 109 | 102 |
| Precision (CV %) | 10.7 | 4.64 | 7.72 | |
| Inter-run | Mean accuracy (%) | 103 | 102 | 101 |
| Precision (CV %) | 10.1 | 8.91 | 6.71 |
| Assessment | Dilution Factor | Statistics | Dilution QC |
|---|---|---|---|
| Dilution integrity | 5-fold | Theoretical concentration (ng/mL) | 6670 |
| Mean concentration (ng/mL) | 7550 | ||
| Mean accuracy (%) | 113 | ||
| Precision (%, CV) | 3.12 | ||
| 10-fold | Theoretical concentration (ng/mL) | 20,000 | |
| Mean concentration (ng/mL) | 19,300 | ||
| Mean accuracy (%) | 96.5 | ||
| Precision (%, CV) | 2.20 | ||
| 30-fold | Theoretical concentration (ng/mL) | 6670 | |
| Mean concentration (ng/mL) | 6220 | ||
| Mean accuracy (%) | 93.2 | ||
| Precision (%, CV) | 4.45 |
| Stability Assessment | Statistics | Low QC (15.0 ng/mL) | Medium QC (165 ng/mL) | High QC (1820 ng/mL) |
|---|---|---|---|---|
| Short-term (4 h, RT) | Mean accuracy (%) | 103 | 102 | 104 |
| Precision (CV %) | 7.22 | 2.54 | 3.08 | |
| Long-term (14 days, −20 °C) | Mean accuracy (%) | 111 | 84.5 | 91.4 |
| Precision (CV %) | 3.71 | 11.7 | 4.17 | |
| Freeze–thaw (3 cycles, −80 °C) | Mean accuracy (%) | 105 | 117 | 116 |
| Precision (CV %) | 5.10 | 5.18 | 2.61 |
| Dose (mg/kg) | T1/2 (min) | Cmax (ng/mL) | AUClast (min∙ng/mL) | Cl (mL/min/kg) | Vss (mL/kg) |
|---|---|---|---|---|---|
| 5 | 52.91 ± 7.30 | 5476.2 ± 426.73 | 171,690.41 ± 19,880.05 | 29.29 ± 3.70 | 897.06 ± 199.49 |
| 10 | 50.49 ± 4.88 | 14,872.07 ± 2653.47 | 357,205.55 ± 36,369.09 | 28.13 ± 2.78 | 646.57 ± 91.48 |
| 30 | 40.76 ± 0.95 | 48,059.1 ± 3920.65 | 1,763,266.5 ± 457,583.63 | 17.88 ± 5.11 | 535.76 ± 110.37 |
| Symbol | Metabolites | m/z | Formula | Retention Time (min) | Error ppm | Mouse Liver Microsome | Human Liver Microsome | Mouse Plasma |
|---|---|---|---|---|---|---|---|---|
| M1 | Amide hydrolysis | 261.1961 | C16H24N2O4 | 7.36 | 0.4 | O | O | O |
| M2 | Desaturation + Oxidation | 406.2125 | C24H27N3O3 | 10.07 | 0.2 | - | - | O |
| M3 | Di-desaturation | 388.2020 | C24H31N3O2 | 11.53 | 1.0 | O | O | O |
| M4 | Oxidation | 408.2282 | C24H29N3O3 | 11.99 | 0.0 | O | O | O |
| M5 | Di-oxidation | 424.2231 | C24H29N3O4 | 12.48 | 0.2 | O | - | O |
| M6 | Di-oxidation | 424.2231 | C24H29N3O4 | 13.46 | 1.9 | - | - | O |
| M7 | Di-oxidation | 424.2231 | C24H29N3O4 | 13.91 | 2.1 | - | - | O |
| M8 | Oxidation | 408.2282 | C24H29N3O3 | 14.00 | 0.2 | O | O | O |
| M9 | Di-desaturation | 388.2020 | C24H31N3O2 | 14.11 | 1.0 | O | O | O |
| M10 | Di-oxidation | 424.2231 | C24H29N3O4 | 14.29 | 0.2 | - | - | O |
| M11 | Tri-oxidation | 440.2180 | C24H29N3O5 | 14.41 | 0.7 | O | - | O |
| M12 | Saturation | 394.2489 | C24H31N3O2 | 15.37 | 1.0 | - | - | O |
| M13 | Di-oxidation | 424.2231 | C24H29N3O4 | 15.87 | 0.7 | - | - | O |
| M14 | Desaturation + Oxidation | 406.2125 | C24H27N3O3 | 16.04 | 1.0 | - | - | O |
| M15 | Tri-oxidation | 440.2180 | C24H29N3O5 | 16.54 | 1.4 | - | - | O |
| M16 | Desaturation | 390.2176 | C24H27N3O2 | 17.27 | 2.8 | O | O | O |
| M17 | Oxidation | 408.2282 | C24H29N3O3 | 17.76 | 2.0 | O | O | O |
| M18 | Desaturation + Oxidation | 406.2125 | C24H27N3O3 | 19.15 | 0.7 | O | - | O |
| M19 | Desaturation + Oxidation | 406.2125 | C24H27N3O3 | 21.02 | 0.7 | - | - | O |
| M20 | Amide hydrolysis followed by carboxylation | 276.1594 | C16H21NO3 | 21.25 | 1.8 | O | O | O |
| Parent | Parent | 392.2333 | C24H29N3O2 | 22.31 | 2.3 | O | O | O |
| M21 | Amide hydrolysis followed by hydroxylation | 262.1802 | C16H23NO2 | 22.76 | 0.4 | O | O | O |
| M22 | Oxidation | 408.2282 | C24H29N3O3 | 24.04 | 1.2 | O | O | O |
| M23 | Desaturation | 390.2176 | C24H27N3O2 | 24.67 | 2.8 | O | O | O |
| M24 | Oxidation | 408.2282 | C24H29N3O3 | 25.90 | 0.7 | O | - | O |
| M25 | Desaturation + Oxidation | 406.2125 | C24H27N3O3 | 26.31 | 0.5 | O | - | O |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, M.; Lee, B.I.; Choi, J.; Park, Y.; Park, S.-J.; Lim, J.-H.; Lee, J.; Shin, Y.G. Quantitative Analysis of Daporinad (FK866) and Its In Vitro and In Vivo Metabolite Identification Using Liquid Chromatography-Quadrupole-Time-of-Flight Mass Spectrometry. Molecules 2022, 27, 2011. https://doi.org/10.3390/molecules27062011
Park M, Lee BI, Choi J, Park Y, Park S-J, Lim J-H, Lee J, Shin YG. Quantitative Analysis of Daporinad (FK866) and Its In Vitro and In Vivo Metabolite Identification Using Liquid Chromatography-Quadrupole-Time-of-Flight Mass Spectrometry. Molecules. 2022; 27(6):2011. https://doi.org/10.3390/molecules27062011
Chicago/Turabian StylePark, Minjae, Byeong Ill Lee, Jangmi Choi, Yuri Park, Seo-Jin Park, Jeong-Hyeon Lim, Jiyu Lee, and Young G. Shin. 2022. "Quantitative Analysis of Daporinad (FK866) and Its In Vitro and In Vivo Metabolite Identification Using Liquid Chromatography-Quadrupole-Time-of-Flight Mass Spectrometry" Molecules 27, no. 6: 2011. https://doi.org/10.3390/molecules27062011