
Stereoisomeric Tris-BINOL-Menthol Bulky Monophosphites: Synthesis, Characterisation and Application in Rhodium-Catalysed Hydroformylation

 , , , , ,
, , , , ,
Abstract
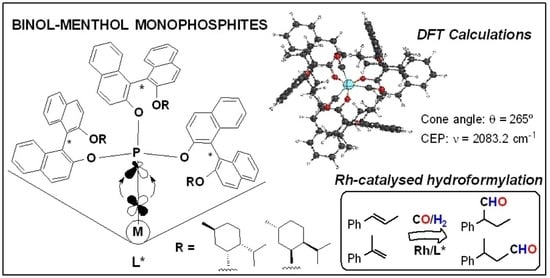
:
1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterisation of Monophosphites
2.2. NMR Studies on Complex Formation in Solution
2.3. Computational Studies: Determination of Electronic and Steric Parameters
2.4. Evaluation in Rh-Catalysed Hydroformylation
3. Materials and Methods
3.1. Reagents and Solvents
3.2. Instrumentation
3.3. Monophosphite Ligands Synthesis
3.3.1. Synthesis and Characterisation of BINOL Monoethers 1–4
(S)-2′-(((1R,2R,5S)-2-isopropyl-5-methylcyclohexyl)oxy)-[1.1′-binaphthalen]-2-ol (1)
(R)-2′-(((1S,2S,5R)-2-isopropyl-5-methylcyclohexyl)oxy)-[1.1′-binaphthalen]-2-ol (2)
(S)-2′-(((1S,2S,5R)-2-isopropyl-5-methylcyclohexyl)oxy)-[1.1′-binaphthalen]-2-ol (3)
(R)-2′-(((1R,2R,5S)- 2-isopropyl-5-methylcyclohexyl)oxy)-[1.1′-binaphthalen]-2-ol (4)
3.3.2. Synthesis and Characterisation of Monophosphites L1–L4
Tris-[(S)-2′-(((1R,2R,5S)-2-isopropyl-5-methylcyclohexyl)oxy)-[1,1′-binaphthalen]-2-yl]-phosphite (L1)
Tris-[(R)-2′-(((1S,2S,5R)-2-isopropyl-5-methylcyclohexyl)oxy)-[1,1′-binaphthalen]-2-yl]-phosphite (L2)
Tris-[(S)-2′-(((1S,2S,5R)-2-isopropyl-5-methylcyclohexyl)oxy)-[1,1′-binaphthalen]-2-yl]-phosphite (L3)
Tris-[(R)-2′-(((1R,2R,5S)-2-isopropyl-5-methylcyclohexyl)oxy)-[1,1′-binaphthalen]-2-yl]-phosphite (L4)
3.4. Rh(I)/Monophosphite Complex Formation in Solution
3.5. DFT Computational Studies
3.6. Catalytic Hydroformylation Procedure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Börner, A.; Franke, R. (Eds.) Hydroformylation. Fundamentals, Processes and Applications in Organic Synthesis; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2016; Volume 1–2, ISBN 9783527677931. [Google Scholar]
- Pereira, M.M.; Carrilho, R.M.B.; Calvete, M.J.F. Organophosphorus Chemistry; Higham, L.J., Allen, D.W., Tebby, J.C., Eds.; Royal Society of Chemistry: London, UK, 2021; Volume 50, pp. 115–149. ISBN 9781839162053. [Google Scholar]
- Kamer, P.C.J.; van Leeuwen, P.W.N.M. (Eds.) Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis; John Wiley & Sons, Ltd.: Chichester, UK, 2012; ISBN 9781118299715. [Google Scholar]
- Börner, A. (Ed.) Phosphorus Ligands in Asymmetric Catalysis; Wiley-VCH: Weinheim, Germany, 2008; ISBN 9783527317462. [Google Scholar]
- Kloß, S.; Selent, D.; Spannenberg, A.; Franke, R.; Börner, A.; Sharif, M. Effects of substitution pattern in phosphite ligands used in rhodium-catalyzed hydroformylation on reactivity and hydrolysis stability. Catalysts 2019, 9, 1036. [Google Scholar] [CrossRef] [Green Version]
- Linnebank, P.R.; Ferreira, S.F.; Kluwer, A.M.; Reek, J.N.H. Regioselective Hydroformylation of Internal and Terminal Alkenes via Remote Supramolecular Control. Chem. Eur. J. 2020, 26, 8214–8219. [Google Scholar] [CrossRef]
- Alsalahi, W.; Trzeciak, A.M. Rhodium-catalyzed hydroformylation under green conditions: Aqueous/organic biphasic, “on water”, solventless and Rh nanoparticle based systems. Coord. Chem. Rev. 2021, 430, 213732. [Google Scholar] [CrossRef]
- Pagar, N.S.; Rajurkar, K.B.; Deshpande, R.M. Kinetics of hydroformylation of camphene using rhodium-phosphite catalyst. Int. J. Chem. Kinet. 2020, 52, 485–495. [Google Scholar] [CrossRef]
- Tang, Y.; Dong, K.; Wang, S.; Sun, Q.; Meng, X.; Xiao, F.S. Boosting the hydrolytic stability of phosphite ligand in hydroformylation by the construction of superhydrophobic porous framework. Mol. Catal. 2019, 474, 110408. [Google Scholar] [CrossRef]
- Hastings, S.D.; Cagle, E.C.; Totsch, T.R.; Tyus, S.D.; Gray, G.M. Comparative Study of Novel Phosphordiamidite and Phosphite Ligands Used in Alkene Hydroformylation; Synthesis, Characterization, Metalation, and Catalytic Evaluation. Eur. J. Inorg. Chem. 2018, 4158–4174. [Google Scholar] [CrossRef]
- Tian, M.; Pang, Z.B.; Li, H.F.; Wang, L.L. Novel MOP-type H8-binaphthyl monodentate phosphite ligands and their applications in transition metal-catalyzed asymmetric 1,4-conjugate additions and hydroformylations. Tetrahedron Asymmetry 2017, 28, 330–337. [Google Scholar] [CrossRef]
- Martin, J.R.; Cagle, E.C.; Lucius, A.L.; Gray, G.M. Correlating the Activity of Rhodium(I)-Phosphite-Lariat Ether Styrene Hydroformylation Catalysts with Alkali Metal Cation Binding through NMR Spectroscopic Titration Methods. Organometallics 2016, 35, 2609–2620. [Google Scholar] [CrossRef]
- Rodrigues, F.M.S.; Carrilho, R.M.B.; Pereira, M.M. Reusable Catalysts for Hydroformylation-Based Reactions. Eur. J. Inorg. Chem. 2021, 2294–2324. [Google Scholar] [CrossRef]
- Van Rooy, A.; Orij, E.N.; Kamer, P.C.J.; van Leeuwen, P.W.N.M. Hydroformylation with a Rhodium/Bulky Phosphite Modified Catalyst. Catalyst Comparison for Oct-1-ene, Cyclohexene, and Styrene. Organometallics 1995, 14, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Muilwijk, K.F.; Kamer, P.C.J.; Van Leeuwen, P.W.N.M. A bulky phosphite-modified rhodium catalyst for the hydroformylation of unsaturated fatty acid esters. J. Am. Oil Chem. Soc. 1997, 74, 223–228. [Google Scholar] [CrossRef]
- Delolo, F.G.; Oliveira, K.C.B.; dos Santos, E.N.; Gusevskaya, E.V. Hydroformylation of biomass-based hydroxyolefins in eco-friendly solvents: New fragrances from myrtenol and nopol. Mol. Catal. 2019, 462, 1–9. [Google Scholar] [CrossRef]
- Van Leeuwen, P.W.N.M.; Claver, C. (Eds.) Rhodium Catalyzed Hydroformylation; Springer: Dordrecht, Germany, 2002; ISBN 9780792365518. [Google Scholar]
- Zhang, B.; Peña Fuentes, D.; Börner, A. Hydroformylation. ChemTexts 2022, 8, 2. [Google Scholar] [CrossRef]
- Diéguez, M. (Ed.) Chiral Ligands. Evolution of Ligand Libraries for Asymmetric Catalysis; CRC Press: Boca Raton, FL, USA, 2021; ISBN 9780367855734. [Google Scholar]
- Margalef, J.; Biosca, M.; de la Cruz Sánchez, P.; Faiges, J.; Pàmies, O.; Diéguez, M. Evolution in heterodonor P-N, P-S and P-O chiral ligands for preparing efficient catalysts for asymmetric catalysis. From design to applications. Coord. Chem. Rev. 2021, 446, 214120. [Google Scholar] [CrossRef]
- Zuccarello, G.; Escofet, I.; Caniparoli, U.; Echavarren, A.M. New-Generation Ligand Design for the Gold-Catalyzed Asymmetric Activation of Alkynes. ChemPlusChem 2021, 86, 1283–1296. [Google Scholar] [CrossRef]
- van Leeuwen, P.W.N.M.; Kamer, P.C.J.; Claver, C.; Pàmies, O.; Diéguez, M. Phosphite-Containing Ligands for Asymmetric Catalysis. Chem. Rev. 2011, 111, 2077–2118. [Google Scholar] [CrossRef]
- Pereira, M.M.; Calvete, M.J.F.; Carrilho, R.M.B.; Abreu, A.R. Synthesis of binaphthyl based phosphine and phosphite ligands. Chem. Soc. Rev. 2013, 42, 6990–7027. [Google Scholar] [CrossRef]
- Reetz, M.T.; Mehler, G. Highly enantioselective Rh-catalyzed hydrogenation reactions based on chiral monophosphite ligands. Angew. Chem. Int. Ed. 2000, 39, 3889–3890. [Google Scholar] [CrossRef]
- Reetz, M.T.; Mehler, G.; Meiswinkel, A.; Sell, T. Enantioselective hydrogenation of enamides catalyzed by chiral rhodium–monodentate phosphite complexes. Tetrahedron Lett. 2002, 43, 7941–7943. [Google Scholar] [CrossRef]
- Jerphagnon, T.; Renaud, J.L.; Bruneau, C. Chiral monodentate phosphorus ligands for rhodium-catalyzed asymmetric hydrogenation. Tetrahedron Asymmetry 2004, 15, 2101–2111. [Google Scholar] [CrossRef]
- Reetz, M.T.; Guo, H.; Jun-An, M.; Goddard, R.; Mynott, R.J. Helical triskelion monophosphites as ligands in asymmetric catalysis. J. Am. Chem. Soc. 2009, 131, 4136–4142. [Google Scholar] [CrossRef]
- Pang, Z.; Tian, M.; Li, H.; Wang, L. Asymmetric Allylic Alkylation and Hydrogenation with Transition Metal Complexes of Diphosphite Ligands Based on (1S,2S)-Trans-1,2-cyclohexanediol. Catal. Lett. 2017, 147, 893–899. [Google Scholar] [CrossRef]
- Park, H.; Kumareswaran, R.; RajanBabu, T.V. Tunable phosphinite, phosphite and phosphoramidite ligands for the asymmetric hydrovinylation reactions. Tetrahedron 2005, 61, 6352–6367. [Google Scholar] [CrossRef]
- Carrilho, R.M.B.; Costa, G.N.; Neves, Â.C.B.; Pereira, M.M.; Grabulosa, A.; Bayón, J.C.; Rocamora, M.; Muller, G. Asymmetric Hydrovinylation and Hydrogenation with Metal Complexes of C3-Symmetric Tris-Binaphthyl Monophosphites. Eur. J. Inorg. Chem. 2014, 1034–1041. [Google Scholar] [CrossRef]
- Borràs, C.; Elías-Rodríguez, P.; Carmona, A.T.; Robina, I.; Pàmies, O.; Diéguez, M. Amino-P Ligands from Iminosugars: New Readily Available and Modular Ligands for Enantioselective Pd-Catalyzed Allylic Substitutions. Organometallics 2018, 37, 1682–1694. [Google Scholar] [CrossRef]
- Clavero, P.; Grabulosa, A.; Rocamora, M.; Muller, G.; Font-Bardia, M. Diphosphorus Ligands Containing a P-Stereogenic Phosphane and a Chiral Phosphite or Phosphorodiamidite—Evaluation in Pd-Catalysed Asymmetric Allylic Substitution Reactions. Eur. J. Inorg. Chem. 2016, 2016, 4054–4065. [Google Scholar] [CrossRef]
- Gavrilov, K.N.; Lyubimov, S.E.; Zheglov, S.V.; Benetsky, E.B.; Davankov, V.A. Enantioselective Pd-catalysed allylation with BINOL-derived monodentate phosphite and phosphoramidite ligands. J. Mol. Catal. A Chem. 2005, 231, 255–260. [Google Scholar] [CrossRef]
- Fuji, K.; Kinoshita, N.; Tanaka, K.; Kawabata, T. Enantioselective allylic substitution catalyzed by an iridium complex: Remarkable effects of the counter cation. Chem. Commun. 1999, 2289–2290. [Google Scholar] [CrossRef]
- Carrilho, R.M.B.; Abreu, A.R.; Petöcz, G.; Bayón, J.C.; Moreno, M.J.S.M.; Kollár, L.; Pereira, M.M. New Binaphthyl-based C3-symmetric Chiral Hemilabile Monophosphite Ligands: Synthesis and Characterization of Their Platinum Complexes. Chem. Lett. 2009, 38, 844–845. [Google Scholar] [CrossRef]
- Carrilho, R.M.B.; Neves, A.C.B.; Lourenço, M.A.O.; Abreu, A.R.; Rosado, M.T.S.; Abreu, P.E.; Eusébio, M.E.S.; Kollár, L.; Bayón, J.C.; Pereira, M.M. Rhodium/tris-binaphthyl chiral monophosphite complexes: Efficient catalysts for the hydroformylation of disubstituted aryl olefins. J. Organomet. Chem. 2012, 698, 28–34. [Google Scholar] [CrossRef]
- Costa, G.N.; Carrilho, R.M.B.; Dias, L.D.; Viana, J.C.; Aquino, G.L.B.; Pineiro, M.; Pereira, M.M. Highly efficient Rh(I)/tris-binaphthyl monophosphite catalysts for hydroformylation of sterically hindered alkyl olefins. J. Mol. Catal. A Chem. 2016, 416, 73–80. [Google Scholar] [CrossRef]
- Mitsunobu, O. The Use of Diethyl Azodicarboxylate and Triphenylphosphine in Synthesis and Transformation of Natural Products. Synthesis 1981, 1981, 1–28. [Google Scholar] [CrossRef]
- Takahashi, M.; Ogasawara, K. An expedient route to some monoalkyl ethers of enantiomerically pure bi-β-naphthol. Tetrahedron Asymmetry 1997, 8, 3125–3130. [Google Scholar] [CrossRef]
- Carrilho, R.M.B.; Pereira, M.M.; Maria, T.M.R.; Eusébio, M.E.S.; Rodrigues, V.H. Crystal structure of (R)-2’-benzyloxy-[1,1’-binaphthalen]-2-yltrifluoromethanesulfonate. Acta Cryst. 2014, 70, o1096–o1097. [Google Scholar] [CrossRef] [Green Version]
- Corvis, Y.; Négrier, P.; Massip, S.; Leger, J.M.; Espeau, P. Insights into the crystal structure, polymorphism and thermal behavior of menthol optical isomers and racemates. CrystEngComm 2012, 14, 7055–7064. [Google Scholar] [CrossRef] [Green Version]
- Spek, A.L. PLATON. A Multipurpose Crystallographic Tool; Utrecht University: Utrecht, The Netherlands, 2002. [Google Scholar]
- Persistence of Vision Pty. Ltd. Persistence of Vision (TM) Raytracer; Persistence of Vision Pty. Ltd.: Williamstown, VIC, Australia, 2004; Available online: http://www.povray.org/ (accessed on 25 February 2022).
- Tolman, C.A. Steric effects of phosphorus ligands in organometallic chemistry and homogeneous catalysis. Chem. Rev. 1977, 77, 313–348. [Google Scholar] [CrossRef]
- Kégl, T.; Pálinkás, N.; Kollár, L.; Kégl, T. Computational Characterization of Bidentate P-Donor Ligands: Direct Comparison to Tolman’s Electronic Parameters. Molecules 2018, 23, 3176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabortty, S.; Almasalma, A.A.; de Vries, J.G. Recent developments in asymmetric hydroformylation. Catal. Sci. Technol. 2021, 11, 5388–5411. [Google Scholar] [CrossRef]
- Diéguez, M.; Pàmies, O.; Ruiz, A.; Claver, C. Asymmetric hydroformylation of styrene catalyzed by carbohydrate diphosphite-Rh(I) complexes. New J. Chem. 2002, 26, 827–833. [Google Scholar] [CrossRef]
- Axtell, A.T.; Klosin, J.; Whiteker, G.T.; Cobley, C.J.; Fox, M.E.; Jackson, M.; Abboud, K.A. Bridging Group Effects in Chelating Bis(2,5-diphenylphospholane) Ligands for Rhodium-Catalyzed Asymmetric Hydroformylation. Organometallics 2009, 28, 2993–2999. [Google Scholar] [CrossRef]
- Nozaki, K.; Sakai, N.; Nanno, T.; Higashijima, T.; Mano, S.; Horiuchi, T.; Takaya, H. Highly Enantioselective Hydroformylation of Olefins Catalyzed by Rhodium(I) Complexes of New Chiral Phosphine-Phosphite Ligands. J. Am. Chem. Soc. 1997, 119, 4413–4423. [Google Scholar] [CrossRef]
- Cobley, C.J.; Klosin, J.; Qin, C.; Whiteker, G.T. Parallel Ligand Screening on Olefin Mixtures in Asymmetric Hydroformylation Reactions. Org. Lett. 2004, 6, 3277–3280. [Google Scholar] [CrossRef] [PubMed]
- Keulemans, A.I.M.; Kwantes, A.; van Bavel, T. The structure of the formylation (OXO) products obtained from olefines and watergas. Recl. Trav. Chim. Pays-Bas 1948, 67, 298–308. [Google Scholar] [CrossRef]
- Pereira, M.M.; Burrows, H.D. (Eds.) Síntese e Estrutura; Escolar Editora: Lisboa, Portugal, 2005. [Google Scholar]
- APEX2, V2014.9; Bruker AXS Inc.: Madison, WI, USA, 2014.
- SAINT, 8.34A; Bruker AXS Inc.: Madison, WI, USA, 2013.
- SADABS, V2014.4; Bruker AXS Inc.: Madison, WI, USA, 2014.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Rackers, J.A.; Wang, Z.; Lu, C.; Laury, M.L.; Lagardère, L.; Schnieders, M.J.; Piquemal, J.P.; Ren, P.; Ponder, J.W. Tinker 8: Software Tools for Molecular Design. J. Chem. Theory Comput. 2018, 14, 5273–5289. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | Ligand (L) | P (bar) | T (°C) | Conversion (%) | Regio. (iso/n ratio) |
| 1 | - | 10 | 80 | 96 | 50/50 |
| 2 | L3-(S)-BINOL-(−)-menthol | 10 | 80 | 99 | 61/39 |
| 3 | L4-(R)-BINOL-(+)-menthol | 10 | 80 | 99 | 59/41 |
| 4 | L4-(R)-BINOL-(+)-menthol | 20 | 80 | 99 | 65/35 |
| 5 | L4-(R)-BINOL-(+)-menthol | 25 | 80 | 99 | 64/36 |
| 6 | L4-(R)-BINOL-(+)-menthol | 20 | 50 | 92 | 94/6 |
| 7 | L4-(R)-BINOL-(+)-menthol | 25 | 50 | 95 | 96/4 |
| 8 | L1-(S)-BINOL-(+)-menthol | 25 | 50 | 93 | 96/4 |
| 9 | L2-(R)-BINOL-(−)-menthol | 25 | 50 | 94 | 96/4 |
| Entry | Substrate | Time (h) | Conversion (%) | Major Product (Regioselectivity, %) |
|---|---|---|---|---|
| 1 |  | 4 | 73 |  80 |
| 2 |  | 18 | 75 |  99 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Felgueiras, A.P.; Rodrigues, F.M.S.; Carrilho, R.M.B.; Cruz, P.F.; Rodrigues, V.H.; Kégl, T.; Kollár, L.; Pereira, M.M. Stereoisomeric Tris-BINOL-Menthol Bulky Monophosphites: Synthesis, Characterisation and Application in Rhodium-Catalysed Hydroformylation. Molecules 2022, 27, 1989. https://doi.org/10.3390/molecules27061989
Felgueiras AP, Rodrigues FMS, Carrilho RMB, Cruz PF, Rodrigues VH, Kégl T, Kollár L, Pereira MM. Stereoisomeric Tris-BINOL-Menthol Bulky Monophosphites: Synthesis, Characterisation and Application in Rhodium-Catalysed Hydroformylation. Molecules. 2022; 27(6):1989. https://doi.org/10.3390/molecules27061989
Chicago/Turabian StyleFelgueiras, Alexandre P., Fábio M. S. Rodrigues, Rui M. B. Carrilho, Pedro F. Cruz, Vitor H. Rodrigues, Tamás Kégl, László Kollár, and Mariette M. Pereira. 2022. "Stereoisomeric Tris-BINOL-Menthol Bulky Monophosphites: Synthesis, Characterisation and Application in Rhodium-Catalysed Hydroformylation" Molecules 27, no. 6: 1989. https://doi.org/10.3390/molecules27061989