
Determination of Methyl Methanesulfonate and Ethyl Methylsulfonate in New Drug for the Treatment of Fatty Liver Using Derivatization Followed by High-Performance Liquid Chromatography with Ultraviolet Detection

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
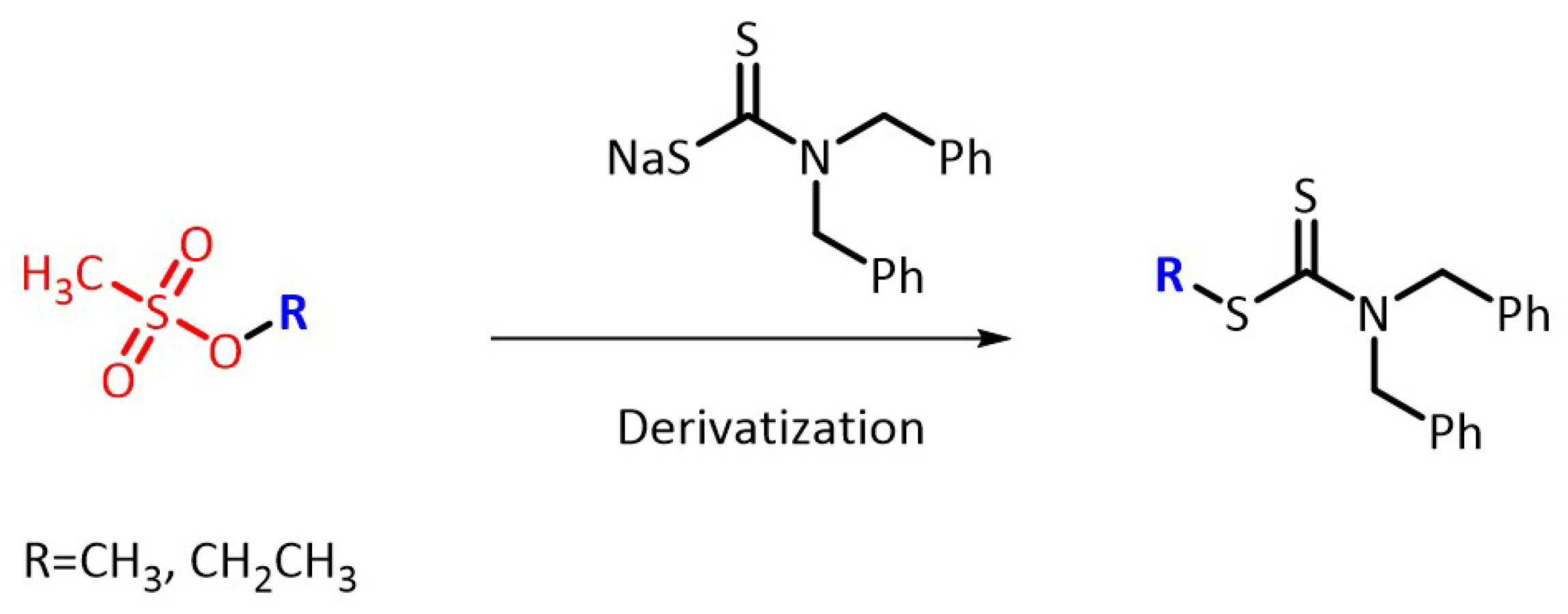
2.1. Optimization of Derivatization Reactions
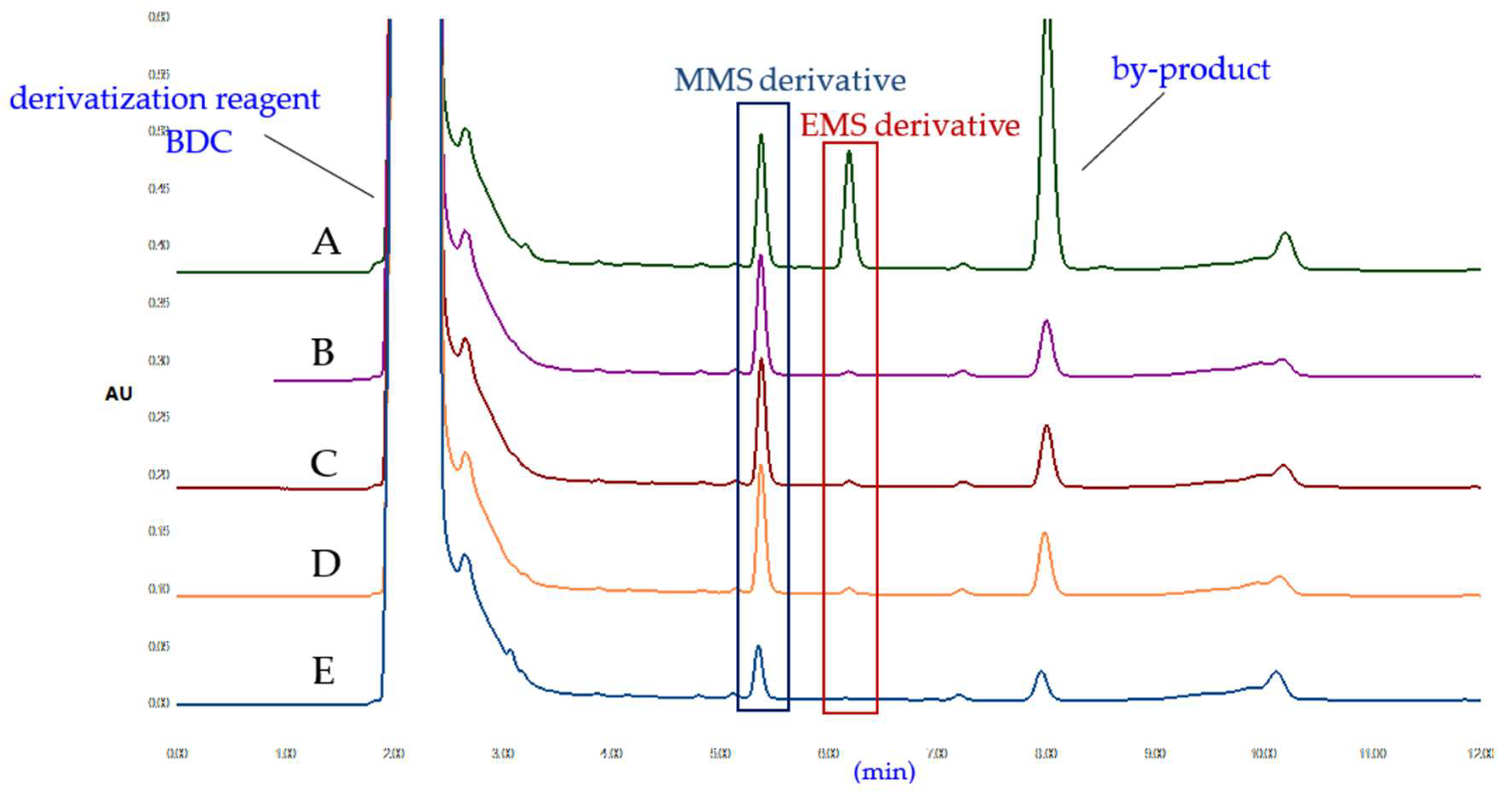
2.1.1. Selection of Derivatization Reagent
2.1.2. Selection of Derivatization Solvent
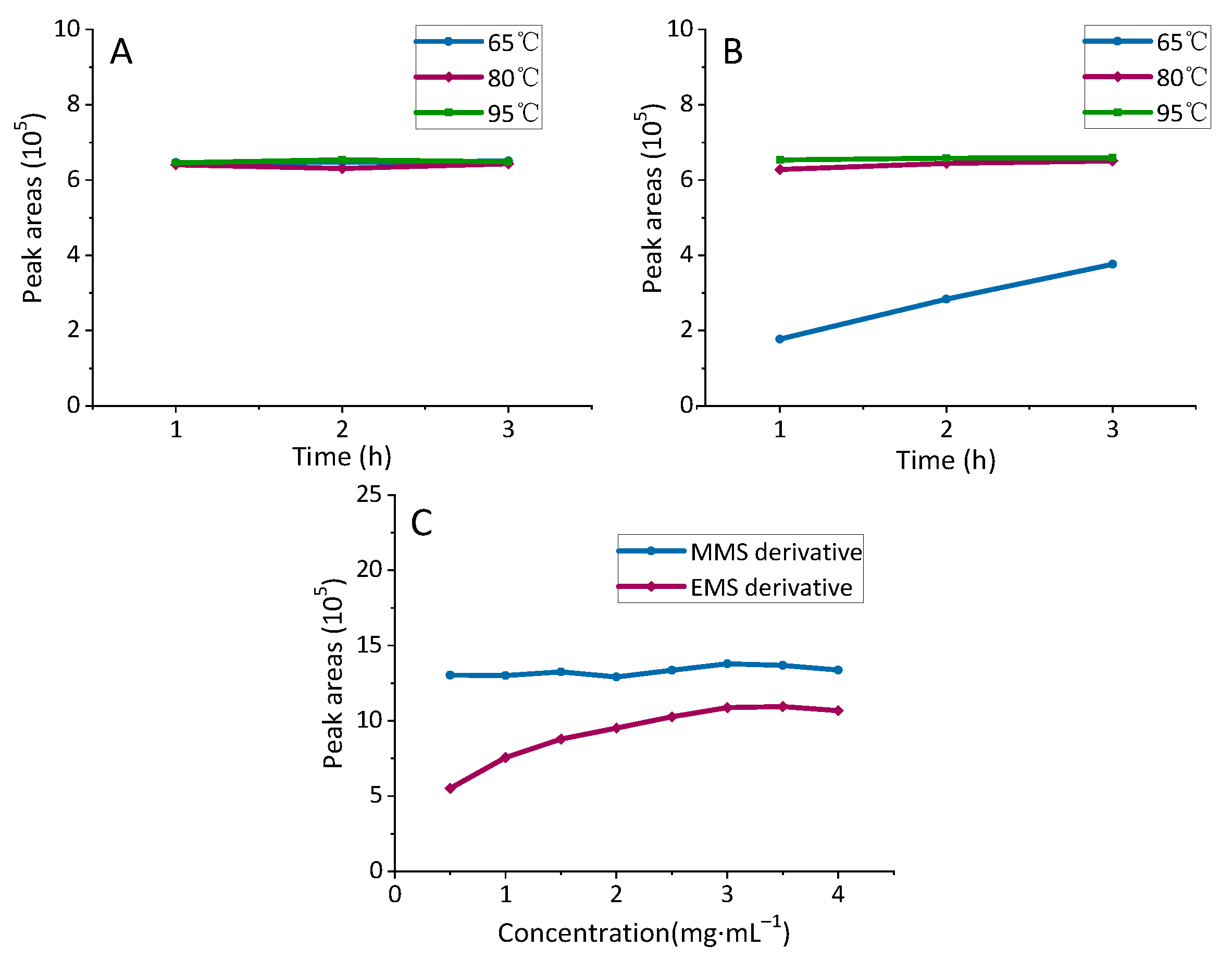
2.1.3. Optimization of Derivatization Reaction Conditions
2.1.4. Optimization of Other Derivatization Parameters
2.2. Method Validation and Application
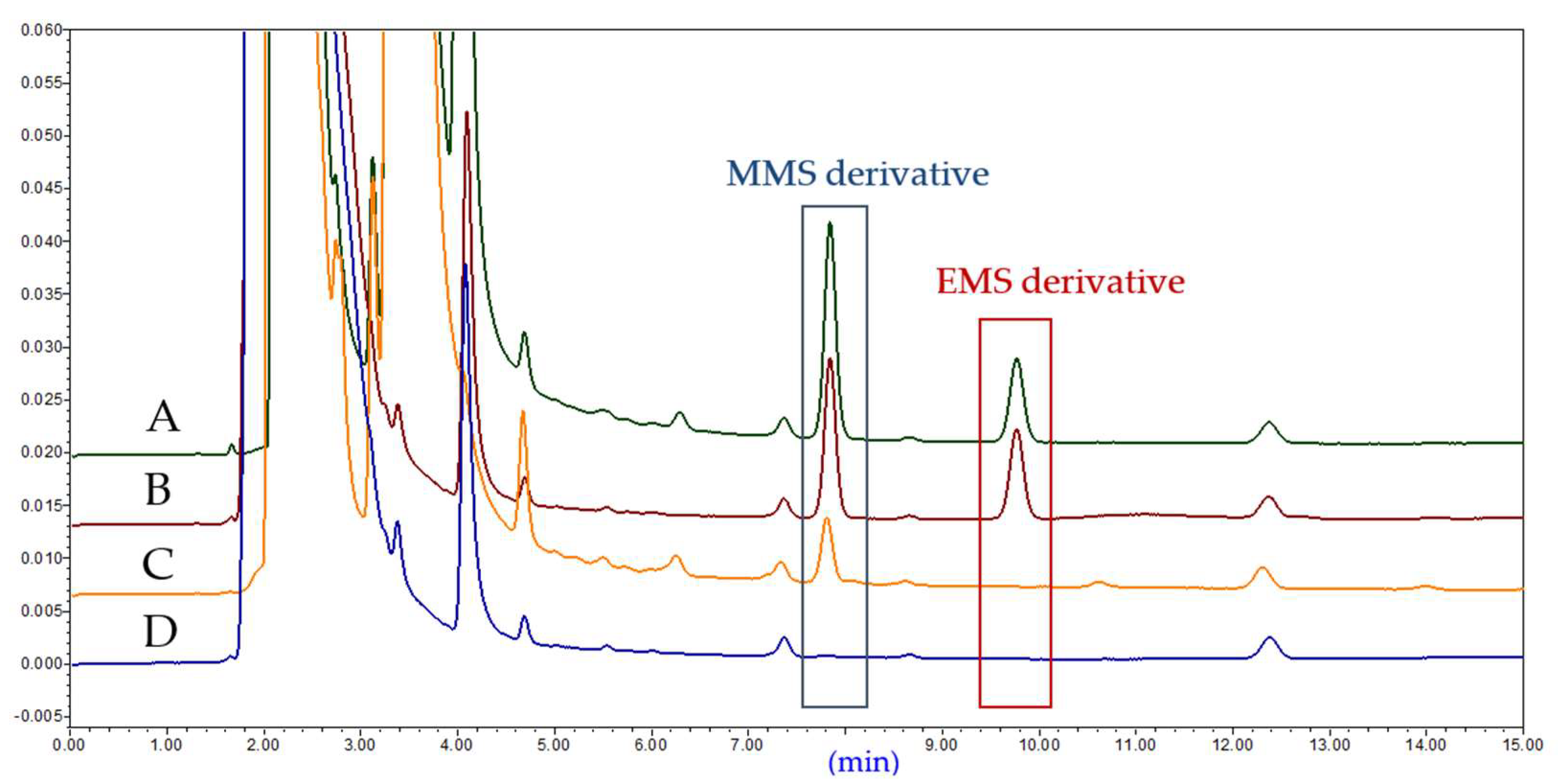
2.2.1. HPLC Development
2.2.2. Method Validation
2.2.3. Sample Analysis
3. Materials and Methods
3.1. Materials, Chemicals, and Reagents
3.2. Instrumentation and Chromatographic Conditions
3.3. Sample Preparation of Derivatization HPLC–UV Method
3.3.1. Standard and Test Solutions
3.3.2. Derivatization Procedure
3.3.3. Method Validation
3.4. Sample Preparations of GC–MS Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Reddy, A.V.B.; Jaafar, J.; Umar, K.; Majid, Z.A.; Aris, A.B.; Talib, J.; Madhavi, G. Identification, control strategies, and analytical approaches for the determination of potential genotoxic impurities in pharmaceuticals: A comprehensive review. J. Sep. Sci. 2015, 38, 764–779. [Google Scholar] [CrossRef] [PubMed]
- Teasdale, A.; Elder, D.P. Analytical control strategies for mutagenic impurities: Current challenges and future opportunities? Trend Anal. Chem. 2018, 101, 66–84. [Google Scholar] [CrossRef]
- Benigni, R.; Bossa, C. Mechanisms of chemical carcinogenicity and mutagenicity: A review with implications for predictive toxicology. Chem. Rev. 2011, 111, 2507–2536. [Google Scholar] [CrossRef] [PubMed]
- Müller, L.; Gocke, E.; Lavé, T.; Pfister, T. Ethyl methanesulfonate toxicity in Viracept—A comprehensive human risk assessment based on threshold data for genotoxicity. Toxicol. Lett. 2009, 190, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Gocke, E.; Bürgin, H.; Müller, L.; Pfister, T. Literature review on the genotoxicity, reproductive toxicity, and carcinogenicity of ethyl methanesulfonate. Toxicol. Lett. 2009, 190, 254–265. [Google Scholar] [CrossRef]
- Eder, E.; Kütt, W.; Deininger, C. On the role of alkylating mechanisms, O-alkylation and DNA-repair in genotoxicity and mutagenicity of alkylating methanesulfonates of widely varying structures in bacterial systems. Chem.-Biol. Interact. 2001, 137, 89–99. [Google Scholar] [CrossRef]
- CHMP Assessment Report for Viracept; EMEA/CHMP/492059/2007; European Medicines Agency: London, UK, 20 September 2007.
- Guidelines on the Limits of Genotoxic Impurities; Committee for Medicinal Products for Human Use (CHMP); European Medicines Agency Evaluation of Medicines for Human Use (EMA): London, UK, 2006.
- Guidance for Industry: Genotoxic and Carcinogenic Impurities in Drug Substances and Products: Recommended Approaches (Draft); U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER); U.S. Food and Drug Administration: Silver Spring, MD, USA, 2008.
- M7(R1): Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk; The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH): Geneva, Switzerland, 2008.
- Eldera, D.P.; Snodin, D.J. Drug substances presented as sulfonic acid salts: Overview of utility, safety and regulation. J. Pharm. Pharmacol. 2009, 61, 269–278. [Google Scholar] [CrossRef]
- Amberg, A.; Harvey, J.S.; Czich, A.; Spirkl, H.; Robinson, S.; White, A.; Elder, D.P. Do Carboxylic/Sulfonic Acid Halides Really Present a Mutagenic and Carcinogenic Risk as Impurities in Final Drug Products? Org. Process Res. Dev. 2015, 19, 1495–1506. [Google Scholar] [CrossRef]
- Ramakrishna, K.; Raman, N.V.V.S.S.; Narayana Rao, K.M.V.; Prasad, A.V.S.S.; Subhaschander Reddy, K. Development and validation of GC-MS method for the determination of methyl methanesulfonate and ethyl methanesulfonate in imatinib mesylate. J. Pharm. Biomed. Anal. 2008, 46, 780–783. [Google Scholar] [CrossRef]
- Zhang, C.; Huang, L.; Wu, Z.; Chang, C.; Yang, Z. Determination of sulfonate ester genotoxic impurities in imatinib mesylate by gas chromatography with mass spectrometry. J. Sep. Sci. 2016, 39, 3558–3563. [Google Scholar] [CrossRef]
- Liu, Z.; Fan, H.; Zhou, Y.; Qian, X.; Tu, J.; Chen, B.; Duan, G. Development and validation of a sensitive method for alkyl sulfonate genotoxic impurities determination in drug substances using gas chromatography coupled to triple quadrupole mass spectrometry. J. Pharm. Biomed. Anal. 2019, 168, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.; Guo, K.; Zhang, T.; Li, T.; Ma, C. Simultaneous Determination of 15 Sulfonate Ester Impurities in Phentolamine Mesylate, Amlodipine Besylate, and Tosufloxacin Tosylate by LC-APCI-MS/MS. J. Anal. Methods. Chem. 2019, 2019, 4059765. [Google Scholar] [CrossRef] [PubMed]
- Kakadiy, P.R.; Pratapa Reddy, B.; Singh, V.; Ganguly, S.; Chandrashekhar, T.G.; Singh, D.K. Low level determinations of methyl methanesulfonate and ethyl methanesulfonate impurities in Lopinavir and Ritonavir Active pharmaceutical ingredients by LC/MS/MS using electrospray ionization. J. Pharm. Biomed. Anal. 2011, 55, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Shi, Y.; Zheng, L.; Feng, F.; Zheng, F.; Liu, W. Rapid and simultaneous determination of sulfonate ester genotoxic impurities in drug substance by liquid chromatography coupled to tandem mass spectrometry: Comparison of different ionization modes. J. Chromatogr. A 2014, 1355, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Methyl, Ethyl and Isopropyl Methanesulfonate in Active Substances; European Directorate for Quality and Medicines & Healthcare (EDQM); European Pharmacopoeia 10.0; Council of Europe: Strasbourg, France, 2019.
- Lee, C.R.; Guivarch, F.; Dau, C.N.V.; Tessier, D.; Krstulovic, A.M. Determination of polar alkylating agents as thiocyanate/isothiocyanate derivatives by reaction headspace gas chromatography. Analyst 2003, 128, 857–863. [Google Scholar] [CrossRef]
- Alzaga, R.; Ryan, R.W.; Taylor-Worth, K.; Lipczynski, A.M.; Szucs, R.; Sandra, P. A generic approach for the determination of residues of alkylating agents in active pharmaceutical ingredients by in situ derivatization-headspace-gas chromatography-mass spectrometry. J. Pharm. Biomed. Anal. 2007, 45, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Xu, J.; Zheng, X.; Liu, W.; Zheng, F. Determination of methyl methanesulfonate and ethyl methanesulfonate in methanesulfonic acid by derivatization followed by high-performance liquid chromatography with ultraviolet detection. J. Sep. Sci. 2017, 40, 3414–3421. [Google Scholar] [CrossRef]
- Li, M.; Gu, C.; Luo, L.; Zhou, J.; Liu, J.; Zheng, F. Determination of trace methanesulfonates in drug matrix using derivatization and headspace single drop microextraction followed by high-performance liquid chromatography with ultraviolet detection. J. Chromatogr. A 2019, 1591, 131–137. [Google Scholar] [CrossRef]
- Wollein, U.; Schramek, N. Simultaneous determination of alkyl mesylates and alkyl besylates in finished drug products by direct injection GC/MS. Eur. J. Pharm. Sci. 2012, 45, 201–204. [Google Scholar] [CrossRef]
- García, A.; Rupérez, F.J.; Ceppa, F.; Pellatib, F.; Barbas, C. Development of chromatographic methods for the determination of genotoxic impurities in cloperastine fendizoate. J. Pharm. Biomed. Anal. 2012, 61, 230–236. [Google Scholar] [CrossRef]
- Yang, X.; Xiong, X.; Cao, J.; Luan, B.; Liu, Y.; Liu, G.; Zhang, L. Matrix precipitation: A general strategy to eliminate matrix interference for pharmaceutical toxic impurities analysis. J. Chromatogr. A 2015, 1379, 16–23. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Derivatization Reagent | Sample | Feature | LOQ of Derivatives | Ref. | |
|---|---|---|---|---|---|
| MMS | EMS | ||||
| DDTC a | Methanesulfonic acid | Simple, reliable, but not applicable to APIs due to matrix interference. | 0.6 ppm | 0.6 ppm | [22] |
| Imatinib mesylate Levofloxacin mesylate | Elimination of drug matrix interference, but it is a complicated operation, and requires a special device. | 40 ppm | 40 ppm | [23] | |
| IMH d | Simple and avoids drug matrix interference. | 1.2 ppm | 2.4 ppm | / | |
| TPO b | IMH | Derivatization reagent interference. | 2.4 ppm | * | / |
| BDC c | IMH | Simple, high sensitivity, and avoids drug matrix interference. | 0.3 ppm | 0.6 ppm | / |
| Parameter | MMS Derivatives | EMS Derivatives |
|---|---|---|
| Linear equation | y = 133,809x + 5669.9 | y = 105,889x − 526.49 |
| R | 0.9998 | 0.9998 |
| Linearity range (μg·mL−1) | 0.03–3.00 | 0.03–3.00 |
| LOQ (ppm) | 0.3 | 0.6 |
| Precision% (n = 6) | 3.23 | 1.66 |
| Intermediate precision% (n = 12) | 3.50 | 2.39 |
| Stability% (24 h) | 2.55 | 2.40 |
| Accuracy at LOQ (n = 3) | ||
| recovery% | 100.95 | 100.17 |
| RSD% | 4.53 | 1.79 |
| Accuracy at 80% level (n = 3) | ||
| recovery% | 100.36 | 99.15 |
| RSD% | 1.88 | 2.77 |
| Accuracy at 100% level (n = 3) | ||
| recovery% | 99.2 | 99.86 |
| RSD% | 1.83 | 1.54 |
| Accuracy at 120% level (n = 3) | ||
| recovery% | 100.03 | 99.73 |
| RSD% | 4.92 | 3.66 |
| Batch No. | PGIs | Derivatization HPLC–UV Method | Derivatization GC–MS Method [19] |
|---|---|---|---|
| ppm | ppm | ||
| 20180608 | MMS | 4.56 | 5.16 |
| EMS | / | / | |
| 20180918 | MMS | 4.84 | 5.30 |
| EMS | / | / | |
| 20181026 | MMS | 5.74 | 5.84 |
| EMS | / | / |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Feng, J.; Wu, S.; Shao, H.; Zhang, W.; Zhang, K.; Zhang, H.; Yang, Q. Determination of Methyl Methanesulfonate and Ethyl Methylsulfonate in New Drug for the Treatment of Fatty Liver Using Derivatization Followed by High-Performance Liquid Chromatography with Ultraviolet Detection. Molecules 2022, 27, 1950. https://doi.org/10.3390/molecules27061950
Wang Y, Feng J, Wu S, Shao H, Zhang W, Zhang K, Zhang H, Yang Q. Determination of Methyl Methanesulfonate and Ethyl Methylsulfonate in New Drug for the Treatment of Fatty Liver Using Derivatization Followed by High-Performance Liquid Chromatography with Ultraviolet Detection. Molecules. 2022; 27(6):1950. https://doi.org/10.3390/molecules27061950
Chicago/Turabian StyleWang, Yue, Jing Feng, Song Wu, Huihui Shao, Wenxuan Zhang, Kun Zhang, Hanyilan Zhang, and Qingyun Yang. 2022. "Determination of Methyl Methanesulfonate and Ethyl Methylsulfonate in New Drug for the Treatment of Fatty Liver Using Derivatization Followed by High-Performance Liquid Chromatography with Ultraviolet Detection" Molecules 27, no. 6: 1950. https://doi.org/10.3390/molecules27061950