
Thiazolidin-4-Ones as Potential Antimicrobial Agents: Experimental and In Silico Evaluation

 , , , , , , ,
, , , , , , ,
Abstract
:
1. Introduction
2. Results and Discussion
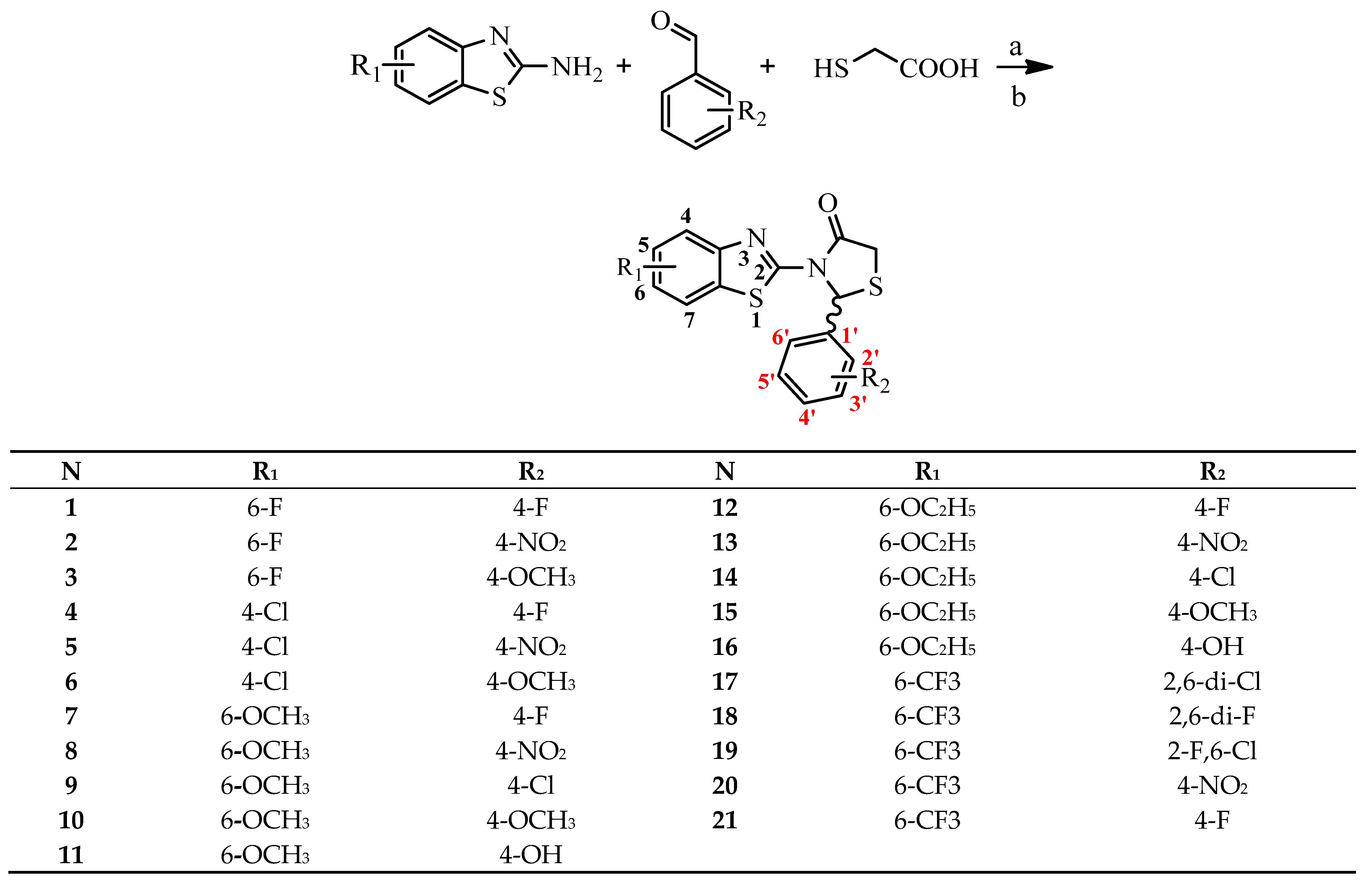
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. Antibacterial Action
2.2.2. Indifferent Effect in Combination with Streptomycin
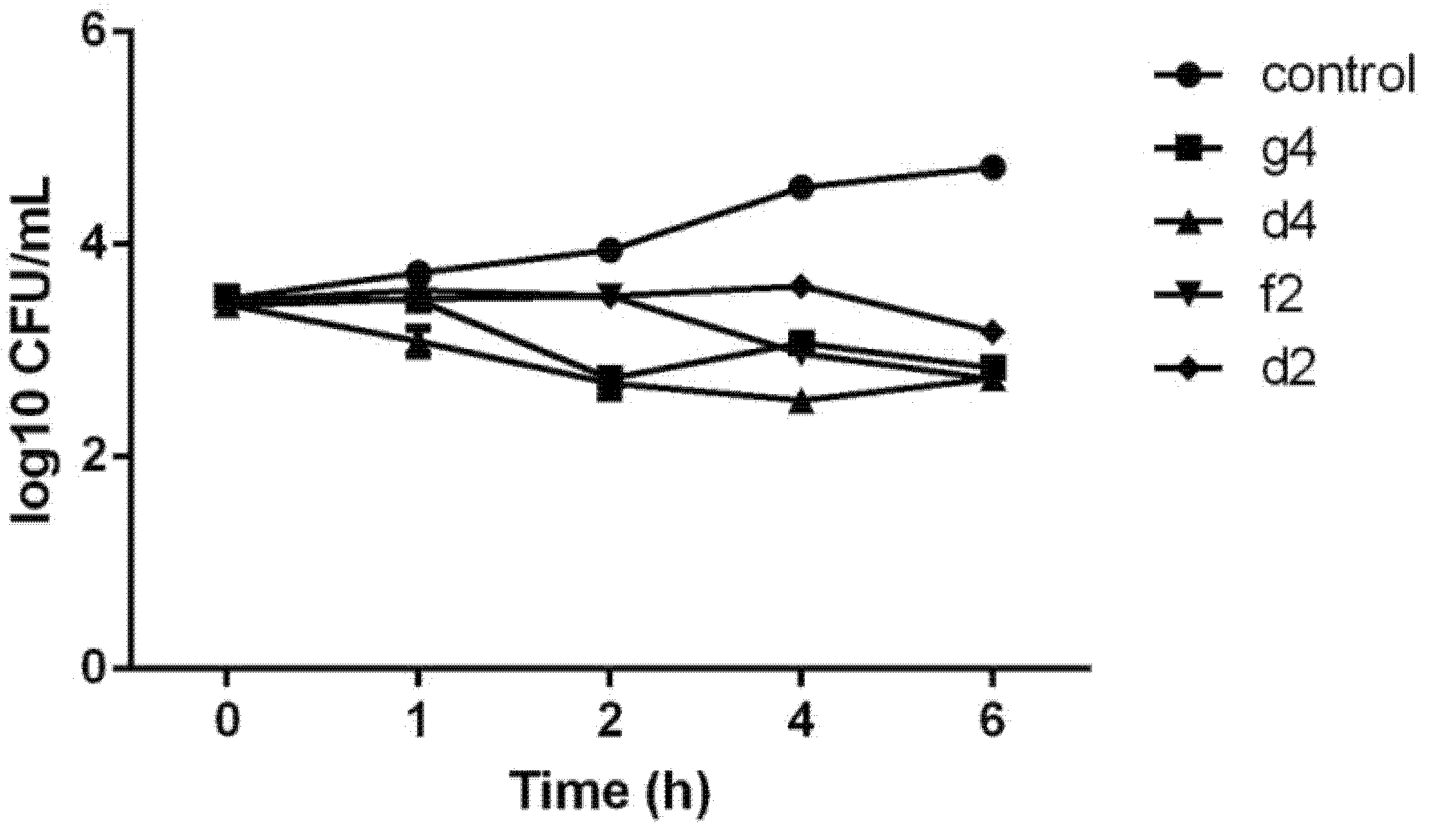
2.2.3. Efficient P. aeruginosa Bactericidal Effect
2.2.4. Antifungal Action
2.3. Docking Studies
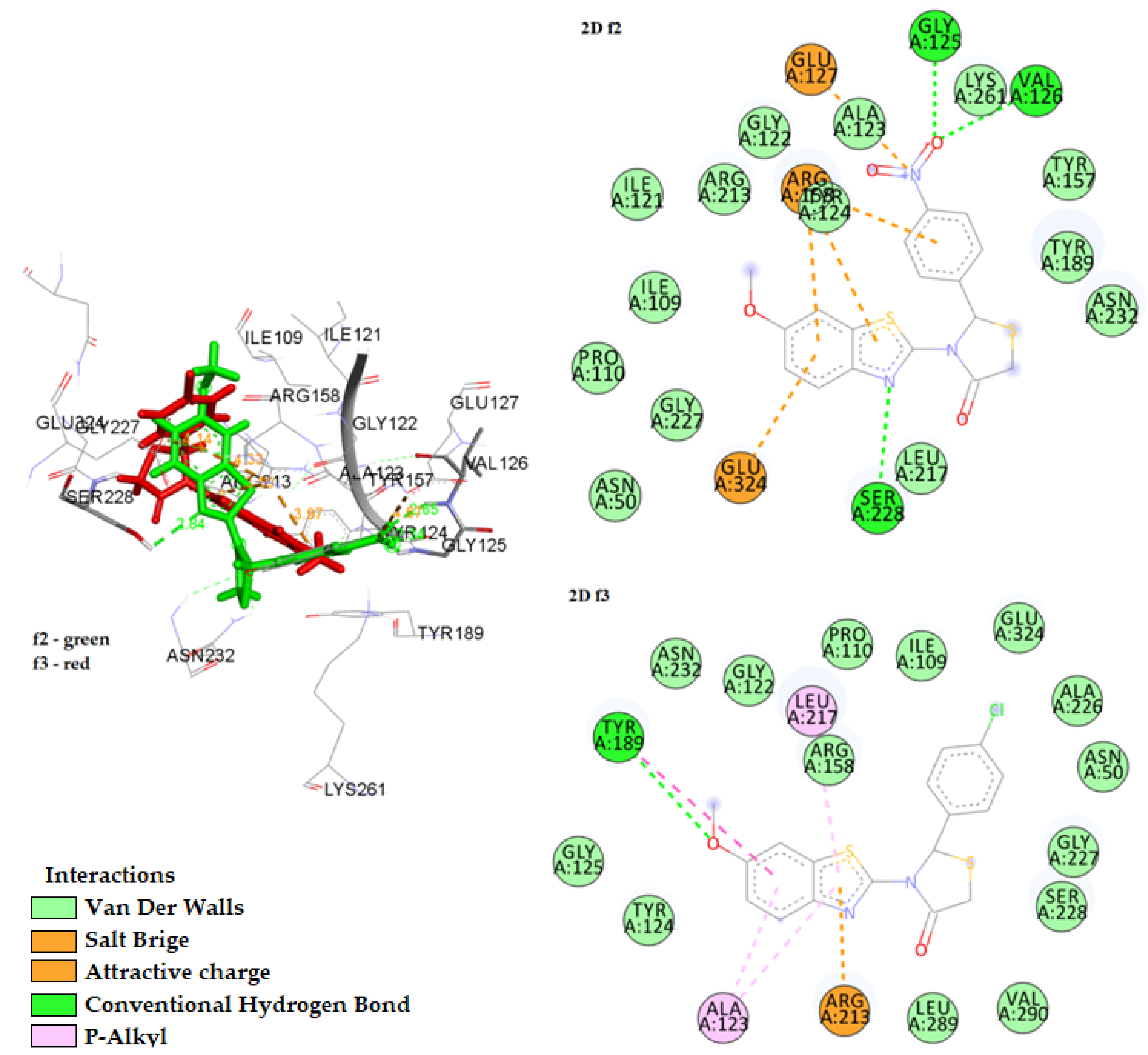
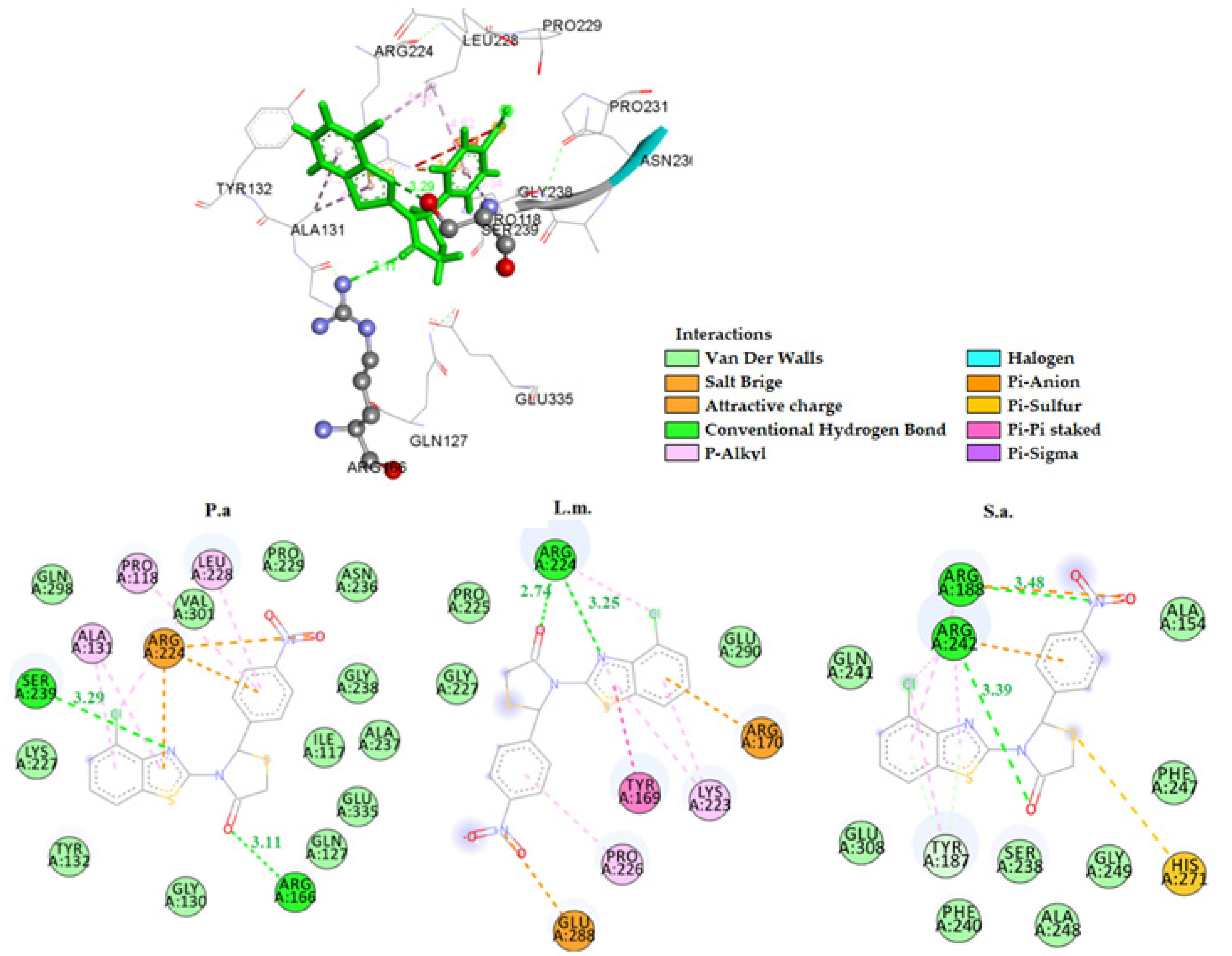
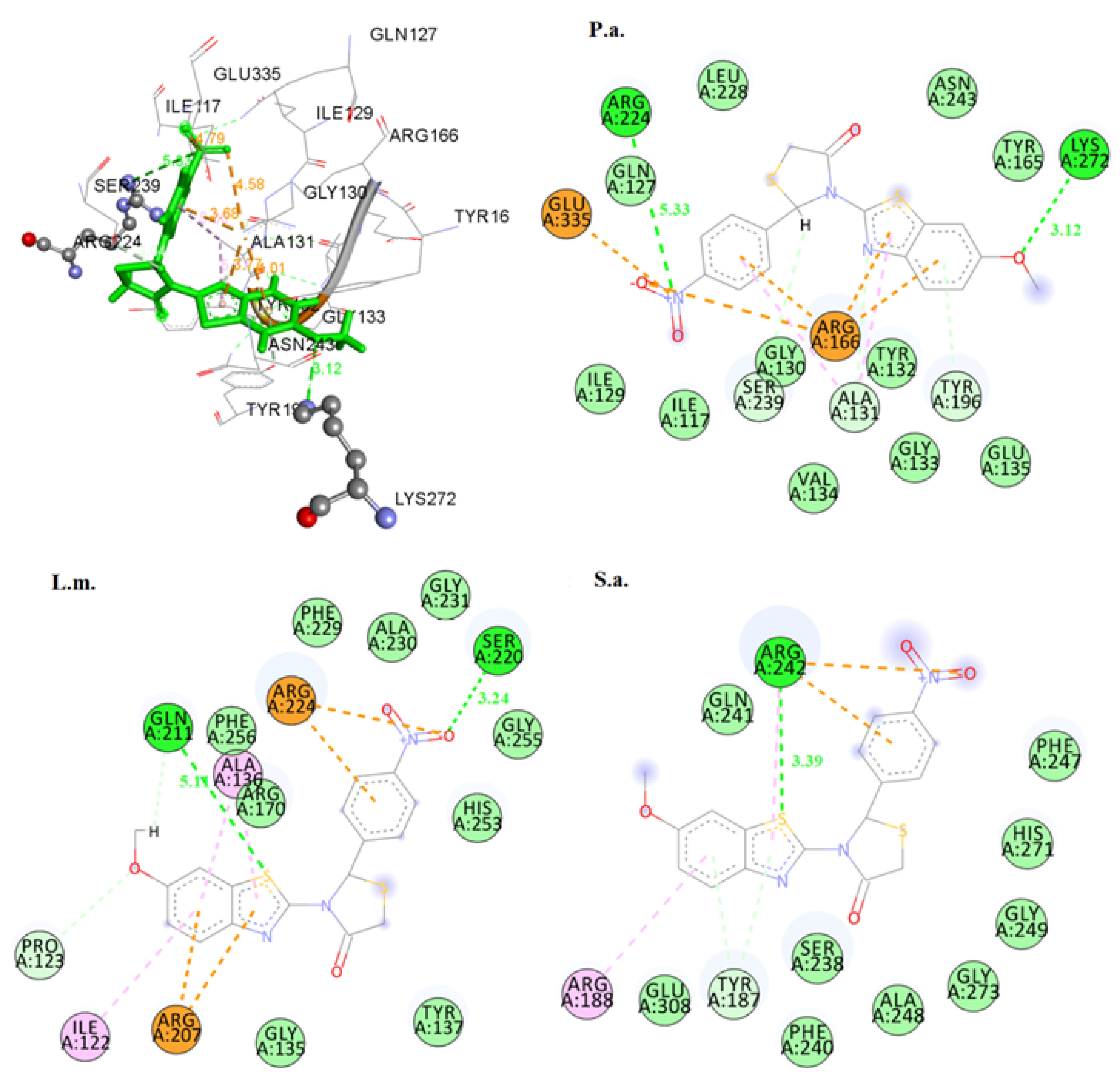
2.3.1. Docking on Antimicrobial Targets
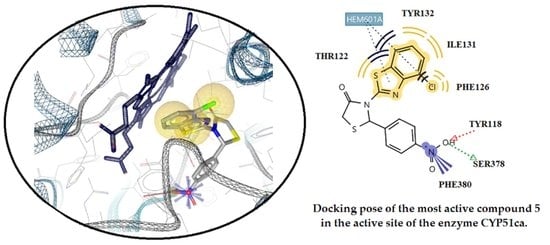
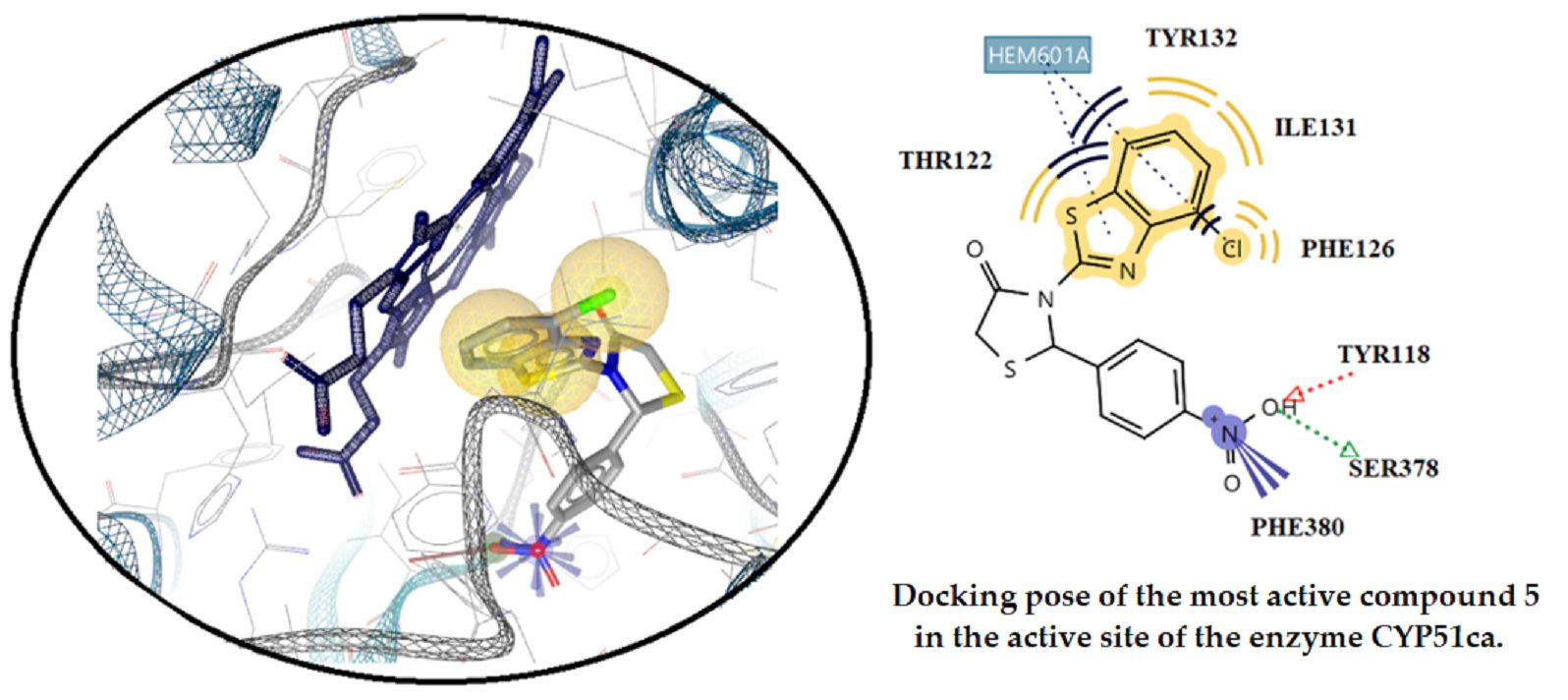
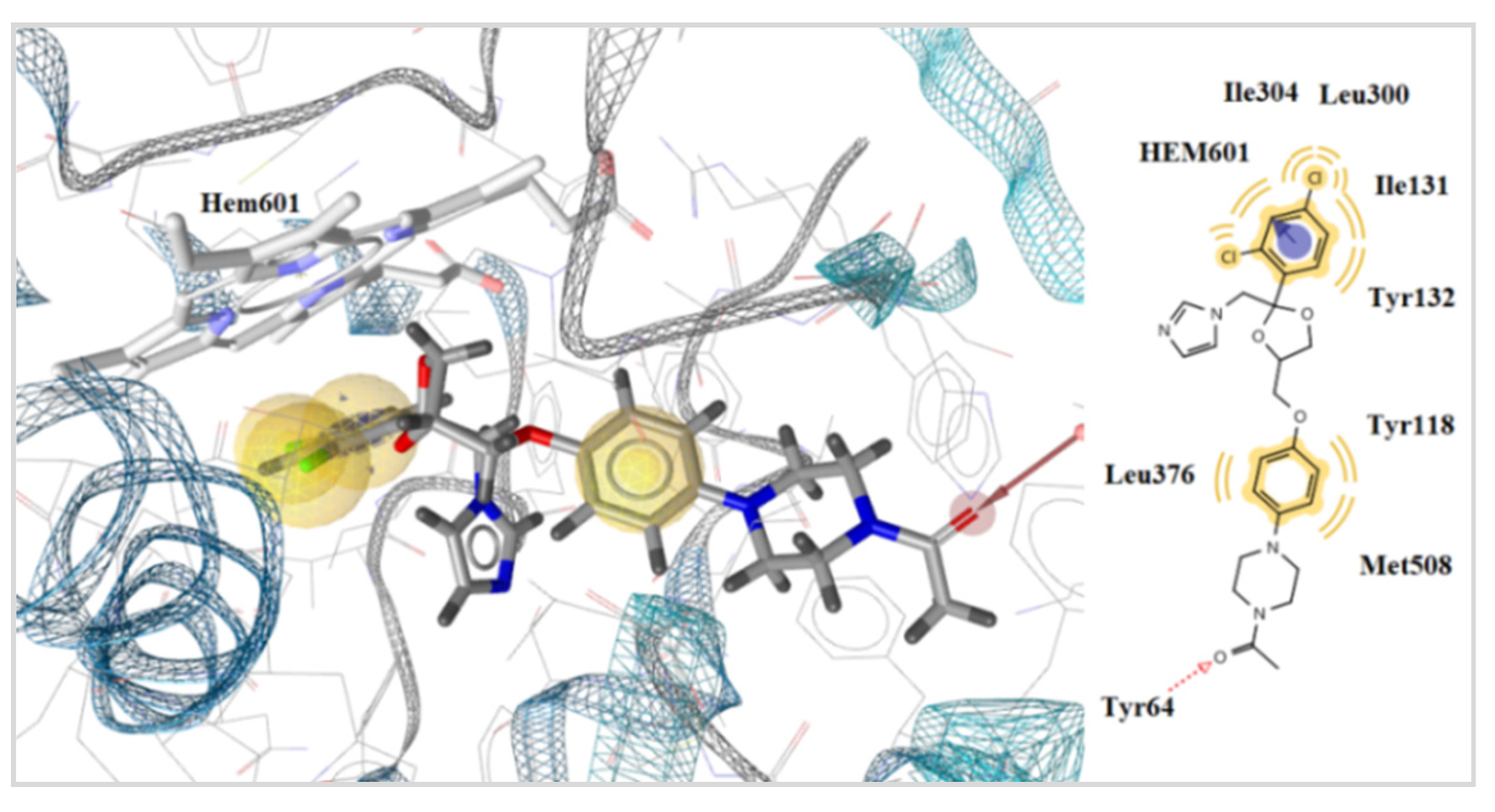
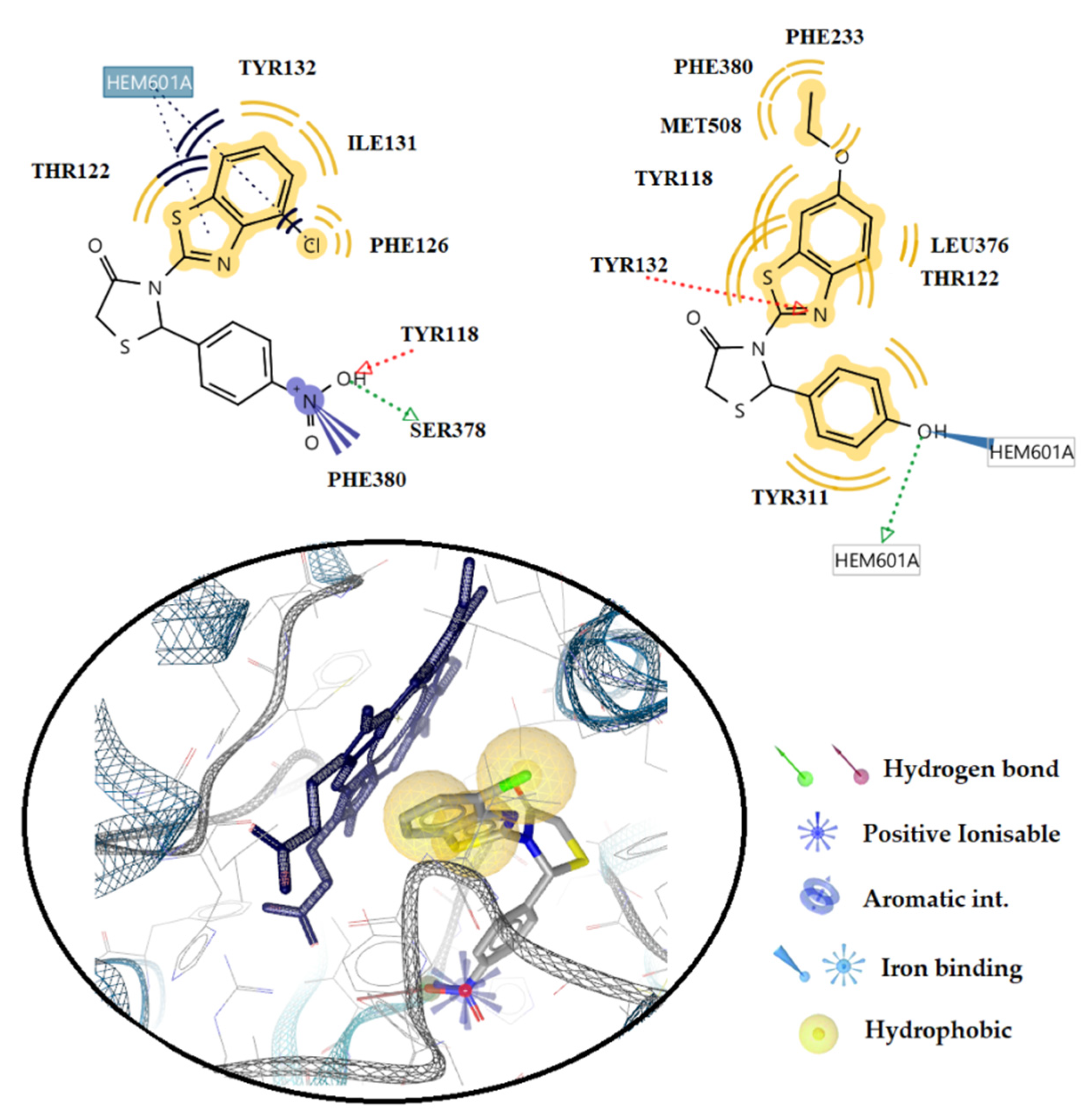
2.3.2. Docking Studies on the Enzyme Lanosterol 14-α Demethylase of Candida albicans
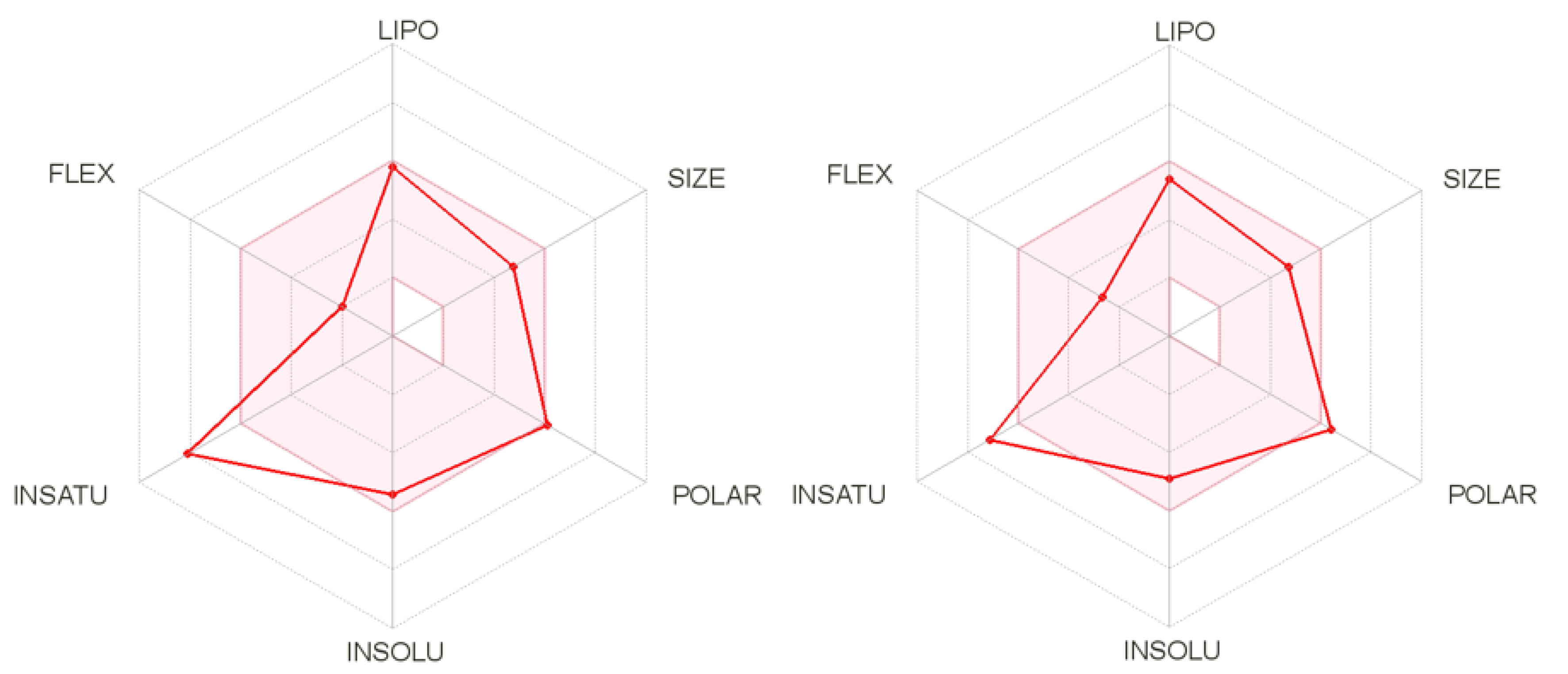
2.4. In Silico Predictive Studies
2.5. Cytotoxicity Assessment
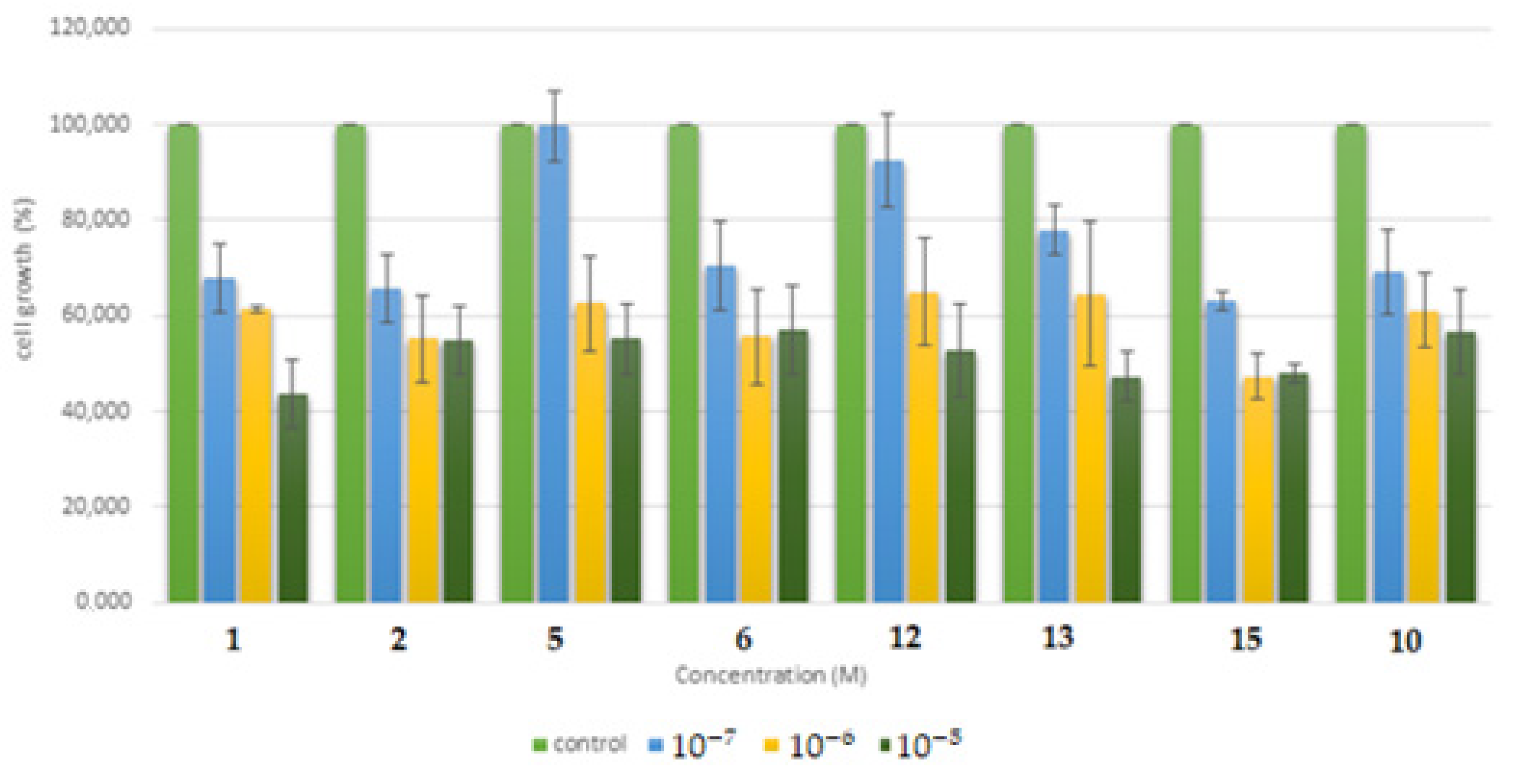
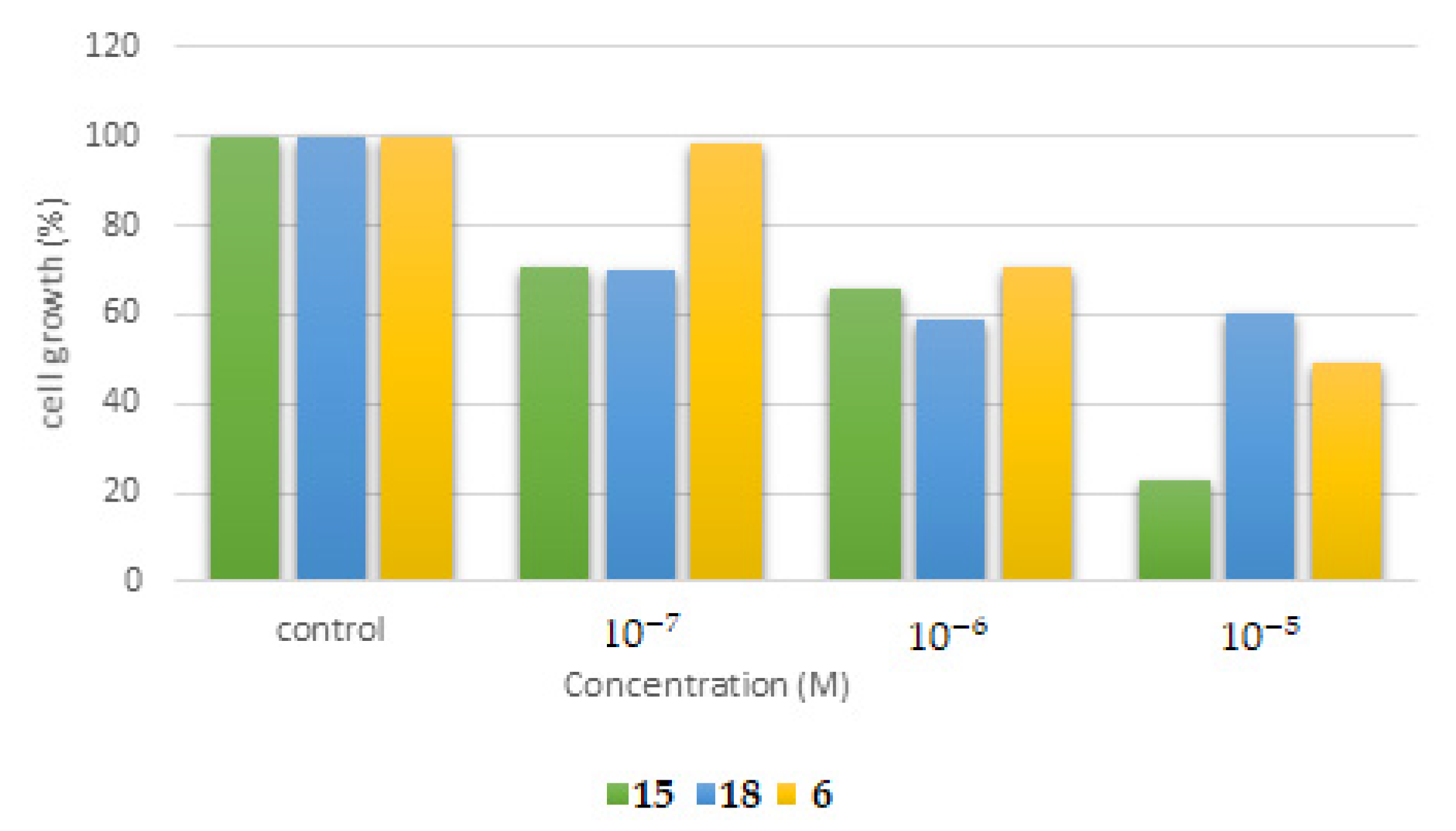
2.5.1. Cytotoxicity Assessment of the Synthesized Molecules in the Human Cancer HSC-3 Cell Line
2.5.2. Cytotoxicity Assessment of the Synthesized Molecules in the Human Normal MRC-5 Cell Line
3. Materials and Methods
3.1. Chemistry
3.2. Biological Evaluation
3.2.1. Antibacterial/Antifungal Action
3.2.2. Inhibition of Biofilm Formation
3.2.3. Checkboard Assay
3.2.4. Time Kill Assay
3.3. Docking Studies
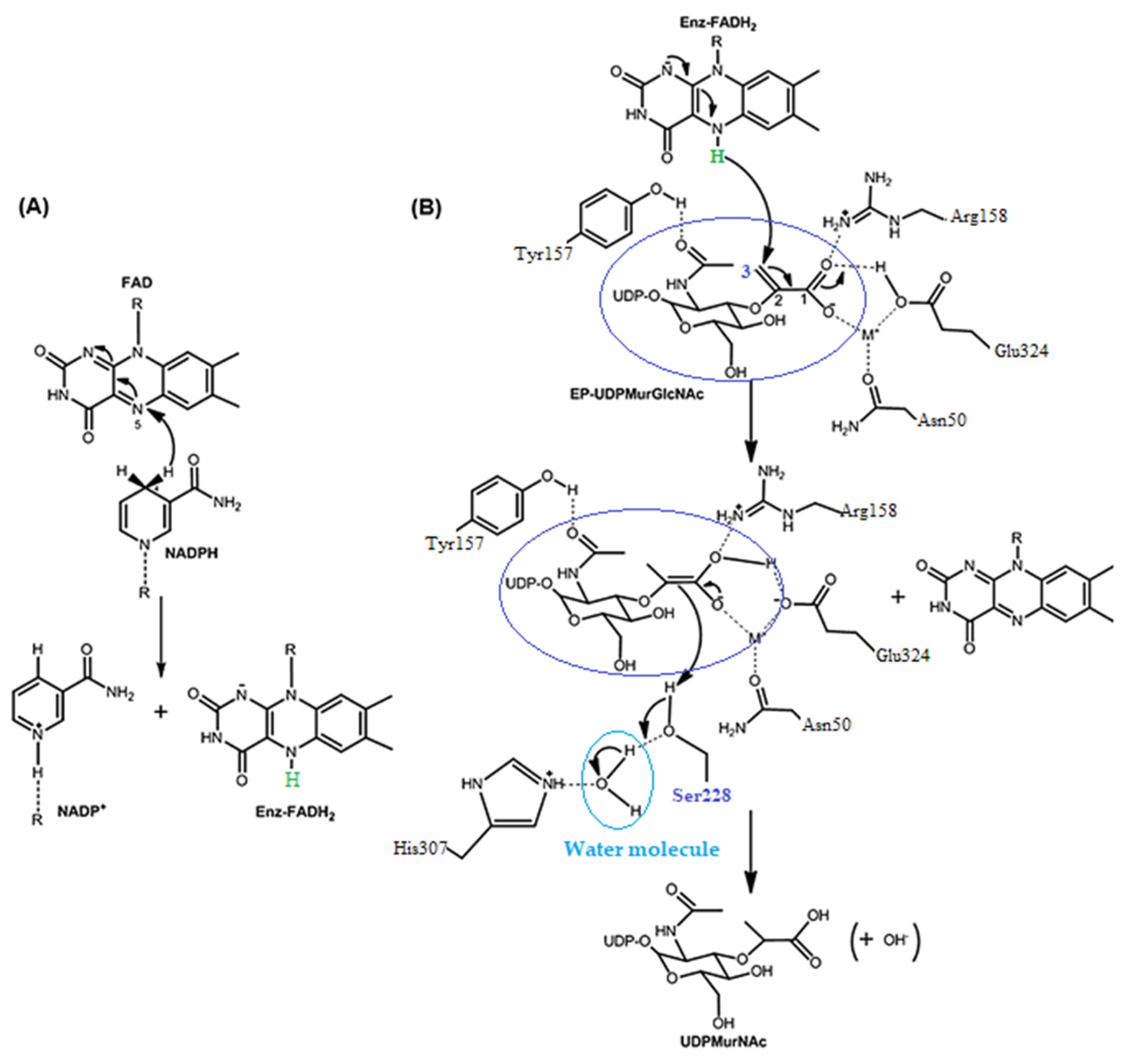
3.3.1. Docking Studies for Prediction of the Mechanism of Antibacterial Activity
3.3.2. Docking Studies for Prediction of the Mechanism of Antifungal Activity
3.4. In Silico Predictive Studies
3.5. Assessment of Cytotoxicity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Davies, D. Understanding biofilm resistance to antibacterial agents. Nat. Rev. Drug. Discov. 2003, 2, 114–122. [Google Scholar] [CrossRef]
- Jamal, M.; Ahmad, W.; Andleeb, S.; Jalil, F.; Imran, M.; Nawaz, M.A.; Hussain, T.; Ali, M.; Rafiq, M.; Kamil, M.A. Bacterial biofilm and associated infections. J. Chin. Med. Assoc. 2018, 81, 7–11. [Google Scholar] [CrossRef] [PubMed]
- RömLing, U.; Balsalobre, C. Biofilm infections, their resilience to therapy and innovative treatment strategies. J. Intern. Med. 2012, 272, 541–561. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Tiwari, M.; Donelli, G.; Tiwari, V. Strategies for combating bacterial biofilms: A focus on anti-biofilm agents and their mechanisms of action. Virulence 2018, 9, 522–555. [Google Scholar] [CrossRef] [PubMed]
- Shenkutie, A.M.; Yao, M.Z.; Siu, G.K.; Wong, B.K.; Leung, P.H. Biofilm-Induced Antibiotic Resistance in Clinical Acinetobacter baumannii Isolates. Antibiotics 2020, 9, 817. [Google Scholar] [CrossRef]
- Burmølle, M.; Webb, J.S.; Rao, D.; Hansen, L.H.; Sørensen, S.J.; Kjelleberg, S. Enhanced biofilm formation and increased resistance to antimicrobial agents and bacterial invasion are caused by synergistic interactions in multispecies biofilms. Appl. Environ. Microbiol. 2006, 72, 3916–3923. [Google Scholar] [CrossRef] [Green Version]
- Simoes, M.; Pereira, M.O.; Vieira, M.J. Effect of mechanical stress on biofilms challenged by diferent chemicals. Water Res. 2005, 39, 5142–5152. [Google Scholar] [CrossRef] [Green Version]
- Morsy, M.A.; Ali, E.M.; Venugopala, K.M.; Nair, A.B.; Greish, K.; El-Daly, M. Screening and Molecular Docking of Novel Benzothiazole Derivatives as Potential Antimicrobial Agents. Antibiotics 2020, 9, 221. [Google Scholar] [CrossRef]
- Haroun, M.; Tratrat, C.; Petrou, A.; Geronikaki, A.; Ivanov, M.; Ciri, A.; Sokovic, M.; Nagaraja, S.; Venugopala, K.; Nair, A.; et al. Exploration of the Antimicrobial Effects of Benzothiazolyl thiazolidin-4-one and In Silico Mechanistic Investigation. Molecules 2021, 26, 61. [Google Scholar] [CrossRef]
- Padalkar, V.; Borse, B.; Gupta, V.; Phatangare, K.; Patil, V.; Sekar, U. Synthesis and antimicrobial activity of novel 2-substituted benzimidazole, benzoxazole and benzothiazole derivatives. Arab. J. Chem. 2016, 9, S1125–S1130. [Google Scholar] [CrossRef] [Green Version]
- Luo, B.; Li, D.; Zhang, A.L.; Gao, J.M. Synthesis, Antifungal Activities and Molecular Docking Studies of Benzoxazole and Benzothiazole Derivatives. Molecules 2018, 23, 2457. [Google Scholar] [CrossRef] [Green Version]
- Ugwu, D.; Okoro, U.; Ukoha, P.; Gupta, A.; Okafor, S. Novel anti-inflammatory and analgesic agents:synthesis, molecular docking and in vivo studies. J. Enzym. Inhib. Med. Chem. 2018, 33, 405–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alizadeh, M.; Jalal, M.; Hamed, K.; Saber, A.; Kheirouri, S.; Tabrizi, F.; Kamari, N. Recent Updates on Anti-Inflammatory and Antimicrobial Effects of Furan Natural Derivatives. J. Inflamm. Res. 2020, 13, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Pérez, L.; Padilla-Martínez, I.; Cruz, A.; Mendieta-Wejebe, J.; Tamay-Cach, F.; Rosales-Hernández, M. Evaluation of a new benzothiazole derivative with antioxidant activity in the initial phase of acetaminophen toxicity. Arab. J. Chem. 2019, 12, 3871–3882. [Google Scholar] [CrossRef]
- Uremis, N.; Uremis, M.M.; Tolun, F.I.; Ceylan, M.; Doganer, A.; Kurt, A.H. Synthesis of 2-Substituted Benzothiazole Derivatives and Their In Vitro Anticancer Effects and Antioxidant Activities Against Pancreatic Cancer Cells. Anticancer Res. 2017, 37, 6381–6389. [Google Scholar]
- Kumar, G.; Singh, N.P. Synthesis, anti-inflammatory and analgesic evaluation of thiazole/oxazole substituted benzothiazole derivatives. Bioorg. Chem. 2021, 107, 104608. [Google Scholar] [CrossRef]
- Kumar, K.R.; Karthik, K.N.S.; Begum, P.R.; Prasada Rao, C.M.M. Synthesis, characterization and biological evaluation of benzothiazole derivatives as potential antimicrobial and analgesic agents. Asian J. Res. Pharm. Sci. 2017, 7, 1–5. [Google Scholar] [CrossRef]
- Irfan, A.; Batool, F.; Zahra Naqvi, S.A.; Islam, A.; Osman, S.M.; Nocentini, A.; Alissa, S.A.; Supuran, C.T. Benzothiazole derivatives as anticancer agents. J. Enzym. Inhib. Med. Chem. 2020, 35, 265–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, M.K.; Baek, A.; Sung, B.; Yang, B.-W.; Choi, G.; Kim, Y.-H.; Kim, M.; Ha, S.; Lee, G.-H.; Kim, H.-K.; et al. Synthesis, Characterization, and Anticancer Activity of Benzothiazole Aniline Derivatives and Their Platinum (II) Complexes as New Chemotherapy Agents. Pharmaceuticals 2021, 14, 832. [Google Scholar] [CrossRef] [PubMed]
- Osmaniye, D.; Levent, S.; Karaduman, A.B.; Ilgın, S.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis of New Benzothiazole Acylhydrazones as Anticancer Agents. Molecules 2018, 23, 54. [Google Scholar] [CrossRef] [Green Version]
- Asiri, Y.I.; Alsayari, A.; Muhsinah, A.B.; Mabkhot, Y.N.; Hassan, M.Z. Benzothiazoles as potential antiviral agents. J. Pharm. Pharmacol. 2020, 72, 1459–1480. [Google Scholar] [CrossRef] [PubMed]
- Elamin, M.B.; Elaziz, A.A.E.S.A.; Abdallah, E.M. Benzothiazole moieties and their derivatives as antimicrobial and antiviral agents: A mini-review. Int. J. Res. Pharm. Sci. 2020, 311, 3309–3315. [Google Scholar] [CrossRef]
- Kumar, M.; Chung, S.-M.; Enkhtaiva, G.; Patel, R.; Shin, H.-S.; Mistry, B. Molecular Docking Studies and Biological Evaluation ofBerberine–Benzothiazole Derivatives as an Anti-Influenza Agent via Blocking of Neuraminidase. Int. J. Mol. Sci. 2021, 22, 2368. [Google Scholar] [CrossRef]
- Azzam, R.A.; Elboshi, H.A.; Elgemeie, G.H. Novel Synthesis and Antiviral Evaluation of New Benzothiazole-Bearing N-Sulfonamide 2-Pyridone Derivatives as USP7 Enzyme Inhibitors. ACS Omega 2020, 5, 30023–30036. [Google Scholar] [CrossRef] [PubMed]
- Najim, N.; Al-Masoudia, A.; Jafarb, N.A.; Abbasc, L.J.; Baqir, S.; Pannecouque, C. Synthesis and anti-HIV Activity of New Benzimidazole, Benzothiazole and Carbohyrazide Derivatives of the anti-Inflammatory Drug Indomethacin. Z. Nat. B 2011, 66, 953–960. [Google Scholar]
- Bhagdev, K.; Sarkar, B. Benzothiazole: As an antidiabetic agent. Ann. Rom. Soc. Cell Biol. 2021, 25, 20269–20285. [Google Scholar]
- Ullah, S.; Mirza, S.; Salar, U.; Hussain, S.; Javaid, K.; Khan, K.M.; Khalil, R.; Atia-Tul-Wahab; Ul-Haq, Z.; Perveen, S.; et al. 2-Mercapto Benzothiazole Derivatives: As Potential Leads for the Diabetic Management. Med. Chem. 2020, 16, 826–840. [Google Scholar] [CrossRef]
- Nath, Ν.; Yar, M.; Pathania, S.; Grover, G.; Debnath, B.; Akhtar, M.J. Synthesis and anticonvulsant evaluation of indoline derivatives of functionalized aryloxadiazole amine and benzothiazole acetamide. J. Mol. Struct. 2021, 1228, 129742. [Google Scholar] [CrossRef]
- Khokra, S.; Arora, K.; Khan, S.; Kaushik, P.; Saini, R.; Husain, A. Synthesis, Computational Studies and Anticonvulsant Activity of Novel Benzothiazole Coupled Sulfonamide Derivatives. Iran. J. Pharm. Res. 2019, 18, 1–15. [Google Scholar]
- Hadanu, R.; Idris, S.; Sutapa, W. Qsar analysis of benzothiazole derivatives of antimalarial compoundsbased on am1 semi-empirical method. Indones. J. Chem. 2015, 15, 86–92. [Google Scholar] [CrossRef]
- Suresh, A.J.; Bharathi, K.; Surya, P.R. Design, Synthesis, Characterization and Biological Evaluation of Some Novel Benzothiazole Derivatives as Anti Tubercular Agents Targeting Glutamine Synthetase-I. J. Pharm Chem. Biol. Sci. 2018, 5, 312–319. [Google Scholar]
- Mehra, R.; Rajput, V.; Gupta, M.; Kumar, A.; Wazir, P.; Khan, I.; Nargotra, A. Benzothiazole Derivative as a Novel Mycobacterium tuberculosis Shikimate Kinase Inhibitor: Identification and Elucidation of Its Allosteric Mode of Inhibition. J. Chem. Inf. Model. 2016, 56, 930–940. [Google Scholar] [CrossRef]
- Haroun, M.; Tratrat, C.; Kositsi, K.; Tsolaki, E.; Petrou, A.; AlDhubiab, B.; Attimarad, M.; Harsha, S.; Elsewedy, H.; Gavalas, A.; et al. New benzothiazole-based thiazolidinones as potent dual anti-inflammatory/antimicrobial agents. Design, synthesis and biological evaluation. Curr. Top. Med. Chem. 2018, 18, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Dincel, E.D.; Ulusoy-Güzeldemirci, N.; Şatana, D.; Küçükbasmacı, Ö. Design, synthesis, characterization and antimicrobial evaluation of some novel hydrazinecarbothioamide, 4-thiazolidinone and 1,2,4-triazole-3-thione derivatives. J. Heterocycl. Chem. 2021, 58, 195–205. [Google Scholar] [CrossRef]
- Desai, N.; Jadeja, K.; Jadeja, D.; Khedkar, V.; Jha, P. Design, synthesis, antimicrobial evaluation, and molecular docking study of some 4-thiazolidinone derivatives containing pyridine and quinazoline moiety. Synth. Commun. 2020, 51, 1–12. [Google Scholar] [CrossRef]
- Bari, S.B.; Firake, S.D. Exploring Anti-inflammatory Potential of Thiazolidinone Derivatives of Benzenesulfonamide via Synthesis, Molecular Docking and Biological Evaluation. AntiInflamm. AntiAllergy Agents Med. Chem. 2016, 15, 44–53. [Google Scholar] [CrossRef]
- Liaras, K.; Fesatidou, M.; Geronikaki, A. Thiazoles and Thiazolidinones as COX/LOX Inhibitors. Molecules 2018, 23, 685. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Kudva, J.; Bharath, B.R.; Ananda, K.; Sadashiva, R.; Kumar, M.S.; Revanasiddappa, B.C.; Kumar, V.; Rekhah, P.D.; Naral, D. Synthesis, structural, biological and in silico studies of new 5-arylidene-4-thiazolidinone derivatives as possible anticancer, antimicrobial and antitubercular agents. New J. Chem. 2019, 43, 1597–1610. [Google Scholar] [CrossRef]
- Ansari, A.F.; Idrees, D.; Hassan, I.; Ahmad, K.; Avecilla, F.; Azam, A. Design, synthesis and biological evaluation of novel pyridine-thiazolidinone derivatives as anticancer agents: Targeting human carbonic anhydrase IX. Eur. J. Med. Chem. 2018, 114, 544–556. [Google Scholar] [CrossRef]
- Geronikaki, A.; Petrou, A.; Kartsev, V.; Eleftheriou, P.; Bogad, R.; Bartolo, B.; Crespane, E.; Francoe, G.; Maga, G. Molecular docking, design, synthesis and biological evaluation of novel 2,3-aryl-thiazolidin-4-ones as potent NNRTIs. SAR QSAR Environ. Res. 2019, 30, 697–714. [Google Scholar] [CrossRef]
- Petrou, A.; Eleftheriou, P.; Geronikaki, A.; Akrivou, M.; Vizirianakis, I. Novel Thiazolidin-4-ones as Potential Non-Nucleoside Inhibitors of HIV-1 Reverse Transcriptase. Molecules 2019, 24, 3821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cihan-Üstündağ, G.; Şatana, D.; Özhan, G.; Çapan, G. Indole-based hydrazide-hydrazones and 4-thiazolidinones: Synthesis and evaluation as antitubercular and anticancer agents. J. Enzym. Inhib. Med. Chem. 2016, 31, 369–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasmin, S.; Capone, F.; Laghezza, A.; Dal Piaz, F.; Loiodice, F.; Vijayan, V.; Devadasan, V.; Mondal, S.; Atlı, O.; Baysal, M.; et al. Novel Benzylidene Thiazolidinedione Derivatives as Partial PPARγ Agonists and their Antidiabetic Effects on Type 2 Diabetes. Sci. Rep. 2017, 7, 14453. [Google Scholar] [CrossRef] [PubMed]
- Rajalakshmi, R.; Santhi, R.; Elakkiya, T. Synthesis and Evaluation of novel oxazinyl thiazolidinone derivatives antioxidant agent as potent antidiabetic. Int. J. Adv. Sci. Technol. 2020, 29, 1724–1773. [Google Scholar]
- Chaban, T.; Ogurtsov, V.; Chaban, I.; Myrko, I.; Harkov, S.; Lelyukh, M. Synthesis of some new 4-iminothiazolidine-2-ones as possible antioxidants agents. Pharmacia 2019, 66, 27–32. [Google Scholar] [CrossRef]
- Saini, S. Synthesis and Anticonvulsant Studies of Thiazolidinone and Azetidinone Derivatives from Indole Moiety. Drug Res. 2019, 69, 445–450. [Google Scholar] [CrossRef]
- Mishchenko, M.; Shtrygol, S.; Kaminskyy, D.; Lesyk, R. Thiazole-Bearing 4-Thiazolidinones as New Anticonvulsant Agents. Sci. Pharm. 2020, 88, 16. [Google Scholar] [CrossRef] [Green Version]
- Kryshchyshyn-Dylevych, A.P.; Zelisko, N.I.; Grellier, P.; Lesyk, R.B. Preliminary evaluation of thiazolidinone- and pyrazoline-related heterocyclic derivatives as potential antimalarial agents. Biopolym. Cell 2020, 36, 48–60. [Google Scholar] [CrossRef]
- Güzel-Akdemir, O.; Angeli, A.; Demir, K.; Supuran, C.T.; Akdemir, A. Novel thiazolidinone-containing compounds, without the well-known sulphonamide zinc-binding group acting as human carbonicanhydrase IX inhibitors. J. Enzym. Inhib. Med. Chem. 2018, 33, 1299–1308. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.P.; Yin, Z.F.; Li, J.Y.; Wang, Z.P.; Wu, Q.J.; Wang, J.; Liu, Y.; Cheng, M.S. Synthesis, Molecular Docking Analysis, and Carbonic Anhydrase Inhibitory Evaluations of Benzenesulfonamide Derivatives containing thiazolidinone. Molecules 2019, 24, 2518. [Google Scholar] [CrossRef] [Green Version]
- Benson, T.E.; Filman, D.J.; Walsh, C.T.; Hogle, J.M. An enzyme–substrate complex involved in bacterial cell wall biosynthesis. Nat. Struct. Mol. Biol. 1995, 2, 644–653. [Google Scholar] [CrossRef]
- Benson, T.E.; Walsh, C.T.; Massey, V. Kinetic characterization of wild-type and S229A mutant MurB: Evidence for the role of Ser 229 as a general acid. Biochemistry 1997, 36, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Benson., T.E.; Marquardt, J.L.; Marquardt, A.C.; Etzkorn, F.A.; Walsh, C.T. Overexpression, purification, and mechanistic study of UDP-N-acetylenolpyruvylglucosamine reductase. Biochemistry 1993, 328, 2024–2030. [Google Scholar] [CrossRef] [PubMed]
- Nishida, S.; Kurokawa, K.; Matsuo, M.; Sakamoto, K.; Ueno, K.; Kita, K.; Sekimizu, K. Identification and Characterization of Amino Acid Residues Essential for the Active Site of UDP-N-acetylenolpyruvylglucosamine Reductase (MurB) from Staphylococcus aureus. J. Biol. Chem. 2005, 281, 1714–1724. [Google Scholar] [CrossRef] [Green Version]
- Fesatidou, M.; Zagaliotis, P.; Camoutsis, C.; Petrou, A.; Eleftheriou, P.; Tratrat, C.; Haroun, M.; Geronikaki, A.; Ciric, A.; Sokovic, M. 5-Adamantan thiadiazole-based thiazolidinones as antimicrobial agents. Design, synthesis, molecular docking and evaluation. Bioorg. Med. Chem. 2018, 26, 4664–4676. [Google Scholar] [CrossRef]
- Juhás, M.; Zitko, J. Molecular Interactions of Pyrazine-Based Compounds to Proteins. J. Med. Chem. 2020, 63, 8901–8916. [Google Scholar] [CrossRef]
- Heravi, M.M.; Zadsirjan, V. Prescribed drugs containing nitrogen heterocycles: An overview. RSC Adv. 2020, 10, 44247–44311. [Google Scholar] [CrossRef]
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications. Molecules 2020, 25, 9. [Google Scholar] [CrossRef]
- Bhutani, P.; Joshi, G.; Raja, N.; Bachhav, N.; Rajanna, P.K.; Bhutani, H.; Paul, A.T.; Kumar, R.U.S. FDA Approved Drugs from 2015–June 2020: A Perspective. J. Med. Chem. 2021, 64, 2339–2381. [Google Scholar] [CrossRef] [PubMed]
- Bootsma, A.N.; Doney, A.C.; Wheeler, S.E. Predicting the Strength of Stacking Interactions between Heterocycles and Aromatic Amino Acid Side Chains. J. Am. Chem. Soc. 2019, 141, 11027–11035. [Google Scholar] [CrossRef]
- Baykov, S.V.; Mikherdov, A.S.; Novikov, A.S.; Geyl, K.K.; Tarasenko, M.V.; Gureev, M.A.; Boyarskiy, V.P. π–π Noncovalent Interaction Involving 1,2,4-and 1,3,4-Oxadiazole Systems: The Combined Experimental, Theoretical, and Database Study. Molecules 2021, 26, 5672. [Google Scholar] [CrossRef] [PubMed]
- Benson, T.E.; Harris, M.S.; Choi, G.H.; Cialdella, J.I.; Herberg, J.T.; Martin, J.P.; Baldwin, E.T. A structural variation for MurB: X-ray crystal structure of Staphylococcus aureus UDP-N-acetylenolpyruvylglucosamine reductase (MurB). Biochemistry 2001, 40, 2340–2350. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Shah, A.H.; Rawal, M.K. Antifungals: Mechanism of Action and Drug Resistance. In Yeast 30 Membrane Transport; Ramos, J., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2016; p. 892. [Google Scholar]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Wars, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Espinel-Ingroff, A. A comparasion of the E-test with the NCCLS M38-P method for antifungal susceptibility testing of common and emerging pathogenic filamentous fungi, J. Clin. Microbiol. 2001, 39, 1360–1367. [Google Scholar] [CrossRef] [Green Version]
- Clinical and Laboratory Standards Institute (CLSI). Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically, Approved Standard, 8th ed.; CLSI Publication M07-A8; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2009. [Google Scholar]
- Kostić, M.; Smiljković, M.; Petrović, J.; Glamočlija, J.; Barros, L.; Ferreira, I.C.F.R.; Ćirić, A.; Soković, M. Chemical, nutritive composition and a wide range of bioactive properties of honey mushroom Armillaria mellea (Vahl: Fr.) Kummer. Food Funct. 2017, 8, 3239–3249. [Google Scholar] [CrossRef] [Green Version]
- Kartsev, V.; Lichitsky, B.; Geronikaki, A.; Petrou, A.; Smiljkovic, M.; Kostic, M.; Radanovic, O.; Soković, M. Design, synthesis and antimicrobial activity of usnic acid derivatives. MedChemComm 2018, 9, 870–882. [Google Scholar] [CrossRef] [Green Version]
- Cady, N.C.; McKean, K.A.; Behnke, J.; Kubec, R.; Mosier, A.P.; Kasper, S.H.; Burz, D.S.; Musah, R.A. Inhibition of biofilm formation, quorum sensing and infection in Pseudomonas aeruginosa by natural products-inspired organosulfur compounds. PLoS ONE 2012, 7, e38492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smiljkovic, M.; Dias, M.; Stojkovic, D.; Barros, L.; Bukvicki, D.; Ferreira, I.C.F.R.; Sokovic, M. Characterization of phenolic compounds in tincture of edible Nepeta nuda: Development of antimicrobial mouthwash. Food Funct. 2018, 9, 5417–5425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aničić, N.; Gašić, U.; Lu, F.; Ćirić, A.; Ivanov, M.; Jevtić, B.; Dimitrijević, M.; Anđelković, B.; Skorić, M.; Nestorović Živković, J.; et al. Antimicrobial and immunomodulating activities of two endemic Nepeta species and their major iridoids isolated from natural sources. Pharmaceuticals 2021, 14, 414. [Google Scholar] [CrossRef] [PubMed]
- Akrivou, M.G.; Demertzidou, V.P.; Theodoroula, N.F.; Chatzopoulou, F.M.; Kyritsis, K.A.; Grigoriadis, N.; Zografos, A.L.; Vizirianakis, I.S. Uncovering the pharmacological response of novel sesquiterpene derivatives that differentially alter gene expression and modulate the cell cycle in cancer cells. Int. J. Oncol. 2018, 53, 2167–2179. [Google Scholar] [CrossRef] [Green Version]
- Tseligka, E.D.; Rova, A.; Amanatiadou, E.P.; Calabrese, G.; Tsibouklis, J.; Fatouros, D.G.; Vizirianakis, I.S. Pharmacological Development of Target-Specific Delocalized Lipophilic Cation-Functionalized Carboranes for Cancer Therapy. Pharm. Res. 2016, 33, 1945–1958. [Google Scholar] [CrossRef] [Green Version]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | B.c. | S.a. | M.l. | L.m. | P.a. | En.cl. | E.c. | S.T. | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | MIC | 0.045 | 0.045 | 0.045 | 0.045 | 0.045 | 0.045 | 0.045 | 0.09 |
| MBC | 0.09 | 0.09 | 0.09 | 0.09 | 0.09 | 0.09 | 0.09 | 0.18 | |
| 2 | MIC | 0.06 | 0.24 | 0.06 | 0.24 | 0.24 | 0.03 | 0.03 | 0.24 |
| MBC | 0.12 | 0.48 | 0.12 | 0.48 | 0.48 | 0.06 | 0.06 | 0.48 | |
| 3 | MIC | 0.06 | 0.24 | 0.06 | 0.06 | 0.06 | 0.06 | 0.06 | 0.24 |
| MBC | 0.12 | 0.48 | 0.12 | 0.12 | 0.12 | 0.12 | 0.12 | 0.48 | |
| 4 | MIC | 0.03 | 0.12 | 0.06 | 0.12 | 0.06 | 0.06 | 0.06 | 0.12 |
| MBC | 0.06 | 0.24 | 0.12 | 0.24 | 0.12 | 0.12 | 0.12 | 0.24 | |
| 5 | MIC | 0.008 | 0.06 | 0.015 | 0.015 | 0.015 | 0.015 | 0.015 | 0.015 |
| MBC | 0.016 | 0.12 | 0.03 | 0.03 | 0.03 | 0.03 | 0.03 | 0.03 | |
| 6 | MIC | 0.03 | 0.06 | 0.06 | 0.06 | 0.06 | 0.03 | 0.06 | 0.06 |
| MBC | 0.06 | 0.12 | 0.12 | 0.12 | 0.12 | 0.06 | 0.12 | 0.12 | |
| 7 | MIC | 0.08 | 0.32 | 0.08 | 0.08 | 0.08 | 0.08 | 0.08 | 0.32 |
| MBC | 0.16 | 0.64 | 0.16 | 0.16 | 0.16 | 0.16 | 0.16 | 0.64 | |
| 8 | MIC | 0.02 | 0.08 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.08 |
| MBC | 0.04 | 0.16 | 0.04 | 0.04 | 0.04 | 0.04 | 0.04 | 0.16 | |
| 9 | MIC | 0.48 | 0.48 | 0.12 | 0.48 | 0.12 | 0.12 | 0.12 | 0.24 |
| MBC | 0.96 | 0.96 | 0.24 | 0.96 | 0.24 | 0.24 | 0.24 | 0.48 | |
| 10 | MIC | 0.03 | 0.24 | 0.06 | 0.03 | 0.06 | 0.03 | 0.03 | 0.24 |
| MBC | 0.06 | 0.48 | 0.12 | 0.06 | 0.12 | 0.06 | 0.06 | 0.48 | |
| 11 | MIC | 0.24 | 0.24 | 0.06 | 0.06 | 0.06 | 0.06 | 0.06 | 0.12 |
| MBC | 0.48 | 0.48 | 0.12 | 0.12 | 0.12 | 0.12 | 0.12 | 0.24 | |
| 12 | MIC | 0.03 | 0.12 | 0.03 | 0.03 | 0.03 | 0.03 | 0.03 | 0.12 |
| MBC | 0.06 | 0.24 | 0.06 | 0.06 | 0.06 | 0.06 | 0.06 | 0.24 | |
| 13 | MIC | 0.03 | 0.24 | 0.06 | 0.24 | 0.06 | 0.06 | 0.06 | 0.24 |
| MBC | 0.06 | 0.48 | 0.12 | 0.48 | 0.12 | 0.12 | 0.12 | 0.48 | |
| 14 | MIC | 0.03 | 0.24 | 0.24 | 0.06 | 0.06 | 0.03 | 0.03 | 0.24 |
| MBC | 0.06 | 0.48 | 0.48 | 0.12 | 0.12 | 0.06 | 0.06 | 0.48 | |
| 15 | MIC | 0.015 | 0.03 | 0.03 | 0.03 | 0.03 | 0.03 | 0.03 | 0.03 |
| MBC | 0.03 | 0.06 | 0.06 | 0.06 | 0.06 | 0.06 | 0.06 | 0.06 | |
| 16 | MIC | 0.015 | 0.12 | 0.12 | 0.03 | 0.03 | 0.03 | 0.03 | 0.12 |
| MBC | 0.03 | 0.24 | 0.24 | 0.06 | 0.06 | 0.06 | 0.06 | 0.24 | |
| 17 | MIC | 0.08 | 0.08 | 0.08 | 0.08 | 0.08 | 0.08 | 0.08 | 0.08 |
| MBC | 0.16 | 0.16 | 0.16 | 0.16 | 0.16 | 0.16 | 0.16 | 0.16 | |
| 18 | MIC | 0.03 | 0.06 | 0.06 | 0.03 | 0.06 | 0.03 | 0.03 | 0.24 |
| MBC | 0.06 | 0.12 | 0.12 | 0.06 | 0.12 | 0.06 | 0.06 | 0.48 | |
| 19 | MIC | 0.1 | 0.4 | 0.2 | 0.1 | 0.1 | 0.1 | 0.1 | 0.4 |
| MBC | 0.2 | 0.8 | 0.4 | 0.2 | 0.2 | 0.2 | 0.2 | 0.8 | |
| 20 | MIC | 0.03 | 0.03 | 0.03 | 0.03 | 0.03 | 0.03 | 0.03 | 0.12 |
| MBC | 0.06 | 0.06 | 0.06 | 0.06 | 0.06 | 0.06 | 0.06 | 0.24 | |
| 21 | MIC | 0.09 | 0.36 | 0.18 | 0.18 | 0.18 | 0.18 | 0.18 | 0.36 |
| MBC | 0.18 | 0.72 | 0.36 | 0.36 | 0.36 | 0.36 | 0.36 | 0.72 | |
| Streptomycin | MIC | 0.025 | 0.1 | 0.05 | 0.15 | 0.1 | 0.025 | 0.1 | 0.1 |
| MBC | 0.05 | 0.2 | 0.1 | 0.3 | 0.2 | 0.05 | 0.2 | 0.2 | |
| Ampicillin | MIC | 0.1 | 0.1 | 0.1 | 0.15 | 0.3 | 0.1 | 0.15 | 0.1 |
| MBC | 0.15 | 0.15 | 0.15 | 0.3 | 0.5 | 0.15 | 0.2 | 0.2 |
| Compounds | MRSA | P.a. | E.c. | |
|---|---|---|---|---|
| 5 | MIC | 0.09 | 0.18 | 0.09 |
| MBC | 0.18 | 0.36 | 0.18 | |
| 6 | MIC | 0.18 | 0.09 | 0.09 |
| MBC | 0.36 | 0.18 | 0.18 | |
| 8 | MIC | 0.36 | 0.18 | 0.09 |
| MBC | 0.72 | 0.36 | 0.18 | |
| 15 | MIC | 0.09 | 0.09 | 0.09 |
| MBC | 0.18 | 0.18 | 0.18 | |
| Streptomycin | MIC | 0.1 | 0.05 | 0.1 |
| MBC | / | 0.1 | 0.2 | |
| Ampicillin | MIC | / | 0.2 | 0.2 |
| MBC | / | / | / |
| Compound | FICI |
|---|---|
| 5 | 3 |
| 6 | 3 |
| 8 | 3 |
| 15 | 3 |
| Compounds | A.f. | A.n. | A.o. | A.v. | T.v. | P.f. | P.v.c. | P.o. | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | MIC | 0.03 | 0.06 | 0.03 | 0.03 | 0.06 | 0.03 | 0.06 | 0.03 |
| MFC | 0.06 | 0.12 | 0.06 | 0.06 | 0.12 | 0.06 | 0.12 | 0.06 | |
| 2 | MIC | 0.03 | 0.015 | 0.03 | 0.03 | 0.06 | 0.06 | 0.06 | 0.06 |
| MFC | 0.06 | 0.03 | 0.06 | 0.06 | 0.12 | 0.12 | 0.12 | 0.12 | |
| 3 | MIC | 0.06 | 0.06 | 0.06 | 0.06 | 0.06 | 0.06 | 0.12 | 0.12 |
| MFC | 0.12 | 0.12 | 0.12 | 0.12 | 0.12 | 0.12 | 0.24 | 0.24 | |
| 4 | MIC | 0.06 | 0.06 | 0.06 | 0.06 | 0.12 | 0.12 | 0.06 | 0.12 |
| MFC | 0.12 | 0.12 | 0.12 | 0.12 | 0.24 | 0.24 | 0.12 | 0.24 | |
| 5 | MIC | 0.01 | 0.01 | 0.01 | 0.01 | 0.02 | 0.02 | 0.02 | 0.02 |
| MFC | 0.02 | 0.02 | 0.02 | 0.02 | 0.04 | 0.04 | 0.04 | 0.04 | |
| 6 | MIC | 0.015 | 0.03 | 0.015 | 0.03 | 0.03 | 0.03 | 0.03 | 0.03 |
| MFC | 0.03 | 0.06 | 0.03 | 0.06 | 0.06 | 0.06 | 0.06 | 0.06 | |
| 7 | MIC | 0.15 | 0.15 | 0.15 | 0.15 | 0.3 | 0.3 | 0.3 | 0.15 |
| MFC | 0.3 | 0.3 | 0.3 | 0.3 | 0.6 | 0.6 | 0.6 | 0.3 | |
| 8 | MIC | 0.01 | 0.02 | 0.01 | 0.01 | 0.01 | 0.01 | 0.08 | 0.02 |
| MFC | 0.02 | 0.04 | 0.02 | 0.02 | 0.02 | 0.02 | 0.16 | 0.04 | |
| 9 | MIC | 0.02 | 0.08 | 0.04 | 0.02 | 0.02 | 0.02 | 0.08 | 0.08 |
| MFC | 0.04 | 0.16 | 0.08 | 0.04 | 0.04 | 0.04 | 0.16 | 0.16 | |
| 10 | MIC | 0.06 | 0.12 | 0.06 | 0.06 | 0.12 | 0.06 | 0.12 | 0.06 |
| MFC | 0.12 | 0.24 | 0.12 | 0.12 | 0.24 | 0.12 | 0.24 | 0.12 | |
| 11 | MIC | 0.03 | 0.06 | 0.06 | 0.06 | 0.12 | 0.12 | 0.12 | 0.12 |
| MFC | 0.06 | 0.12 | 0.12 | 0.12 | 0.24 | 0.24 | 0.24 | 0.24 | |
| 12 | MIC | 0.06 | 0.06 | 0.06 | 0.06 | 0.03 | 0.03 | 0.03 | 0.03 |
| MFC | 0.12 | 0.12 | 0.12 | 0.12 | 0.06 | 0.06 | 0.06 | 0.06 | |
| 13 | MIC | 0.06 | 0.06 | 0.03 | 0.06 | 0.03 | 0.06 | 0.12 | 0.06 |
| MFC | 0.12 | 0.12 | 0.06 | 0.12 | 0.06 | 0.12 | 0.24 | 0.12 | |
| 14 | MIC | 0.06 | 0.06 | 0.06 | 0.06 | 0.12 | 0.06 | 0.12 | 0.06 |
| MFC | 0.12 | 0.12 | 0.12 | 0.12 | 0.24 | 0.12 | 0.24 | 0.12 | |
| 15 | MIC | 0.03 | 0.03 | 0.03 | 0.06 | 0.06 | 0.03 | 0.12 | 0.03 |
| MFC | 0.06 | 0.06 | 0.06 | 0.12 | 0.12 | 0.06 | 0.24 | 0.06 | |
| 16 | MIC | 0.02 | 0.02 | 0.02 | 0.02 | 0.01 | 0.01 | 0.02 | 0.02 |
| MFC | 0.04 | 0.04 | 0.04 | 0.04 | 0.02 | 0.02 | 0.04 | 0.04 | |
| 17 | MIC | 0.02 | 0.08 | 0.02 | 0.02 | 0.08 | 0.04 | 0.08 | 0.04 |
| MFC | 0.04 | 0.16 | 0.04 | 0.04 | 0.16 | 0.08 | 0.16 | 0.08 | |
| 18 | MIC | 0.04 | 0.08 | 0.04 | 0.08 | 0.16 | 0.08 | 0.16 | 0.08 |
| MFC | 0.08 | 0.16 | 0.08 | 0.16 | 0.32 | 0.16 | 0.32 | 0.16 | |
| 19 | MIC | 0.24 | 0.24 | 0.24 | 0.24 | 0.48 | 0.48 | 0.48 | 0.48 |
| MFC | 0.48 | 0.48 | 0.48 | 0.48 | 0.96 | 0.96 | 0.96 | 0.96 | |
| 20 | MIC | 0.03 | 0.03 | 0.03 | 0.03 | 0.06 | 0.06 | 0.06 | 0.06 |
| MFC | 0.06 | 0.06 | 0.06 | 0.06 | 0.12 | 0.12 | 0.12 | 0.12 | |
| 21 | MIC | 0.09 | 0.09 | 0.09 | 0.09 | 0.09 | 0.18 | 0.18 | 0.18 |
| MFC | 0.18 | 0.18 | 0.18 | 0.18 | 0.18 | 0.36 | 0.36 | 0.36 | |
| Bifonazole | MIC | 0.15 | 0.15 | 0.15 | 0.1 | 0.15 | 0.2 | 0.1 | 0.2 |
| MFC | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 0.25 | 0.2 | 0.25 | |
| Ketoconazole | MIC | 0.2 | 0.2 | 0.15 | 0.2 | 1 | 0.2 | 0.2 | 1 |
| MFC | 0.5 | 0.5 | 0.2 | 0.5 | 1.5 | 0.5 | 0.3 | 1.5 |
| N. | Est. Binding Energy (kcal/mol) | I-H E. coli MurB | Residues E. coli MurB | |||
|---|---|---|---|---|---|---|
| Gyrase 1KZN (R-isomer) | Thymidylate Kinase 4QGG (R-isomer) | E. coli MurB 2Q85 (S-isomer) | E. coli MurB 2Q85 (R-isomer) | |||
| 1 | −5.81 | −3.11 | −7.52 | −8.25 | 2 | Gly122, Ser228 |
| 2 | −1.79 | −2.51 | −5.26 | −6.15 | 1 | Ser228 |
| 3 | −4.36 | - | −6.14 | −7.11 | 2 | Gly122, Ser228 |
| 4 | −4.22 | - | −7.06 | −7.68 | 2 | Arg158 |
| 5 | −5.26 | −2.55 | −8.66 | −10.21 | 3 | Arg158, Ser228, Gln287 |
| 6 | −4.19 | −1.25 | −6.98 | −8.27 | 2 | Arg158, Ser228 |
| 7 | −1.46 | - | −5.24 | −6.11 | 1 | Ser228 |
| 8 | −5.87 | −3.11 | −7.83 | −9.15 | 3 | Gly125, Val126, Ser228 |
| 9 | −1.96 | −1.11 | −4.28 | −5.17 | 1 | Tyr189 |
| 10 | −4.09 | - | −6.52 | −7.70 | 2 | Gly122, Ser228 |
| 11 | −3.32 | −1.22 | −5.49 | −6.55 | 2 | Gly122, Ser228 |
| 12 | −4.79 | −2.16 | −7.26 | −8.22 | 2 | Arg158, Ser228 |
| 13 | −2.71 | −1.27 | −5.18 | −6.88 | 2 | Ser228, Arg213 |
| 14 | −1.25 | - | −3.28 | −6.62 | 1 | Ser228 |
| 15 | −4.27 | −2.51 | −8.80 | −10.08 | 3 | Arg158, Ser228, Asn232 |
| 16 | −3.34 | −1.28 | −6.29 | −8.07 | 2 | Ser228, Ala226 |
| 17 | −2.47 | −1.63 | −6.33 | −7.52 | 2 | Arg158, Asn232 |
| 18 | −3.18 | - | −6.17 | −7.79 | 2 | Arg158, Tyr189 |
| 19 | −2.87 | - | −4.11 | −5.16 | 1 | Arg158 |
| 20 | −1.27 | - | −3.03 | −5.33 | 1 | Asn232 |
| 21 | −2.10 | −1.25 | −4.57 | −5.74 | 1 | |
| N. | Bacteria (Structure) | Est. Binding Energy (kcal/mol) | I-H | Residues | Pi-Interactions |
|---|---|---|---|---|---|
| P.a. (4JAY) | −10.18 | 2 | Arg166, Ser239 | Ala131, Pro118, Leu228, Arg224 | |
| 5 | L.m. (3TX1) | −10.03 | 2 | Arg224 | Tyr169, Lys223, Arg224, Pro226, Glu228 |
| S.a. (1HSK) | −10.95 | 2 | Arg188, Arg242 | Arg188, Tyr187, Arg242, His271 | |
| P.a. (4JAY) | −9.02 | 2 | Arg224, Lys272 | Ala131, Arg166, Ser239, Tyr196, Glu335 | |
| 8 | L.m. (3TX1) | −8.77 | 2 | Gln211, Ser220 | Ile122, Ala136, Arg207, Arg224 |
| S.a. (1HSK) | −7.13 | 1 | Arg242 | Arg188, Tyr187, Arg242 | |
| P.a. (4JAY) | −9.11 | 2 | Gly164, Lys242 | Arg166, Leu245, Asp268, Arg270 | |
| 15 | L.m. (3TX1) | −8.23 | 2 | Arg170, Ser220 | Met132, Ala134, Arg170, Glu290 |
| S.a. (1HSK) | −10.14 | 3 | Ser238, Lys250, His271 | Ala155, Tyr155, Arg188, Phe247, Lys250, His271 |
| Est. Binding Energy | |||||
|---|---|---|---|---|---|
| N. | Dihydrofolate Reductase 4HOF | CYP51 C. albicans 5V5Z | I-H | Residues | Interactions with Heme HEM601 |
| 1 | −3.61 | −7.39 | 1 | Tyr132 | Hydrophobic |
| 2 | −5.19 | −10.17 | 1 | Ser378 | Hydrophobic |
| 3 | −3.11 | −8.58 | 1 | Tyr64 | Hydrophobic |
| 4 | −4.15 | −7.58 | 1 | Tyr132 | Hydrophobic |
| 5 | −6.28 | −13.93 | 2 | Tyr118, Ser378 | p. ionizable, Hydrophobic |
| 6 | −4.27 | −11.05 | 1 | Tyr118 | Hydrophobic |
| 7 | −1.25 | −7.22 | 1 | Tyr118 | Hydrophobic |
| 8 | −5.36 | −11.13 | 1 | Tyr132 | Hydrophobic |
| 9 | - | −8.72 | 1 | Tyr64 | Hydrophobic |
| 10 | −2.69 | −8.21 | 1 | Tyr118 | Hydrophobic |
| 11 | −3.55 | −9.06 | 1 | Hem601 | H-bond, Hydrophobic |
| 12 | −5.15 | −9.75 | 1 | Tyr132 | Hydrophobic, aromatic |
| 13 | −5.22 | −9.22 | 1 | Ser378 | Hydrophobic |
| 14 | −4.16 | −9.15 | 1 | Tyr132 | Hydrophobic |
| 15 | −4.28 | −9.23 | 1 | Tyr118 | Hydrophobic |
| 16 | −6.12 | −1.98 | 2 | Tyr132, Hem601 | H-bond, Hydrophobic |
| 17 | −2.88 | −10.48 | 1 | Tyr132 | Hydrophobic, Aromatic |
| 18 | −1.24 | −8.6 | 1 | Tyr118 | Hydrophobic |
| 19 | −1.87 | −5.14 | - | - | Hydrophobic |
| 20 | −2.14 | −6.17 | 1 | Tyr118 | Hydrophobic |
| 21 | −1.34 | −5.82 | - | - | Hydrophobic |
| Ketoconazole | −6.75 | −8.23 | 1 | Tyr64 | Hydrophobic, Aromatic |
| No. | MW | Number of HBA a | Number of HBD b | Log Po/w (iLOGP) c | Log S d | TPSA e | Lipinski, Ghose, Veber, Egan, and Muegge Violations | Bioavailability Score | Drug-Likeness Model Score |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 348.39 | 4 | 0 | 3.09 | Moderately soluble | 86.74 | 0 | 0.55 | −0.18 |
| 2 | 375.40 | 5 | 0 | 2.28 | Moderately soluble | 132.56 | 0 | 0.55 | −0.22 |
| 3 | 360.43 | 4 | 0 | 3.19 | Moderately soluble | 95.97 | 0 | 0.55 | 0.09 |
| 4 | 364.84 | 3 | 0 | 3.10 | Moderately soluble | 86.74 | 0 | 0.55 | 0.38 |
| 5 | 391.85 | 4 | 0 | 2.02 | Moderately soluble | 132.56 | 0 | 0.55 | 0.03 |
| 6 | 376.88 | 3 | 0 | 3.23 | Poorly soluble | 103.23 | 0 | 0.55 | 0.34 |
| 7 | 360.43 | 4 | 0 | 3.02 | Moderately soluble | 95.97 | 0 | 0.55 | 0.28 |
| 8 | 387.43 | 5 | 0 | 2.49 | Moderately soluble | 141.79 | 0 | 0.55 | −0.17 |
| 9 | 376.88 | 3 | 0 | 3.10 | Poorly soluble | 95.97 | 0 | 0.55 | 0.39 |
| 10 | 372.46 | 4 | 0 | 3.45 | Moderately soluble | 105.20 | 0 | 0.55 | −0.06 |
| 11 | 358.43 | 4 | 1 | 2.56 | Moderately soluble | 116.30 | 0 | 0.55 | 0.13 |
| 12 | 374.45 | 4 | 0 | 3.25 | Poorly soluble | 95.97 | 0 | 0.55 | 0.30 |
| 13 | 401.46 | 5 | 0 | 4.32 | Moderately soluble | 141.79 | 0 | 0.55 | −0.17 |
| 14 | 390.19 | 3 | 0 | 3.41 | Moderately soluble | 95.97 | 0 | 0.55 | 0.42 |
| 15 | 403.50 | 5 | 1 | 3.27 | Moderately soluble | 125.43 | 0 | 0.55 | 0.03 |
| 16 | 372.46 | 4 | 1 | 2.84 | Moderately soluble | 116.20 | 0 | 0.55 | 0.14 |
| 17 | 449.30 | 5 | 0 | 3.19 | Moderately soluble | 86.74 | 1 | 0.55 | −0.31 |
| 18 | 416.39 | 7 | 0 | 3.03 | Moderately soluble | 86.74 | 0 | 0.55 | −0.38 |
| 19 | 432.84 | 6 | 0 | 3.13 | Moderately soluble | 86.74 | 0 | 0.55 | −0.32 |
| 20 | 425.40 | 7 | 0 | 2.44 | Moderately soluble | 132.56 | 0 | 0.55 | −0.40 |
| 21 | 398.40 | 6 | 0 | 3.18 | Moderately soluble | 86.74 | 0 | 0.55 | −0.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tratrat, C.; Petrou, A.; Geronikaki, A.; Ivanov, M.; Kostić, M.; Soković, M.; Vizirianakis, I.S.; Theodoroula, N.F.; Haroun, M. Thiazolidin-4-Ones as Potential Antimicrobial Agents: Experimental and In Silico Evaluation. Molecules 2022, 27, 1930. https://doi.org/10.3390/molecules27061930
Tratrat C, Petrou A, Geronikaki A, Ivanov M, Kostić M, Soković M, Vizirianakis IS, Theodoroula NF, Haroun M. Thiazolidin-4-Ones as Potential Antimicrobial Agents: Experimental and In Silico Evaluation. Molecules. 2022; 27(6):1930. https://doi.org/10.3390/molecules27061930
Chicago/Turabian StyleTratrat, Christophe, Anthi Petrou, Athina Geronikaki, Marija Ivanov, Marina Kostić, Marina Soković, Ioannis S. Vizirianakis, Nikoleta F. Theodoroula, and Michelyne Haroun. 2022. "Thiazolidin-4-Ones as Potential Antimicrobial Agents: Experimental and In Silico Evaluation" Molecules 27, no. 6: 1930. https://doi.org/10.3390/molecules27061930