
Fullerenes against COVID-19: Repurposing C60 and C70 to Clog the Active Site of SARS-CoV-2 Protease


Abstract
:1. Introduction
2. Results
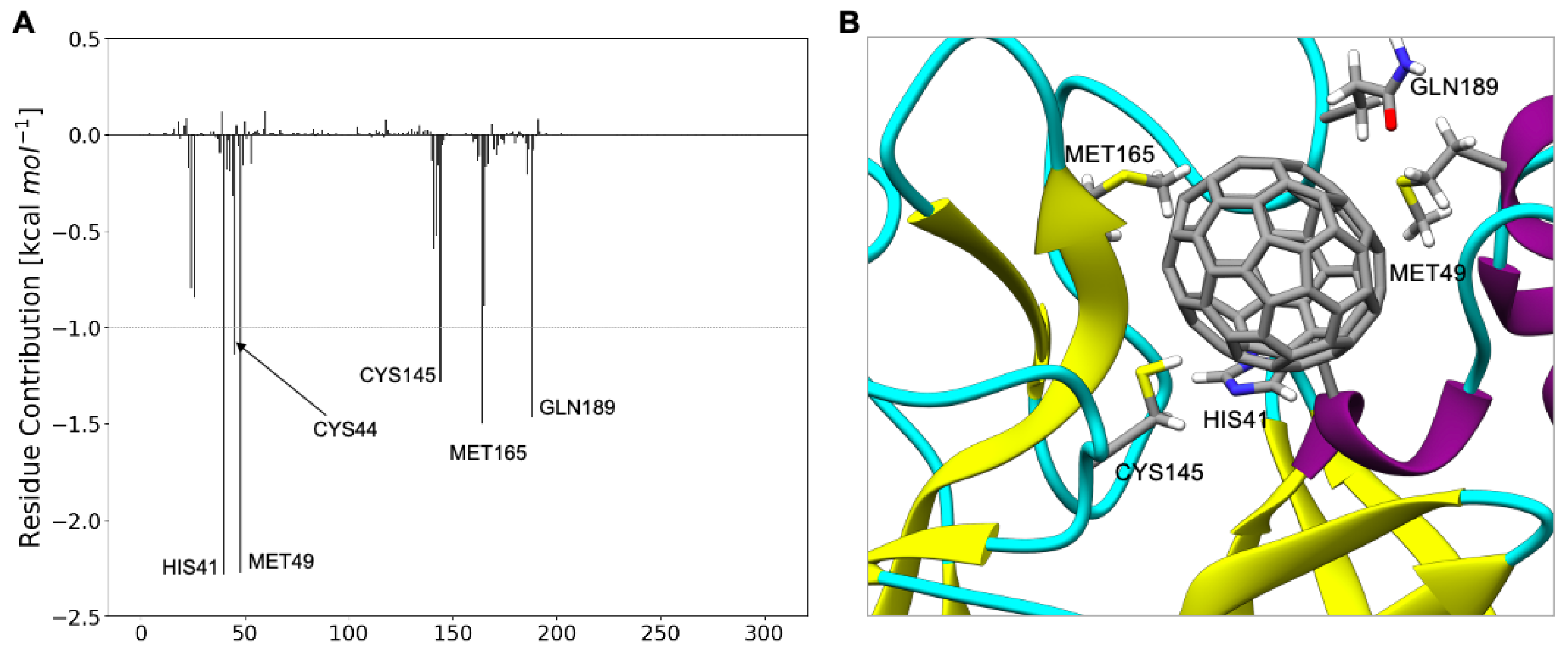
2.1. Determining the C60 Binding Pocket in SARS-CoV-2 Mpro
2.2. Determining the Binding Energy between C60 and SARS-CoV-2 Mpro
2.3. Comparing the Binding of C60 in SARS-CoV-2 Mpro and HIV Protease
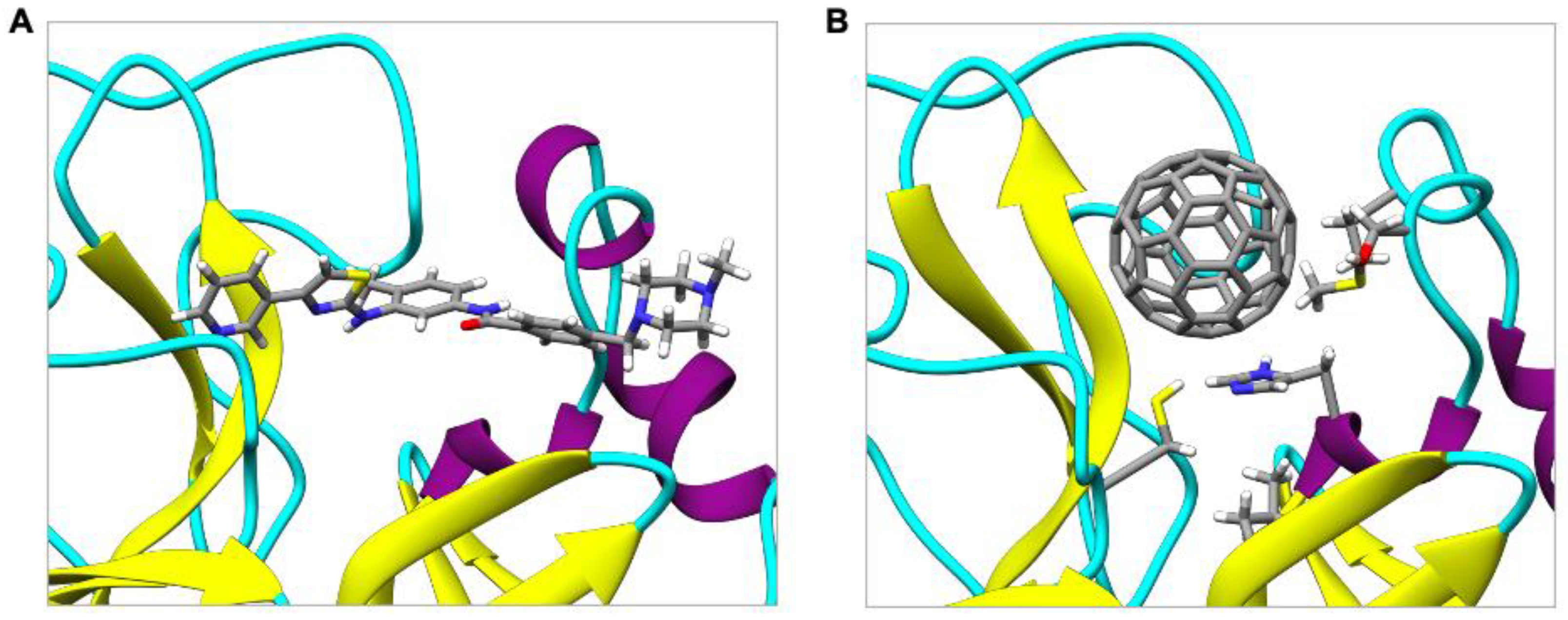
2.4. Comparing the Binding of C60 in SARS-CoV-2 Mpro with Masitinib
2.5. Determining the Binding Energy between C70 and SARS-CoV-2 Mpro
2.6. Evaluating the Binding Enrgy of Masitinib, C60, and C70 to SARS-CoV-2 Mpro in Different Protonation States of the Catalytic Dyad
3. Conclusions
4. Materials and Methods
4.1. System Setup
4.2. Docking
4.3. MD Simulations
4.4. Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) Analysis
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Li, J.; Lai, S.; Gao, G.F.; Shi, W. The emergence, genomic diversity and global spread of SARS-CoV-2. Nature 2021, 600, 408–418. [Google Scholar] [CrossRef]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Kneller, D.W.; Phillips, G.; O’Neill, H.M.; Jedrzejczak, R.; Stols, L.; Langan, P.; Joachimiak, A.; Coates, L.; Kovalevsky, A. Structural plasticity of SARS-CoV-2 3CL Mpro active site cavity revealed by room temperature X-ray crystallography. Nat. Commun. 2020, 11, 7–12. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved a-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Yang, L.; Zhao, X.E. Co-crystallization and structure determination: An effective direction for anti-SARS-CoV-2 drug discovery. Comput. Struct. Biotechnol. J. 2021, 19, 4684–4701. [Google Scholar] [CrossRef]
- Harrison, C. Coronavirus puts drug repurposing on the fast track. Nat. Biotechnol. 2020, 38, 379–381. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, A.; Mandal, K. Repurposing an Antiviral Drug against SARS-CoV-2 Main Protease. Angew. Chem. Int. Ed. 2021, 60, 23492–23494. [Google Scholar] [CrossRef]
- Gupta, A.; Zhou, H.X. Profiling SARS-CoV-2 Main Protease (MPRO) Binding to Repurposed Drugs Using Molecular Dynamics Simulations in Classical and Neural Network-Trained Force Fields. ACS Comb. Sci. 2020, 22, 826–832. [Google Scholar] [CrossRef]
- Kuzikov, M.; Costanzi, E.; Reinshagen, J.; Esposito, F.; Vangeel, L.; Wolf, M.; Ellinger, B.; Claussen, C.; Geisslinger, G.; Corona, A.; et al. Identification of Inhibitors of SARS-CoV-2 3CL-Pro Enzymatic Activity Using a Small Molecule in Vitro Repurposing Screen. ACS Pharmacol. Transl. Sci. 2021, 4, 1096–1110. [Google Scholar] [CrossRef]
- Hasan, M.; Parvez, M.S.A.; Azim, K.F.; Imran, M.A.S.; Raihan, T.; Gulshan, A.; Muhit, S.; Akhand, R.N.; Ahmed, S.S.U.; Uddin, M.B. Main protease inhibitors and drug surface hotspots for the treatment of COVID-19: A drug repurposing and molecular docking approach. Biomed. Pharmacother. 2021, 140, 111742. [Google Scholar] [CrossRef]
- Pinzi, L.; Tinivella, A.; Caporuscio, F.; Rastelli, G. Drug Repurposing and Polypharmacology to Fight SARS-CoV-2 Through Inhibition of the Main Protease. Front. Pharmacol. 2021, 12, 636989. [Google Scholar] [CrossRef]
- Macip, G.; Garcia-Segura, P.; Mestres-Truyol, J.; Saldivar-Espinoza, B.; Ojeda-Montes, M.J.; Gimeno, A.; Cereto-Massagué, A.; Garcia-Vallvé, S.; Pujadas, G. Haste makes waste: A critical review of docking-based virtual screening in drug repurposing for SARS-CoV-2 main protease (M-pro) inhibition. Med. Res. Rev. 2022, 42, 744–769. [Google Scholar] [CrossRef]
- Zhu, W.; Shyr, Z.; Lo, D.C.; Zheng, W. Viral proteases as targets for coronavirus disease 2019 drug development. J. Pharmacol. Exp. Ther. 2021, 378, 166–172. [Google Scholar] [CrossRef]
- Di Giosia, M.; Nicolini, F.; Ferrazzano, L.; Soldà, A.; Valle, F.; Cantelli, A.; Marforio, T.D.; Bottoni, A.; Zerbetto, F.; Montalti, M.; et al. Stable and Biocompatible Monodispersion of C 60 in Water by Peptides. Bioconjug. Chem. 2019, 30, 808–814. [Google Scholar] [CrossRef]
- Rozhin, P.; Charitidis, C.; Marchesan, S. Self-assembling peptides and carbon nanomaterials join forces for innovative biomedical applications. Molecules 2021, 26, 4084. [Google Scholar] [CrossRef]
- Calvaresi, M.; Zerbetto, F. Baiting proteins with C60. ACS Nano 2010, 4, 2283–2299. [Google Scholar] [CrossRef]
- Calvaresi, M.; Zerbetto, F. Fullerene sorting proteins. Nanoscale 2011, 3, 2873–2881. [Google Scholar] [CrossRef]
- De Leo, F.; Magistrato, A.; Bonifazi, D. Interfacing proteins with graphitic nanomaterials: From spontaneous attraction to tailored assemblies. Chem. Soc. Rev. 2015, 44, 6916–6953. [Google Scholar] [CrossRef]
- Calvaresi, M.; Zerbetto, F. The Devil and Holy Water: Protein and Carbon Nanotube Hybrids. Acc. Chem. Res. 2013, 46, 2454–2463. [Google Scholar] [CrossRef]
- Marchesan, S.; Prato, M. Under the lens: Carbon nanotube and protein interaction at the nanoscale. Chem. Commun. 2015, 51, 4347–4359. [Google Scholar] [CrossRef]
- Di Giosia, M.; Valle, F.; Cantelli, A.; Bottoni, A.; Zerbetto, F.; Fasoli, E.; Calvaresi, M. High-throughput virtual screening to rationally design protein—Carbon nanotube interactions. Identification and preparation of stable water dispersions of protein—Carbon nanotube hybrids and efficient design of new functional materials. Carbon N. Y. 2019, 147, 70–82. [Google Scholar] [CrossRef]
- Di Giosia, M.; Marforio, T.D.; Cantelli, A.; Valle, F.; Zerbetto, F.; Su, Q.; Wang, H.; Calvaresi, M. Inhibition of α-chymotrypsin by pristine single-wall carbon nanotubes: Clogging up the active site. J. Colloid Interface Sci. 2020, 571, 174–184. [Google Scholar] [CrossRef]
- Rozhin, P.; Abdel Monem Gamal, J.; Giordani, S.; Marchesan, S. Carbon Nanomaterials (CNMs) and Enzymes: From Nanozymes to CNM-Enzyme Conjugates and Biodegradation. Materials 2022, 15, 1037. [Google Scholar] [CrossRef]
- Di Costanzo, L.; Geremia, S. Atomic details of carbon-based nanomolecules interacting with proteins. Molecules 2020, 25, 3555. [Google Scholar] [CrossRef]
- Reina, G.; Peng, S.; Jacquemin, L.; Andrade, A.F.; Bianco, A. Hard Nanomaterials in Time of Viral Pandemics. ACS Nano 2020, 14, 9364–9388. [Google Scholar] [CrossRef]
- Serrano-Aroca, Á.; Takayama, K.; Tuñón-Molina, A.; Seyran, M.; Hassan, S.S.; Pal Choudhury, P.; Uversky, V.N.; Lundstrom, K.; Adadi, P.; Palù, G.; et al. Carbon-Based Nanomaterials: Promising Antiviral Agents to Combat COVID-19 in the Microbial-Resistant Era. ACS Nano 2021, 15, 8069–8086. [Google Scholar] [CrossRef]
- Mallakpour, S.; Azadi, E.; Hussain, C.M. Fight against COVID-19 pandemic with the help of carbon-based nanomaterials. New J. Chem. 2021, 45, 8832–8846. [Google Scholar] [CrossRef]
- Innocenzi, P.; Stagi, L. Carbon-based antiviral nanomaterials: Graphene, C-dots, and fullerenes. A perspective. Chem. Sci. 2020, 11, 6606–6622. [Google Scholar] [CrossRef]
- Sengupta, J.; Hussain, C.M. Carbon nanomaterials to combat virus: A perspective in view of COVID-19. Carbon Trends 2021, 2, 100019. [Google Scholar] [CrossRef]
- Khan, A.; Ahsan, O.; Wei, D.Q.; Ansari, J.K.; Najmi, M.H.; Muhammad, K.; Waheed, Y. Computational evaluation of abrogation of hbx-bcl-xl complex with high-affinity carbon nanotubes (Fullerene) to halt the hepatitis b virus replication. Molecules 2021, 26, 6433. [Google Scholar] [CrossRef]
- Friedman, S.H.; DeCamp, D.L.; Kenyon, G.L.; Sijbesma, R.P.; Srdanov, G.; Wudl, F. Inhibition of the HIV-1 Protease by Fullerene Derivatives: Model Building Studies and Experimental Verification. J. Am. Chem. Soc. 1993, 115, 6506–6509. [Google Scholar] [CrossRef]
- Sijbesma, R.; Srdanov, G.; Wudl, F.; Castoro, J.A.; Wilkins, C.; Friedman, S.H.; Decamp, D.L.; Kenyon, G.L. Synthesis of a Fullerene Derivative for the Inhibition of HIV Enzymes. J. Am. Chem. Soc. 1993, 6510–6512. [Google Scholar] [CrossRef]
- Schinazi, R.F.; Sijbesma, R.; Srdanov, G.; Hill, C.L.; Wudl, F. Synthesis and virucidal activity of a water-soluble, configurationally stable, derivatized C60 fullerene. Antimicrob. Agents Chemother. 1993, 37, 1707–1710. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, E.; Tokuyama, H.; Yamago, S.; Shiraki, T.; Sugiura, Y. Biological activity of water-soluble fullerenes. Structural dependence of DNA cleavage, cytotoxicity, and enzyme inhibitory activities including HIV-protease inhibition. Bull. Chem. Soc. Jpn. 1996, 69, 2143–2151. [Google Scholar] [CrossRef]
- Friedman, S.H.; Ganapathi, P.S.; Rubin, Y.; Kenyon, G.L. Optimizing the Binding of Fullerene Inhibitors of the HIV-1 Protease through Predicted Increases in Hydrophobic Desolvation. J. Med. Chem. 1998, 41, 2424–2429. [Google Scholar] [CrossRef]
- Luca Marcorin, G.; Da Ros, T.; Castellano, S.; Stefancich, G.; Bonin, I.; Miertus, S.; Prato, M. Design and synthesis of novel [60] fullerene derivatives as potential HIV aspartic protease inhibitors. Org. Lett. 2000, 2, 3955–3957. [Google Scholar] [CrossRef]
- Bosi, S.; Da Ros, T.; Spalluto, G.; Balzarini, J.; Prato, M. Synthesis and anti-HIV properties of new water-soluble bis- functionalized[60]fullerene derivatives. Bioorg. Med. Chem. Lett. 2003, 13, 4437–4440. [Google Scholar] [CrossRef]
- Marchesan, S.; Da Ros, T.; Spalluto, G.; Balzarini, J.; Prato, M. Anti-HIV properties of cationic fullerene derivatives. Bioorg. Med. Chem. Lett. 2005, 15, 3615–3618. [Google Scholar] [CrossRef]
- Troshina, O.A.; Troshin, P.A.; Peregudov, A.S.; Kozlovskiy, V.I.; Balzarini, J.; Lyubovskaya, R.N. Chlorofullerene C60Cl6: A precursor for straightforward preparation of highly water-soluble polycarboxylic fullerene derivatives active against HIV. Org. Biomol. Chem. 2007, 5, 2783–2791. [Google Scholar] [CrossRef]
- Strom, T.A.; Durdagi, S.; Ersoz, S.S.; Salmas, R.E.; Supuran, C.T.; Barron, A.R. Fullerene-based inhibitors of HIV-1 protease. J. Pept. Sci. 2015, 21, 862–870. [Google Scholar] [CrossRef]
- Yasuno, T.; Ohe, T.; Kataoka, H.; Hashimoto, K.; Ishikawa, Y.; Furukawa, K.; Tateishi, Y.; Kobayashi, T.; Takahashi, K.; Nakamura, S.; et al. Fullerene derivatives as dual inhibitors of HIV-1 reverse transcriptase and protease. Bioorg. Med. Chem. Lett. 2021, 31, 127675. [Google Scholar] [CrossRef]
- Kobayashi, T.; Yasuno, T.; Takahashi, K.; Nakamura, S.; Mashino, T.; Ohe, T. Novel pyridinium-type fullerene derivatives as multitargeting inhibitors of HIV-1 reverse transcriptase, HIV-1 protease, and HCV NS5B polymerase. Bioorg. Med. Chem. Lett. 2021, 49, 128267. [Google Scholar] [CrossRef]
- Hurmach, V.V.; Platonov, M.O.; Prylutska, S.V.; Scharff, P.; Prylutskyy, Y.I.; Ritter, U. C60 fullerene against SARS-CoV-2 coronavirus: An in silico insight. Sci. Rep. 2021, 11, 17748. [Google Scholar] [CrossRef]
- Suárez, M.; Makowski, K.; Lemos, R.; Almagro, L.; Rodríguez, H.; Herranz, M.Á.; Molero, D.; Ortiz, O.; Maroto, E.; Albericio, F.; et al. An Androsterone-H2@C60 hybrid: Synthesis, Properties and Molecular Docking Simulations with SARS-Cov-2. Chempluschem 2021, 86, 972–981. [Google Scholar] [CrossRef]
- Skariyachan, S.; Gopal, D.; Deshpande, D.; Joshi, A.; Uttarkar, A.; Niranjan, V. Carbon fullerene and nanotube are probable binders to multiple targets of SARS-CoV-2: Insights from computational modeling and molecular dynamic simulation studies. Infect. Genet. Evol. 2021, 96, 105155. [Google Scholar] [CrossRef]
- Drayman, N.; DeMarco, J.K.; Jones, K.A.; Azizi, S.A.; Froggatt, H.M.; Tan, K.; Maltseva, N.I.; Chen, S.; Nicolaescu, V.; Dvorkin, S.; et al. Masitinib is a broad coronavirus 3CL inhibitor that blocks replication of SARS-CoV-2. Science 2021, 373, 931–936. [Google Scholar] [CrossRef]
- Calvaresi, M.; Arnesano, F.; Bonacchi, S.; Bottoni, A.; Calò, V.; Conte, S.; Falini, G.; Fermani, S.; Losacco, M.; Montalti, M.; et al. C60@Lysozyme: Direct observation by nuclear magnetic resonance of a 1:1 fullerene protein adduct. ACS Nano 2014, 8, 1871–1877. [Google Scholar] [CrossRef]
- Calvaresi, M.; Bottoni, A.; Zerbetto, F. Thermodynamics of Binding between Proteins and Carbon Nanoparticles: The Case of C60@Lysozyme. J. Phys. Chem. C 2015, 119, 28077–28082. [Google Scholar] [CrossRef] [Green Version]
- Calvaresi, M.; Furini, S.; Domene, C.; Bottoni, A.; Zerbetto, F. Blocking the passage: C60 geometrically clogs K+ channels. ACS Nano 2015, 9, 4827–4834. [Google Scholar] [CrossRef] [Green Version]
- Trozzi, F.; Marforio, T.D.; Bottoni, A.; Zerbetto, F.; Calvaresi, M. Engineering the Fullerene-protein Interface by Computational Design: The Sum is More than its Parts. Isr. J. Chem. 2017, 57, 547–552. [Google Scholar] [CrossRef]
- di Giosia, M.; Valle, F.; Cantelli, A.; Bottoni, A.; Zerbetto, F.; Calvaresi, M. C60 bioconjugation with proteins: Towards a palette of carriers for all pH ranges. Materials 2018, 11, 691. [Google Scholar] [CrossRef] [Green Version]
- Bologna, F.; Mattioli, E.J.; Bottoni, A.; Zerbetto, F.; Calvaresi, M. Interactions between Endohedral Metallofullerenes and Proteins: The Gd@C60–Lysozyme Model. ACS Omega 2018, 3, 13782–13789. [Google Scholar] [CrossRef] [Green Version]
- Ganazzoli, F.; Raffaini, G. Classical atomistic simulations of protein adsorption on carbon nanomaterials. Curr. Opin. Colloid Interface Sci. 2019, 41, 11–26. [Google Scholar] [CrossRef]
- Marforio, T.D.; Calza, A.; Mattioli, E.J.; Zerbetto, F.; Calvaresi, M. Dissecting the supramolecular dispersion of fullerenes by proteins/peptides: Amino acid ranking and driving forces for binding to c60. Int. J. Mol. Sci. 2021, 22, 11567. [Google Scholar] [CrossRef]
- Di Giosia, M.; Zerbetto, F.; Calvaresi, M. Incorporation of Molecular Nanoparticles Inside Proteins: The Trojan Horse Approach in Theranostics. Acc. Mater. Res. 2021, 8, 594–605. [Google Scholar] [CrossRef]
- Di Giosia, M.; Bomans, P.H.H.; Bottoni, A.; Cantelli, A.; Falini, G.; Franchi, P.; Guarracino, G.; Friedrich, H.; Lucarini, M.; Paolucci, F.; et al. Proteins as supramolecular hosts for C60: A true solution of C60 in water. Nanoscale 2018, 10, 9908–9916. [Google Scholar] [CrossRef] [Green Version]
- Soldà, A.; Cantelli, A.; Di Giosia, M.; Montalti, M.; Zerbetto, F.; Rapino, S.; Calvaresi, M. C60@lysozyme: A new photosensitizing agent for photodynamic therapy. J. Mater. Chem. B 2017, 5, 6608–6615. [Google Scholar] [CrossRef]
- Di Giosia, M.; Soldà, A.; Seeger, M.; Cantelli, A.; Arnesano, F.; Nardella, M.I.; Mangini, V.; Valle, F.; Montalti, M.; Zerbetto, F.; et al. A Bio-Conjugated Fullerene as a Subcellular-Targeted and Multifaceted Phototheranostic Agent. Adv. Funct. Mater. 2021, 31, 2101527. [Google Scholar] [CrossRef]
- Martínez-Ortega, U.; Figueroa-Figueroa, D.I.; Hernández-Luis, F.; Aguayo-Ortiz, R. In Silico Characterization of Masitinib Interaction with SARS-CoV-2 Main Protease. ChemMedChem 2021, 16, 2339–2344. [Google Scholar] [CrossRef]
- Wu, X.; Yang, S.T.; Wang, H.; Wang, L.; Hu, W.; Cao, A.; Liu, Y. Influences of the size and hydroxyl number of fullerenes/fullerenols on their interactions with proteins. J. Nanosci. Nanotechnol. 2010, 10, 6298–6304. [Google Scholar] [CrossRef]
- Luo, S.; Huang, K.; Zhao, X.; Cong, Y.; Zhang, J.Z.H.; Duan, L. Inhibition mechanism and hot-spot prediction of nine potential drugs for SARS-CoV-2 Mproby large-scale molecular dynamic simulations combined with accurate binding free energy calculations. Nanoscale 2021, 13, 8313–8332. [Google Scholar] [CrossRef]
- Pavlova, A.; Lynch, D.L.; Daidone, I.; Zanetti-Polzi, L.; Smith, M.D.; Chipot, C.; Kneller, D.W.; Kovalevsky, A.; Coates, L.; Golosov, A.A.; et al. Inhibitor binding influences the protonation states of histidines in SARS-CoV-2 main protease. Chem. Sci. 2021, 12, 1513–1527. [Google Scholar] [CrossRef]
- Kneller, D.W.; Phillips, G.; Weiss, K.L.; Zhang, Q.; Coates, L.; Kovalevsky, A. Direct Observation of Protonation State Modulation in SARS-CoV-2 Main Protease upon Inhibitor Binding with Neutron Crystallography. J. Med. Chem. 2021, 64, 4991–5000. [Google Scholar] [CrossRef]
- Chan, H.T.H.; Moesser, M.A.; Walters, R.K.; Malla, T.R.; Twidale, R.M.; John, T.; Deeks, H.M.; Johnston-Wood, T.; Mikhailov, V.; Sessions, R.B.; et al. Discovery of SARS-CoV-2 Mpropeptide inhibitors from modelling substrate and ligand binding. Chem. Sci. 2021, 12, 13686–13703. [Google Scholar] [CrossRef]
- Kneller, D.W.; Phillips, G.; Weiss, K.L.; Pant, S.; Zhang, Q.; O’Neill, H.M.; Coates, L.; Kovalevsky, A. Unusual zwitterionic catalytic site of SARS-CoV-2 main protease revealed by neutron crystallography. J. Biol. Chem. 2020, 295, 17365–17373. [Google Scholar] [CrossRef]
- Ramos-Guzmán, C.A.; Ruiz-Pernía, J.J.; Tuñón, I. Unraveling the SARS-CoV-2 Main Protease Mechanism Using Multiscale Methods. ACS Catal. 2020, 10, 12544–12554. [Google Scholar] [CrossRef]
- Arafet, K.; Serrano-Aparicio, N.; Lodola, A.; Mulholland, A.J.; González, F.V.; Świderek, K.; Moliner, V. Mechanism of inhibition of SARS-CoV-2 MprobyN3peptidyl Michael acceptor explained by QM/MM simulations and design of new derivatives with tunable chemical reactivity. Chem. Sci. 2021, 12, 1433–1444. [Google Scholar] [CrossRef]
- Maier, J.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Betz, R.M.; Botello-Smith, W.; Cerutti, D.S.; Cheatham, I.T.E.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; et al. Amber 16; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, 363–367. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | ΔH | VDW | EEl | EGB | Enon-polar | TΔS | ΔGbind |
|---|---|---|---|---|---|---|---|
| C60@Mpro | −34.5 | −44.8 | 0.0 | 12.8 | −2.5 | −15.7 | −18.8 |
| C60@ProHIV | −38.0 | −51.6 | 0.0 | 16.0 | −2.4 | −18.3 | −19.7 |
| Complex | ΔH | VDW | EEl | EGB | Enon-polar | TΔS | ΔGbind |
|---|---|---|---|---|---|---|---|
| C60@Mpro | −34.5 | −44.8 | 0.0 | 12.8 | −2.5 | −15.7 | −18.8 |
| masitinib@Mpro | −41.2 | −51.4 | −18.1 | 34.0 | −5.8 | −24.7 | −16.5 |
| Complex | ΔH | VDW | EEl | EGB | Enon-polar | TΔS | ΔGbind |
|---|---|---|---|---|---|---|---|
| C60@Mpro | −34.5 | −44.8 | 0.0 | 12.8 | −2.5 | −15.7 | −18.8 |
| C70@Mpro | −45.6 | −59.2 | 0.0 | 16.8 | −3.2 | −17.6 | −28.0 |
| Complex | His41-Cys145 ΔGbinding | His41+-Cys145-ΔGbinding |
|---|---|---|
| masitinib@Mpro | −16.5 | −5.1 |
| C60@Mpro | −18.8 | −24.3 |
| C70@Mpro | −28.0 | −29.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marforio, T.D.; Mattioli, E.J.; Zerbetto, F.; Calvaresi, M. Fullerenes against COVID-19: Repurposing C60 and C70 to Clog the Active Site of SARS-CoV-2 Protease. Molecules 2022, 27, 1916. https://doi.org/10.3390/molecules27061916
Marforio TD, Mattioli EJ, Zerbetto F, Calvaresi M. Fullerenes against COVID-19: Repurposing C60 and C70 to Clog the Active Site of SARS-CoV-2 Protease. Molecules. 2022; 27(6):1916. https://doi.org/10.3390/molecules27061916
Chicago/Turabian StyleMarforio, Tainah Dorina, Edoardo Jun Mattioli, Francesco Zerbetto, and Matteo Calvaresi. 2022. "Fullerenes against COVID-19: Repurposing C60 and C70 to Clog the Active Site of SARS-CoV-2 Protease" Molecules 27, no. 6: 1916. https://doi.org/10.3390/molecules27061916