
The Use of Constituent Spectra and Weighting in Extended Multiplicative Signal Correction in Infrared Spectroscopy

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. The Effect of Different Normalization Procedures and the Importance of Weighting in the EMSC
2.2. The Effect of Different Normalization Procedures—Case Study I
2.3. Using Constituent Spectra in the EMSC: Analyte and Interferent Spectra
2.4. Orthogonality and Pitfalls When Using Constituent Spectra
Orthogonality between Model Spectra
2.5. Using Constituent Spectra for Estimating Chemical Compounds
2.5.1. Case Study II: Estimation of Glucose in ATR Spectra of Growth Media
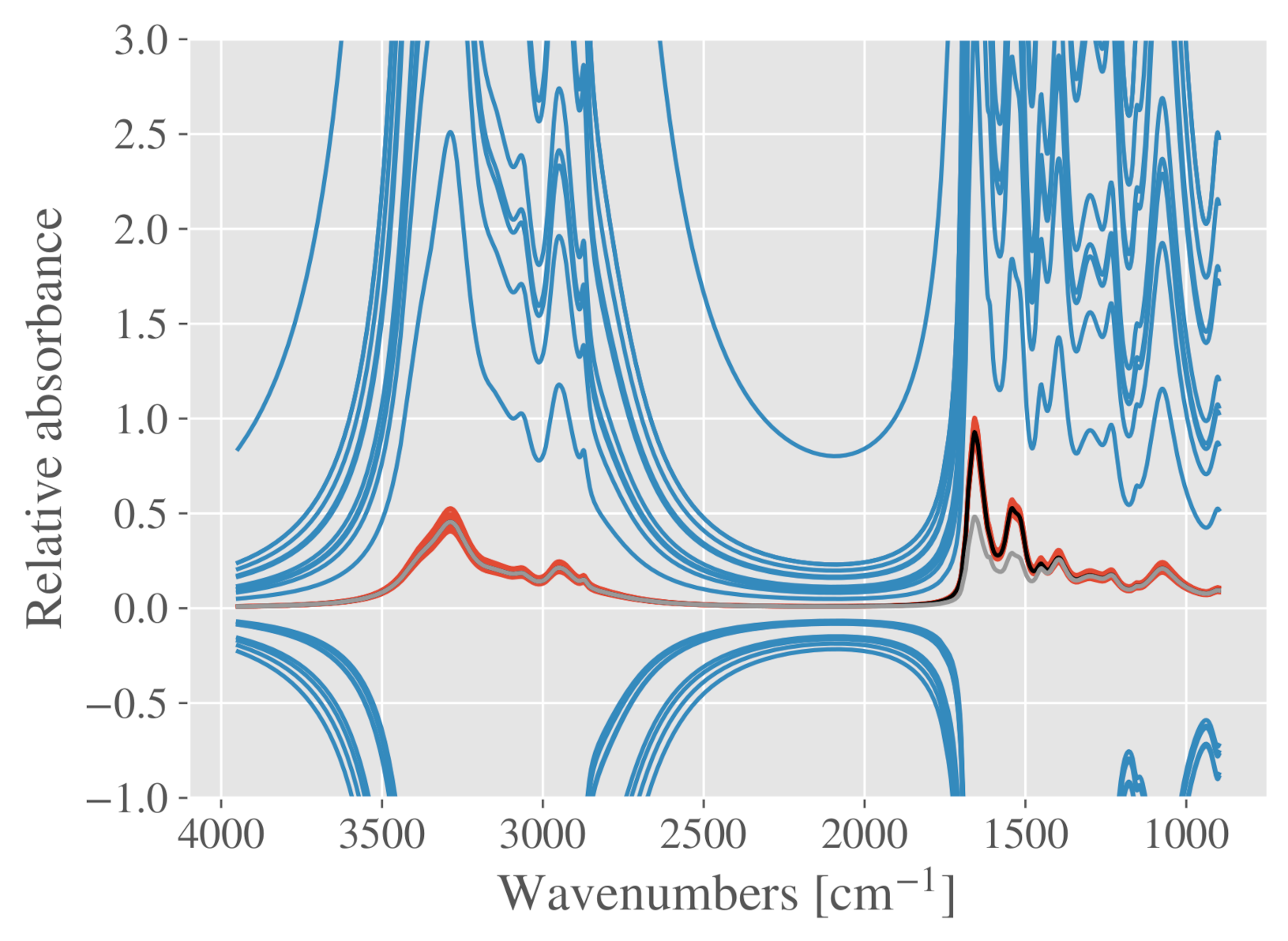
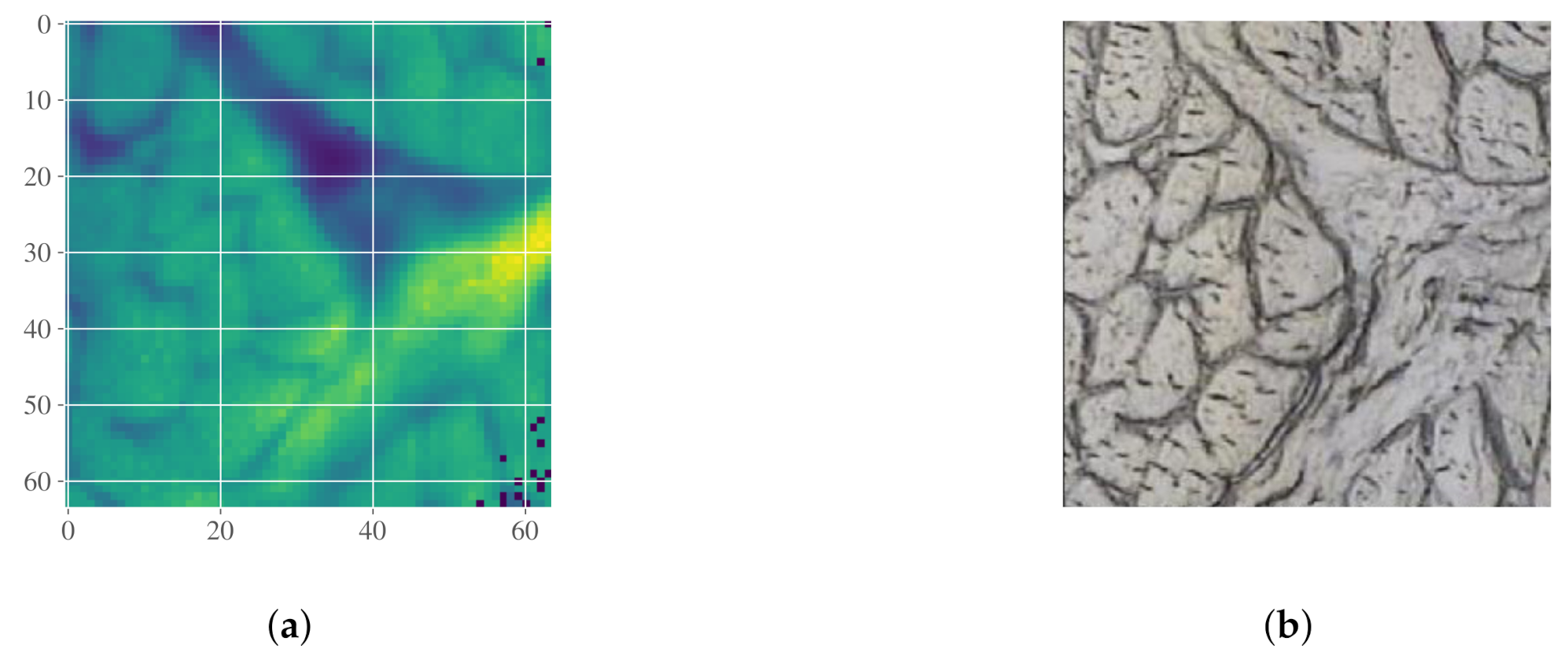
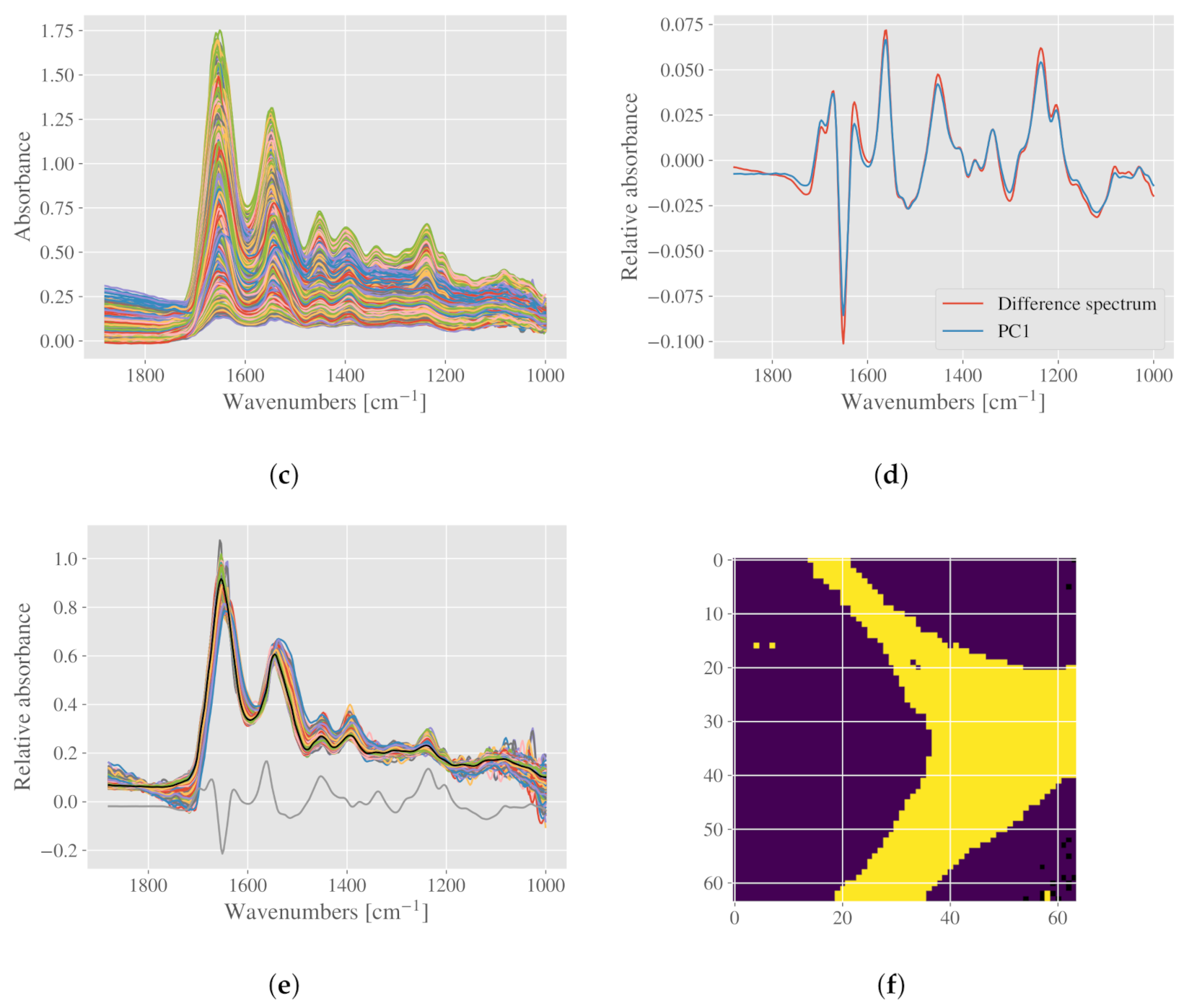
2.5.2. Case Study III: Detection of Connective Tissue and Myofibers in Infrared Microspectroscopy of Beef Loin Sections
3. Theory and Methods
3.1. Datasets
3.1.1. Filamentous Fungi Dataset
3.1.2. Growth Media Dataset
3.1.3. Beef Loin Dataset
3.2. Baseline and Multiplicative Effects in Infrared Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
| EMSC | Extended multiplicative signal correction |
| FTIR | Fourier transform infrared |
| HTS-XT | High throughput screening extension |
| MSC | Multiplicative signal correction |
| PLSR | Partial least squares regression |
| PUFA | Polyunsaturated fatty acid |
| RMSE | Root mean square error |
| SR-ATR | Single-reflection attenuated total reflectance |
References
- van Soest, J.J.; Tournois, H.; de Wit, D.; Vliegenthart, J.F. Short-range structure in (partially) crystalline potato starch determined with attenuated total reflectance Fourier-transform IR spectroscopy. Carbohydr. Res. 1995, 279, 201–214. [Google Scholar] [CrossRef] [Green Version]
- Blazhko, U.; Shapaval, V.; Kovalev, V.; Kohler, A. Comparison of augmentation and pre-processing for deep learning and chemometric classification of infrared spectra. Chemom. Intell. Lab. Syst. 2021, 215, 104367. [Google Scholar] [CrossRef]
- Zimmermann, B.; Kohler, A. Optimizing Savitzky-Golay parameters for improving spectral resolution and quantification in infrared spectroscopy. Appl. Spectrosc. 2013, 67, 892–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, R.; Dhanoa, M.S.; Lister, S.J. Standard normal variate transformation and de-trending of near-infrared diffuse reflectance spectra. Appl. Spectrosc. 1989, 43, 772–777. [Google Scholar] [CrossRef]
- Martens, H.; Jensen, S.; Geladi, P. Multivariate linearity transformation for near-infrared reflectance spectrometry. In Proceedings of the Nordic Symposium on Applied Statistics; Stokkand Forlag Publishers: Stavanger, Norway, 1983; pp. 205–234. [Google Scholar]
- Kohler, A.; Solheim, J.; Tafintseva, V.; Zimmermann, B.; Shapaval, V. Model-Based Pre-Processing in Vibrational Spectroscopy. In Comprehensive Chemometrics, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar] [CrossRef]
- Zimmermann, B.; Tkalčec, Z.; Mešić, A.; Kohler, A. Characterizing aeroallergens by infrared spectroscopy of fungal spores and pollen. PLoS ONE 2015, 10, e0124240. [Google Scholar] [CrossRef] [Green Version]
- Zimmerman, B.; Tafintseva, V.; Bagcıoglu, M.; Høegh Berdahl, M.; Kohler, A. Analysis of allergenic pollen by FTIR microspectroscopy. Anal. Chem. 2016, 88, 803–811. [Google Scholar] [CrossRef]
- Tafintseva, V.; Shapaval, V.; Smirnova, M.; Kohler, A. Extended multiplicative signal correction for FTIR spectral quality test and pre-processing of infrared imaging data. J. Biophotonics 2020, 13, e201960112. [Google Scholar] [CrossRef]
- Diehn, S.; Zimmermann, B.; Tafintseva, V.; Bağcıoğlu, M.; Kohler, A.; Ohlson, M.; Fjellheim, S.; Kneipp, J. Discrimination of grass pollen of different species by FTIR spectroscopy of individual pollen grains. Anal. Bioanal. Chem. 2020, 412, 6459–6474. [Google Scholar] [CrossRef]
- Martens, H.; Stark, E. Extended multiplicative signal correction and spectral interference subtraction: New preprocessing methods for near infrared spectroscopy. J. Pharm. Biomed. Anal. 1991, 9, 625–635. [Google Scholar] [CrossRef]
- Afseth, N.K.; Kohler, A. Extended multiplicative signal correction in vibrational spectroscopy, a tutorial. Chemom. Intell. Lab. Syst. 2012, 117, 92–99. [Google Scholar] [CrossRef]
- Solheim, J.H.; Gunko, E.; Petersen, D.; Großerüschkamp, F.; Gerwert, K.; Kohler, A. An open-source code for Mie extinction extended multiplicative signal correction for infrared microscopy spectra of cells and tissues. J. Biophotonics 2019, 12, e201800415. [Google Scholar] [CrossRef]
- Bağcıoğlu, M.; Kohler, A.; Seifert, S.; Kneipp, J.; Zimmermann, B. Monitoring of plant–environment interactions by high-throughput FTIR spectroscopy of pollen. Methods Ecol. Evol. 2017, 8, 870–880. [Google Scholar] [CrossRef] [Green Version]
- Bertram, H.C.; Kohler, A.; Böcker, U.; Ofstad, R.; Andersen, H.J. Heat-induced changes in myofibrillar protein structures and myowater of two pork qualities. A combined FT-IR spectroscopy and low-field NMR relaxometry study. J. Agric. Food Chem. 2006, 54, 1740–1746. [Google Scholar] [CrossRef]
- Martens, H.; Nielsen, J.P.; Engelsen, S.B. Light scattering and light absorbance separated by extended multiplicative signal correction. Application to near-infrared transmission analysis of powder mixtures. Anal. Chem. 2003, 75, 394–404. [Google Scholar] [CrossRef]
- Solheim, J.H.; Borondics, F.; Zimmermann, B.; Sandt, C.; Muthreich, F.; Kohler, A. An automated approach for fringe frequency estimation and removal in infrared spectroscopy and hyperspectral imaging of biological samples. J. Biophotonics 2021, 14, e202100148. [Google Scholar] [CrossRef]
- Kohler, A.; Böcker, U.; Warringer, J.; Blomberg, A.; Omholt, S.; Stark, E.; Martens, H. Reducing inter-replicate variation in Fourier transform infrared spectroscopy by extended multiplicative signal correction. Appl. Spectrosc. 2009, 63, 296–305. [Google Scholar] [CrossRef]
- Tafintseva, V.; Shapaval, V.; Blazhko, U.; Kohler, A. Correcting replicate variation in spectroscopic data by machine learning and model-based pre-processing. Chemom. Intell. Lab. Syst. 2021, 215, 104350. [Google Scholar] [CrossRef]
- Kohler, A.; Kirschner, C.; Oust, A.; Martens, H. Extended multiplicative signal correction as a tool for separation and characterization of physical and chemical information in Fourier transform infrared microscopy images of cryo-sections of beef loin. Appl. Spectrosc. 2005, 59, 707–716. [Google Scholar] [CrossRef]
- Tafintseva, V.; Lintvedt, T.A.; Solheim, J.H.; Zimmermann, B.; Rehman, H.U.; Virtanen, V.; Shaikh, R.; Nippolainen, E.; Afara, I.; Saarakkala, S.; et al. Preprocessing Strategies for Sparse Infrared Spectroscopy: A Case Study on Cartilage Diagnostics. Molecules 2022, 27, 873. [Google Scholar] [CrossRef]
- Dzurendová, S.; Shapaval, V.; Tafintseva, V.; Kohler, A.; Byrtusová, D.; Szotkowski, M.; Márová, I.; Zimmermann, B. Assessment of biotechnologically important filamentous fungal biomass by Fourier transform Raman spectroscopy. Int. J. Mol. Sci. 2021, 22, 6710. [Google Scholar] [CrossRef]
- Langseter, A.M.; Dzurendova, S.; Shapaval, V.; Kohler, A.; Ekeberg, D.; Zimmermann, B. Evaluation and optimisation of direct transesterification methods for the assessment of lipid accumulation in oleaginous filamentous fungi. Microb. Cell Factories 2021, 20, 59. [Google Scholar] [CrossRef] [PubMed]
- Kosa, G.; Shapaval, V.; Kohler, A.; Zimmermann, B. FTIR spectroscopy as a unified method for simultaneous analysis of intra-and extracellular metabolites in high-throughput screening of microbial bioprocesses. Microb. Cell Factories 2017, 16, 195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, B.J.; Carney, P.S.; Bhargava, R. Theory of midinfrared absorption microspectroscopy: I. Homogeneous samples. Anal. Chem. 2010, 82, 3474–3486. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.J.; Carney, P.S.; Bhargava, R. Theory of mid-infrared absorption microspectroscopy: II. Heterogeneous samples. Anal. Chem. 2010, 82, 3487–3499. [Google Scholar] [CrossRef] [PubMed]
- Mayerich, D.; Van Dijk, T.; Walsh, M.J.; Schulmerich, M.V.; Carney, P.S.; Bhargava, R. On the importance of image formation optics in the design of infrared spectroscopic imaging systems. Analyst 2014, 139, 4031–4036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohlenhoff, B.; Romeo, M.; Diem, M.; Wood, B.R. Mie-type scattering and non-Beer–Lambert absorption behaviour of human cells in infrared micro-spectroscopy. Biophys. J. 2005, 88, 3635–3640. [Google Scholar] [CrossRef] [Green Version]
- Bassan, P.; Kohler, A.; Martens, H.; Lee, J.; Byrne, H.; Dumas, P.; Gazi, E.; Brown, M.; Clarke, N.; Gardner, P. Resonant Mie Scattering (RMieS) correction of infrared spectra from highly scattering biological samples. Analyst 2010, 135, 268–277. [Google Scholar] [CrossRef]
- Konevskikh, T.; Ponossov, A.; Blümel, R.; Lukacs, R.; Kohler, A. Fringes in FTIR spectroscopy revisited: Understanding and modelling fringes in infrared spectroscopy of thin films. Analyst 2015, 140, 3969–3980. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Normalization Strategy | Predicting % Fat of Total Biomass | Predicting % PUFA of Total Fat |
|---|---|---|
| Strategy A | (2) , RMSE = 5.24 | (4) , RMSE = 3.79 |
| Strategy B | (7) , RMSE = 8.87 | (2) , RMSE = 2.37 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solheim, J.H.; Zimmermann, B.; Tafintseva, V.; Dzurendová, S.; Shapaval, V.; Kohler, A. The Use of Constituent Spectra and Weighting in Extended Multiplicative Signal Correction in Infrared Spectroscopy. Molecules 2022, 27, 1900. https://doi.org/10.3390/molecules27061900
Solheim JH, Zimmermann B, Tafintseva V, Dzurendová S, Shapaval V, Kohler A. The Use of Constituent Spectra and Weighting in Extended Multiplicative Signal Correction in Infrared Spectroscopy. Molecules. 2022; 27(6):1900. https://doi.org/10.3390/molecules27061900
Chicago/Turabian StyleSolheim, Johanne Heitmann, Boris Zimmermann, Valeria Tafintseva, Simona Dzurendová, Volha Shapaval, and Achim Kohler. 2022. "The Use of Constituent Spectra and Weighting in Extended Multiplicative Signal Correction in Infrared Spectroscopy" Molecules 27, no. 6: 1900. https://doi.org/10.3390/molecules27061900