
Sulforaphane and Its Bifunctional Analogs: Synthesis and Biological Activity


Faculty of Chemistry, Institute of Organic Chemistry, Lodz University of Technology, Zeromskiego 116, 90-924 Lodz, Poland
Molecules 2022, 27(5), 1750; https://doi.org/10.3390/molecules27051750
Submission received: 9 February 2022
/
Revised: 4 March 2022
/
Accepted: 5 March 2022
/
Published: 7 March 2022
(This article belongs to the Special Issue Featured Reviews in Organic Chemistry)
Abstract
:For decades, various plants have been studied as sources of biologically active compounds. Compounds with anticancer and antimicrobial properties are the most frequently desired. Cruciferous plants, including Brussels sprouts, broccoli, and wasabi, have a special role in the research studies. Studies have shown that consumption of these plants reduce the risk of lung, breast, and prostate cancers. The high chemopreventive and anticancer potential of cruciferous plants results from the presence of a large amount of glucosinolates, which, under the influence of myrosinase, undergo an enzymatic transformation to biologically active isothiocyanates (ITCs). Natural isothiocyanates, such as benzyl isothiocyanate, phenethyl isothiocyanate, or the best-tested sulforaphane, possess anticancer activity at all stages of the carcinogenesis process, show antibacterial activity, and are used in organic synthesis. Methods of synthesis of sulforaphane, as well as its natural or synthetic bifunctional analogues with sulfinyl, sulfanyl, sulfonyl, phosphonate, phosphinate, phosphine oxide, carbonyl, ester, carboxamide, ether, or additional isothiocyanate functional groups, and with the unbranched alkyl chain containing 2–6 carbon atoms, are discussed in this review. The biological activity of these compounds are also reported. In the first section, glucosinolates, isothiocyanates, and mercapturic acids (their metabolites) are briefly characterized. Additionally, the most studied anticancer and antibacterial mechanisms of ITC actions are discussed.
1. Introduction
1.1. Isothiocyanates—General Properties
1.1.1. Glucosinolates
The Brassicaceae family [1] also called Cruciferae, includes more than 2000 plant species. Among them, many are edible plants, such as Brussels sprouts, broccoli, radish, horse radish, cabbage, wasabi, and rocket [2,3]. These vegetables are characterized by high chemopreventive activity. International research [4,5] shows that their consumption decreases the risk of lung [6], breast [7], colon [8], and prostate cancers [9]. The high chemopreventive effects of Cruciferae, compared to other plants, are associated with the high content of glucosinolates (GSLs). GSLs [10,11,12,13,14,15,16,17] were discovered more than 200 years ago and they contain sulfur secondary metabolites. In 1956, Ettlinger and Lundeen [18] proposed general structures of GSLs (Figure 1). GSLs are composed of three elements: β-D-thioglucose group (in red), sulfonated oxime moiety (in blue), and side chain R (in green), whose structures correspond to the α-amino acids used during biosynthesis (Figure 1). The structures of GSLs were confirmed by synthesis in 1957 [19]. To date, more than 200 GSLs have been identified, and due of the structure of side chain R, they are divided into three main groups: aliphatic (1–11), aromatic (12–14), and indole derivatives (15–16). Aliphatic GSLs covering more than 50% of glucosinolates are divided into: alkenyl (1–3), hydroxyalkenyl (4–5), and those containing sulfur on II (6–7), IV (8–10), and VI (11) oxidation states [20]. In addition, GSLs can be found in other plants—different from Brassicaceae—such as Moringaceae [21,22,23], of which, the most widely cultivated is Moringa oleifera. GSLs with glycosylated R-groups belong to this group (17–20) (Figure 1).
In plants, GSLs are stored in vacuoles, which protect them against degradation caused by myrosinase (β-D-thioglucosidase) (EC 3.2.1.1) [24,25]. In plants, the glucosinolate–isothiocyanate system has defense functions against insects, pathogens, and herbivores [26]. Cell damage, e.g., while chewing the plants, results in the formation of a variety of glucosinolate breakdown products (Figure 2). In mammals, GSLs are converted to isothiocyanates, through the bacteria present in the digestive tract. Reducing the amount of bacterial flora, as a result of using antibiotics, eliminates this pathway [27].
The breakdown of GSLs initiated by enzymatic hydrolysis of the thioglucoside bond leads to the elimination of β-D-glucose and the formation of an unstable thiohydroximate-O-sulfonate, which, depending on the pH, undergoes the so-called Lossen rearrangement to isothiocyanate (pH 7), or a decomposition to nitrile (pH 4). It was shown that a low pH inhibits the Lossen rearrangement [28]. For unstable β-hydroxyalkenyl isothiocyanates, subsequent cyclization to oxazolidine-2-thione takes place (Figure 2). Under certain conditions, plants modify the direction of aglucon degradation in the presence of specific proteins, such as the epithiospecifier protein (ESP) [29], the nitrile-specifier protein (NSP) [30], and the thiocyanate-forming protein (TFP) [31], catalyzing formation of epithionitrile, nitrile, or thiocyanate, respectively. On the other hand, the epithiospecifier modifier protein (ESM) inhibits the formation of nitrile and favors the formation of isothiocyanates [32] (Figure 2). Detailed information about enzymatic hydrolysis of glucosinolates and the functions of specifier proteins were reviewed by Burow and Wittstock [33].
1.1.2. Isothiocyanates
Biologically inactive GSLs undergo enzymatic transformations to biologically active isothiocyanates (ITCs). ITCs [34,35,36,37] are low molecular weight heterocummulenes with a general formula R–N=C=S (R-NCS), where R may be a structurally diverse aliphatic, aromatic or heterocyclic substituent. They are characterized by sharp and pungency odors [38] and relatively high volatility. Due to the presence of a reactive electrophilic carbon atom in the isothiocyanate group (–NCS), these compounds easily react in physiological conditions, in a reversible way with thiols, resulting in sensitivity to pH dithiocarbamates or forming thioureas in an irreversible reaction with amines. It was shown that ITCs react 1000 times faster with thiols than with amines [39]. Hydrolysis of ITCs leads to amines (Figure 3) [40].
Natural isothiocyanates, such as benzyl isothiocyanate (21, BITC) [41], phenethyl isothiocyanate (22, PEITC) [42,43,44], allyl isothiocyanate (23, AITC) [45], and the best known and the studied sulforaphane (24, SFN) [46,47,48] (Figure 4) have shown chemopreventive properties.
ITCs inhibit the carcinogenesis process and tumor development in vitro and in vivo by inhibiting the activity of carcinogens, inhibiting the cell cycle, activating apoptosis, and inhibiting metastasis. These compounds (due to their reactivities) also modify many proteins involved in cancer processes [49,50]. They inhibit cytochrome P450 enzymes, activate phase II enzymes by activating the Nrf2 factor, affect cell cycle regulators and Bcl-2 proteins, activate caspases, and inhibit Nf-κB factor [51,52]. The anticancer mechanisms of isothiocyanates, including SFN, are described in detail in Section 2.1. The large number of proteins potentially reacting with ITCs shows that ITCs do not have one particular molecular target. This is the advantage of ITCs, because it makes it more difficult for cancer cells to become resistant to ITCs. On the other hand, this feature can be a disadvantage, as it makes studying the anticancer mechanisms of isothiocyanate much more difficult. In addition to chemopreventive properties, ITCs have antibacterial properties [53,54] (described in detail in Section 2.2). They are also used as herbicides and fungicides [55]. Isothiocyanates also play an important role in organic synthesis as substrates in the synthesis of heterocyclic compounds [56,57], thioamides [58], and thiourea organocatalysts [59]. ITCs are also exploited as molecular probes [60,61].
SFN–1-isothiocyanato-4-(methylsulfinyl)butane (24) was, for the first time, “obtained” by Schmid and Karrer in 1948 [62]. It is the best known (and studied) isothiocyanate. It was, for the first time, isolated by Zhang [63] in 1992, from broccoli, where its concentration ranged between 0.8 and 21.7 μmol/g d wt [64]. Isolation of SFN led to increased interest in this compound, as well as other isothiocyanates, as confirmed by a large number of scientific papers on this subject. Analysis of the Web of Science database shows that, since 1992, about 3890 articles have been published on SFN, and over 5600 on isothiocyanates [65].
SFN, due to the presence of the chiral center on the sulfur atom, occurs as two enantiomers—natural (R)-SFN and synthetic (S)-SFN. Most tests used racemic SFN; however, studies confirm that (R)-SFN has biological activity [66]. The name of the “most popular isothiocyanate”, SFN, is due to its ability to simultaneously modify many cellular targets associated with cancer development, including DNA protection, by inhibiting the activity of mutagenic factors (phase I) and activation of phase II factors responsible for detoxification, inhibiting the proliferation of cancer cells and activating apoptosis, thereby limiting the process of multiplication of mutated cancer cells, and inhibiting the process of neogenesis and metastasis. SFN is able to prevent, remove, and reverse preneoplastic lesions [67]. Recent research by Chlopicki et al. [68] shows that L-SFN also exhibits antioxidant and protective effects on endothelial cells. All of these features mean that SFN, according to the US National Cancer Institute, is one of the 40 most promising anticancer compounds [69].
When writing about SFN—its natural analogues, possessing a sulfur atom on II, IV, and VI oxidation states, and an alkyl chain containing 3 to 5 carbon atoms, should also be mentioned (Figure 5).
These include: iberin (25) and alyssin (26) [70], with methylsulfinyl group, iberverin (27) [71], erucin (ER) (28) [72] and berteroin (29) [73] with the methylsulfanyl group, and cheirolin (30) [74], and erysolin (31) [75] with the methylsulfonyl group. The α,β-unsaturated analog of sulforaphene (32) is also known [76] (Figure 5).
1.1.3. Mercapturic Aid
After entering ITCs into the cell, glutathione (GSH, 33) [77,78,79,80,81,82] is the first target of ITCs. In the cell, under the influence of glutathione S-transferase (GST), an immediate reaction of ITCs occurs with the -SH group of the cysteine residue of GSH. The S-(N-alkyl/arylthiocarbamoyl)glutathione (ITC–GSH) is formed, initiating the process of isothiocyanate metabolism, called the mercapturic acid pathway. The ITC–GSH under the influence of γ-glutamyl transferase (GT) dipeptidase (cysteinoglycinase (CG)) and N-acetyltransferase (AT) (transformation to conjugate with cysteinylglycine, cysteine, and N-acetylcysteine), giving mercapturic acids (ITC–NACs) as the final products of intracellular metabolism of ITCs (Figure 6) [40].
Compared to conjugates with cysteinylglycine and cysteine, which are formed in the intercellular space, mercapturic acid is formed in the liver, is transported to the kidneys, and removed with urine [40]. Studies have shown that in the urine of people who consume cruciferous plants, mercapturic acid is the main metabolite [83]. It was found that after 8 h of ingestion of broccoli sprouts, about 60% of the ITCs are removed in the urine [84]. Detailed studies show that 7% is pure SFN, less than 1% an SFN–GSH conjugate and SFN conjugate with cysteinylglycine, about 28% an SFN conjugate with cysteine, and about 65% ITC–NAC formed from SFN [85,86].
After diffusing into the cell, ITCs in the presence of GST undergo rapid reactions with GSH (33), leading the formation of the ITC–GSH conjugate. This compound, under the influence of efflux pumps, moves to the intercellular space, where it breaks down in the hydrolysis reaction with recovery of the original ITCs, which again enter the cell. The result of this process is the depletion of intracellular GSH and high accumulation of the ITCs in the cell (ITC concentration in the cell, compared to the concentration in the intercellular area, is 100–200 times larger) [88]. Glutathione depletion in the cell allows ITCs to react with other proteins containing cysteine residues (the presence of a thiol group). The consequences of this process are the increase of ROS in the cell, leading to the induction of various biological responses, including short-term, or a complete arrest of the cell proliferation process, as well as the activation of apoptosis or necrosis [89,90]. The formation via the mercapturic acid pathway leads to other biological active conjugates causing, e.g., inhibition of histone deacetylase activity [91], and assures the possibility of ITC reaction with other intercellular and intracellular proteins.
2. Mechanism Determining Biological Activity of ITCs
2.1. The Mechanism of Anticancer Activity
There are many excellent reviews on anticancer activities, as well as the mechanisms of action of ITCs [92,93,94,95,96] and SFN [97,98,99,100]. In vitro and in vivo tested ITCs, including SFN, show their biological activities at all stages of carcinogenesis. Generally, carcinogenesis consists of three stages: (i) initiation—a rapid and irreversible process leading to genotypic changes and DNA damage due to interaction with the carcinogen; (ii) promotion—the selective proliferation of initiated cells leading to the formation of pre-cancerous lesions; (iii) progression—transformation of benign pre-cancerous lesions into cancer. This section briefly summarizes the most studied ITC anticancer mechanisms.
2.1.1. Initiation Stage
At the initiation stage, isothiocyanates exhibit chemopreventive activities by regulating xenobiotics metabolism. In the conversion of carcinogens and their elimination, two groups of enzymes participate: phase I enzymes and phase II enzymes.
Inhibition Phase I Enzymes and Activation Phase II Enzymes
Phase I enzymes belonging to the cytochrome P-450 (CYP) family catalyze oxidation, reduction, and hydrolysis reactions, preparing the carcinogen for further changes, which leads to metabolic activation of some procancerogens and the formation of metabolites capable of interacting with DNA and causing mutations [101]. ITCs inhibit and reduce the activity of CYP 1A1, 1A2, 2A6, 3A4, 2B1, 2D6, and 2E1 in cancer tissues [52]. Yokoi et al. [102] confirmed that PEITC (22) completely inhibits CYP 1A2, 2A6, 2B6, 2C9, 2C19, 2D6, 2E1, and 3A4 activity.
In phase II metabolism of carcinogens, a particular role is played by glutathione S-transferase (GST), NAD(P)H: quinone oxidoreductase (NQO1), UDP-glucuronyltransferase (UGT), quinone reductase (QR), and nicotinamide N-methyltransferase [103,104], enzymes responsible for increase the solubility of xenobiotics in water and facilitate their excretion [105]. In vitro studies on the HepG2 human hepatocyte line showed that the use of SFN (24) caused an increase in the concentration of mRNA UGT 1A1 and GST A1 [106], and an increase in the activity of NQO1 [107], accompanied by bilirubin glucuronidation [108].
One of the proposed mechanisms of stimulation of phase II enzymes by SFN concerns the activation of the Nrf2 [109] allowing the interaction with the antioxidant response element (ARE) encoding the detoxification enzyme genes. Under normal conditions, Nrf2 occurs in the cellular cytosol, and it is binding to the Kelch-like ECH-associated protein 1 (Keap1) leading the Nrf2–Keap1 complex. SFN, or other ITCs after entering the cell, react with thiol groups present on the surface of the Keap1, resulting in the degradation of the Nrf2–Keap1 complex. The released and activated Nrf2 migrates to the cell nucleus, where it binds to ARE and stimulates transcription of genes encoding phase II enzymes [109]. Studies have shown that the activation of Nrf2, apart from what is described above, can also take place due to the activation of the MAPK protein kinase pathway [46].
2.1.2. Promotion Stage
At the promotion stage, SFN (and other ITCs) show antiproliferative activity against tumor cells by, e.g., inhibiting the cell cycle, inducing apoptosis or inhibiting histone deacetylase (HDAC).
Inhibition of the Cell Cycle
Tumor cells are characterized by rapid growth resulting from the loss or disturbed functioning of mechanisms of cell cycle regulation. Cyclin-dependent kinases (CDKs), cyclins, and cyclin-dependent kinase inhibitors are involved in controlling the cycle of healthy cells. The formation of a cyclin and CDK complexes permit the cell to pass through the next phases of the cycle, while the CDK inhibitors stop the cycle in a specific phase. The research has shown that SFN acts as a cell cycle regulator, inhibiting the cell cycle in the G2/M phase [110]. Crossing the G2/M point requires activation of a complex of the cyclin B and CDK1. Phosphorylation of CDK1 by Wee1 and Myt1 kinases leads to inactivation of the complex, while the activity of the Cdc25 protein phosphatase conditions the activity of the complex, allowing to go to the M phase. The Cdc25 protein phosphatase is regulated by Chk1 and Chk2 kinases, and phosphorylation of Cdc25 inactivates it and leads to the deactivation of the cyclin B and CDK1 complex, finally stopping the cell cycle in the G2/M phase [111]. Studies have shown that SFN prevents the transfer of PC-3 cancer cells to the M phase of the cell cycle by decreasing the levels of cyclin B1, Cdk25B, and Cdk25C, and increasing phosphorylation of Cdk25C by Chk2 [112]. In addition, it was shown that one of the mechanisms of action of SFN on the HT-29 colorectal tumor cell line is the expression of protein p21 [113]. Studies on the same cell line have shown that ITCs also inhibits the cell cycle in the G1 phase by reducing regulation of cyclin A, D, and E [114]. The apoptotic effect depends mainly on the dose of the compound and the time of exposure to it [115]. Studies on the Caco-2 cell line have shown that SFN at a concentration of 20 μM inhibits the cell cycle in the G2/M phase, while at a concentration above 20 μM, it inhibits the G1 phase. In addition, the short-term exposure of tumor cells to SFN resulted in reversible inhibition of the cell cycle in the G2/M phase, while complete inhibition of the cell cycle required more than 12 h of incubation [116].
Inducing Apoptosis
Apoptosis, programmed cell death, is one of the most commonly used strategies to fight cancer cells. In some cases, the low susceptibility of cancer cells to signals triggering the process of apoptosis contributes to the development of tumors. SFN is involved in activating signals that lead to apoptosis in many types of cancer cells. SFN stimulates the formation of apoptotic bodies, reduces the concentration of anti-apoptotic Bcl-2 and Bcl-XL proteins, increases the expression of the proapoptotic Bax protein, the activation of caspase 3, and the degradation of the poly(ADP-ribose) polymerase, and thus interacts on the mitochondrial-dependent apoptosis factors [117]. In addition, SFN is responsible for decreasing the activity of apoptosis inhibitors (IAP: cIAP1, cIAP2, and XIAP) and the induction of the Apaf-1 protein [118]. In addition to SFN, other natural isothiocyanates participate in the process of apoptosis. BITC activates procaspases-8 and -9 [119]. AITC and PEITC increase the level of the t-Bid proapoptotic protein in HL-60 leukemia cells [120], and AITC further reduces the Bcl-XL protein concentration in LNCaP prostate cancer cells [121]. Mechanistic studies suggest that the generation of the reactive oxygen species (ROS) by SFN induces the mechanism of cancer cell death. Tests performed on the human prostate tumor cell PC-3 line showed that ROS generated in the presence of SFN changed the potential of the mitochondrial membrane, leading to the release of cytochrome C from the intermembrane space to the cytoplasm [122]. Cytochrome C, together with the Apaf-1 protein and ATP, form an apoptosome, activating procaspase-9. The apoptosome and caspase-9 complex recruits and activates procaspase-3 and/or procaspase-7, leading to apoptosis [123].
Inhibiting Histone Deacetylase (HDAC)
Histones undergo reversible acetylation of selected N-terminus lysine residues. The modification occurring in the presence of acetyltransferase leads to serious changes at all levels of chromatin structures, causing disorders of the chromatin, folding into higher-order structures [124], increasing its solubility under physiological conditions and, most importantly, favor transcription [125]. Unlike acetylation, deacetylation of histones leads to the blockage of chromatin structures and, as a consequence, to the termination of the transcription process. Histone deacetylase is associated with many cancers, it causes suppression of transcription and affects the dysregulation of mechanisms controlling the cell cycle and apoptosis. In addition, HDAC by deacetylation of tumor suppressor genes, e.g., the p21 gene, leads to silencing of their transcriptions or their complete deactivation. In vitro studies performed on the HCT 116 cell line showed that SFN at concentrations of 3–15 μM causes a decrease in HDAC activity [126]. The process of inhibiting histone deacetylase is combined with the inhibition of the cell cycle and induction of apoptosis. Studies show that inhibition of HDAC with SFN contribute to inhibition of the cell cycle in PC-3 cells in the G2/M phase [127], while inhibition of HDAC using BITC causes the deactivation of NF-κB, which leads to decreased activity of cyclin D1 and, consequently, to the inhibition of the cell cycle [128]. In addition, as a consequence of the inhibition of histone deacetylase with SFN, researchers observed an increase in the concentration of p21 and Bax proteins [126], which are involved in the process of apoptosis.
2.1.3. Progression Stage
At the stage of progression, isothiocyanates, as well as SFN, are responsible for inhibiting the process of angiogenesis and metastasis.
Inhibition of Angiogenesis and Metastasis
Angiogenesis is the process of formation of new blood vessels [129]. It is claimed that it is an important stage in tumor growth and metastasis, as a result of which, oxygen and nutrients are supplied to the formed cancer. For this reason, the stage of angiogenesis has become the target of anticancer therapies. The main cytokinin initiating the process of angiogenesis is the vascular endothelial growth factor (VEGF), responsible for an increase in vascular permeability and stimulating proteolytic enzymes. In addition to VEGF, fibroblast growth factor 2 (FGF-2) and epidermal growth factor (EGF) are important factors involved in angiogenesis [130].
Metastasis is the ability to spread cancer cells from the primary outbreak to lymph nodes, and to tissues and organs, and is a hallmark of malignant tumors. The metastasis process requires the activation of proteolytic enzymes, e.g., metalloproteinases (MMPs). These enzymes belong to the family of zinc-containing enzymes and are capable of degrading the basement membrane, which is necessary to penetrate endothelial cells into new places and create new vessels. MMPs are overexpressed in cancer cells [131]. Studies have shown that PEITC has an anti-angiogenic effect by inhibiting the activity of VEGF and EGF [132]. Additionally, tests on human umbilical vein endothelial cells (HUVECs) with SFN have demonstrated that this compound is involved in the regulation of the various stages of angiogenesis, by reducing vascular formation and propagation of endothelial cells [133]. In addition, SFN inhibits metalloproteinase-9 activity and reduces the metastatic ability of MDA-MB-231, a triple-negative breast cancer cell line.
2.2. Antibacterial Activity
Antibacterial properties of ITCs are not as wide tested as anticancer properties. To date, only two extensive reviews have described the antibacterial activities of ITCs. Dufour et al. [53] in 2015 described, in detail, the antibacterial modes of action, e.g., the effects on influencing the membrane, inhibition of enzymic or regulatory activities, the effect of ITCs on respiratory enzymes, the induction of heat shock and oxidative stress responses, and the induction of a stringent response of natural ITCs (SFN, BITC, PEITC, etc.), as well as mechanisms of resistance to ITCs. Moreover, Romeo et al. [54], in 2018, described the antibacterial properties of natural ITCs against Gram-positive (H. pylori, S. aureus, etc.) and Gram-negative (P. aeruginosa, E. coli, etc.) bacteria. For this reason, in this review, the antibacterial activities of ITCs are generally and briefly characterized.
In Japan, ITCs are used as natural food additives to protect them against microorganisms [134]. Highly volatile allyl isothiocyanate (AITC) plays a special role. It is used in antimicrobial food packaging to reduce, inhibit, and delay the growth of microorganisms in packed food. ITCs obtained from cruciferous vegetables, most often from horseradish, wasabi, or radish, are also added to food as spices [135]. The bacteriostatic and bactericidal effect of ITCs depend on the dose, and the concentration responsible for the above-mentioned effects is comparable or lower than the concentration of classical antibiotics used for the same purposes. ITCs show synergism with commonly used antibiotics. For example, a solution of p-hydroxyphenethyl isothiocyanate in glucose exhibits synergism with aminoglycoside antibiotics and streptomycin in relation to E. coli and S. aureus [136]. Moreover, AITC and phenethyl isothiocyanate (PEITC) show synergism with streptomycin against some Gram-negative bacteria (E. coli or P. aeruginosa) [137]. However, it was shown that even a small change in the concentration of isothiocyanate or antibiotic can cause the opposite effect and suppress antibacterial effect [138]. The mechanism of synergism, or its quenching caused by ITCs, is not yet known. Bacteriostatic properties of ITCs have also been noticed in agriculture, and ITCs are used to reduce the population of bacteria found in soil; for this purpose—genetically modified Arabidopsis thaliana (radish) plants using a transgenic A. thaliana that overexpress glucosinolate with p-hydroxybenzyl substituent (14, sinalbin, Figure 1) [139]. Glucosinolates, by diffusion, are transported from the roots to the rhizosphere, where under the action of extracellular myrosinase, they are transformed into ITCs [140].
Mechanism of Antibacterial Activity
The mechanism of antibacterial activity of ITCs is not as well understood. It is claimed that the antibacterial activity of ITCs is associated with disintegration of the cell membrane (causing the outflow of all metabolites), inhibition of bacterial quorum sensing—a system of communication between bacteria through autoinducers, inhibition of biofilm production, inhibition of enzymes necessary for the proper functioning of bacteria, induction of thermal shock, or induction of oxidative stress. The most commonly studied Gram-negative and Gram-positive bacteria strains and the antibacterial mechanisms of ITCs are briefly discussed in this section.
Regarding H. pylori (Gram-negative) [141], SFN has the highest activity in relation to this strain, as well as to resistant strains of H. pylori (an inhibition of SFN is similar to the inhibition of antibiotics—clarithromycin and metronidazole) [142]. It is known that stomach infections by H. pylori are possible due to the ureases [143] converting urea into ammonia, resulting in the neutralization of acidic gastric juices [144]. SFN and other ITCs likely inhibit the activity of H. pylori urease, but this is still the subject of many studies [145].
Regarding Ca. jejuni (Gram-negative), benzyl isothiocyanate (BITC) is the most active natural isothiocyanate in relation to both antibiotic-resistant and -sensitive strains, Ca. jejuni [146]. Studies show that antibacterial activity of BITC is associated with the activation of metabolic pathways responsible for thermal shock and oxidative stress, leading to protein aggregation, energy metabolism disorders and, finally, bacterial death [147].
BITC also has the highest activity against S. enterica [148] (Gram-negative) and disintegrates cell membranes [149].
Enterohemorrhagic E. coli (EHEC) (Gram-negative) [150], whose strain O157:H7 produces the Shiga toxin, is the most studied, enteric, pathogenic E. coli strain. AITC is characterized by antibacterial activity on the E. coli O157:H7 strain, similar to polymyxin B, causing cell membrane disintegration and metabolite efflux and, consequently, bacterial death [151]. AITC inhibits two key enzymes in the metabolism of bacteria: thioredoxin reductase, involved in the synthesis of ribonucleotides, and acetate kinase, which is associated with energy metabolism [152]. From the group of natural ITCs (SFN, AITC, BITC, phenyl, and isopropyl isothiocyanate), the highest comparable to AITC activity in inhibiting E. coli EHEC, including E. coli O157:H7, showed BITC. In comparison to conventional antibiotics, the tested ITCs better inhibited the production of the Shiga toxin. Detailed studies have shown that the aforementioned ITCs affect penta/tetraphosphate (p)ppGpp, which influences RNA polymerase activity, bacteriophages development, and Shiga toxin production. ITCs increase the levels of penta/tetraphosphate (p)ppGpp, decrease the synthesis of RNA, and inhibit the development of prophageand the production of the Shiga toxin [153].
P. aeruginosa (Gram-negative) aerobic bacterium is able to colonize various environments and produce biofilm [154,155]. Many bacteria, including Pseudomonas strains, are equipped with a quorum sensing (QS) [156] system, by which bacteria communicate to coordinate gene expression. This allows them to control the expression of genes that are important for the entire population, by secreting and receiving signal molecules called autoinducers. This system controls biofilm formation, bioluminescence generation, the production of antibiotics and siderophores, and bacterial motility [157]. These coordinated behaviors allow bacteria to compete with multicellular organisms and survive sharp and sudden environmental changes. In P. aeruginosa, there are two main QS systems—las and rhl. The las system consists of synthesis LasI, the “autoinduction” gene responsible for the synthesis of autoinducers N-[3-oxo-dodecanoyl]-L-homoserine lactones (3-oxo-C12-HSL) and lasR, genes encoding transcription regulators. The rhl system consists of the pair RhlI/RhlR, which respond to N-butyryl homoserine lactones (C4-HSL) [158,159,160]. Studies on natural isothiocyanates (AITC, BITC, and PEITC) and the mixtures of those ITCs have shown that AITC and the “cocktail” of ITCs were characterized by the highest activity. However, only PEITC inhibited biofilm production [161]. In other studies, AITC and PEITC caused cell membrane disintegration and an increase in the hydrophilic nature of the membrane, changing its physicochemical properties [162]. Meijler et al. [163], with the P. aeruginosa strain, showed that by using SFN and its sulfide analog erucin (28, Figure 5), it is possible to affect bacterial QS. The consequence of this was the inhibition of the production of biofilm and pyocyanin—a cytotoxic dye that affects the central nervous system, urological system, and vascular system, causing inflammation [164]. Both biofilm and pyocyanin production are virulence factors controlled by QS. Iberin (25, Figure 5), an SFN analogue, also affects bacterial QS, by reducing the activity of the RhlI/RhlR expression [165].
Studies on the activities of natural ITCs (AITC, BITC, PEITC, and SFN) and mixtures of these ITCs (AITC, BITC, and PEITC) have shown that the mixtures show the highest activities in relation to the S. aureus [166,167] (Gram-positive) strain. Among individual ITCs, BITC had the best activity, while AITC was inactive. It was also shown that BITC, PEITC, and SFN, and their mixtures showed higher activity than vancomycin [168]. Contrary to earlier research suggesting that AITC is inactive against S. aureus, Lu et al. [169], in 2016, proved that AITC causes growth inhibition of S. aureus, and similar to PEITC, causes cell membrane disintegration and bacterial death [162].
2.3. Clinical Trials of SFN
In addition to in vitro and in vivo tests, SFN has also been selected for clinical trials; however, there is a limited number of these results in the literature. Talalay et al. [170] described a placebo-controlled, double-blind phase I clinical trial of healthy volunteers using extracts of sprouts containing either glucosinolates (principally glucoraphanin, the precursor of SFN) or ITCs (principally SFN). After 7 days of trials, no significant toxicities associated with taking the extracts at the doses employed were observed. In 2007, Visvanathan et al. [171] published research, where eight healthy women took an oral dose of broccoli sprout preparations containing 200 μmol of SFN. Studies demonstrated that sulforaphane distributed to the breast epithelial cells in vivo and exerted pharmacodynamic action in these target cells, consistent with its mechanism of chemoprotective efficacy. Fahey et al. [172] showed that administration of sulforaphane (100 μmol/day on 14 days) improved the bronchoprotection response in asthmatics who had an increase in NQO1 gene expression and did not have a decrease in their initial response to the MCh challenge. Therefore, SFN administration was able to improve a major defect of even mild asthma. Yanaka and co-workers [173] conducted research in which they fed forty-eight H. pylori-infected patients with broccoli sprouts (70 g/d; containing 420 μmol of SFN precursor) for 8 weeks. Results showed antibacterial effects of SFN on H. pylori, leading to reduced gastritis, as well as an indirect (systemic) effect by increasing the mammalian cytoprotective (phase II) response. More information about the clinical trials of SFN are in the review articles [174,175].
Although SFN was tested in phase I and phase II clinical trials, where it had good anticancer and antibacterial properties, it was never qualified as a drug. This may be due to the polymorphism of the genes for the GSTM1 and GSTT1 glutathione S-transferase isoenzymes. GST is involved in the detoxification of many chemical carcinogens and is responsible for the metabolism of ingested ITCs. Cohort studies indicate a protective effect of a diet rich in cruciferous vegetables against cancers of the lung, colon and breast only in patients with GSTM1 and GSTT1-null genotype [176]. Due to the fact that GST participates in the metabolism of SFN and thus affects its excretion, lower enzyme activity in people with GST gene polymorphism may result in slower elimination and longer exposure of cancer cells to this compound provided with the diet. In people with gene GSTT1 a greater excretion of ITCs outside the body was observed, and thus a shorter time of exposure to cancer cells [177].
3. Methods of Synthesis of ITCs
Many ITC synthesis methods are described in the literature. The choice of the synthetic method depends on the availability of the starting substrates as well as on the sensitivity of other functional groups present in the substrates to the reaction conditions. Methods on the synthesis of ITCs (known thus far) are described in the Houben-Weyl encyclopedia [178]; new aspects of the synthesis were described by Mukerjee and Ashare [56], Wentrup et al. [179], and Singh et al. [180]. Primary amines are used as starting materials in the synthesis of isothiocyanates (Figure 8).
The synthesis of ITCs from primary amines, or their salts, involve the use of thiocarbonyl reagents, such as thiophosgene (34) and its substitutes, or carbon disulfide (35). They “enable” either directly or indirectly via the intermediate dithiocarbamates transformation of amines into ITCs (Figure 8, Path a and b). Alternatively, azides may be applied as substrates. These in turn allow the preparation of ITCs via a tandem Staudinger/aza-Wittig reaction, through the intermediate iminophosphoranes (Figure 8, Path c).
3.1. Synthesis ITCs Using Thiophosgene and Its Substitutes
The reaction of primary amines, or their salts with thiophosgene (34), is presently one of the oldest (but most commonly used) methods in the synthesis of ITCs [181,182]. The original product of the reaction is unstable thiocarbonyl chloride derivative [183], which, after elimination of hydrogen chloride, in an alkaline medium is transformed into the target isothiocyanate (Figure 9). Thiophosgene enables efficient conversion of both aliphatic and aromatic amines into ITCs. This method, however, is not suitable for bifunctional amines with reactive nucleophilic groups in the vicinal position because of subsequent cyclization of the original product.
A small excess of thiophosgene is recommended for use to prevent the formation of symmetrical thioureas as a side product. In contrast, the presence of an organic or inorganic base facilitates the formation of isothiocyanate and serves to neutralize hydrogen chloride. The use of concentrated strong bases (e.g., NaOH) is not recommended in this case because of the easy hydrolysis of ITCs in an alkaline medium. Reactions using thiophosgene can be carried out in a homogeneous—as well as a two-phase—system. In a two-phase system, water-organic solvents, both thiophosgene and the forming ITC, do not undergo subsequent reactions. Chloroform [184] and dichloromethane [185] are the most commonly used organic solvents and, as a base, calcium carbonate [186], sodium bicarbonate [187], or diluted sodium hydroxide solution [188] are usually used. The reaction is carried out at room temperature, and due to the heterogeneity system, intensive stirring is necessary [189]. In a homogeneous system, toluene [190] or acetone [191] are the most often used solvents, and triethylamine is used as a base.
Despite the relatively high toxicity of thiophosgene and its unpleasant smell, the aforementioned method is still popular due to its simplicity, versatility, and mild reactive conditions. For these reasons, thiophosgene is sometimes replaced by less toxic and less reactive reagents. Among them, the most widely used are thiocarbonyldiimidazole (36) [192], 1,1′-thiocarbonyldi-2,2′-pyridone (37) [193], and di-2-pyridyl thionocarbonate (38) [194] (Figure 10).
Although the use of these reagents guarantees high efficiency of thiocarbonylation, their use increases the cost-effectiveness of the method compared to the thiophosgene method.
3.2. Synthesis ITCs with a Desulfurizing Agent
The second most commonly used approach in the synthesis of ITCs is a two-step reaction, involving the conversion of amine or its salt and carbon disulfide (35) into the intermediate dithiocarbamate, which, in the presence of a desulfurizing reagent, undergoes transformation to the target ITCs [195] (Figure 11).
Most often, these two-stage reactions are performed in a “one-pot” version. The method is compatible with various functional groups and the range of its applicability is limited only by the nucleophilicity of the starting amines. In most cases, dithiocarbamates are formed quickly and in quantitative yield at room temperature. Only for aromatic amines, with strong electron-withdrawing substituents in the ring, does the formation of dithiocarbamates require the use of strong bases, such as sodium hydride, increasing the reaction time and elevating temperature. The above-mentioned method was described for the first time in 1886 by Hofmann [196]. Since then, there has been rapid development of ITC synthesis using various desulfurizing reagents. Unfortunately, the use of many of them involve drastic reaction conditions; byproducts formed in these reactions, such as heavy metal salts or carbodiimides, are difficult to remove [56]. Therefore, in recent years, effort has been made to find such desulfurizing reagents that would make in situ conversions of dithiocarbamates into the target ITCs quick, easy, and efficient.
Many desulfurizing reagents are known. Among them, the most widely used are ethyl chloroformate (39) [195,197], hydrogen peroxide (40) [198], peptide coupling reagents such as HBTU (41), PyBOP (42) [199], TBTU (43) [200], TFFH (44) [201], DCC (45) [202], and T3P® (46) [203], tosyl chloride (47) [204], mesyl chloride (48) [205], molecular iodine (49) [206], with (diacetoxyiodo)benzene (50) [207], di-tert-butyl dicarbonate (51) [208], methyl acrylate (52) [209], 2,4,6-trichloro-1,3,5-triazine (53) [210], coper (II) sulfate (54) [211], cobalt (II) chloride (55) [212], sodium persulfate (56) [213], and 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium toluene-4-sulfonate (DMT/NMM/TsO−) (57) [214] (Figure 12). Eschliman and Bossmann [215] described some of the aforementioned reagents.
The most commonly used desulfurizing reagents are briefly characterized below, because each of these reagents has different properties. Hydrogen peroxide (40) allows obtaining target alkyl ITCs in a short time at room temperature. The main disadvantages are the moderate yields and limitations to the synthesis of aliphatic ITCs. PyBOP (42) and TFFH (44) coupling reagents, in turn, enable the synthesis of ITC in the solid phase; however, the thiourea side-product is formed under the reaction’s conditions. The use of tosyl chloride (47) leads to aliphatic and aromatic ITCs in high yields. The use of di-tert-butyl dicarbonate (51), in the presence of a catalytic amount of DMAP or DABCO, allows obtaining aryl and aliphatic ITCs with high purity and in high yields within a few minutes. However, the synthesis of aryl ITC requires an extended reaction time. The most important advantage of this reagent is that the by-products formed in the reaction are easily removable gases (CO2, COS) or volatile liquids (tert-butyl alcohol). The application of (diacetoxyiodo)benzene (50) allows the preparation of aryl ITCs in high yields; however the high price of 50 precludes its use in a large-scale synthesis. For alkyl and cycloalkyl, ITCs yields are lower. Cheaper and non-toxic molecular iodine (49) also enables preparation of aromatic ITCs in high yields, and aliphatic isothiocyanates are formed in higher yields than in the reaction with (diacetoxyiodo)benzene (50). The reaction is environmentally friendly because it occurs in water in the presence of sodium bicarbonate [216]. The use of cyanuric chloride (53) allows obtaining high yields of aliphatic and aromatic ITCs, with both electron-donating and electron-withdrawing substituents. The advantage of this reaction is that it is performed in water. Propane phosphonic acid anhydride (T3P®) (46) enables synthesis of aliphatic, aromatic, and bifunctional ITCs, as well as isothiocyanates derived from esters of α-amino acids in high yields and with high purity. The method is compatible with a variety of protecting groups, and the reactions occur without racemization. A recently described coupling reagent 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium toluene-4-sulfonate (DMT/NMM/TsO−) (57) also enables synthesis of aliphatic and aromatic isothiocyanates, in a short time, in organic solvent as well as in water, and in microwave or in normal conditions, with very good yields. Additionally, DMT/NMM/TsO− enables synthesis of isothiocyanate derivatives of natural and unnatural amino acids without racemization.
In addition to the synthesis of isothiocyanates performed under conventional conditions, microwave-assisted synthesis of ITCs using primary amines and carbon disulfide as substrates were recently developed [217]. This approach enables synthesis of aliphatic and aromatic ITCs in high yields. Transformation of intermediate dithiocarbamates into ITCs occurs without the addition of any desulfurizing agent.
3.3. Synthesis ITCs via the Tandem Staudinger/aza-Wittig Reaction
The tandem Staudinger/aza-Wittig reaction is an alternative and equally efficient method used for the preparation of ITCs. The Staudinger reaction is the reaction of organic azide with a tricoordinated organophosphorus compound (triphenylphosphine (Ph3P), triphenyl phosphite, or triethyl phosphite) leading an iminophosphorane with the losses of nitrogen (Figure 13a) [218].
The use of the aza-Wittig reaction for ITC synthesis was first reported by Molina et al. [219]. It involves the conversion of primary amines and dibromotriphenylphosphorane (triphenylphosphine dibromide) (PPh3PBr2) into iminophosphoranes, followed by their reactions, with carbon disulfide (35) giving target ITCs (Figure 13b). The tandem Staudinger/aza-Wittig reaction was first described by Tsuge et al. [220] in 1984 (Figure 13c). The advantage of the tandem reaction over the two-stage process, and the mechanistic aspects of this transformation were reported in 2006 by Isoda et al. [221]. In a tandem reaction, iminophosphorane formed from azide and Ph3P reacts in situ with CS2, “giving” the target ITCs in high yield (Figure 13c). However, problems may occasionally occur with the separation of ITCs from an equimolar amount of triphenylphosphine sulfide formed in the reaction, and small amounts of triphenylphosphine oxide (a by-product of Ph3P oxidation).
3.4. The Latest Approaches to ITC Syntheses
In the past few years, several efficient approaches to the syntheses of ITCs were developed. They utilize primary amines as starting materials, and the appropriate fluorine reagents as desulfurizing agents, replacing toxic thiophosgene or carbon disulfide (Figure 14).
Liao et al. [222] used primary amines as substrates, and the Langlois reagent (CF3SO2Na) [223] in the presence of copper iodide and diethyl phosphonate, to obtain a library of structurally diverse aromatic as well as aliphatic ITCs in high yields (Figure 14, path a). The replacement of toxic thiophosgene with the Langlois reagent resulted in the development of a simple, safe, and environmentally friendly method of ITC synthesis. The disadvantages of this method include long reaction times, high temperatures, and low functional group tolerance (e.g., pyridinyl).
A highly efficient, selective, and fast method for the synthesis of ITCs from primary amines, by using a bench-stable, solid reagent (Me4N)SCF3 (tetramethylammonium trifluoromethane thiosulfate), was described by Schoenebeck et al. [224] (Figure 14, path b). The target ITCs are easily separated from the solid side products, and the method is compatible with several functional groups. In addition, the use of (Me4N)SCF3 enables the transformation of secondary diamines into cyclic thioureas.
An alternative method of ITC preparation involves the reaction of thiocarbonyl fluoride (CF2=S), generated from difluorocarbene and elemental sulfur (S8), with primary amines (Figure 14, path c) [225]. Difluorocarbene is valuable and versatile, intermediate in organic synthesis, and is generated from Ph3P+CF2CO2− (PDFA) [226]. Synthesis of ITCs is very fast, and the series of aromatic isothiocyanates with electron-donating and electron-withdrawing groups in the aromatic ring, as well as aliphatic ITCs, have been obtained in high yields. The only disadvantage of this method is that PDFA is relatively expensive, and in the reaction of o-phenylenediamines with the PDFA/S8 system, difluoromethylthiolated heterocycles are formed.
The methodology using thiocarbonyl fluoride was also reported by Zhen et al. [227]. The authors obtained a pool of aromatic and aliphatic ITCs, in moderate to good yields, in the reaction of thiocarbonyl fluoride generated from CF3SiMe3 (the Ruppert–Prakash reagent), elemental sulfur, and KF or AgSCF3 at mild conditions (Figure 14, path d). The Ruppert–Prakash reagent is a stable, relatively cheap, easy to handle and widely used reagent [228].
In 2019, Feng and Zhang [229] established an organophosphine-free, one-pot, copper-catalyzed, three-component synthesis of ITCs from primary amines, sodium bromodifluoroacetate (BrCF2CO2Na), and elemental sulfur (S8) in the presence of K3PO4, as a base (Figure 14, path e). According to the authors, isothiocyanation of amines takes place through the intermediate thiocarbonyl fluoride, or alternatively, via intermediate isocyanide, followed by its reaction with sulfur—this despite the fact that the reaction requires a prolonged time and a high temperature for completion, compatible with different functional groups, and a series of aromatic as well as aliphatic isothiocyanates have been obtained in moderate to good yields. For o-phenylenediamine and o-hydroxyaniline, as substrates, the appropriate 1-difluoromethyl benzimidazole and benzoxazole have been obtained, respectively. This and other methods of synthesis isothiocyanates using element sulfur have been described in a recently published review article [230].
Recently, Wei et al. [231] published an efficient method synthesis of a library of isothiocyanates from primary amines using trifluoromethanesulfonyl chloride in the presence of reducing the agent triphenylphosphine and sodium iodide (Figure 14, path f). Trifluoromethanesulfonyl chloride (CF3SO2Cl) is commercially available, cheap, easy to handle, and widely used (e.g., in electrophilic chlorination [232], trifluoromethylation [233], or in chloro-trifluoromethylthiolation of alkenes and alkyne [234]) reagents. The authors obtained aromatic ITCs with electron-donating or electron-withdrawing groups as well as aliphatic ITCs with good yields. Except for ITCs, the authors synthesized thiocarbamoyl fluorides using secondary amines with good yields.
4. Synthesis of Bifunctional Analogs of Sulforaphane and Their Properties
This section focuses on the most commonly used methods for the synthesis of SFN and its difunctional analogues. This involves isothiocyanation of amines with thiophosgene or carbon disulfide/desulfurizing agents system, or the tandem Staudinger/aza-Wittig method using azides as substrates. The methods where SFN and its analogues are obtained by myrosinase catalyzed hydrolysis of glucosinolates isolated from Cruciferae [235,236,237], or obtained by other methods (e.g., reaction with KSCN [238]), are not included. The choice is restricted to SFN and its natural or synthetic bifunctional analogues with an unbranched alkyl chain containing two to six carbon atoms and having sulfinyl, sulfanyl, sulfonyl, phosphonate, phosphinate, phosphine oxide, carbonyl, ester, amide, ether, or a second isothiocyanate group. Except for the synthesis, the biological activity of SFN and its analogues are also discussed.
4.1. Synthesis of Sulforaphane and Its Sulfur Analogues and Their Properties
One of the most commonly used synthetic pathways of SFN (24) utilizing thiophosgene (34) for isothiocyanation of amines was described by Vermeulen et al. [239]. The authors started from 1,4-dibromobutane (58) and potassium phthalimide to form 1-bromo-4-N-phthalimido)butane (59). Next, compound 59 was converted into N-(4-methylsulfanyl-butyl)phthalimide (60) in the reaction with sodium methyl mercaptide. Deprotection of the amino group in 60 with hydrazine hydrate provided 4-(methylsulfanyl)butan-1-amine (61), a key intermediate in the synthesis of SFN and its analogues. Thus, erucin (28) was obtained in an 80% yield by the reaction of amine 61 with thiophosgene (34) in a two-phase system (chloroform/water) using sodium hydroxide as a base. Oxidation of 28 with m-chloroperbenzoic acid (MCPBA) afforded SFN (24) in a 90% yield. In the final stage, the authors converted SFN (24) into the N-acetyl-S-(N-4-methylsulfinylbutylthiocarbamoyl)-L-cysteine (sulforaphane mercapturic acid, 62) in the reaction with N-acetyl-L-cysteine (NAC) in a 77% yield (Figure 15).
The same approach to SFN preparation was used by Mays et al. [240]. The authors transformed 4-(methylsulfanyl)butan-1-amine (61) into erucin (28) in a 84% yield using thiophosgene (34) and sodium hydroxide as a base. Oxidation of 28 with an equimolar amount of MCPBA resulted in SFN (24) in a 84% yield, and the use of excess MCPBA led to erysolin (31) in a 60% yield (Figure 16).
The cytotoxicity of erucin, SFN and erysolin, as well as other synthesized ITCs, were examined on eight human cancer cell lines representing a broad range of carcinomas, including breast, colon, CNS, livery, ovary, prostate, and a mouse mammary normal epithelial cell (NmuMG) control line (Table 1).
SFN (24) had the lowest IC50 values for Hep3B (human liver carcinoma), erucin (28) for SF-268 (human CNS glioblastoma), and erysolin (31) for MCF7 (women’s breast cancer ER+ (Luminal A). SFN and erysolin presented the highest IC50 for NCI/ADR RES (human breast carcinoma), and erucin for HCI-H460 (human breast carcinoma). IC50 on NmuMG for SFN and erysolin was almost similar; however, for erucin, it was unquestionably higher (Table 1).
In addition to thiophosgene, its substitute (1,1′-thiocarbonyldi-2,2′-pyridone (37)) was used for SFN synthesis. Conaway et al. [241] oxidized starting N-(4-methylsulfanyl-butyl)phthalimide (60) to N-(4-methylsulfinyl-butyl)phthalimide (63) by MCPBA, followed by the amino group deprotection in 63 with hydrazine monohydrate to obtain 4-(methylsulfinyl)butan-1-amine (64) in a 60% yield. In the final step, isothiocyanation of amine 64 with 37 afforded SFN (24) in a 50% yield (Figure 17).
The authors tested tumor-inhibitory activities of phenethyl isothiocyanate (PEITC, 22), SFN (24), and their N-acetylcysteine conjugates (65 and 62) (Figure 17) on the development of malignancy from benign tumors in the lung of A/J mice after administration of NNK and B(a)P, two potent carcinogens of cigarette smoke involved in lung cancer in smokers. The results show that PEITC, SFN, and their N-acetylcysteine conjugates added to the diet after lung adenomas, inhibiting the progression to adenocarcinomas. The inhibitory effects of these compounds are likely to be associated with a combination of reduced cell proliferation and induced apoptosis.
SFN (24), its homologues iberin (25), and alyssin (26), as well as erucin (28), iberverin (27), and berteroin (29) were synthesized by Moon and co-workers [242]. The authors converted difunctional amines with sulfanyl (61, 66 and 67) and sulfinyl moieties (64, 68 and 69) and with unbranched alkyl chains containing 3 to 5 carbon atoms into the corresponding ITCs (24–29), using thiophosgene (34) and sodium hydroxide as a base, with moderate to good yields (Figure 18).
The bactericidal activity against H. pylori of synthesized ITCs was tested. All tested ITCs (24–29) showed strong anti-Helicobacter activity at the level of a 5 mg/disk exhibiting >5 cm inhibitory zones.
Based on the methodology presented in Figure 15, Ernst and co-workers [243] obtained iberverin (27) from 4-(methylsulfanyl)propan-1-amine (66) and thiophosgene (34) in a 78% yield. Oxidation of iberverin (27), with equimolar or excess amounts of MCPBA, provided iberin (25) or cheirolin (30) with 71% and 56% yields, respectively (Figure 19).
It was found that iberin (25), iberverin (27), and cheirolin (30) significantly induced Nrf2 nuclear translocation in NIH3T3 fibroblasts. The increase of nuclear Nrf2 levels was accompanied by an increase of heme oxygenase (HO-1) and γ-glutamylcysteine synthetase (γGCS) mRNA and other protein levels. Iberverin (27), iberin (25), and cheirolin (30) exhibited a similar potency to SFN (24) in terms of their Nrf2-dependent gene expression. Induction of Nrf2 by iberverin, iberin, and cheirolin may have occurred via the extracellular signal-related kinase (ERK)-dependent signal-transduction pathway.
A large group of ITCs can be considered as SFN analogues, having non-methyl substituents on the sulfinyl group. These compounds include fluorine derivatives of SFN, synthesized by Kiełbasiński and co-workers [244]. Thus, substrate 59 was converted into a trifluoromethyl derivative 70, and transformed the target product 73 via two independent routes. In the first one, amine hydrochloride 71, obtained after deprotection of amino group of compound 70, was converted into isothiocyanate 72 with a yield of 40% by the reaction with thiophosgene (34) or di-2-pyridyl thiocarbonate (38). Oxidation of the trifluoromethylsulfanyl group of 72, using MCPBA, gave the trifluoromethyl analog of SFN 73 a 50% yield. In the second synthetic pathway, phthalimido-sulfide 70 was oxidized to sulfoxide 74, followed by hydrazinolysis and subsequent isothiocyanation; thus, forming amine hydrochloride with thiophosgene (34) to give the target isothiocyanate 73 in a 87% yield. Using the first synthetic route, the authors also obtained trifluoroethyl analogues of SFN 75 and alyssin 76 with high yields (Figure 20).
The ITCs 73, 75–76, obtained as racemates, were separated on preparative chiral HPLC to the enantiomerically pure products. All three pairs of enantiomers of fluorine-containing analogs 73, 75–76 were tested in vitro for their cytotoxicity against malignant melanoma cell lines Malme-3M and normal skin fibroblast Malme-3. In Table 2, the activity of the most promising compound 75 is presented.
After 48 h of incubation, the optically active fluorine analogs (R)-75 and (S)-75 exhibited higher cytotoxicity than SFN; however, after 72 h of incubation, the cytotoxicity was comparable. These results could suggest that the anticancer mechanism of SFN and its fluorine analogs are different. As seen from Table 2, the most promising was (S)-1-isothiocyanato-4-((2,2,2-trifluoroethyl)sulfinyl)butane (S)-75).
Recently, the same research group described synthesis and biological activity of fluoroaryl analogs of SFN with 4 and 5 carbon atoms in the unbranched alkyl chain [245]. Thus, the starting ω-aminoalk-1-yl fluoroaryl or fluoroarylmethyl sulfides 77 were converted to the final fluoroaryl or fluoroarylmethyl analogs of SFN 80a–h by two alternative pathways (Figure 21). In the first path, amines 77 were transformed in the reaction with thiophosgene (34) into fluoroaryl or fluoroarylmethyl ω-isothiocyanatoalk-1-yl sulfides 78, followed by oxidation with MCPBA, to afford the target isothiocyanates 80a–h. In the second approach, sulfides 77 were oxidized to sulfoxides 79, followed by the treatment with thiophosgene, to provide 80a–h. Final α-(fluorosulfinyl)- and α-(fluoroarylmethylsulfinyl)-ω-isothiocyanatoalkanes 80a–h were obtained in good and very good yields.
All fluoroaryl and fluoroarylmethyl analogs 80a–h were tested in vitro for their anticancer, antibacterial, antifungal, and antiviral properties. The anticancer activity on the skin cancer cell line (MALME-3M), colon cancer cell line (HT-29), and breast cancer cell lines (MCF-7 and MDA-MB-231), as well as their normal cell lines, were studied. After 72 h of incubation, all compounds presented higher activity than SFN. The most active on all cancer cell lines were ITCs 80d and 80e (Table 3).
It was found that fluoroaryl or fluoroarylmethyl analogs 80a–h, as well as SFN, were inactive against Gram-negative bacteria (E. coli and P. aeruginosa). However, SFN and its fluoroaryl or fluoroarylmethyl analogs 80b–f had antibacterial activity against Gram-positive bacteria, including methicillin-resistant S. aureus (MARS), except for B. subtilis and E. hirae strains. In particular, ITC 80e was the most active (MIC values were in the range of 0.031–0.0625). Concerning anti-HIV activity, only ITC 80e showed similar activity to SFN (0.5 μM inhibited HIV replication in 9% of cases). Other ITCs were inactive.
In 2016, Shi et al. [246] synthesized a series of analogues of SFN, in which the methyl group adjacent to sulfur was replaced with heterocyclic moieties, such as furan (a), 5-methoxy-3H-imidazo[4,5-b]pyridine (b), 6-methoxy-1H-benzo[d]imidazole (c), 5-phenyl-1H-tetrazole (d), or benzo[d]thiazole (e). The sulfides 81a–e were transformed into amines 82a–e, and then were converted into sulfide derivatives of ITCs 83a–e with thiophosgene (34), in good and very good yields. In the next step, compounds 83d–e were oxidized with MCPBA into sulfoxide analogues of SFN 84d–e with good yields (Figure 22).
Sulfoxide and sulfone analogues of SFN with heterocyclic moieties 84a–c and 89a–c were prepared in a modified way. Compounds 81a–c were oxidized by tert-butyl hydroperoxide (TBHP) to sulfoxides 85a–c and sulfones 86a–c and then transformed to amines 87a–c and 88a–c using methylamine. In the final step, isothiocyanation of amines 87a–c and 88a–c with thiophosgene afforded ITCs 84a–c and 89a–c in good yields (Figure 23).
Sulfone analogues with tetrazole and thiazole moieties 89d–e were obtained from amines 82d–e prior to Boc protection of amino groups, and oxidation formed N-Boc amines to sulfone analogues 90d–e. Removal of the Boc protecting group in 90d–e, followed by the reaction free amines 88d–e with thiophosgene, allowed to obtain the final sulfone analogues 89d–e in low yields (Figure 24).
The series of SFN analogues with heterocyclic moieties 83a–e, 84a–e, and 89a–e were evaluated for their anticancer activities (breast cancer cell lines MCF-7 and SUM-159, acute leukemia stem cell-like cell line, and KG-1a). Among all synthesized analogues, tetrazole analogues 83d, 84d, and 89d were generally the most potent—and significantly more active—than SFN against the tree cancer cell lines (Table 4).
Moreover, compound 83d, as well as SFN, induced apoptosis in the SUM-159 cell line by increasing caspase-3 activity and significantly reducing the ALDH+ subpopulation in the SUM-159 cell line from 3.10% to 0.16%.
Recently Sestito et al. [247] designed and synthesized a new class of multitarget H2S-donor hybrids 99–104, combining the rivastigmine-scaffold, an acetylcholinesterase inhibitor with brain region selectivity [248], a well-known drug approved for Alzheimer’s disease, with SFN and erucin. Thus, mercaptobutyl derivative 91 was alkylated with the appropriate chloroacetamide 92a–c under the basic conditions to give the corresponding thioethers 93–95 with moderate yields. Hydrazinolysis of derivatives 93–95 provided amines 96–98, which, after the reaction with thiophosgene, gave ITCs 99–101 in 25–90% yields. Oxidation of ITCs 99–101 by Oxone® led to sulfoxide 102–104 in moderate yield (Figure 25).
Studies on the murine microglia cell line (BV-2) showed that all synthesized ITC hybrids 99–104 exhibited a H2S-donor profile in vitro. Compounds 99–104 showed significantly anti-inflammatory and antioxidant activities and induced the expression of proteins (i.e., GSH) involved in the antioxidant defense in the neuronal cell line. All hybrids produced a significant decrease in ROS production elicited by pro-inflammatory stimulus compared to the rivastigmine, which completely lacks antioxidant activity. The new hybrids were also able to reduce NO release in BV-2 cells, whereas rivastigmine showed no effect. Moreover, the most active compounds 99 and 100 increased the GSH level in the human neuroblastoma cell line SH-SY5Y.
In 2013, Hu et al. [249] obtained an extensive library of sulfanyl and sulfinyl analogs of SFN with an alkyl or phenyl substituent on a sulfur atom, and with an alkyl linker containing 3 to 6 carbon atoms using the dithiocarbamate approach and applying mesyl chloride (48) as a desulfurizing agent. Sulfide derivatives of ITCs 28, 128–147, were prepared, in the presence of Et3N, from the corresponding ω-(alkylthio)alkanamines 61, 108–127, carbon disulfide (35), and mesyl chloride (48) in 43–65% yields. The key intermediates, the appropriate ω-(alkylthio)alkanamines 61, and 108–127, were obtained from ω-bromoalkylphthalimides 59 and 105–107 in a standard procedure. Oxidation of sulfides 28, and 128–147 with MCPBA, led to sulfoxide analogues 24 and 148–167, with very good yields (Figure 26).
All synthesized ITCs 28, 24, 148–167 were evaluated in vitro for their cytotoxicity against liver hepatocellular carcinoma (HepG2), human lung adenocarcinoma (A549), woman breast cancer ER+ (Luminal A)(MCF-7), human colon cancer cell line (HCT-116) and human neuroblastoma cell line (SH-SY5Y). All tested compounds exhibited more potent inhibitory against five cancer cell lines than SFN. The IC50 for the most active ITCs are presented in Table 5.
For HepG2, the most active was ITC 151 with the IC50 value 2.05 μM. ITC 161 had the strongest inhibition against A549 with IC50 5.64 μM, and ITC 155 possess significant cytotoxicity for MCF-7 with IC50 3.3 μM. Derivative 152 against HCT-116 had an IC50 value of 2.06 μM, and isothiocyanate 157 against SH-SY5Y ITC had an IC50 value 2.79. Based on the tested ITCs 28, 24, 148–167, it was found that compounds with sulfanyl moiety 135 showed weaker inhibitory effects than most derivatives; however, compound 168 with a sulfone group showed a higher inhibitory activity against all cancer cell lines than SFN. It may indicate that replacing the sulfoxide group with a sulfone group results in higher biological activity. The studies on anticancer mechanisms examined on the HepG2 cancer cell line showed that SFN, as well as model ITC 155, could induce the S or G2/M phase cycle arrest, and 155 has more potent inhibitory activity than SFN. Moreover, ITC 155 exhibited greater induction of apoptosis upon treatment than SFN. ITC 155 presented a time- and dose-dependent activation on the Nrf2 transcription factor. Moreover, 155 acted as a more potent Nrf2 inducer than SFN.
Meijer and co-workers [163] synthesized SFN (24) and its analogue erucin (28) via the tandem Staudinger/aza-Wittig reaction. Thus, the starting 4-bromo-butanol (169) was transformed using a standard procedure into azido thioether 170. Then, this compound, in the reaction with triphenylphosphine and carbon disulfide, was converted into erucin (28) in an 81% yield. SFN (24) was obtained by oxidation of erucin (28) with MCPBA in 77% yield (Figure 27).
Studies on P. aeruginosa show that SFN and erucin strongly inhibit quorum sensing (QS) and virulence (biofilm formation and pyocyanin production). The assays on P. aeruginosa and in E. coli strongly indicate that SFN and erucin effectively bind LasR, resulting in inhibition of QS activation at concentrations that can be found in broccoli.
The Staudinger/aza-Wittig tandem reaction was used in the synthesis of enantiomerically pure (R)-SFN ((R)-24) [250] using diacetone-D-glucofuranose (171) (DAG)-methodology [251]. The stereoselective synthesis of SFN was based on the reaction of 1-azidobutanesulfinyl chloride (172) with DAG (171), using Hünig’s base as a catalyst, and affording the sulfinate ester (S)-173 in a 90% yield and in 94% diastereomeric excess. Reaction of methylmagnesium bromide with the sulfinate ester (S)-173 provided 4-azidobutyl methyl sulfoxide ((R)-174) with inversion of configuration, which, in turn, under the Staudinger/aza-Wittig reaction with triphenylphosphine and carbon disulfide, gave enantiopure (R)-SFN ((R)-24) in a 71% yield. The same methodology was applied in the synthesis of the other analogues. Thus, condensation of the selected Grignard reagents with sulfinate ester (S)-173 led to the desired azido sulfoxides, which, in a two-step Staudinger/aza-Wittig reaction, allowed obtaining enantiopure (R)-sulfinyl ITCs 175–178 in 47–90% yields (Figure 28).
The enantiopure analogues of SFN 175–178 were assayed in the activation of the cytoprotective transcription factor Nrf2. The obtained results indicate that there is a close relationship of Nrf2 activation and the steric demand of the substituent at the sulfinyl sulfur. The SFN analogues with an alkyl side chain were more active than the analogue with the aromatic sulfinyl group (R)-177. Within the dialkyl sulfoxides (R)-175, (R)-176, and (R)-178, the most active one is the pentyl sulfoxide (R)-175, while the dialkyl sulfoxide (R)-178, with an extended alkyl chain, is the least active.
The same research group in 2014 described synthesis of both enantiomers of SFN homologues with alkyl chains of different lengths between the isothiocyanate group and the sulfinyl group, with the chiral auxiliary derived from diacetone-D-glucofuranose (DAG) [252]. Thus, azidoalkanesulfinyl chlorides 179a–c in reaction with diacetone-D-glucofuranose (171), in the presence of DIPEA as a base, afforded the (S)-sulfinate esters 173 and (S)-180–181 with high yields, and good diastereomeric excesses. However, when pyridine was used as a base, (R)-sulfinate esters 173 and (R)-180–181 were obtained in high yields, although in lower diastereomeric excesses (Figure 29).
The replacement of chiral auxiliary in sulfinates 173 and 180–181 by the Grignard reagents takes place with the inversion of configuration at the sulfinyl sulfur. Condensation of methylmagnesium bromide with the sulfinate ester (S)-173, (S)-180, and (S)-181 afforded the corresponding azidoalkyl methyl sulfoxides (R)-174, (R)-182, and (R)-183 in high yields. Subsequent reactions of azides with triphenylphosphine and carbon disulfide led to enantiomerically pure (R)-SFN ((R)-24), (R)-alyssin ((R)-26), and their homologue (R)-184 with high yields (Figure 30).
Using the same methodology, the enantiomerically pure (S)-SFN ((S)-24), (S)-alyssin ((S)-26), and their homologue (S)-184 were obtained from the sulfinate ester (R)-173, (R)-180 and (R)-181 with high yields (Figure 31).
In the same way, using ethylmagnesium bromide or butylmagnesium bromide as a Grignard reagent, the authors synthesized analogues of SFN, in which the methyl group was replaced by ethyl or butyl substituents. Enantiopure sulfinyl analogues (R)-187 and (R)-188 were prepared with high yields (Figure 32).
The efficiency of the synthesized compounds as the inductors of phase II detoxifying enzymes was evaluated by studying their ability to activate the cytoprotective transcription factor Nrf2. It was shown that homologues containing 5 and 6 carbon atoms in the alkyl chain showed beneficial effects on the activation of Nrf2, increasing the steric size of the substituent on the sulfur has negative effects on the biological activity. Which sulfur stereochemistry has no effect on the ability of these analogues to activate the cytoprotective transcription factor Nrf2 is additionally noted (Table 6).
All synthesized (R) and (S) analogues of SFN were evaluated in vitro for their cytotoxicity against human lung adenocarcinoma (A549) and the fetal lung fibroblast (MRC-5) cell line as normal cells. Synthesized analogues were more effective against lung cancer than (R)-SFN, and among them, (S)-184 was the most promising compound that showed slight selectivity than SFN towards the cancer cell (Table 6).
4.2. Synthesis of Phosphorus Analogues of Sulforaphane and Their Properties
In this section, phosphorus analogues of sulforaphane (P-SFN) compounds, where methylsulfinyl moiety was replaced by phosphonate, phosphinate, or phosphine oxide groups, are described.
The first type of information about phosphorus analogues of SFN came from Posner et al. [188]. The synthesis and biological activity of (4-isothiocyanatobutyl)dimethylphosphine oxide (191), the phosphine oxide analogue of SFN, was described. The target (4-isothiocyanatobutyl)dimethylphosphine oxide (191) was obtained under basic conditions by isothiocyanation of (4-aminobutyl)dimethylphosphine oxide (190) with thiophosgene (34) in a 68% yield (Figure 33). The key intermediate 190 was prepared via a standard procedure for the synthesis of phosphine oxides from diethyl phosphite (189).
Phosphorus analogues of SFN 191, as well as sulforaphane (24), were evaluated in vitro as inducer potencies of NAD(P)H:quinine oxidoreductase (QR) in murine hepatoma cells (Hepa 1c1c7) (Table 7).
As seen in Table 7, the potency of dimethylphosphine oxide 191 and SFN (24) were almost equal.
In 2011, Oleksyszyn et al. [200] described the synthesis of a series of α- and β-dialkoxyphosphoryl isothiocyanates. Dialkyl α-(isothiocyanatoalkyl)phosphonates 204–215 were obtained in two ways, as shown in Figure 34. In the first way (method A) for the transformation of dialkyl α-azidoalkylphosphonates 192–198 into isothiocyanates 204–210, the tandem Staudinger/aza-Wittig reaction was used, and the final P-SFN were obtained in 35–75% yields. In the second way (method B), the targets P-SFN 211–215 were prepared with good and very good yields via desulfuration of dithiocarbamates formed in situ from aminophosphonates hydrochlorides 199–203 and carbon disulfide with TBTU. Method B was also used for the synthesis of β-dialkoxyphosphoryl isothiocyanates 216–218 (Figure 34).
The obtained P-SFN were tested for cytotoxicity on five cancer lines: lung cancer (A549), breast cancer (T47D and MCF-7), colon cancer (LoVo), as well as its doxorubicin-resistant variant–LoVo/DX. All tested α- and β-dialkoxyphosphoryl isothiocyanates showed very good antiproliferative activities in vitro comparable to the most active of natural isothiocyanates BITC (21) and PEITC (22). Moreover, the mechanism of anticancer activity was evaluated. Isothiocyanate 205 showed inhibition of the cell cycle in the subG0/G1 phase, and compound 216 inhibited the cell cycle in the G2/M phase.
Recently, Janczewski and co-workers [203] developed a one-pot, two-step procedure for the synthesis of phosphorous analogues of SFN 223–226 from aminophosphonate hydrochlorides 219–222 and carbon disulfide using propane phosphonic acid anhydride (T3P®) (46) as a desulfurizing agent. Reaction occurred in the presence of triethylamine via the intermediate dithiocarbamates and the target analogues 223–226 with the isothiocyanato group in the α- and β-positions, in relation to phosphorus, and with alkyl and aryl substituents obtained in moderate yields (Figure 35).
A series of diaryl (1-isothiocyanoalkyl)phosphonates 237–246 were prepared and tested for inhibition of human tumor proliferation by Oleksyszyn and co-workers [253]. The starting aminophosphonate hydrobromides 227–236 were transformed in an alkaline environment in the presence of CS2, and HBTU or H2O2 as desulfurizing agents into isothiocyanates 237–246 in 57–90% yields (with HBTU) or 55–93% (with H2O2) (Figure 36). Alkyl substituted analogs 237–241, and 3,4-dimethoxyphenyl derivative 246 were obtained in better yields in the reaction with H2O2, while for the other compounds, 242–245 HBTU resulted in slightly higher yields.
The in vitro antiproliferative activities of synthesized ITCs 237–246 against LoVo, LoVo/DX, A549, and MCF-7 cell lines were in the range of natural isothiocyanates despite the significant differences in their structures. Among them, ITC 246 was the most active (Table 8).
In 2017, Gajda and Wietrzyk and co-workers [254] designed and synthesized a library of novel bifunctional SFN analogues 282–316, structurally diverse dialkyl, and diphenyl ω-(isothiocyanato)alkylphosphonates (P-ITCs) with an unbranched alkyl side chain containing 2 to 6 carbon atoms. The synthesis of P-ITCs was based on the conversion of dialkyl [255] as well as diphenyl ω-azidoalkylphosphonates 247–281 into the target dialkyl and diphenyl ω-(isothiocyanato)alkylphosphonates 282–316, using the tandem Staudinger/aza-Wittig reaction with triphenylphosphine and carbon disulfide with good and very good yields (Figure 37).
The authors developed a one-pot strategy enabling the direct conversion of ω-(isothiocyanato)alkylphosphonates 287–291 into the selected alkyl, or phenyl ω-(isothiocyanato)alkylphosphonates 282–286, 297, 302, 316–325 in moderate and very high yields (Figure 38).
Dealkylation of diethyl ω-(isothiocyanato)alkylphosphonates 287–291 by bromotrimethylsilane (BTMS) generated intermediate bis(trimethylsilyl)alkylphosphonates 326. Next, crude 326 was converted into the appropriate ω-(isothiocyanato)alkylphosphonic dichlorides 327 treated with oxalyl chloride (COCl)2 in the presence of a catalytic amount of DMF. The subsequent reaction of crude dichlorides 327 with the appropriate alcohol or phenol, in the presence of triethylamine, and a catalytic amount of DMAP, provided the target P-ITCs 282–286, 297, 302, 316–325 in moderate yields (Figure 38).
All synthesized P-ITCs 282–325 were evaluated in vitro for antiproliferative activity against the colorectal adenocarcinoma cell line LoVo and its doxorubicin-resistant subline LoVo/DX. SFN, and other natural isothiocyanates, such as BITC or AITC, were used as references. All tested compounds 282–325 showed high activity on LoVo and LoVo/DX, higher than natural ITCs (SFN and BITC). The most active were ω-(isothiocyanato)alkylphosphonates with branched isopropyl 293–297 and isobutyl 298–302 groups on phosphorus. The activity was, for some compounds, more than 10 times higher than SFN activity (Table 9).
In addition, the antiproliferative activity of selected P-ITCs 291, 297, and 316 were tested on seven cancer cell lines: murine mammary gland cancer (4T1), leukemia (HL60) and its subline resistant for mitoxantrone (HL60/MX2), non-small lung cancer (A549), uterus sarcoma (MESSA and MESSA/Dx-5), and normal murine fibroblast (BALB/3T3). Compounds 291 (IC50 = 0.5 ± 0.2 μM) and 316 (IC50= 0.8 ± 0.2 μM) showed similar activity to cisplatin (CDDP) (IC50 = 0.4 ± 0.2 μM), relative to HL60 cells. Research with the selected P-ITCs 291, 297, and 316, demonstrated that P-ITCs inhibited the G2/M cell cycle on LoVo and LoVo/DX, where P-ITC 316 was more active than SFN, as well as P-ITCs 291, 297, and 316-induced apoptosis. In vivo studies of the selected P-ITCs showed slightly lower anticancer and antimetastatic activity in comparison to naturally occurring BITC. However, recent studies in vivo on Zebrafish have shown that P-SFN 291 was characterized by high anticancer activity and low toxicity [256].
The same research group described the synthesis of phosphinates and phosphine oxides analogues of SFN with an unbranched alkyl side chain containing 2 to 6 carbon atoms and with aliphatic and phenyl substituents on the phosphorus atom [257]. The (ω-Isothiocyanatoalkyl)dimethylphosphine oxides 191, 328–330, were obtained in a three-step reaction following the procedure described by Posner et al. [188]. Thus, crude (ω-bromoalkyl)dimethylphosphine oxides 331–334, prepared in the reaction of diethyl phosphite 189, with methylmagnesium chloride and 1,n-dibromoalkanes, were subsequently converted into (ω-azidoalkyl)dimethylphosphine oxides 335–338 in the microwave-assisted reaction with an aqueous solution of sodium azide. Next, crudes 335–338 were converted to (ω-isothiocyanatoalkyl)dimethylphosphine oxides 191, 328–330 in the tandem Staudinger/aza-Wittig reaction with triphenylphosphine and carbon disulfide in low yields (Figure 39).
A different approach was applied for the synthesis of (ω-isothiocyanatoalkyl)diphenylphosphine oxides 349–353. Thus, starting N-Boc derivatives 339–343 were deprotected under the acidic conditions to afford (ω-aminoalkyl)diphenylphosphine oxides 344–348, quantitatively. The subsequent isothiocyanation of amines 344–348 with thiophosgene provided the target (ω-isothiocyanatoalkyl)diphenylphosphine oxides 349–353 in good and very good yields (Figure 40).
The next group of P-ITCs, such as ethyl (ω-isothiocyanatoalkyl)(diethoxymethyl)phosphinates 358–361, and methyl and ethyl (ω-isothiocyanatoalkyl)(phenyl)phosphinates 370–377, were synthesized from the appropriate (ω-azidoalkyl)phosphinates 354–357 and 362–369 using the Staudinger/aza-Wittig reaction, with triphenylphosphine and carbon disulfide, in moderate and good yields (Figure 41 and Figure 42).
All P-ITCs 191, 328–330, 349–353, 358–361, and 370–377 were evaluated in vitro for antiproliferative activity against LoVo and LoVo/DX cancer cell lines, and SFN, as well as other natural isothiocyanates, such as BITC or AITC, were used as a reference. Almost all tested compounds, except (ω-isothiocyanatoalkyl)dimethylphosphine oxides 191, 328–330, showed high activity on LoVo and LoVo/DX, higher than natural ITCs (SFN and BITC). The most active were (ω-isothiocyanatoalkyl)diphenylphosphine oxides 349–353 and methyl and ethyl (ω-isothiocyanatoalkyl)(phenyl)phosphinates 370–377, which IC50 on LoVo were between 1.8 ± 0.4 and 4.7 ± 1.3 μM and were much more active than SFN and BITC. The antiproliferative activity of the selected P-ITCs 353 and 377 were also tested on 4T1, HL60 and HL60/MX2, A549, MESSA, and MESSA/Dx-5, and BALB/3T3 cell lines, where both P-ITCs showed similar activity. Compounds 353 and 377 were assessed for their mechanisms of action as inducers of the G2/M cell cycle arrest and apoptosis. Ethyl (6-isothiocyanatohexyl)(phenyl)phosphinate (377) was tested in vivo on the 4T1 cell line and demonstrated moderate antitumor activity, similar to that BITC.
The last paper devoted to the synthesis and biological activity of P-ITCs is from 2019 [258]. The authors synthesized a series of diaryl ω-(isothiocyanato)alkylphosphonates, with a chlorine atom and methoxy, dimethoxy, methylsulfanyl, or methoxycarbonyl groups at ortho, meta, or para positions of the phenyl ring, and with an unbranched alkyl chain 378–411 (n = 2–6). Using the same methodology [254] as shown in Figure 38, diethyl ω-(isothiocyanato)alkylphosphonates 287–291 were converted in a one-pot reaction with bromotrimethylsilane, oxalyl chloride, and the appropriate phenol derivatives to target diaryl ω-(isothiocyanato)alkylphosphonates 378–411 with good yields (Figure 43).
Diaryl ω-(isothiocyanato)alkylphosphonates 378–411 were evaluated in vitro for antibacterial activity on S. aureus and P. aeruginosa strains in comparison with natural PEITC and with gentamicin used as a reference antibiotic. All synthesized P-ITCs characterized high antibacterial activity, higher than natural PEITC. Against the S. aureus strain, the most active were 379 (IC50 = 1.5 ± 0.1 μM) and 383 (IC50 = 2.5 ± 0.2 μM) with activity similar to the gentamicin (IC50 = 1.0 ± 0.1 μM). Against the P. aeruginosa strain, the most active were 381, 383–384, 388–389, with IC50 between 3.0 ± 2.3 and 4.0 ± 1.6 μM. The selected compounds were three- and four-times more active than gentamicin (IC50 = 12 ± 1.0 μM).
Except for antibacterial activity, all P-ITCs were evaluated for antiproliferative activity against LoVo and LoVo/DX cancer call lines. All compounds presented high antiproliferative activity, higher or similar to SFN. The most active was compound 407 (IC50= 1.0 ± 0.1 μM), which was 3 times higher than cytostatic CDDP, and more than 22 times higher than SFN.
Selected diaryl ω-(isothiocyanato)alkylphosphonates 379, 383, 388, and 393 were also converted into mercapturic acid 412–415 derivatives in the reaction with N-acetyl-L-cysteine (NAC) and sodium bicarbonate, followed by acidification in good yields (Figure 44).
Preliminary evaluation of the in vitro antibacterial activity on the S. aureus strain showed that mercapturic acid 412–415 derivatives exhibited moderate antibacterial activity, lower or similar to the parent isothiocyanates 379, 383, 388, and 393. More accurate research concerning synthesis and antiproliferative activity of phosphonates, phosphinates, and phosphine oxide isothiocyanate-derived mercapturic acids, were described by Psurski and co-workers [259].
4.3. Synthesis of Carbonyl and Amide Analogues of Sulforaphane and Their Properties
The next group of SFN analogues are molecules containing carbonyl, ester, and amide moieties. Posner et al. [188] used the same methodology with thiophosgene, shown in Figure 33 for synthesis of carbonyl analogue of SFN, where methylsulfinyl moiety was replaced by carbonyl group (419). In this way, δ-valerolactam (416), in the reaction with di-tert-butyl dicarbonate and methylmagnesium iodide. was transformed into 4-N-Boc-aminobutyl methyl ketone (417). Ketone 417 under hydrolytic conditions was converted to the 4-aminobutyl-methyl ketone (418), followed by the reaction with thiophosgene to afford the 2-oxohexyl isothiocyanate (419) with an overall yield of 6% from lactam 413 (Figure 45).
Carbonyl analogue 419 of SFN, as well as SFN (24), were evaluated in vitro as inducer potencies of NAD(P)H:quinine oxidoreductase (QR) in murine hepatoma cells (Hepa 1c1c7) (Table 10).
As seen in Table 10, the 2-oxohexyl isothiocyanate (419) and SFN (24) were the same in potency as the inducer of NAD(P)H:quinine oxidoreductase.
In 2009, Amara et al. [260] synthesized carbonyl analogues of SFN with a longer alkyl chain containing 8 to 10 carbon atoms, being inhibitors of quorum sensing in P. aeruginosa. As a template, the authors used a structure of a natural autoinducer of P. aeruginosa–3-oxo-C12-N-acyl homoserine lactone (3-oxo-C12-HSL, 420). Final ITCs 424–427 were obtained with good and very good yields from the appropriate azides 421–423 in the tandem Staudinger/aza-Wittig reaction with triphenylphosphine and carbon disulfide (Figure 46).
The inhibitions of quorum sensing of P. aeruginosa by ITCs 424–426 was evaluated using the luminescent PAO1-luxABCDE wild type strain. All synthesized ITCs 424–426 strongly inhibited luminescence in this wild type strain. The strongest inhibitor of luminescence appeared to be 426 (IC50 = 45.2 ± 0.7 μM), followed by 425 (IC50 = 113 ± 19 μM) and 424 (~300 μM). Additionally, in the assay with the wild type PAO1 strain, ITC 425 presented significant inhibition of the quorum sensing-controlled virulence factor expression, as well as biofilm formation. The authors showed that the obtained isothiocyanate-based probes covalently and selectively bound Cys79, found in the LasR-binding pocket.
The same research group synthesized the second generation of ITCs based on ITC 425 with halogen moieties (fluorine, bromine, and chlorine) at the β-position [261]. As in previous studies, azides 427–429 were transformed into ITCs 430–432 in the reaction with triphenylphosphine and carbon disulfide with good yields (Figure 47).
The bioactivity of new ITCs 430–432 were evaluated using P. aeruginosa PAO1-UW and PAO-JP2 (lasl/rhll double mutant) strains carrying the luxCDABE cassette as reporters for LasR activation. In this tests, ITC 430 showed more complete inhibition of LasR than parental ITC 425. On the other hand, ITCs 431 and 432 had higher inhibition of LasR than fluorine-analogue of ITC. Additionally, ITC 430 inhibited pyocyanin production by almost 40%, and its inhibition was stronger than in the presence of ITC 425. Moreover, ITC 430 caused a swarming inhibition of 44% on agar plates, a stronger effect than that observed for ITC 425. Other ITCs, 431 and 432, did not inhibit swarming at all. The authors also showed that, in vivo, ITC 430 increased survival of C. elegans from bacterial infections over the course of four days.
The Staudinger/aza-Wittig reaction was also applied to the synthesis of analogues of 6-(methylsulfinyl)hexyl isothiocyanate (6-MITC, 433) with the methyl sulfinyl group replaced by another functional group [262]. For this purpose, 1,6-hexanediol (434) was transformed in a multi-stage reaction into THP-protected iodide 435. Then iodide 435, after prior azidation with NaN3, was turned into a THP derivative of ITC 436 with triphenylphosphine and carbon disulfide in an 82% yield. After, deprotection of the tetrahydropyranyl (THP) group with TsOH gave 6-isothiocyanatohexan-1-ol (437) in a 71% yield. ITC 437 was used as the starting material in the divergent synthesis of 6-isothiocyanatohexyl acetate (438), 6-isothiocyanatohexyl 2,2,2-trifluoroacetate (439), and 6-isothiocyanatohexanal (440) (Figure 48).
The same sequence of reactions: azidation and reaction with triphenylphosphine/carbon disulfide system, was used for the efficient synthesis of 1-isothiocyanato-6-methoxyhexane (443) and 1-isothiocyanato-6-(methylthio)hexane (444) from 1-chloro-6-methoxyhexane (441) and (6-chlorohexyl)(methyl)sulfane (442) (Figure 49).
All synthesized ITCs 437–440 and 443–444, as well as thiocyanate 445, were screened for their antiproliferative and anti-NO production activities, in vitro, using mouse macrophage-like cell line J774.1 cells (Table 11).
Compounds 437–439 and 443 had almost similar antiproliferative and anti-NO production activities at those of ITC 433. ITCs 440 and 444 were less active than ITC 443. Thiocyanate 445, compared with ITC 433, had no activity, which shows that the isothiocyanate group is responsible for biological activity.
Milelli, Minarini, and co-workers [263] described synthesis of novel quinazoline derivatives with polymethylene-amide linkers of different lengths, and possessing an isothiocyanate group, 451–455, or derivatives, such as 457, where NCS moiety was directly connected to the aromatic ring. In substrates 446–450 and 456, isothiocyanation of amino groups was accomplished with 1,1′-thiocarbonyldi-2,2′-pyridone (37) in satisfactory yields (Figure 50).
ITCs 451–455 and 457 were evaluated for their ability to inhibit the proliferation of the highly epidermal growth factor receptor (EGFR-TK) expressed in human epithelial cancer cells A431 and HaCaT cancer cells. ITCs 451–455 had similar activity on the A431 cell line (IC50 between 14.38 ± 1.5 μM and 17.06 ± 1.87 μM) to SFN (IC50 = 15.76 ± 1.89 μM), and also similar activity on the HaCaT cell line. The most active was ITC 457, which IC50 on the A431 cell line was much higher than SFN and other ITCs (IC50= 2.04 ± 0.22 μM) and also higher on HaCaT (IC50 = 2.62 ± 0.31 μM). Which ITC 457 did not affect the normal cell proliferation of human fibroblasts (HGFs) is noteworthy. The inhibitory effect of new ITCs 451–455 and 457 on EGFR-TK activity evaluated on A431 cell lysates showed that ITC 457 had inhibition effects at the nanomolar concentrations with a maximum inhibition of 89% at 10 μM. ITCs 451–455 were weaker inhibitors. Additionally, treatment of A431 cells with ITC 457 at 2 μM led to apoptosis, in terms of DNA fragmentation.
In 2019, Boehm and co-workers [264] synthesized crystalline SFN analogues 459 and 461. Both isothiocyanates 459 and 461 were obtained in good yields, in a two-step reaction of the appropriate amine hydrochlorides 458 or 460, and carbon disulfide in the presence of triethylamine, using hydrogen peroxide or tosyl chloride as desulfurizing agents (Figure 51).
The dialkyl tertiary squaramide 461 isolated as a crystalline solid with good solid-state stability was shown to be a covalent Nrf2 activator that binds to the BTB domain of KEAP1. Additionally, ITC 461 has demonstrated efficiency for activation of the Nrf2 pathway in human bronchial epithelial (BEAS2B) cells, translating to a dose-dependent inhibition of lung inflammation in an in vivo model of pulmonary oxidative stress.
Recently, Wang and co-workers [265] designed and synthesized a series of cyclin-dependent kinase 9 (CDK9) inhibitors with cancer stem cell (CSC) inhibition activity for non-small-cell lung cancer (NSCLC) therapy. The structures of the inhibitors were based on the combination of the pyrrolo-[2,3-d]pyrimidines-2-amine from ribociclib as CDK9 pharmacophore, and analogues of SFN with alkyl chains of different lengths as targeting CSC pharmacophore. Briefly, the key N-Boc amines 462a–e and 463aa–de were converted into the final ITCs 464a–e and 465aa–de after prior deprotection of the amino group by trifluoroacetic acid, followed by the reaction with carbon disulfide and N,N’-dicyclohexylcarbodiimide (DCC, 45) (Figure 52). The same methodology was used to transform amines 466, 467a–g, and 468 into ITCs 469, 470a–g, and 471 (Figure 53).
All ITCs 464a–e, 465aa–de, 469, 470a–g, and 471 were evaluated regarding inhibition activity against CDK4, CDK6, and CDK9, with ribociclib as the positive control. Some compounds showed high potent activity against CDK9 as well as high selectivity for CDK9. The inhibition of tested ITCs was similar or higher than inhibition of ribociclib. In particular, ITC 470e with an 8-isothiocyanatooctanamide linker, exhibited high enzymatic inhibition (IC50 = 11 nM vs. IC50 of ribociclib = 197 nM). ITC 470e was tested against several tumor cell lines, including NSCLC, breast cancer, hepatocarcinoma, cervical cancer, leukemia, and lymphoma, using CCK8 assay and SFN as positive controls. ITC 470e had the highest activity against NSCLC cell lines, especial A549 and H1299, with IC50 values less than 0.5 μM. For comparison, IC50 of SFN on A549 was more than 10 μM and IC50 of ribociclib was 7.455 μM. For other cell lines, ITC 470e exhibited lower anti-viability activity. These results demonstrated good cellular selectivity of ITC 470e. Additionally, studies on A549 and H1299 showed that ITC 470e dose-dependently inhibited the cell cycle at G2/M phase and induced apoptosis of A549 cells in a concentration-dependent manner. Moreover, ITC 470e decreased the formation of colonies in the two NSCLC cell lines. In vivo studies on H1299 xenograft mouse models showed that ITC 470e displayed potent anti-tumor activities.
4.4. Synthesis of Ether-Linked Analogues of Sulforaphane and Their Properties
Perez and co-workers [266] synthesized ether-linked isothiocyanate as the LasR antagonist of quorum sensing in P. aeruginosa. Briefly, etherification of phenol 472, followed by amino group deprotection with TFA and a two-step reaction with carbon disulfide and molecular iodine, afforded ether-linked isothiocyanates 473 and 474 in low yields (Figure 54). A LasR antagonist bioassay exhibited low IC50 values (ITC 473 IC50 > 200 μM and ITC 474 IC50 = 145 ± 105 μM) with no effect on bacterial growth.
In 2019, Bussolo and Minutolo et al. [267] synthesized novel glycoconjugated H2S donors with the isothiocyanate group. For this purpose, the starting N-Boc-glycoconjugates 475a–b were deprotected with TFA, followed by the reaction of amino derivatives 476a–b with an excess of carbon disulfide, triethylamine, and hydrogen peroxide to afford final, fully acetylated ITCs 478a–b, with low yields. On the other hand, deacetylation of O-acetylated amines 476a–b with sodium methanolate, followed by the isothiocyanation of amines 477a–b, performed in the same manner as above, gave isothiocyanate glycopyranosides 479a–b in 21% and 22% yields, respectively (Figure 55).
ITCs 478b and 479a were evaluated for their cell inhibitory effects on viability, using pancreatic adenocarcinoma tumor cells (AsPC-1). Studies showed that glucose-derivative 479a was totally ineffective at inhibiting cell viability. However acetyl-galactosamine-derivative 478b exhibited a marked cytotoxicity against AsPC-1 tumor cells in a concentration-dependent manner with a pIC50 value of 4.45 ± 0.02. ITC 478b efficiently released H2S intracellularly with desirable slow kinetics and inhibition of cell cycle progression G0/G1 phase.
He and co-workers [268] designed and synthesized artemisinin–isothiocyanate derivatives with anti-glioblastoma effects. The artemisinin–isothiocyanate derivatives 481a–c were obtained in good yields from amines 480a–c in a two-step reaction with carbon disulfide and triethylamine used as a base, followed by desulfurization with acyl chloride (Figure 56).
All ITCs 481a–c showed higher anti-tumor effects in vitro than dihydroartemisinin (DHA) against human glioblastoma U87 cells. The most active was ITC 481b (IC50 = 7.41 ± 1.56 μM) (IC50 DHA = 118.95 ± 12.39 μM). ITC 481b reduced the viability of glioblastoma multiforme (GBM) in a concentration-dependent manner. ITC 481b also inhibited migration and induced apoptosis in U87 cells. Pyknosis and nuclear shrinkages were observed in ITC 481b-treated cells. Additionally, caspase 9 and cytochrome c were induced in U87 cells treated with ITC 481b and cleaved caspase 3 was increased. On the other hand, the anti-apoptotic protein Bcl-2 was downregulated, and pro-apoptotic protein BAX was upregulated.
4.5. Synthesis of Diisothiocyanates
Oleksyszyn and co-workers [269] synthesized a library of diisothiocyanates (diITCs) as well as their mercapturic acid derivatives conjugated with N-acetyl cysteine. Both isothiocyanate functional groups in diITCs were separated with unbranched alkyl linkers containing three and four carbon atoms (484 and 485). DiITCs 484 and 4825 were obtained from the corresponding diamines 482 and 483 in a two-step procedure with carbon disulfide and triethylamine as a base, followed by the reaction with HBTU as desulfurizing reagents (Figure 57).
Synthesized diITC were evaluated in vitro on LoVo and LoVo/DX cancer cell lines. Their IC50 values on the LoVo cancer cell line were very high and were lower than 2 μM. Additionally, their biological activities were higher than their mercapturic acid derivatives.
Diisothiocyanates were also obtained by Mustaev and co-workers [55]. The authors synthesized diITC 485, 490–493, with unbranched alkyl linker containing four to eight carbon atoms from the corresponding diamines 483, 486–489, and thiophosgene in the presence of triethylamine as a base (Figure 58).
The growth-inhibitory activity of synthesized diITCs 485, 490–493, against pathogenic bacteria, fungi, and molds were evaluated. DiITCs 485, 490–493, were more active against Gram-positive bacteria (B. cereus; MIC = 2 μg/mL) than Gram-negative (E. coli; MIC > 64 μg/mL). Studies on wide selections of Gram-negative as well as Gram-positive strains showed that the most active were diITCs 492 and 493, which, on Gram-positive strains, such as Streptococcus, Staphylococcus, and Bacillus, exhibited high activity (MIC = 1–2 μg/mL). DiITCs 492 and 493 presented high activity against mycobacteria. The most active were against M. bovis and M. tuberculosis (MIC = 2–4 μg/mL) than against nontuberculous mycobacteria. DiITC 492 efficiently inhibited the growth of the pathogens, such as Candida albicans or Candida glabrata, for which MIC < 1 μg/mL.
4.6. Summary of the Synthetic Routs of SFN and Its Bifunctional Analogs and Their Biological Activity
The described above methods of the synthesis of SFN and its bifunctional analogs are presented in Table 12, divided into various functional groups.
Based on the Table 12, it can be seen that the synthesis of SFN, and its sulfur analogs substituted with the methyl group, but also by varied aliphatic, aromatic, fluorine, or heterocyclic substituents, are the most described in the literature. Among them, the method using thiophosgene (CSCl2) is dominant. The next often used method is the tandem Staudinger/aza-Wittig reaction, and the least frequently used is the two-step method using a desulfurizing agent. Another group involves phosphorus analogues of SFN (P-SFN) with phosphonate, phosphinate, or phosphine oxide moiety with aliphatic or aromatic substituents. For this class of compounds, both the tandem Staudinger/aza-Wittig reaction and the method using a desulfurizing agent were equally frequently used. The reaction with CSCl2 for synthesis P-SFN was used less frequently. Carbonyl and amide analogs of SFN were synthesized by all three methods. The synthesis of ether-linked analogs of SFN was completely performed by the two-step methodology with a desulfurizing agent, while the synthesis of diisothiocyanates was also performed with the desulfurizing agent and thiophosgene. The tandem Staudinger/aza-Wittig reaction, to the synthesis of ether-linked analogs of SFN and diisothiocyanates, was not used.
Table 13, summarizes the relationship between the biological activity of analogs of SFN and various functional groups. The table presents the most anticancer-active analogs of SFN containing sulfinyl, phosphonate, phosphinate, phosphine oxide, carbonyl, carboxamide, or additional isothiocyanate functional groups. Due to the fact that there are no differences in biological activity between SFN and its natural analogs, such as erucin or alyssin (see Figure 5), these compounds are not placed in Table 13. Only the analogs of SFN are placed in Table 13 for which biological activity is compared with SFN.
Based on Table 13, it can be seen that replacing the methyl group in sulfinyl moiety in SFN with structurally diverse fluorine ((S)-75, 80d), heterocyclic (84d), or aliphatic (e.g., iso-butyl) (152) substituents result in increases of the biological activities of analogs of SFN on different cancer cell lines, such as Malme-3M, MCF-7, or A549. For phosphorus analogs of SFN (P-SFN), the presence of phosphine oxide moiety with methyl substituents (191) does not affect biological activity. However, replacement of the methyl sulfinyl group by phosphonate moiety with aliphatic (298) or aromatic (407) substituents or phosphinate moiety (372) results in an increase in the anticancer activity on LoVo and LoVo/DX cancer cell lines. Carbonyl or amide analogs of SFN (419 and 455) can by characterized by biological activities similar to SFN, but on the other hand, for some analogs (e.g., 470e), biological activity could be higher than for SFN. The presence of the second isothiocyanato group (484) also results in an increase in the biological activity on LoVo and LoVo/DX cancer cell lines.
5. Conclusions
There are many examples describing the synthesis of SFN and its natural or synthetic difunctional analogues, with methyl sulfinyl group replaced by sulfinyl, sulfanyl, sulfonyl, phosphonate, phosphinate, phosphine oxide, carbonyl, ester, amide, ether, or a second isothiocyanate group. The most common strategies involve the use of thiophosgene as a thiocarbonyl transfer reagent, utilization of a carbon disulfide/desulfurizing agent system (dithiocarbamates approach), or use of the tandem Staudinger/aza-Wittig reaction (triphenylphosphine/carbon disulfide system). In the first two approaches, primary amines are utilized as substrates; the last comprises azides as starting materials. The choice of method depends on the availability of starting substrates and sensitivity of functional groups present in the substrates to the reaction conditions. Several ITCs are characterized by high biological activity (anticancer or antibacterial), usually higher than parental SFN. Which modifications of the structure of SFN seem to have important impacts on the biological activities of the new analogues of SFN.
As SFN is biologically active, natural ITC is found in cruciferous vegetables, and it is non-toxic—it has been selected for phase I and II clinical trials, where it is administered in the form of an extract or broccoli sprouts. The results of these studies are promising and indicate which SFN, in the future, may be considered as an anticancer drug. The situation is different for synthetic analogs of SFN, which have been described in this review, having sulfinyl, sulfanyl, sulfonyl, phosphonate, phosphinate, phosphine oxide, carbonyl, ester, amide, ether, or a second isothiocyanate group. The vast majority of these compounds have only been tested for anticancer or antibacterial activity in vitro, and a few analogs, additionally in vivo, on mice or Zebrafish. None of the synthetic analogs of SFN described in this review qualified for clinical trials. This is likely due to the toxicity of these compounds in higher doses, often seen in in vitro or in vivo studies. SFN, which is a natural product, does not show toxicity, even in higher doses. Another reason is the financial constraints of many research teams.
One interesting solution would be to synthesize a conjugate—a combination of SFN and a drug or peptide, with anticancer properties, characterized by no (or low) toxicity. This conjugate could separate, in cancer cells, on two drugs, causing increased anticancer activity.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available upon request from the corresponding author.
Acknowledgments
The author expresses his gratitude to Beata Kolesińska and Tadeusz Gajda for their helpful discussions.
Conflicts of Interest
The author declares no conflict of interest.
References and Notes
- Avato, P.; Argentieri, M.P. Brassicaceae: A rich source of health improving phytochemicals. Phytochem. Rev. 2015, 14, 1019–1033. [Google Scholar] [CrossRef]
- Verhoeven, D.T.H.; Verhagen, H.; Goldbohm, R.A.; van den Brandt, P.A.; van Poppel, G. A review of mechanism underlying anticarcinogenicity by brassica vegetables. Chem. Biol. Interact. 1997, 103, 79–129. [Google Scholar] [CrossRef]
- Jeffery, E.H.; Araya, M. Physiological effects of broccoli consumption. Phytochem. Rev. 2009, 8, 283–298. [Google Scholar] [CrossRef]
- Ambrosone, C.B.; McCann, S.E.; Freudenheim, J.L.; Marshall, J.R.; Zhang, Y.; Shields, P.G. Breast cancer risk in premenopausal women is inversely associated with consumption of broccoli, a source of isothiocyanates, but is not modified by GST genotype. J. Nutr. 2004, 134, 1134–1138. [Google Scholar] [CrossRef] [PubMed]
- Brennan, P.; Hsu, C.C.; Moullan, N.; Szeszenia-Dabrowska, N.; Lissowska, J.; Zaridze, D.; Rudnai, P.; Fabianova, E.; Mates, D.; Bencko, V.; et al. Effect of cruciferous vegetables on lung cancer in patients stratified by genetic status: A mendelian randomisation approach. Lancet 2005, 366, 1558–1560. [Google Scholar] [CrossRef]
- Wang, L.I.; Giovannucci, E.L.; Hunter, D.; Neuberg, D.; Su, L.; Christiani, D.C. Dietary intake of Cruciferous vegetables, Glutathione S-transferase (GST) polymorphisms and lung cancer risk in a Caucasian population. Cancer Causes Control 2004, 15, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Fowke, J.H.; Chung, F.L.; Jin, F.; Qi, D.; Cai, Q.; Conaway, C.; Cheng, J.R.; Shu, X.O.; Gao, Y.T.; Zheng, W. Urinary isothiocyanate levels, brassica, and human breast cancer. Cancer Res. 2003, 63, 3980–3986. [Google Scholar] [PubMed]
- Lin, H.J.; Probst-Hensch, N.M.; Louie, A.D.; Kau, I.H.; Witte, J.S.; Ingles, S.A.; Frankl, H.D.; Lee, E.R.; Haile, R.W. Glutathione transferase null genotype, broccoli, and lower prevalence of colorectal adenomas. Cancer Epidemiol. Biomark. Prev. 1998, 7, 647–652. [Google Scholar]
- Joseph, M.A.; Moysich, K.B.; Freudenheim, J.L.; Shields, P.G.; Bowman, E.D.; Zhang, Y.; Marshall, J.R.; Ambrosone, C.B. Cruciferous vegetables, genetic polymorphisms in glutathione S-transferases M1 and T1, and prostate cancer risk. Nutr. Cancer 2004, 50, 206–213. [Google Scholar] [CrossRef]
- Verhoeven, D.T.H.; Goldbohm, R.A.; van Poppel, G.; Verhagen, H.; van den Brandt, P.A. Epidemiological studies on brassica vegetables and cancer risk. Cancer Epidermiol. Biomark. Prev. 1996, 5, 733–748. [Google Scholar]
- Rungapamestry, V.; Duncan, A.J.; Fuller, Z.; Ratcliffe, B. Effect of cooking brassica vegetables on the subsequent hydrolysis and metabolic fate of glucosinolates. Proc. Nutr. Soc. 2007, 66, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Kostov, R.V. Glucosinolates and isothiocyanates in health and disease. Trends. Mol. Med. 2012, 18, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Ishida, M.; Hara, M.; Fukino, N.; Kakizaki, T.; Morimitsu, Y. Glucosinolate metabolism, functionality and breeding for the improvement of Brassicaceae vegetables. Breed. Sci. 2014, 64, 48–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahey, J.W.; Zalcmann, A.T.; Talalay, P. The chemical diversity and distribution of glucosinolates and isothiocyanates among plants. Phytochemistry 2001, 56, 5–51. [Google Scholar] [CrossRef]
- Rollin, P.; Tatibouët, A. Glucosinolates: The synthetic approach. C. R. Chimie 2011, 14, 194–210. [Google Scholar] [CrossRef]
- Bell, L.; Wagstaff, C. Glucosinolates, myrosinase hydrolysis products, and flavonols found in rocket (Eruca sativa and Diplotaxis tenuifolia). J. Agric. Food Chem. 2014, 62, 4481–4492. [Google Scholar] [CrossRef] [PubMed]
- Traka, M.; Mithen, R. Glucosinolates, isothiocyanates and human health. Phytochem. Rev. 2009, 8, 269–282. [Google Scholar] [CrossRef]
- Ettlinger, M.G.; Lundeen, A.J. The structures of sinigrin and sinalbin; an enzymatic rearrangement. J. Am. Chem. Soc. 1956, 78, 4172–4173. [Google Scholar] [CrossRef]
- Ettlinger, M.G.; Lundeen, A.J. First synthesis of a mustard oil glucoside; the enzymatic lossen rearrangement. J. Am. Chem. Soc. 1957, 79, 1764–1765. [Google Scholar] [CrossRef]
- Hanschen, F.S.; Lamy, E.; Schreiner, M.; Rohn, S. Reactivity and stability of glucosinolates and their breakdown products in foods. Angew. Chem. Int. Ed. 2014, 53, 11430–11450. [Google Scholar] [CrossRef]
- Fahey, J.W.; Olson, M.E.; Stephenson, K.K.; Wade, K.L.; Chodur, G.M.; Odee, D.; Nouman, W.; Massiah, M.; Alt, J.; Egner, P.A.; et al. The diversity of chemoprotective glucosinolates in Moringaceae (Moringa spp.). Sci. Rep. 2018, 8, 7994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahey, J.W. Moringa oleifera: A review of the medical evidence for its nutritional, therapeutic, and prophylactic properties. Trees Life J. 2015, 1, 5. [Google Scholar]
- Olson, M.E.; Fahey, J.W. Moringa oleifera: A multipurpose tree for the dry tropics. Rev. Mex. Biodivers. 2011, 82, 1071–1082. [Google Scholar]
- Kissen, R.; Rossiter, J.; Bones, A.M. The ‘mustard oil bomb’: Not so easy to assemble?! Localization, expression and distribution of the components of the myrosinase enzyme system. Phytochem. Rev. 2009, 8, 69–86. [Google Scholar] [CrossRef]
- Halkier, B.A.; Gershenzon, J. Biology and biochemistry of glucosinolates. Annu. Rev. Plant Biol. 2006, 57, 303–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, J.R.; van Dam, N.M.; van Loon, J.J.A. Role of glucosinolates in insect-plant relationships and multitrophic interactions. Annu. Rev. Entomol. 2009, 54, 57–83. [Google Scholar] [CrossRef]
- Shapiro, T.A.; Fahey, J.W.; Wade, K.L.; Stephenson, K.K.; Talalay, P. Human metabolism and excretion of cancer chemoprotective glucosinolates and isothiocyanates of cruciferous vegetables. Cancer Epidemiol. Biomark. Prev. 1998, 7, 1091–1100. [Google Scholar]
- Uda, Y.; Kurata, T.; Arakawa, N. Effects of pH and ferrous ion on the degradation of glucosinolates by myrosinase. Agric. Biol. Chem. 1986, 50, 2735–2740. [Google Scholar]
- Foo, H.L.; Grønning, M.; Goodenough, L.; Bones, A.M.; Danielsen, B.E.; Whiting, D.A.; Rossiter, J.T. Purification and characterisation of epithiospecifier protein from Brassica napus: Enzymic intramolecular sulphur addition within alkenyl thiohydroximates derived from alkenyl glucosinolate hydrolysis. FEBS Lett. 2000, 468, 243–246. [Google Scholar] [CrossRef] [Green Version]
- Burow, M.; Losansky, A.; Müller, R.; Plock, A.; Kliebenstein, D.J.; Wittstock, U. The genetic basis of constitutive and herbivore-induced ESP-independent nitrile formation in Arabidopsis. Plant Physiol. 2009, 149, 561–574. [Google Scholar] [CrossRef] [Green Version]
- Burow, M.; Bergner, A.; Gershenzone, J.; Wittstock, U. Glucosinolate hydrolysis in Lepidium sativum-identification of the thiocyanate-forming protein. Plant Mol. Biol. 2007, 63, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Wentzell, A.M.; Kliebestein, D.J. Genotype, age, tissue, and environment regulate the structural outcome of glucosinolate activation. Plant Physiol. 2008, 147, 415–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burow, M.; Wittstock, U. Regulation and function of specifier proteins in plants. Phytochem. Rev. 2009, 8, 87–99. [Google Scholar] [CrossRef]
- Mitsiogianni, M.; Koutsidis, G.; Mavroudis, N.; Trafalis, D.T.; Botaitis, S.; Franco, R.; Zoumpourlis, V.; Amery, T.; Galanis, A.; Pappa, A.; et al. The role of isothiocyanates as cancer chemo-preventive, chemo-therapeutic and anti-melanoma agents. Antioxidants 2019, 8, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palliyaguru, D.L.; Yuan, J.M.; Kensler, T.W.; Fahey, J.W. Isothiocyanates: Translating the power of plants to people. Mol. Nutr. Food Res. 2018, 62, e1700965. [Google Scholar] [CrossRef]
- Fimognari, C.; Turrini, E.; Ferruzzi, L.; Lenzi, M.; Hrelia, P. Natural isothiocyanates: Genotoxic potential versus chemoprevention. Mutat. Res. 2012, 750, 107–131. [Google Scholar] [CrossRef]
- Oliviero, T.; Verkerk, R.; Dekker, M. Isothiocyanates from brassica vegetables—Effects of processing, cooking, mastication, and digestion. Mol. Nutr. Food Res. 2018, 62, e1701069. [Google Scholar] [CrossRef] [Green Version]
- Bell, L.; Oloyede, O.O.; Lignou, S.; Wagstaff, C.; Methven, L. Taste and flavor perceptions of glucosinolates, isothiocyanates, and related compounds. Mol. Nutr. Food Res. 2018, 62, e170990. [Google Scholar] [CrossRef] [Green Version]
- Drobnica, L.; Kristian, P.; Augustin, J. The chemistry of the -NCS group. In Chemistry of Cyanates and Their Thio Derivatives; John Wiley & Sons: Chichester, UK, 1977; Volume 2, p. 1003. [Google Scholar]
- Wu, X.; Zhou, Q.H.; Xu, K. Are isothiocyanates potential anti-cancer drugs? Acta Pharmacol. Sin. 2009, 30, 501–512. [Google Scholar] [CrossRef] [Green Version]
- Rao, C.V. Benzyl isothiocyanate: Double trouble for breast cancer cells. Cancer Prev. Res. 2013, 6, 760–763. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Wright, S.E.; Kim, S.H.; Srivastava, S.K. Phenethyl isothiocyanate: A comprehensive review of anti-cancer mechanism. Biochim. Biophys. Acta 2014, 1846, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, C.Z.; Zhang, X.; Wu, L.X.; Wen, C.J.; Hu, L.; Lv, Q.L.; Shen, D.Y.; Zhou, H.H. Advances in molecular signaling mechanisms of β-phenethyl isothiocyanate antitumor effects. J. Agric. Food Chem. 2015, 63, 3311–3322. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.G.; Chiao, J.W. Prostate cancer chemopreventive activity of phenethyl isothiocyanate through epigenetic regulation (Review). Int. J. Oncol. 2010, 37, 533–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y. Allyl isothiocyanate as a cancer chemopreventive phytochemical. Mol. Nutr. Food Res. 2010, 54, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Myzak, M.C.; Dashwood, R.H. Chemoprotection by sulforaphane: Keep one eye beyond Keap1. Cancer Lett. 2006, 233, 208–218. [Google Scholar] [CrossRef] [Green Version]
- Juge, N.; Mithen, R.F.; Traka, M. Molecular basis for chemoprevention by sulforaphane: A comprehensive review. Cell. Mol. Life Sci. 2007, 64, 1105–1127. [Google Scholar] [CrossRef]
- Briones-Herrera, A.; Eugenio-Pérez, D.; Reyes-Ocampo, J.G.; Rivera-Mancía, S.; Pedraza-Chaverri, J. New highlights on the health-improving effects of sulforaphane. Food Funct. 2018, 9, 2589–2606. [Google Scholar] [CrossRef]
- Mi, L.; Di Pasqua, A.J.; Chung, F.L. Proteins as binding targets of isothiocyanates in cancer prevention. Carcinogenesis 2011, 32, 1405–1413. [Google Scholar] [CrossRef] [Green Version]
- Brown, K.K.; Hampton, M.B. Biological targets of isothiocyanates. Biochim. Biophys. Acta 2011, 1810, 888–894. [Google Scholar] [CrossRef]
- Zhang, Y. The molecular basis that unifies the metabolism, cellular uptake and chemopreventive activities of dietary isothiocyanates. Carcinogenesis 2012, 33, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Hecht, S.S. Inhibition of carcinogenesis by isothiocyanates. Drug Metab. Rev. 2000, 32, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Dufour, V.; Stahl, M.; Baysse, C. The antibacterial properties of isothiocyanates. Microbiology 2015, 161, 229–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romeo, L.; Iori, R.; Rollin, P.; Bramanti, P.; Mazzon, E. Isothiocyanates: An overview of their antimicrobial activity against human infections. Molecules 2018, 23, 624. [Google Scholar] [CrossRef] [Green Version]
- Kurepina, N.; Kreiswirth, B.N.; Mustaev, A. Growth-inhibitory activity of natural and synthetic isothiocyanates against representative human microbial pathogens. J. Appl. Microbiol. 2013, 115, 943–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukerjee, A.K.; Ashare, R. Isothiocyanates in the chemistry of heterocycles. Chem. Rev. 1991, 91, 1–24. [Google Scholar] [CrossRef]
- Brandsma, L.; Nedolya, N.A.; Tarasova, O.A.; Trofimov, B.A. Synthesis of heterocyclic compounds from metallated unsaturated compounds and isothiocyanates. (Review). Chem. Heterocycl. Compd. 2000, 36, 1241–1260. [Google Scholar] [CrossRef]
- Pace, V.; Monticelli, S.; de la Vega-Hernández, K.; Castoldi, L. Isocyanates and isothiocyanates as versatile platforms for accessing (thio)amide-type compounds. Org. Biomol. Chem. 2016, 14, 7848–7854. [Google Scholar] [CrossRef]
- Koutoulogenis, G.; Kaplaneris, N.; Kokotos, C.G. (Thio)urea-mediated synthesis of functionalized six-membered rings with multiple chiral centers. Beilstein J. Org. Chem. 2016, 12, 462–495. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Mi, L.; Sanda, M.; Silverstein, S.; Aggarwal, M.; Wang, D.; Gupta, P.; Goldman, R.; Appella, D.H.; Chung, F.L. A click chemistry approach to identify protein targets of cancer chemopreventive phenethyl isothiocyanate. RSC Adv. 2014, 4, 3920–3923. [Google Scholar] [CrossRef]
- Clulow, J.A.; Strock, E.M.; Lanyonn-Hogg, T.; Kalesh, K.A.; Jones, L.H.; Tate, E.W. Competition-based, quantitative chemical proteomics in breast cancer cells identifies new target profiles for sulforaphane. Chem. Commun. 2017, 53, 5182–5185. [Google Scholar] [CrossRef] [Green Version]
- Schmid, H.; Karrer, P. Synthese der racemischen und der optisch aktiven formen des Sulforaphans. Helv. Chim. Acta 1948, 31, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Talalay, P.; Cho, C.H.; Posner, G.H. A major inducer of anticarcinogenic protective enzymes from broccoli: Isolation and elucidation of structure. Proc. Natl. Acad. Sci. USA 1992, 89, 2399–2403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kushad, M.M.; Brown, A.F.; Kurilich, A.C.; Juvik, J.A.; Klein, B.P.; Wallig, M.A.; Jeffery, E.H. Variation of glucosinolates in vegetable crops of Brassica oleracea. J. Agric. Food Chem. 1999, 47, 1541–1548. [Google Scholar] [CrossRef] [PubMed]
- Information Based on Web of Science Datebase of 3 January 2022.
- Abdull Razis, A.F.; Iori, R.; Ioannides, C. The natural chemopreventive phytochemical R-sulforaphane is a far more potent inducer of the carcinogen-detoxifying enzyme systems in rat liver and lung than the S-isomer. Int. J. Cancer 2011, 128, 2775–2782. [Google Scholar] [CrossRef] [PubMed]
- Fimognari, C.; Hrelia, P. Sulforaphane as a promising molecule for fighting cancer. Mutat. Res. 2007, 635, 90–104. [Google Scholar] [CrossRef]
- Szczesny-Malysiak, E.; Stojak, M.; Campagna, R.; Grosicki, M.; Jamrozik, M.; Kaczara, P.; Chlopnicki, S. Bardxolone metyl displays detrimental effects on endothelial bioenergetics, suppresses endothelial ET-1 release, and increases endothelial permeability in human microvascular endothelium. Oxid. Med. Cell. Longev. 2020, 2020, 4678252. [Google Scholar] [CrossRef]
- Kelloff, G.J.; Crowell, J.A.; Steele, V.E.; Lubet, R.A.; Malone, W.A.; Boone, C.W.; Kopelovich, L.; Hawk, E.T.; Lieberman, R.; Lawrence, J.A.; et al. Progress in cancer chemoprevention: Development of diet-derived chemopreventive agents. J. Nutr. 2000, 130, 467–471. [Google Scholar] [CrossRef]
- Karrer, P.; Scheitlin, E.; Siegrist, H. Über homologe des sulforaphans und über ω-aminoalkyl-sulfoxyde. Helv. Chim. Acta 1950, 33, 1237–1241. [Google Scholar] [CrossRef]
- Kjær, A.; Gmelin, R.; Larsen, I. Isothiocyanates XII. 3-Methylthiopropyl isothiocyanate (Iberverin), a new naturally occurring mustard oil. Acta Chem. Scand. 1955, 9, 1143–1147. [Google Scholar] [CrossRef] [Green Version]
- Kjær, A.; Gmelin, R. Isothiocyanates XI. 4-Methylthiobutyl isothiocyanate, a new naturally occurring mustard Oil. Acta Chem. Scand. 1955, 9, 542–544. [Google Scholar] [CrossRef]
- Kjær, A.; Larsen, I.; Gmelin, R.; Prydz, H. Isothiocyanates XIV. 5-Methylthiopentyl isothiocyanate, a new mustard oil present in nature as a glucoside (glucoberteroin). Acta Chem. Scand. 1955, 9, 1311–1316. [Google Scholar] [CrossRef]
- Schneider, W. Zur Kenntnis des schwefelhaitigen Alkaloid aus dem Goldlack-Samen. Chem. Ber. 1908, 41, 4469. [Google Scholar] [CrossRef] [Green Version]
- Schneider, W.; Kaufmann, H. Untersuchungen über Senföle. II. Erysolin, ein Sulfonsenföl aus Erysimum perowskianum. Justus Liebig’s Ann. Chem. 1912, 392, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Balenović, K.; Deljac, A.; Monković, I.; Štefanac, Z. Synthesis of (±) sulphoraphene. Tetrahedron 1966, 22, 2139–2143. [Google Scholar] [CrossRef]
- Bilska, A.; Kryczyk, A.; Włodek, L. Różne oblicza biologicznej roli glutationu. The different aspects of the biological role of glutathione. Postepy Hig. Med. Dosw. 2007, 61, 438–453. [Google Scholar]
- Pastore, A.; Federici, G.; Bertini, E.; Piemonte, F. Analysis of glutathione: Implication in redox and detoxification. Clin. Chim. Acta 2003, 333, 19–39. [Google Scholar] [CrossRef]
- Cotgreave, I.A.; Gerdes, R.G. Recent trends in glutathione biochemistry—Glutathione–protein interactions: A molecular link between oxidative stress and cell proliferation? Biochem. Biophys. Res. Commun. 1998, 242, 1–9. [Google Scholar] [CrossRef]
- Ghibelli, L.; Fanelli, C.; Rotilio, G.; Lafavia, E.; Coppola, S.; Colussi, C.; Civitareale, P.; Ciriolo, M.R. Rescue of cells from apoptosis by inhibition of active GSH extrusion. FASEB J. 1998, 12, 479–486. [Google Scholar] [CrossRef] [Green Version]
- Poot, M.; Teubert, H.; Rabinovitch, P.S.; Kavanagh, T.J. De novo synthesis of glutathione is required for both entry into and progression through the cell cycle. J. Cell. Physiol. 1995, 163, 555–560. [Google Scholar] [CrossRef]
- Hall, A.G. Glutathione and the regulation of cell death. Adv. Exp. Med. Biol. 1999, 457, 199–203. [Google Scholar]
- Chung, F.L.; Jiao, D.; Getahun, S.M.; Yu, M.C. A urinary biomarker for uptake of dietary isothiocyanates in humans. Cancer Epidemiol. Biomark. Prev. 1998, 7, 103–108. [Google Scholar]
- Ye, L.; Dinkova-Kostova, A.T.; Wade, K.L.; Zhang, Y.; Shapiro, T.A.; Talalay, P. Quantitative determination of dithiocarbamates in human plasma, serum, erythrocytes and urine: Pharmacokinetics of broccoli sprout isothiocyanates in humans. Clin. Chim. Acta 2002, 316, 43–53. [Google Scholar] [CrossRef]
- Gasper, A.V.; Al-Janobi, A.; Smith, J.A.; Bacon, J.R.; Fortun, P.; Atherton, C.; Taylor, M.A.; Hawkey, C.J.; Barrett, D.A.; Mithen, R.F. Glutathione S-transferase M1 polymorphism and metabolism of sulforaphane from standard and high-glucosinolate broccoli. Am. J. Clin. Nutr. 2005, 82, 1283–1291. [Google Scholar] [CrossRef] [PubMed]
- Egner, P.A.; Kensler, T.W.; Chen, J.G.; Gange, S.J.; Groopman, J.D.; Friesen, M.D. Quantification of sulforaphane mercapturic acid pathway conjugates in human urine by high-performance liquid chromatography and isotope-dilution tandem mass spectrometry. Chem. Res. Toxicol. 2008, 21, 1991–1996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamy, E.; Scholtes, C.; Herz, C.; Mersch-Sundermann, V. Pharmacokinetics and pharmacodynamics of isothiocyanates. Drug Metab. Rev. 2011, 43, 387–407. [Google Scholar] [CrossRef] [PubMed]
- Cavell, B.E.; Syed Alwi, S.S.; Donlevy, A.; Packham, G. Anti-angiogenic effects of dietary isothiocyanates: Mechanisms of action and implications for human health. Biochem. Pharmacol. 2011, 81, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Trachootham, D.; Zhou, Y.; Zhang, H.; Demizu, Y.; Chen, Z.; Pelicano, H.; Chiao, P.J.; Achanta, G.; Arlinghaus, R.B.; Liu, J.; et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by β-phenylethyl isothiocyanate. Cancer Cell 2006, 10, 241–252. [Google Scholar] [CrossRef] [Green Version]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell. Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef]
- Dashwood, R.H.; Ho, E. Dietary agents as histone deacetylase inhibitors: Sulforaphane and structurally related isothiocyanates. Nutr. Rev. 2008, 66, S36–S38. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.V.; Singh, K. Cancer chemoprevention with dietary isothiocyanates mature for clinical translational research. Carcinogenesis 2012, 33, 1833–1842. [Google Scholar] [CrossRef] [Green Version]
- Milelli, A.; Fimognari, C.; Ticchi, N.; Neviani, P.; Minarini, A.; Tumiatti, V. Isothiocyanate synthetic analogs: Biological activities, structure-activity relationships and synthetic strategies. Mini Rev. Med. Chem. 2014, 14, 963–977. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.; Tuli, H.S.; Mittal, S.; Shandilya, J.K.; Tiwari, A.; Sandhu, S.S. Isothiocyanates: A class of bioactive metabolites with chemopreventive potential. Tumor Biol. 2015, 36, 4005–4016. [Google Scholar] [CrossRef] [PubMed]
- Gründemann, C.; Huber, R. Chemoprevention with isothiocyanates—From bench to bedside. Cancer Lett. 2018, 414, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Navarro, S.L.; Li, F.; Lampe, J.W. Mechanism of action of isothiocyanates in cancer chemoprevention: An update. Food Funct. 2011, 2, 579–587. [Google Scholar] [CrossRef]
- Clarke, J.D.; Dashwood, R.H.; Ho, E. Multi-target prevention of cancer by sulforaphane. Cancer. Res. 2008, 269, 291–304. [Google Scholar]
- Tomczyk, J.; Olejnik, A. Sulforafan—Potencjalny czynnik w prewencji i terapii chorób nowotworowych. Sulforaphane—A possibile agent in prevention and therapy of cancer. Postepy Hig. Med. Dosw. 2010, 64, 590–603. [Google Scholar]
- Jiang, X.; Liu, Y.; Ma, L.; Ji, R.; Qu, Y.; Xin, Y.; Lv, G. Chemopreventive activity of sulforaphane. Drug Des. Dev. Ther. 2018, 12, 2905–2913. [Google Scholar] [CrossRef] [Green Version]
- Leone, A.; Diorio, G.; Sexton, W.; Schell, M.; Alexandrow, M.; Fahey, J.W.; Kumar, N.B. Sulforaphane for the chemoprevention of bladder cancer: Molecular mechanism targeted approach. Oncotarget 2017, 8, 35412–35424. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.S.; Smith, T.J.; Hong, J.Y. Cytochrome P-450 enzymes as targets for chemoprevention against chemical carcinogenesis and toxicity: Opportunities and limitations. Cancer. Res. 1994, 54, 1982s–1986s. [Google Scholar]
- Nakajima, M.; Yoshida, R.; Shimada, N.; Yamazaki, H.; Yokoi, T. Inhibition and inactivation of human cytochrome P450 isoforms by phenethyl isothiocyanate. Drug Metab. Dispos. 2001, 29, 1110–1113. [Google Scholar]
- Campagna, R.; Pozzi, V.; Sartini, D.; Salvolini, E.; Brisigotti, V.; Molinelli, E.; Campanati, A.; Offidani, A.; Emanuelli, M. Beyond nicotinamide metabolism: Potential role of nicotinamide N-methyltransferase as a biomarker in skin cancer. Cancers 2021, 13, 4943. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, V.; Salvolini, E.; Lucarini, G.; Salvucci, A.; Campagna, R.; Rubini, C.; Sartini, D.; Emanuelli, M. Cancer stem cell enrichment is associated with enhancement of nicotinamide N-methyltransferase expression. IUBMB Life 2020, 72, 1415–1425. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.S. Chemoprevention of cancer by isothiocyanates, modifiers of carcinogen metabolism. J. Nutr. 1999, 129, 768S–774S. [Google Scholar] [CrossRef] [PubMed]
- Bacon, J.R.; Williamson, G.; Garner, R.C.; Lappin, G.; Langouet, S.; Bao, Y. Sulforaphane and quercetin modulate PhIP–DNA adduct formation in human HepG2 cells and hepatocytes. Carcinogenesis 2003, 24, 1903–1911. [Google Scholar] [CrossRef]
- Jiang, Z.Q.; Chen, C.; Yang, B.; Hebbar, V.; Tony Kong, A.N. Differential responses from seven mammalian cell lines to the treatments of detoxifying enzyme inducers. Life Sci. 2003, 72, 2243–2253. [Google Scholar] [CrossRef]
- Basten, G.P.; Bao, Y.; Wiliamson, G. Sulforaphane and its glutathione conjugate but not sulforaphane nitrile induce UDP-glucuronosyl transferase (UGT1A1) and glutathione transferase (GSTA1) in cultured cells. Carcinogenesis 2002, 23, 1399–1404. [Google Scholar] [CrossRef] [Green Version]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef] [Green Version]
- Fimognari, C.; Nusse, M.; Cesari, R.; Iori, R.; Cantelli-Forti, G.; Hrelia, P. Growth inhibition, cell-cycle arrest and apoptosis in human T-cell leukemia by the isothiocyanate sulforaphane. Carcinogenesis 2002, 23, 581–586. [Google Scholar] [CrossRef]
- Garrett, M.D. Cell cycle control and cancer. Curr. Sci. 2001, 5, 515–522. [Google Scholar]
- Singh, S.V.; Herman-Antosiewicz, A.; Singh, A.V.; Lew, K.L.; Srivastava, S.K.; Kamath, R.; Brown, K.D.; Zhang, L.; Baskaran, R. Sulforaphane-induced G2/M phase cell cycle arrest involves checkpoint kinase 2-mediated phosphorylation of cell division cycle 25C. J. Biol. Chem. 2004, 279, 25813–25822. [Google Scholar] [CrossRef] [Green Version]
- Rose, P.; Huang, Q.; Ong, C.N.; Wheiteman, M. Broccoli and watercress suppress matrix metalloproteinase-9 activity and invasiveness of human MDA-MB-231 breast cancer cells. Toxicol. Appl. Pharmacol. 2005, 209, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.L.; Khor, T.O.; Yu, S.; Kong, A.N. PEITC induces G1 cell cycle arrest on HT-29 cells through the activation of p38 MAPK signaling pathway. AAPS J. 2008, 10, 277–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakubíková, J.; Sedlák, J.; Mithen, M.; Bao, Y. Role of PI3K/Akt and MEK/ERK signaling pathways in sulforaphane- and erucin-induced phase II enzymes and MRP2 transcription, G2/M arrest and cell death in Caco-2 cells. Biochem. Pharmacol. 2005, 69, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Pappa, G.; Bartsch, H.; Gerhauser, C. Biphasic modulation of cell proliferation by sulforaphane at physiologically relevant exposure times in a human colon cancer cell line. Mol. Nutr. Food Res. 2007, 51, 977–984. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, G.Y.; Bae, S.J.; Yoo, Y.H.; Choi, Y.H. Induction of apoptosis by isothiocyanate sulforaphane in human cervical carcinoma HeLa and hepatocarcinoma HepG2 cells through activation of caspase-3. Oncol. Rep. 2007, 18, 181–187. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.; Lew, K.L.; Xiao, H.; Herman-Antosiewicz, A.; Xiao, D.; Brown, C.K.; Singh, S.V. D,L-Sulforaphane-induced cell death in human prostate cancer cells is regulated by inhibitor of apoptosis family proteins and Apaf-1. Carcinogenesis 2007, 28, 151–162. [Google Scholar] [CrossRef]
- Basu, A.; Halder, S. Dietary isothiocyanate mediated apoptosis of human cancer cells is associated with Bcl-XL phosphorylation. Int. J. Oncol. 2008, 33, 657–663. [Google Scholar]
- Xu, K.; Thornalley, P.J. Signal transduction activated by the cancer chemopreventive isothiocyanates: Cleavage of BID protein, tyrosine phosphorylation and activation of JNK. Br. J. Cancer 2001, 84, 670–673. [Google Scholar] [CrossRef] [Green Version]
- Xiao, D.; Srivastava, S.K.; Lew, K.L.; Zeng, Y.; Hershberger, P.; Johnson, C.S.; Trump, D.L.; Singh, S.V. Allyl isothiocyanate, a constituent of cruciferous vegetables, inhibits proliferation of human prostate cancer cells by causing G2/M arrest and inducing apoptosis. Carcinogenesis 2003, 24, 891–897. [Google Scholar] [CrossRef]
- Singh, S.V.; Srivastava, S.K.; Choi, S.; Lew, K.L.; Antosiewicz, J.; Xiao, D.; Zeng, Y.; Watkins, S.C.; Johnson, C.S.; Trump, D.L.; et al. Sulforaphane-induced cell death in human prostate cancer cells is initiated by reactive oxygen species. J. Biol. Chem. 2005, 280, 19911–19924. [Google Scholar] [CrossRef] [Green Version]
- Marek, Ł. Rola apoptosomu w aktywacji prokaspazy 9. The role of the apoptosome in the activation of procaspase-9. Postepy Hig. Med. Dosw. 2013, 67, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ramirez, M.; Rocchini, C.; Ausio, J. Modulation of chromatin folding by histone acetylation. J. Biol. Chem. 1995, 270, 17923–17928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walia, H.; Chen, H.Y.; Sun, J.M.; Holtz, L.T.; Davie, J.R. Histone acetylation is required to maintain the unfolded nucleosome structure associated with transcribing DNA. J. Biol. Chem. 1998, 273, 14516–14522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myzak, M.C.; Karplus, P.A.; Chung, F.L.; Dashwood, R.H. A novel mechanism of chemoprotection by sulforaphane: Inhibition of histone deacetylase. Cancer Res. 2004, 64, 5767–5774. [Google Scholar] [CrossRef] [Green Version]
- Ho, E.; Dashwood, R.H. Dietary manipulation of histone structure and function. World Rev. Nutr. Diet 2010, 101, 95–102. [Google Scholar]
- Batra, S.; Sahu, R.P.; Kandala, P.K.; Srivastava, S.K. Benzyl isothiocyanate–mediated inhibition of histone deacetylase leads to NF-κB turnoff in human pancreatic carcinoma cells. Mol. Cancer Ther. 2010, 9, 1596–1608. [Google Scholar] [CrossRef] [Green Version]
- Risau, W. Mechanisms of angiogenesis. Nature 1997, 386, 671–674. [Google Scholar] [CrossRef]
- Zielonka, T.M. Angiogeneza—Część 1. Mechanizm powstawania nowych naczyń krwionośnych. Angiogenesis—Part 1. Mechanism of neovascularization. Alerg. Astma Immun. 2003, 8, 169–174. [Google Scholar]
- Wideł, M.S.; Wideł, M. Mechanizmy przerzutowania i molekularne markery progresji nowotworów złośliwych. I. Rak jelita grubego. Mechanisms of metastasis and molecular markers of malignant tumor progression. I. Colorectal cancer. Postepy Hig. Med. Dosw. 2006, 60, 453–470. [Google Scholar]
- Xiao, D.; Singh, S.V. Phenethyl isothiocyanate inhibits angiogenesis In vitro and Ex vivo. Cancer Res. 2007, 67, 2239–2246. [Google Scholar] [CrossRef] [Green Version]
- Asakage, M.; Tsuno, N.T.; Kitayama, J.; Tsuchiya, T.; Yoneyama, S.; Yamada, J.; Okaji, Y.; Kaisaki, S.; Osada, T.; Takahashi, K.; et al. Sulforaphane induces inhibition of human umbilical vein endothelial cells proliferation by apoptosis. Angiogenesis 2006, 9, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Nadarajah, D.; Han, J.H.; Holley, R.A. Use of mustard flour to inactivate Escherichia coli O157:H7 in ground beef under nitrogen flushed packaging. Int. J. Food Microbiol. 2005, 99, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Delaquis, P.J.; Sholberg, P.L. Antimicrobial activity of gaseous allyl isothiocyanate. J. Food Prot. 1997, 60, 943–947. [Google Scholar] [CrossRef] [PubMed]
- Tajima, H.; Kimoto, H.; Taketo, A. Specific antimicrobial synergism of synthetic hydroxy isothiocyanates with aminoglycoside antibiotics. Biosci. Biotechnol. Biochem. 2001, 65, 1886–1888. [Google Scholar] [CrossRef]
- Saavedra, M.J.; Borges, A.; Dias, C.; Aires, A.; Bennett, R.N.; Rosa, E.S.; Simões, M. Antimicrobial activity of phenolics and glucosinolate hydrolysis products and their synergy with streptomycin against pathogenic bacteria. Med. Chem. 2010, 6, 174–183. [Google Scholar] [CrossRef]
- Tajima, H.; Kimoto, H.; Taketo, A. Paradoxical effect of synthetic hydroxy isothiocyanates on antimicrobial action of aminoglycosides. Biosci. Biotechnol. Biochem. 2003, 67, 1844–1846. [Google Scholar] [CrossRef]
- Bressan, M.; Roncato, M.A.; Bellvert, F.; Comte, G.; Haichar, F.Z.; Achouak, W.; Berge, O. Exogenous glucosinolate produced by Arabidopsis thaliana has an impact on microbes in the rhizosphere and plant roots. ISME J. 2009, 3, 1243–1257. [Google Scholar] [CrossRef]
- Borek, V.; Morra, M.J.; McCaffrey, J.P. Myrosinase activity in soil extracts. Soil. Sci. Soc. Am. J. 1996, 60, 1792–1797. [Google Scholar] [CrossRef]
- Björkholm, N.; Zhukhovitsky, V.; Löfman, C.; Hultén, K.; Enroth, H.; Block, M.; Rigo, R.; Falk, P.; Engstrand, L. Helicobacter pylori entry into human gastric epithelial cells: A potential determinant of virulence, persistence, and treatment failures. Helicobacter 2000, 5, 148–154. [Google Scholar] [CrossRef]
- Fahey, J.W.; Haristoy, X.; Dolan, P.M.; Kensler, T.W.; Scholtus, I.; Stephenson, K.K.; Talalay, P.; Lozniewski, A. Sulforaphane inhibits extracellular, intracellular, and antibiotic-resistant strains of Helicobacter pylori and prevents benzo[a]pyrene-induced stomach tumors. Proc. Natl. Acad. Sci. USA 2002, 99, 7610–7615. [Google Scholar] [CrossRef] [Green Version]
- Kafarski, P.; Talma, M. Recent advances in design of new urease inhibitors: A review. J. Adv. Res. 2018, 13, 101–112. [Google Scholar] [CrossRef]
- Ha, N.C.; Oh, S.T.; Sung, J.Y.; Cha, K.A.; Lee, M.H.; Oh, B.H. Supramolecular assembly and acid resistance of Helicobacter pylori urease. Nat. Struct. Biol. 2001, 8, 505–509. [Google Scholar] [CrossRef]
- Fahey, J.W.; Stephenson, K.K.; Wade, K.L.; Talalay, P. Urease from Helicobacter pylori is inactivated by sulforaphane and other isothiocyanates. Biochem. Biophys. Res. Commun. 2013, 435, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Dufour, V.; Alazzam, B.; Ermel, G.; Thepaut, M.; Rossero, A.; Tresse, O.; Baysse, C. Antimicrobial activities of isothiocyanates against Campylobacter jejuni isolates. Front. Cell. Infect. Microbiol. 2012, 2, 53. [Google Scholar] [CrossRef] [Green Version]
- Dufour, V.; Stahl, M.; Rosenfeld, E.; Stintzi, A.; Baysse, C. Insights into the Mode of Action of Benzyl Isothiocyanate on Campylobacter jejuni. Appl. Environ. Microbiol. 2013, 79, 6958–6968. [Google Scholar] [CrossRef] [Green Version]
- Feasey, N.A.; Dougan, G.; Kingsley, R.A.; Heyderman, R.S.; Gordon, M.A. Invasive non-typhoidal salmonella disease: An emerging and neglected tropical disease in Africa. Lancet 2012, 379, 2489–2499. [Google Scholar] [CrossRef]
- Sofrata, A.; Santangelo, E.M.; Azeem, M.; Borg-Karlson, A.K.; Gustafsson, A.; Pütsep, K. Benzyl isothiocyanate, a major component from the roots of Salvadora persica is highly active against Gram-negative bacteria. PLoS ONE 2011, 6, e23045. [Google Scholar] [CrossRef]
- Nataro, J.P.; Kaper, J.B. Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 1998, 11, 142–201. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.M.; Preston III, J.F.; Wei, C.I. Antibacterial mechanism of allyl isothiocyanate. J. Food Prot. 2000, 63, 727–734. [Google Scholar] [CrossRef]
- Luciano, F.B.; Holley, R.A. Enzymatic inhibition by allyl isothiocyanate and factors affecting its antimicrobial action against Escherichia coli O157:H7. Int. J. Food Microbiol. 2009, 131, 240–245. [Google Scholar] [CrossRef]
- Nowicki, D.; Rodzik, O.; Herman-Antosiewicz, A.; Szalewska-Pałasz, A. Isothiocyanates as effective agents against enterohemorrhagic Escherichia coli: Insight to the mode of action. Sci. Rep. 2016, 6, 22263. [Google Scholar] [CrossRef] [Green Version]
- Donlan, R.M. Biofilm formation: A clinically relevant microbiological process. Clin. Infect. Dis. 2001, 33, 1387–1392. [Google Scholar] [CrossRef] [Green Version]
- Defez, C.; Fabbro-Peray, P.; Bouziges, N.; Gouby, A.; Mahamat, A.; Daurès, J.P.; Sotto, A. Risk factors for multidrug-resistant Pseudomonas aeruginosa nosocomial infection. J. Hosp. Infect. 2004, 57, 209–216. [Google Scholar] [CrossRef]
- Bassler, B.L. Small talk. Cell-to-cell communication in bacteria. Cell 2002, 109, 421–424. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.B.; Bassler, B.L. Quorum sensing in bacteria. Annu. Rev. Microbiol. 2001, 55, 165–199. [Google Scholar] [CrossRef] [Green Version]
- Van Delden, C.; Iglewski, B.H. Cell-to-cell signaling and Pseudomonas aeruginosa infections. Emerg. Infect. Dis. 1998, 4, 551–560. [Google Scholar] [CrossRef]
- Li, Y.H.; Tian, X.L. Quorum sensing and bacterial social interactions in biofilms. Sensors 2012, 12, 2519–2538. [Google Scholar] [CrossRef]
- Whiteley, M.; Lee, K.M.; Greenberg, E.P. Identification of genes controlled by quorum sensing in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 1999, 96, 13904–13909. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, S.J.; Mutters, N.T.; Blessing, B.; Günther, F. Natural isothiocyanates express antimicrobial activity against developing and mature biofilms of Pseudomonas aeruginosa. Fitoterapia 2017, 119, 57–63. [Google Scholar] [CrossRef]
- Borges, A.; Abreu, A.C.; Ferreira, C.; Saavedra, M.J.; Simões, L.C.; Simões, M. Antibacterial activity and mode of action of selected glucosinolate hydrolysis products against bacterial pathogens. J. Food Sci. Technol. 2015, 52, 4737–4748. [Google Scholar] [CrossRef] [Green Version]
- Ganin, H.; Rayo, J.; Amara, N.; Levy, N.; Krief, P.; Meijler, M.M. Sulforaphane and erucin, natural isothiocyanates from broccoli, inhibit bacterial quorum sensing. Med. Chem. Commun. 2013, 4, 175–179. [Google Scholar] [CrossRef]
- Hall, S.; McDermott, C.; Anoopkumar-Dukie, S.; McFarland, A.J.; Forbes, A.; Perkins, A.V.; Davey, A.K.; Chess-Williams, R.; Kiefel, M.J.; Arora, D.; et al. Cellular effects of pyocyanin, a secreted virulence factor of Pseudomonas aeruginosa. Toxins 2016, 8, 236. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, T.H.; Bragason, S.K.; Phipps, R.K.; Christensen, L.D.; van Gennip, M.; Alhede, M.; Skindersoe, M.; Larsen, T.O.; Hoiby, N.; Bjarnsholt, T.; et al. Food as a source for Quorum Sensing inhibitors: Iberin from horseradish revealed as a Quorum Sensing inhibitor of Pseudomonas aeruginosa. Appl. Environ. Microbiol. 2012, 78, 2410–2421. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, C.P.; Daniels, M.; Zhao, F.; Alegre, M.L.; Chong, A.S.; Daum, R.S. Protective immunity against recurrent Staphylococcus aureus skin infection requires antibody and interleukin-17A. Infect. Immun. 2014, 82, 2125–2134. [Google Scholar] [CrossRef] [Green Version]
- Libert, M.; Elkholti, M.; Massaut, J.; Karmali, R.; Mascart, G.; Cherifi, S. Risk factors for meticillin resistance and outcome of Staphylococcus aureus bloodstream infection in a Belgian university hospital. J. Hosp. Infect. 2008, 68, 17–24. [Google Scholar] [CrossRef]
- Aires, A.; Mota, V.R.; Saavedra, M.J.; Rosa, E.A.S.; Bennett, R.N. The antimicrobial effects of glucosinolates and their respective enzymatic hydrolysis products on bacteria isolated from the human intestinal tract. J. Appl. Microbiol. 2009, 106, 2086–2095. [Google Scholar] [CrossRef]
- Lu, Z.J.; Dockery, C.R.; Crosby, M.; Chavarria, K.; Patterson, B.; Giedd, M. Antibacterial activities of wasabi against Escherichia coli O157:H7 and Staphylococcus aureus. Front. Microbiol. 2016, 7, 1403. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, T.A.; Fahey, J.W.; Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Stephenson, K.K.; Wade, K.L.; Ye, L.; Talalay, P. Safety, tolerance, and metabolism of broccoli sprout glucosinolates and isorhiocyanates: A clinical phase I study. Nutr. Cancer. 2006, 55, 53–62. [Google Scholar] [CrossRef]
- Cornblatt, B.S.; Ye, L.; Dinkova-Kostova, A.T.; Erb, M.; Fahey, J.W.; Singh, N.K.; Chen, M.-S.A.; Stierer, T.; Garrett-Mayer, E.; Argani, P.; et al. Preclinical and clinical evaluation of sulforaphane for chemoprevention in the breast. Carcinogesis 2007, 28, 148–1490. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.H.; Reynolds, C.; Brookre, A.; Talalay, P.; Fahey, J.W. Sulforaphane improves the bronchoprotective response in asthmatics through Nrf2-mediated gene pathways. Respir. Res. 2015, 16, 106. [Google Scholar] [CrossRef] [Green Version]
- Yanaka, A.; Fahey, J.W.; Fukumoto, A.; Nakayama, M.; Inoue, S.; Zhang, S.; Tauchi, M.; Suzuki, H.; Hyodo, I.; Yamamoto, M. Dietary sulforaphane-rich broccoli sprouts reduce colonization and attenuate gastritis in Helicobacter pylori-infected mice and humans. Cancer Prev. Res. 2009, 2, 353–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houghton, C.A. Sulforaphane: Its “coming of age” as a clinically relevant nutraceutical in the prevention and treatment of chronic disease. Oxid. Med. Cell. Longev. 2019, 2019, 2716870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagishita, Y.; Fahey, J.W.; Dinkova-Kostova, A.T.; Kensler, T.W. Broccoli or sulforaphane: Is it the source or dose that matters? Moleclues 2019, 24, 3593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lampe, J.W.; Peterson, S. Brassica, biotransformation and cancer risk: Genetic polymorphism after the preventive effects of cruciferous vegetables. J. Nutr. 2002, 132, 2991–2994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seow, A.; Shi, C.Y.; Chung, F.L.; Jiao, D.; Hankin, J.H.; Lee, H.P.; Coetzee, G.A.; Yu, M.C. Urinary total isothiocyanate (ITC) in a population-based sample of middle-aged and older Chinese in Singapore: Relationship with dietary total ITC and glutathione S-transferase M1/T1/P1 genotypes. Cancer Epidemiol. Biomark. Prev. 1998, 7, 775–781. [Google Scholar]
- Knight, J.G. Science of Synthesis, 18: Houben-Weyl Methods of Molecular Transformation, Compounds with Four and Three Carbon Heteroatom Bond; Georg Thieme Verlag: Stuttgart, Germany, 2005; Volume 18.2.8, pp. 189–243. [Google Scholar]
- Wentrup, C.; Finnerty, J.J.; Koch, R. Amino-, alkoxy-, and alkylthio-isocyanates and -isothiocyanates, RX-NCY, their isomers RX-YCN and RX-CNY, and their rearrangements. Curr. Org. Chem. 2011, 15, 1745–1759. [Google Scholar] [CrossRef]
- Bedane, K.G.; Singh, G.S. Reactivity and diverse synthetic applications of acyl isothiocyanates. Arkivoc 2015, 6, 206–245. [Google Scholar] [CrossRef] [Green Version]
- Rathke, A. Die Chemie auf der 45. Versammlung deutscher Naturforscher und Aerzte (12.—18. August). Ber. Dtsch. Chem. Ges. 1872, 5, 799. [Google Scholar]
- Rathke, A. Ueber Chlorschwefelkohlenstoffe. Ann. Chim. 1873, 167, 218. [Google Scholar] [CrossRef] [Green Version]
- Dyson, G.M.; George, H.J. CCXX.—The reactions of thiocarbonyl chloride. Part I. Reaction with aromatic primary aminocompounds. J. Chem. Soc. 1924, 1702–1708. [Google Scholar] [CrossRef]
- Linders, J.T.M.; Monn, J.A.; Mattson, M.V.; George, C.; Jacobson, A.E.; Rice, K.C. Synthesis and binding properties of MK-801 isothiocyanates; (+)-3-isothiocyanato-5-methyl-10,11-dihydro-5H dibenzo[a,d]cyclohepten-5,10-imine hydrochloride: A new, potent and selective electrophilic affinity ligand for the NMDA receptor-coupled phencyclidine binding site. J. Med. Chem. 1993, 36, 2499–2507. [Google Scholar] [PubMed]
- Kutschy, P.; Dzurilla, M.; Takasugi, M.; Torok, M.; Achbergerova, I.; Homzova, R.; Racova, M. New syntheses of indole phytoalexins and related compounds. Tetrahedron 1998, 54, 3549–3566. [Google Scholar] [CrossRef]
- Kutschy, P.; Achbergerova, I.; Dzurilla, M.; Takasugi, M. Synthesis of indole phytoalexins brassinin and cyclobrassinin via [1-(tert-butoxycarbonyl)indol-3-yl]-methyl isothiocyanate as the Key biomimetic intermediate. Synlett 1997, 289–290. [Google Scholar] [CrossRef]
- Yamada, K.; Rice, K.C.; Flippen-Anderson, J.L.; Eissenstat, M.A.; Ward, S.J.; Johnson, M.R.; Howlett, A.C. (Aminoalkyl)indole isothiocyanates as potential electrophilic affinity ligands for the brain cannabinoid receptor. J. Med. Chem. 1996, 39, 1967–1974. [Google Scholar] [CrossRef] [PubMed]
- Posner, G.H.; Cho, C.G.; Green, J.V.; Zhang, Y.; Talalay, P. Design and synthesis of bifunctional isothiocyanate analogs of sulforaphane: Correlation between structure and potency as inducers of anticarcinogenic detoxication enzymes. J. Med. Chem. 1994, 37, 170–176. [Google Scholar] [CrossRef]
- Dyson, G.M.; George, H.J.; Hunter, R.F. CCCCVI.—The interaction of thiocarbonyl chloride and chloro-substituted anilines and the inhibitory action of ortho-substituents. J. Chem. Soc. 1926, 3041–3044. [Google Scholar] [CrossRef]
- Hallenbach, W.; Horner, L. Die Synthese eines fluoreszierenden isothiocyanats als nachweisreagenz für die NH-Funktion. Synthesis 1985, 8, 791. [Google Scholar] [CrossRef]
- Ares, J.J.; Kador, P.F.; Miller, D.D. Synthesis and biological evaluation of irreversible inhibitors of aldose reductase. J. Med. Chem. 1986, 29, 2384–2389. [Google Scholar] [CrossRef]
- Larsen, C.; Harpp, D.N. Thiocarbonyl transfer reagent chemistry. 3. Selective displacements with formaldehyde hydrazones and other nucleophiles. J. Org. Chem. 1981, 46, 2465–2466. [Google Scholar] [CrossRef]
- Kim, S.; Yi, K.Y. 1,1’-Thiocarbonyldi-2,2’-pyridone. A new useful reagent for functional group conversions under essentially neutral conditions. J. Org. Chem. 1986, 56, 2613–2615. [Google Scholar] [CrossRef]
- Kim, S.; Yi, K.Y. Di-2-Pyridyl thionocarbonate. A new reagent for the preparation of isothiocyanates and carbodiimides. Tetrahedron Lett. 1985, 26, 1661–1664. [Google Scholar] [CrossRef]
- Hodgkins, J.E.; Ettlinger, M.G. The synthesis of isothiocyanates from amines. J. Org. Chem. 1956, 21, 404–405. [Google Scholar] [CrossRef]
- Hofmann, A.W. Ueber die dem Senföl entsprechenden Isomeren der Schwefelcyanwasserstoffäther. Chem. Ber. 1868, 1, 170. [Google Scholar] [CrossRef]
- Hodgkins, J.E.; Reeves, W.P. The modified kaluza synthesis. III. The synthesis of some aromatic isothiocyanates. J. Org. Chem. 1964, 29, 3098–3099. [Google Scholar] [CrossRef]
- Li, G.; Tajima, H.; Ohtani, T. An improved procedure for the preparation of isothiocyanates from primary amines by using hydrogen peroxide as the dehydrosulfurization reagent. J. Org. Chem. 1997, 62, 4539–4540. [Google Scholar] [CrossRef]
- Boas, U.; Gertz, H.; Christensen, J.B.; Heegaard, P.M.H. Facile synthesis of aliphatic isothiocyanates and thioureas on solid phase using peptide coupling reagents. Tetrahedron Lett. 2004, 45, 269–272. [Google Scholar] [CrossRef]
- Psurski, M.; Błażewska, K.; Gajda, A.; Gajda, T.; Wietrzyk, J.; Oleksyszyn, J. Synthesis and antiproliferative activity of novel α- and β-dialkoxyphosphoryl isothiocyanates. Bioorg. Med. Chem. Lett. 2011, 21, 4572–4576. [Google Scholar] [CrossRef]
- Boas, U.; Pedersen, B.; Christensen, J.B. Tetramethyl fluoro formamidinium hexafluorophoshate—An improved synthesis and some new uses. Synth. Commun. 1998, 28, 1223–1231. [Google Scholar] [CrossRef]
- Liu, S.; Tang, C.; Ho, B.; Ankersen, M.; Stidsen, C.E.; Crider, A.M. Nonpeptide somatostatin agonists with sst4 selectivity: synthesis and structure–activity relationships of thioureas. J. Med. Chem. 1998, 41, 4693–4705. [Google Scholar] [CrossRef]
- Janczewski, Ł.; Gajda, A.; Frankowski, S.; Goszczyński, T.M.; Gajda, T. T3P®—A benign desulfurating reagent in the synthesis of isothiocyanates. Synthesis 2018, 50, 1141–1151. [Google Scholar]
- Wong, R.; Dolman, S.J. Isothiocyanates from tosyl chloride mediated decomposition of in situ generated dithiocarbamic acid salts. J. Org. Chem. 2007, 72, 3969–3971. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, Z.; Sun, X.; Ma, H.; Chen, X.; Ren, J.; Hu, K. New method for the synthesis of sulforaphane and related isothiocyanates. Synthesis 2011, 24, 3991–3996. [Google Scholar] [CrossRef]
- Nath, J.; Ghosh, H.; Yella, R.; Patel, B.K. Molecular iodine mediated preparation of isothiocyanates from dithiocarbamic acid salts. Eur. J. Org. Chem. 2009, 1849–1851. [Google Scholar] [CrossRef]
- Ghosh, H.; Yella, R.; Nath, J.; Patel, B.K. Desulfurization mediated by hypervalent iodine(III): A novel strategy for the construction of heterocycles. Eur. J. Org. Chem. 2008, 6189–6196. [Google Scholar] [CrossRef]
- Munch, H.; Hansen, J.S.; Pittelkow, M.; Christensen, J.B.; Boas, U. A new efficient synthesis of isothiocyanates from amines using di-tert-butyl dicarbonate. Tetrahedron Lett. 2008, 49, 3117–3119. [Google Scholar] [CrossRef]
- Jamir, L.; Ali, A.R.; Ghosh, H.; Chipem, F.A.S.; Patel, B.K. The thiocarbonyl ‘S’ is softer than thiolate ‘S’: A catalyst-free one-pot synthesis of isothiocyanates in water. Org. Biomol. Chem. 2010, 8, 1674–1678. [Google Scholar] [CrossRef]
- Sun, N.; Li, B.; Shao, J.; Mo, W.; Hu, B.; Shen, Z.; Hu, X. A general and facile one-pot process of isothiocyanates from amines under aqueous conditions. Beilstein J. Org. Chem. 2012, 8, 61–70. [Google Scholar] [CrossRef]
- Mandapati, U.; Pinapati, S.; Rudraraju, R. Copper promoted desulfurization towards the synthesis of isothiocyanates. Tetrahedron Lett. 2017, 58, 125–128. [Google Scholar] [CrossRef]
- Seelam, M.; Shaik, B.; Kammela, P.R. Cobalt mediated by desulfurization toward the synthesis of isothiocyanates. Synth. Commun. 2016, 46, 1759–1765. [Google Scholar] [CrossRef]
- Fu, Z.; Yuan, W.; Chen, N.; Yang, Z.; Xu, J. Na2S2O8-mediated efficient synthesis of isothiocyanates from primary amines in water. Green Chem. 2018, 20, 4484–4491. [Google Scholar] [CrossRef]
- Janczewski, Ł.; Kręgiel, D.; Kolesińska, B. Synthesis of isothiocyanates using DMT/NMM/TsO− as a new desulfurization reagent. Molecules 2021, 26, 2740. [Google Scholar] [CrossRef] [PubMed]
- Eschliman, K.; Bossmann, S.H. Synthesis of isothiocyanates: An update. Synthesis 2019, 51, 1746–1752. [Google Scholar] [CrossRef]
- Nath, J.; Jamir, L.; Patel, B.K. Improved procedure for the preparation of isothiocyanates via iodine-mediated desulfurization of dithiocarbamic acid salts. Green Chem. Lett. Rev. 2011, 4, 1–34. [Google Scholar] [CrossRef] [Green Version]
- Janczewski, Ł.; Gajda, A.; Gajda, T. Direct, microwave-assisted synthesis of isothiocyanates. Eur. J. Org. Chem. 2019, 2528–2532. [Google Scholar] [CrossRef]
- Staudinger, H.; Hauser, E. Über neue organische Phosphorverbindungen IV† Phosphinimine. Helv. Chim. Acta 1921, 4, 1353. [Google Scholar] [CrossRef] [Green Version]
- Molina, P.; Alajarin, M.; Arques, A. Convenient improved syntheses of isocyanates or isothiocyanates from amines. Synthesis 1982, 596–597. [Google Scholar] [CrossRef]
- Tsuge, O.; Kanemasa, S.; Matsuda, K. One-pot synthesis of N-[(trimethylsilyl)methyl]imines and (trimethylsilyl)methyl-substituted heterocumulenes from (trimethylsilyl)methyl azide. J. Org. Chem. 1984, 49, 2688–2691. [Google Scholar] [CrossRef]
- Isoda, T.; Hayashi, K.; Tamai, S.; Kumagai, T.; Nagao, Y. Efficient synthesis of isothiocyanates based on the tandem Staudinger/aza-Wittig reactions and mechanistic consideration of the tandem reactions. Chem. Pharm. Bull. 2006, 54, 1616–1619. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.Y.; Deng, J.C.; Ke, Y.P.; Zhong, X.L.; Xu, L.; Tang, R.Y.; Zheng, W. Isothiocyanation of amines using the Langlois reagent. Chem. Commun. 2017, 53, 6073–6076. [Google Scholar] [CrossRef]
- Langlois, B.R.; Laurent, E.; Roidot, N. Trifluoromethylation of aromatic compounds with sodium trifluoromethanesulfinate under oxidative conditions. Tetrahedron Lett. 1991, 32, 7525–7528. [Google Scholar] [CrossRef]
- Scattolin, T.; Klein, A.; Schoenebeck, F. Synthesis of isothiocyanates and unsymmetrical thioureas with the bench-stable solid reagent (Me4N)SCF3. Org. Lett. 2017, 19, 1831–1833. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Lin, J.H.; Xiao, J.C. Reaction of thiocarbonyl fluoride generated from difluorocarbene with amines. Angew. Chem. Int. Ed. 2017, 56, 16669–16673. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Cai, J.; Lin, J.H.; Guo, Y.; Xiao, J.C. Synthesis and decarboxylative Wittig reaction of difluoromethylene phosphobetaine. Chem. Commun. 2013, 49, 7513–7515. [Google Scholar] [CrossRef]
- Zhen, L.; Fan, H.; Wang, X.; Jiang, L. Synthesis of thiocarbamoyl fluorides and isothiocyanates using CF3SiMe3 and elemental sulfur or AgSCF3 and KBr with amines. Org. Lett. 2019, 21, 2106–2110. [Google Scholar] [CrossRef]
- Liu, X.; Xu, C.; Wang, M.; Liu, Q. Trifluoromethyltrimethylsilane: Nucleophilic trifluoromethylation and beyond. Chem. Rev. 2015, 115, 683–730. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Zhang, X.G. Organophosphine-free copper-catalyzed isothiocyanation of amines with sodium bromodifluoroacetate and sulfur. Chem. Commun. 2019, 55, 1144–1147. [Google Scholar] [CrossRef] [PubMed]
- Németh, A.G.; Ábrányi-Balogh, P. Recent advances in the synthesis of isothiocyanates using elemental sulfur. Catalyst 2021, 11, 1081. [Google Scholar] [CrossRef]
- Wei, J.; Liang, S.; Jiang, L.; Yi, W. Synthesis of thiocarbamoyl fluorides and isothiocyanates using amines with CF3SO2Cl. J. Org. Chem. 2020, 85, 12374–12381. [Google Scholar] [CrossRef]
- Hamashima, Y.; Nagi, T.; Shimizu, R.; Tsuchimoto, T.; Sodeoka, M. Catalytic asymmetric α-chlorination of 3-acyloxazolidin-2-one with a trinary catalytic system. Eur. J. Org. Chem. 2011, 2011, 3675–3678. [Google Scholar] [CrossRef]
- Nagib, D.A.; MacMillan, D.W.C. Trifluoromethylation of arenes and heteroarenes by means of photoredox catalysis. Nature 2011, 480, 224–228. [Google Scholar] [CrossRef]
- Jiang, L.; Ding, T.; Yi, W.-b.; Zeng, X.; Zhang, W. Fluoroalkylsulfonyl chlorides promoted vicinal chloro-fluoroalkylthiolation of alkenes and alkynes. Org. Lett. 2018, 20, 2236–2240. [Google Scholar] [CrossRef] [PubMed]
- De Nicola, G.R.; Rollin, P.; Mazzon, E.; Iori, R. Novel gram-scale production of enantiopure R-sulforaphane from Tuscan black kale seeds. Molecules 2014, 19, 6975–6986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iori, R.; Bernardi, R.; Gueyrard, D.; Rollin, P.; Palmieri, S. Formation of glucoraphanin by chemoselective oxidation of natural glucoerucin: A chemoenzymatic route to sulforaphane. Bioorg. Med. Chem. Lett. 1999, 9, 1047–1048. [Google Scholar] [CrossRef]
- Papi, A.; Orlandi, M.; Bartolini, G.; Barillari, J.; Iori, R.; Paolini, M.; Ferroni, F.; Fumo, M.G.; Pedulli, G.F.; Valgimigli, L. Cytotoxic and antioxidant activity of 4-methylthio-3-butenyl isothiocyanate from Raphanus sativus L. (Kaiware Daikon) sprouts. J. Agric. Food Chem. 2008, 56, 875–883. [Google Scholar] [CrossRef]
- Terada, Y.; Masuda, H.; Watanabe, T. Structure−activity relationship study on isothiocyanates: Comparison of TRPA1-activating ability between allyl isothiocyanate and specific flavor components of wasabi, horseradish, and white mustard. J. Nat. Prod. 2015, 78, 1937–1941. [Google Scholar] [CrossRef]
- Vermeulen, M.; Zwanenburg, B.; Chittenden, G.J.F.; Verhagen, H. Synthesis of isothiocyanate-derived mercapturic acids. Eur. J. Med. Chem. 2003, 38, 729–737. [Google Scholar] [CrossRef]
- Mays, J.R.; Roska, R.L.W.; Sarfaraz, S.; Mukhtar, H.; Rajski, S.R. Identification, synthesis, and enzymology of non-natural glucosinolate chemopreventive candidates. ChemBioChem 2008, 9, 729–747. [Google Scholar] [CrossRef]
- Conaway, C.C.; Wang, C.X.; Pittman, B.; Yang, Y.M.; Schwartz, J.E.; Tian, D.; McIntee, E.J.; Hecht, S.S.; Chung, F.L. Phenethyl isothiocyanate and sulforaphane and their N-acetylcysteine conjugates inhibit malignant progression of lung adenomas induced by tobacco carcinogens in A/J mice. Cancer Res. 2005, 65, 8548–8557. [Google Scholar] [CrossRef] [Green Version]
- Moon, J.K.; Kim, J.R.; Ahn, Y.J.; Shibamoto, T. Analysis and anti-Helicobacter activity of sulforaphane and related compounds present in broccoli (Brassica oleracea L.) sprouts. J. Agric. Food Chem. 2010, 58, 6672–6677. [Google Scholar] [CrossRef]
- Ernst, I.M.A.; Palani, K.; Esatbeyoglu, T.; Schwarz, K.; Rimbach, G. Synthesis adn Nrf2-inducing activity of the isothiocyanates iberverin, iberin and cheirolin. Pharmacol. Res. 2013, 70, 155–162. [Google Scholar] [CrossRef]
- Kiełbasiński, P.; Łuczak, J.; Cierpiał, T.; Błaszczyk, J.; Sieroń, L.; Wiktorska, K.; Lubelska, K.; Milczarek, M.; Chilmończyk, Z. New enantiomeric fluorine-containing derivatives of sulforaphane: Synthesis, absolute configurations and biological activity. Eur. J. Med. Chem. 2014, 76, 332–342. [Google Scholar] [CrossRef]
- Cierpiał, T.; Kiełbasiński, P.; Kwiatkowska, M.; Łyżwa, P.; Lubelska, K.; Kuran, D.; Dąbrowska, A.; Kruszewska, H.; Mielczarek, L.; Chilmonczyk, Z.; et al. Fluoroaryl analogs of sulforaphane—A group of compounds of anticancer and antimicrobial activity. Bioorg. Chem. 2020, 94, 103454. [Google Scholar] [CrossRef]
- Shi, Y.H.; Dai, D.F.; Li, J.; Dong, Y.W.; Jiang, Y.; Li, H.G.; Gao, Y.; Chong, C.K.; Li, H.Y.; Chu, X.Q.; et al. Sulforaphane analohues with heterocyclic moieties: Synthesis and inhibitory activities against cancer cell lines. Molecules 2016, 21, 514. [Google Scholar] [CrossRef] [Green Version]
- Setito, S.; Pruccoli, L.; Runfola, M.; Citi, V.; Martelli, A.; Saccomanni, G.; Calderone, V.; Tarozzi, A.; Rapposelli, S. Design and synthesis of H2S-donor hybrids: A new treatment for Alzheimer’s disease? Eur. J. Med. Chem. 2019, 184, 111745. [Google Scholar] [CrossRef]
- Polinsky, R.J. Clinical pharmacology of rivastigmine: A new-generation acetylcholinesterase inhibitor for the treatment of Alzheimer’s disease. Clin. Ther. 1998, 20, 634–647. [Google Scholar] [CrossRef]
- Hu, K.; Qi, Y.J.; Zhao, J.; Jiang, H.F.; Chen, X.; Ren, J. Synthesis and biological evaluation of sulforaphane derivatives as potential antitumor agents. Eur. J. Med. Chem. 2013, 64, 529–539. [Google Scholar] [CrossRef]
- Khiar, N.; Werner, S.; Mallouk, S.; Lieder, F.; Alcudia, A.; Fernández, I. Enantiopure sulforaphane analogues with various substituents at the sulfinyl sulfur: Asymmetric synthesis and biological activities. J. Org. Chem. 2009, 74, 6002–6009. [Google Scholar] [CrossRef]
- Fernández, I.; Khiar, N.; Llera, J.M.; Alcudia, F. Asymmetric synthesis of alkane- and arenesulfinates of diacetone-D-glucose (DAG): An improved and general route to both enantiomerically pure sulfoxides. J. Org. Chem. 1992, 57, 6789–6796. [Google Scholar] [CrossRef]
- Elhalem, E.; Recio, R.; Werner, S.; Lieder, F.; Calderón-Montaño, J.M.; López-Lázaro, M.; Fernández, I.; Khiar, N. Sulforaphane homologues: Enantiodivergent synthesis of both enantiomers, activation of the Nrf2 transcription factor and selective cytotoxic activity. Eur. J. Med. Chem. 2014, 87, 552–563. [Google Scholar] [CrossRef] [Green Version]
- Psurski, M.; Piguła, M.; Ciekot, J.; Winiarski, Ł.; Wietrzyk, J.; Oleksyszyn, J. Convenient syntheses of novel 1-isothiocyano-alkylphosphonate diphenyl ester derivatives with potential biological activity. Tetrahedron Lett. 2012, 53, 5845–5847. [Google Scholar] [CrossRef]
- Psurski, M.; Janczewski, Ł.; Świtalska, M.; Gajda, A.; Goszczyński, T.M.; Oleksyszyn, J.; Wietrzyk, J.; Gajda, T. Novel phosphonate analogs of sulforaphane: Synthesis, in vitro and in vivo activity. Eur. J. Med. Chem. 2017, 132, 63–80. [Google Scholar] [CrossRef]
- Janczewski, Ł.; Gajda, A.; Braszczyńska, J.; Gajda, T. Microwave-assisted synthesis of dialkyl ω-azidoalkylphosphonates. Synth. Commun. 2016, 46, 1625–1633. [Google Scholar] [CrossRef]
- Rudzinska-Radecka, M.; Janczewski, Ł.; Gajda, A.; Godlewska, M.; Chmielewska-Krzesinska, M.; Waskowicz, K.; Podlasz, P. The anti-tumoral potential of phosphonate analog of sulforaphane in Zebrafish xenograft model. Cells 2021, 10, 3219. [Google Scholar] [CrossRef]
- Janczewski, Ł.; Psurski, M.; Świtalska, M.; Gajda, A.; Goszczyński, T.M.; Oleksyszyn, J.; Wietrzyk, J.; Gajda, T. Design, synthesis, and evaluation of ω-(isothiocyanato)alkylphosphinates and phosphine oxides as antiproliferative agents. ChemMedChem 2018, 13, 105–115. [Google Scholar] [CrossRef] [Green Version]
- Janczewski, Ł.; Burchacka, E.; Psurski, M.; Ciekot, J.; Gajda, A.; Gajda, T. New diaryl ω-(isothiocyanato)alkylphosphonates and their mercapturic acids as potential antibacterial agents. Life Sci. 2019, 219, 264–271. [Google Scholar] [CrossRef]
- Psurski, M.; Janczewski, Ł.; Świtalska, M.; Gajda, A.; Goszczyński, T.M.; Ciekot, J.; Winiarski, Ł.; Oleksyszyn, J.; Wietrzyk, J.; Gajda, T. Phosphorus-containing isothiocyanate-derived mercapturic acids as a useful alternative for parental isothiocyanates in experimental oncology. Bioorg. Med. Chem. Lett. 2018, 28, 2611–2615. [Google Scholar] [CrossRef]
- Amara, N.; Mashiach, R.; Amar, D.; Krief, P.; Spieser, S.A.H.; Bottomley, M.J.; Aharoni, A.; Meijler, M.M. Covalent inhibition of bacterial quorum sensing. J. Am. Chem. Soc. 2009, 131, 10610–10619. [Google Scholar] [CrossRef]
- Amara, N.; Gregor, R.; Rayo, J.; Dandela, R.; Daniel, E.; Liubin, N.; Willems, H.M.E.; Ben-Zvi, A.; Krom, B.P.; Meijler, M.M. Fine-tuning covalent inhibition of bacterial quorum sensing. ChemBioChem 2016, 17, 825–835. [Google Scholar] [CrossRef]
- Noshita, T.; Kidachi, Y.; Funayama, H.; Kiyota, K.; Yamaguchi, H.; Ryoyama, K. Anti-nitric oxide production activity of isothiocyanates correlates with their polar surface area rather than their lipophilicity. Eur. J. Med. Chem. 2009, 44, 4931–4936. [Google Scholar] [CrossRef]
- Tarozzi, A.; Marchetti, C.; Nicolini, B.; D’Amico, M.; Ticchi, N.; Pruccoli, L.; Tumiatti, V.; Simoni, E.; Lodola, A.; Mor, M.; et al. Combined inhibition of the EGFR/AKT pathways by a novel conjugate of quinazoline with isothiocyanate. Eur. J. Med. Chem. 2016, 117, 283–291. [Google Scholar] [CrossRef]
- Boehm, J.; Davis, R.; Murar, C.E.; Li, T.; McCleland, B.; Dong, S.; Yan, H.; Kerns, J.; Moody, C.J.; Wilson, A.J.; et al. Discovery of a crystalline sulforaphane analog with good solid-state stability and engagement of the Nrf2 pathway in vitro and in vivo. Bioorg. Med. Chem. 2019, 27, 579–588. [Google Scholar] [CrossRef]
- Wang, X.; Yu, C.; Wang, C.; Ma, Y.; Wang, T.; Li, Y.; Huang, Z.; Zhou, M.; Sun, P.; Zheng, J.; et al. Novel cyclin-dependent kinase 9 (CDK9) inhibitor with suppression of cancer stemness activity against non-small-cell lung cancer. Eur. J. Med. Chem. 2019, 181, 111535. [Google Scholar] [CrossRef]
- O’Brien, K.T.; Noto, J.G.; Nichols-O’Neill, L.; Perez, L.J. Potent irreversible inhibitors of LasR quorum sensing in Pseudomonas aeruginosa. ACS Med. Chem. Lett. 2015, 6, 162–167. [Google Scholar] [CrossRef] [Green Version]
- Fortunato, S.; Lenzi, C.; Granchi, C.; Citi, V.; Martelli, A.; Calderone, V.; Di Pietro, S.; Signore, G.; Di Bussolo, V.; Minutolo, F. First examples of H2S-releasing glycoconjugates: Stereoselective synthesis and anticancer activities. Bioconjugate Chem. 2019, 30, 614–620. [Google Scholar] [CrossRef]
- Nyein, C.M.; Zhong, X.; Lu, J.; Luo, H.; Wang, J.; Rapposelli, S.; Li, M.; Ou-yang, Y.; Pi, R.; He, X. Synthesis and anti-glioblastoma effect of artemisinin-isothiocyanate derivatives. RSC Adv. 2018, 8, 40974–40983. [Google Scholar] [CrossRef] [Green Version]
- Grzywa, R.; Winiarski, Ł.; Psurski, M.; Rudnicka, A.; Wietrzyk, J.; Gajda, T.; Oleksyszyn, J. Synthesis and biological activity of diisothiocyanate-derived mercapturic acids. Bioorg. Med. Chem. Lett. 2016, 26, 667–671. [Google Scholar] [CrossRef]
Figure 1.
Structures of the main groups of glucosinolates.

Figure 2.
Enzymatic degradation of glucosinolates.

Figure 3.
Reactions of isothiocyanates.

Figure 4.
Structures of selected natural isothiocyanates.

Figure 5.
Structures of natural analogs of SFN.

Figure 6.
Metabolism of ITCs via the mercapturic acid pathway. R, an aliphatic or aromatic substituent.
Figure 6.
Metabolism of ITCs via the mercapturic acid pathway. R, an aliphatic or aromatic substituent.

Figure 7.
Accumulation of ITCs in the cell.

Figure 8.
Synthesis of ITCs using amines (Path a and b) or azides (Path c) as substrates.

Figure 9.
Synthesis of ITCs with thiophosgene.

Figure 10.
Thiophosgene surrogates.

Figure 11.
Synthesis of isothiocyanates using a desulfurizing agent.

Figure 12.
Selected desulfurizing agents.

Figure 13.
ITCs synthesis via Staudinger/aza-Wittig reaction.

Figure 14.
Synthesis of ITCs using fluorine-containing reagents.

Figure 15.
Synthesis of SFN (24) and its mercapturic acid 62.

Figure 16.
Synthesis of erucin (28), SFN (24), and erysolin (31).

Figure 17.
Synthesis of SFN using 1,1′-thiocarbonyldi-2,2′-pyridone (37).

Figure 18.
Synthesis SFN (24), erucin (28), and their analogs using thiophosgene.

Figure 19.
Synthesis of iberverin (27), iberin (25), and cheirolin (30).

Figure 20.
Synthesis of trifluoromethyl (73) and trifluoroethyl (75–76) analogs of SFN.

Figure 21.
Synthesis of fluoroaryl and fluoroarylmethyl analogs 80a–h.

Figure 22.
Synthesis of heterocyclic analogues 84d–e of SFN.

Figure 23.
Synthesis of sulfoxide 84a–c and sulfone 89a–c analogues of SFN.

Figure 24.
Synthesis of sulfone analogues of SFN 89d–e.

Figure 25.
Synthesis of erucin and SFN-rivastigmine hybrids 99–104.

Figure 26.
Synthesis of SFN (24) and its derivatives 148–167.

Figure 27.
Synthesis of SFN and erucin via Staudinger/aza-Wittig reaction.

Figure 28.
Synthesis of enantiopure (R)-SFN ((R)-24) and its enantiopure 175–178 analogues.

Figure 29.
Enantiodivergent synthesis of (S)- and (R)-sulfinate esters 173, 180, and 181 using the DAG-methodology.
Figure 29.
Enantiodivergent synthesis of (S)- and (R)-sulfinate esters 173, 180, and 181 using the DAG-methodology.

Figure 30.
Synthesis of enantiopure (R)-SFN ((R)-24), (R)-alyssin ((R)-26), and their homologue (R)-184.
Figure 30.
Synthesis of enantiopure (R)-SFN ((R)-24), (R)-alyssin ((R)-26), and their homologue (R)-184.

Figure 31.
Synthesis of enantiopure (S)-SFN ((S)-24), (S)-alyssin ((S)-26), and their homologue (S)-184.
Figure 31.
Synthesis of enantiopure (S)-SFN ((S)-24), (S)-alyssin ((S)-26), and their homologue (S)-184.

Figure 32.
Synthesis of enantiopure analogues of (R)-SFN-(R)-187 and (R)-188.

Figure 33.
Synthesis of (4-isothiocyanatobutyl)dimethylphosphine oxide (191).

Figure 34.
Synthesis of α- and β-dialkoxyphosphoryl isothiocyanates 204–218.

Figure 35.
Synthesis of α- and β-dialkoxyphosphoryl isothiocyanates 223–226.

Figure 36.
Synthesis of diaryl (1-isothiocyanoalkyl)phosphonates 237–246.

Figure 37.
Synthesis of dialkyl and diphenyl ω-(isothiocyanato)alkylphosphonates 282–316.

Figure 38.
Conversion of diethyl ω-(isothiocyanato)alkylphosphonates 287–291 into dialkyl or diphenyl ω-(isothiocyanato)alkylphosphonates 282–286, 297, 302, 316–325.
Figure 38.
Conversion of diethyl ω-(isothiocyanato)alkylphosphonates 287–291 into dialkyl or diphenyl ω-(isothiocyanato)alkylphosphonates 282–286, 297, 302, 316–325.

Figure 39.
Synthesis of (ω-isothiocyanatoalkyl)dimethylphosphine oxides 191 and 328–330.

Figure 40.
Synthesis of (ω-isothiocyanatoalkyl)diphenylphosphine oxides 349–353.

Figure 41.
Synthesis of (ω-isothiocyanatoalkyl)(diethoxymethyl)phosphinates 358–361.

Figure 42.
Synthesis of methyl and ethyl (ω-isothiocyanatoalkyl)(phenyl)phosphinates 370–377.

Figure 43.
Synthesis of diaryl ω-(isothiocyanato)alkylphosphonates 378–411.

Figure 44.
Preparation of phosphonates isothiocyanate-derived mercapturic acids 412–415.

Figure 45.
Synthesis of 2-oxohexyl isothiocyanate (419).

Figure 46.
Synthesis of ITCs 424–426.

Figure 47.
Synthesis of β-halo-ITCs 430–432.

Figure 48.
Synthesis of analogues of 6-MITC: ITCs 436–440.

Figure 49.
Synthesis of 1-isothiocyanato-6-methoxyhexane (443) and 1-isothiocyanato-6-(methylthio)hexane (444).
Figure 49.
Synthesis of 1-isothiocyanato-6-methoxyhexane (443) and 1-isothiocyanato-6-(methylthio)hexane (444).

Figure 50.
Synthesis of ITCs 451–455 and 457.

Figure 51.
Synthesis of SFN analogues 459 and 461.

Figure 52.
Synthesis of ITCs 464a–e and 465aa–de.

Figure 53.
Synthesis of ITCs 469, 470a–g, and 471.

Figure 54.
Synthesis of ether-linked isothiocyanate analogues 473 and 474.

Figure 55.
Synthesis of the target ITCs derivatives 478a–b and 479a–b.

Figure 56.
Synthesis of artemisinin–isothiocyanate derivatives 481a–c.

Figure 57.
Synthesis of diisothiocyanates 484 and 485.

Figure 58.
Synthesis of diisothiocyanates 485, 490–493.


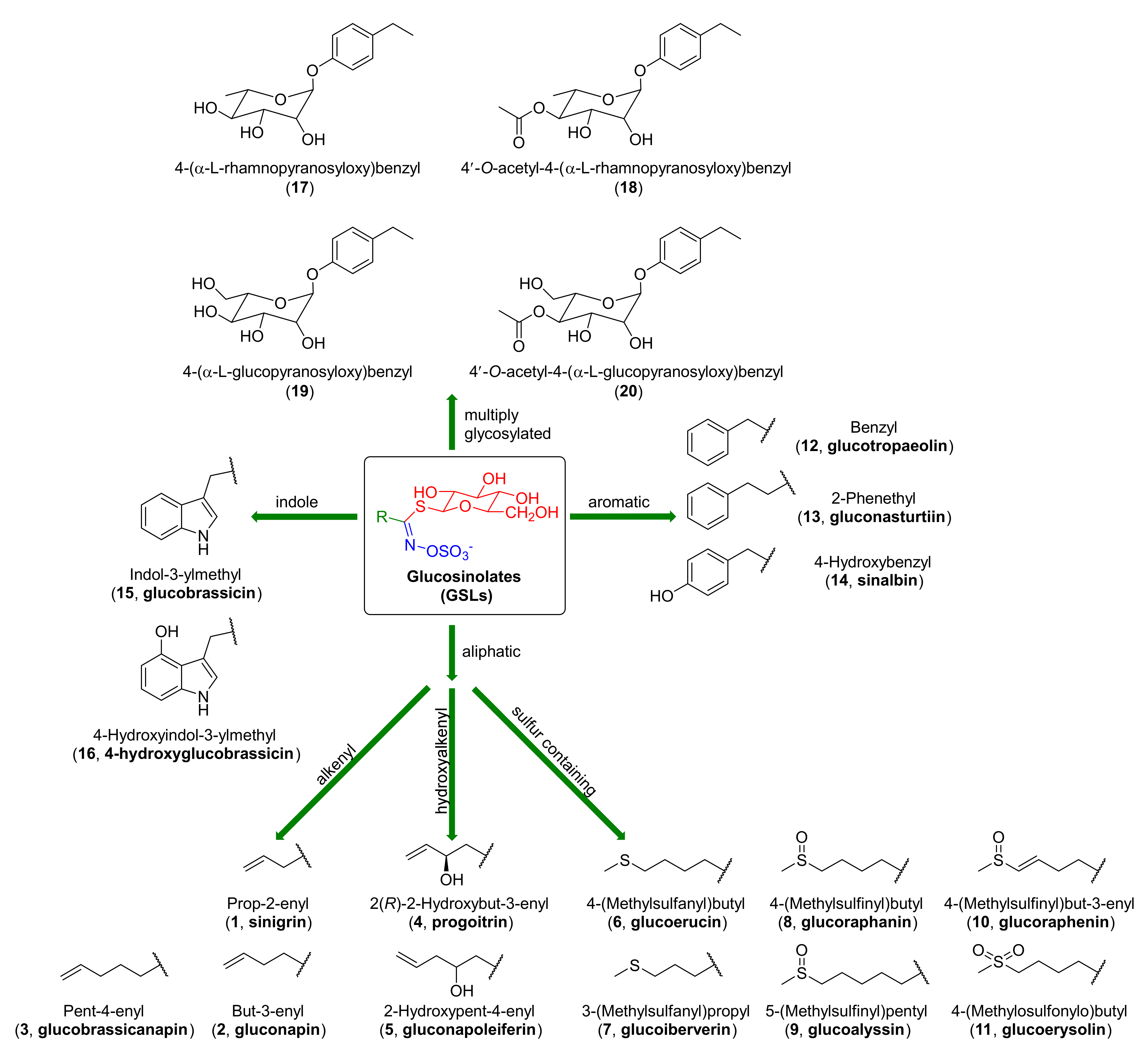{kind=link}
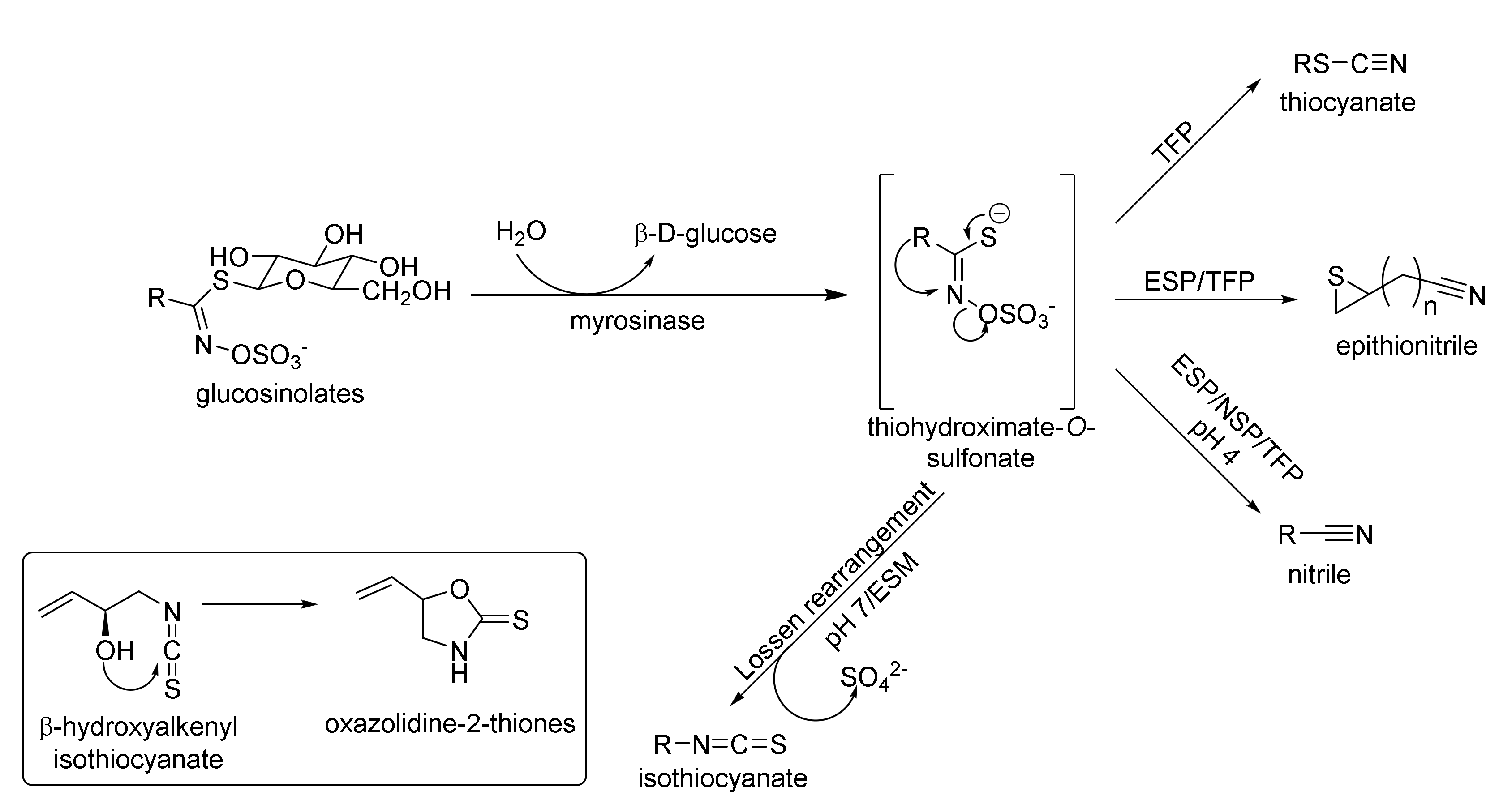{kind=link}
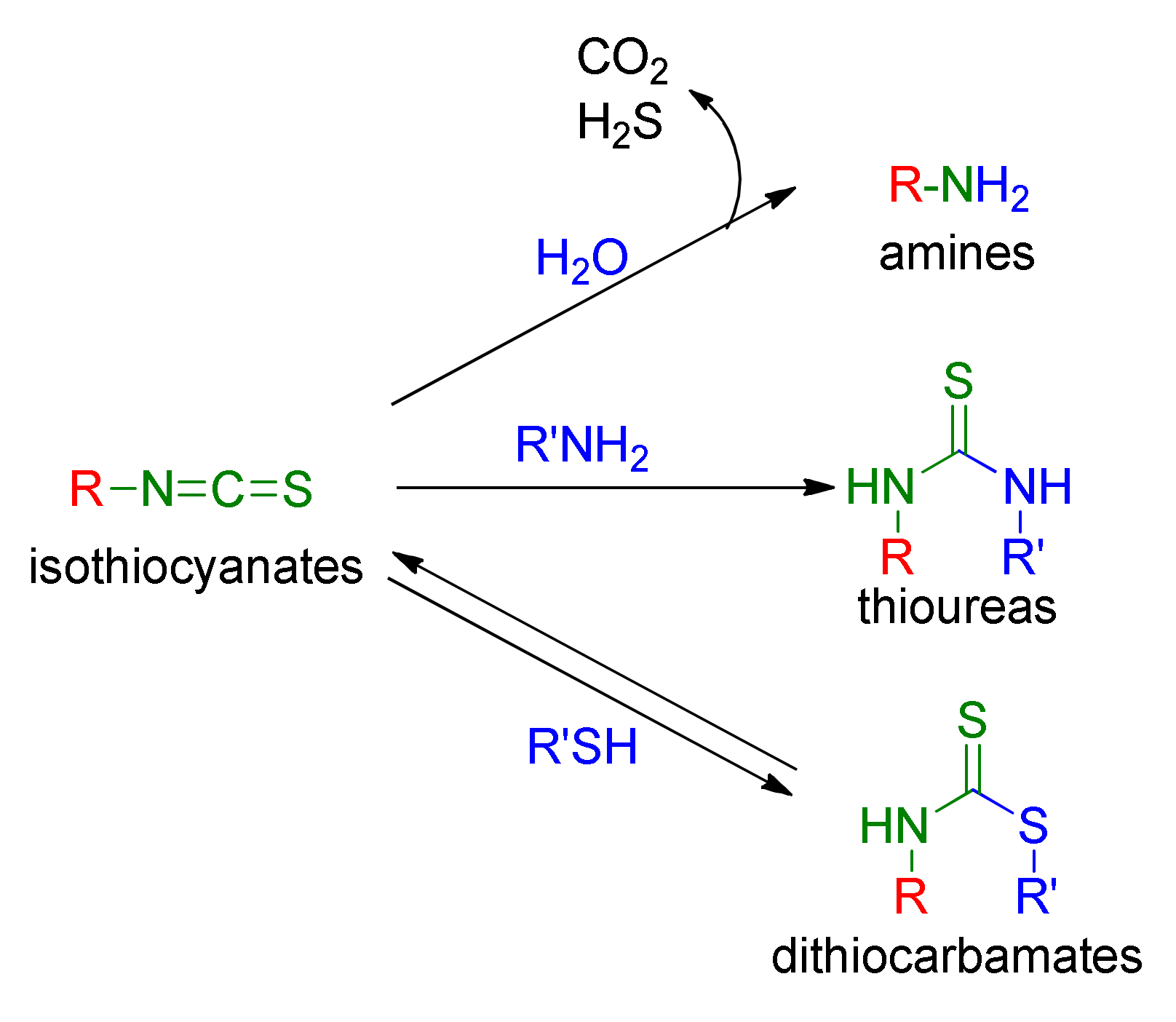{kind=link}
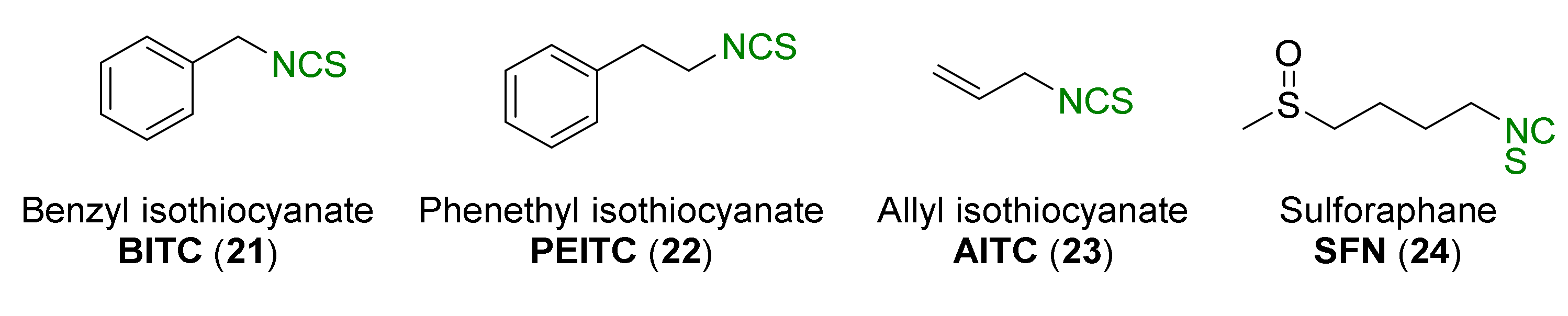{kind=link}
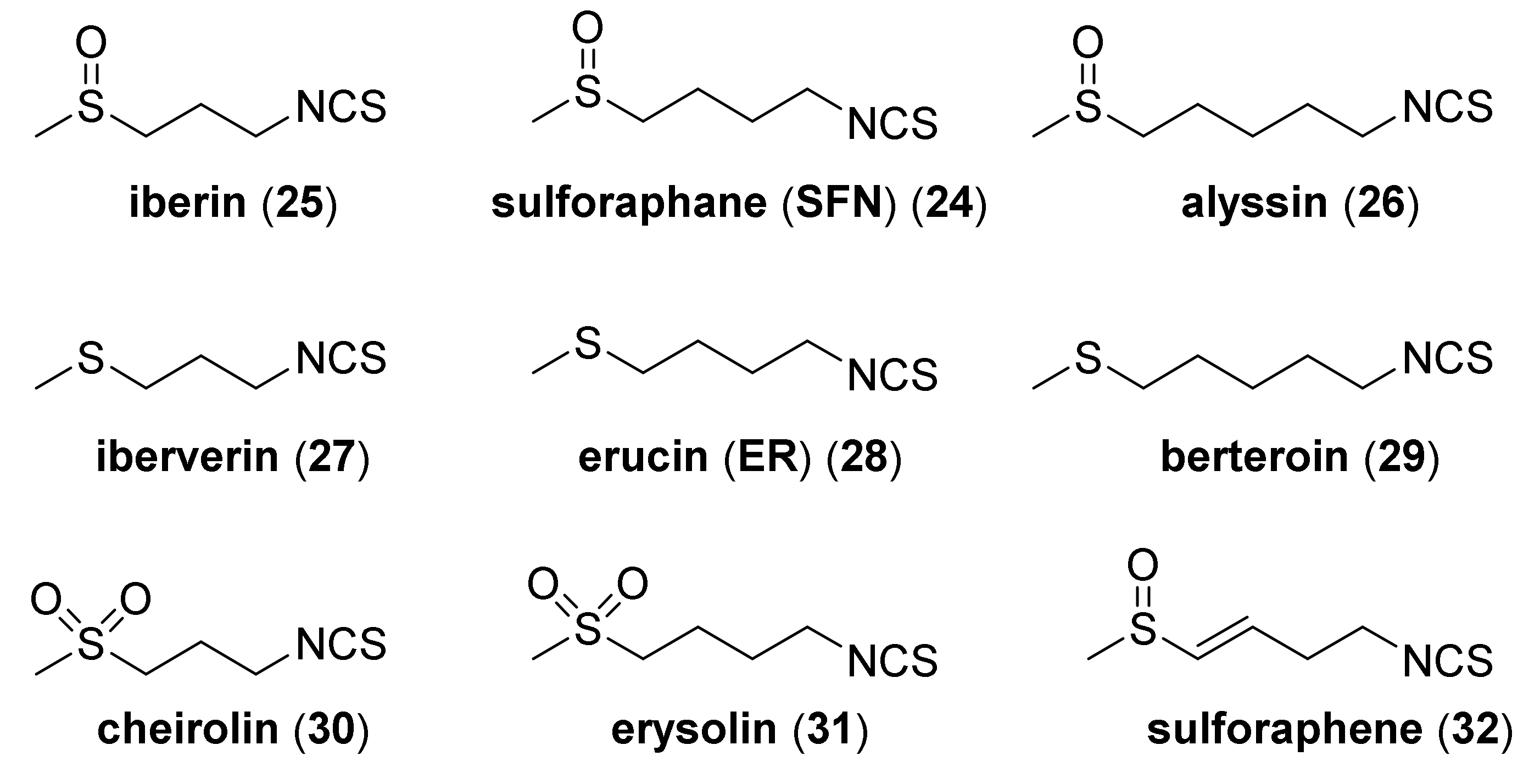{kind=link}
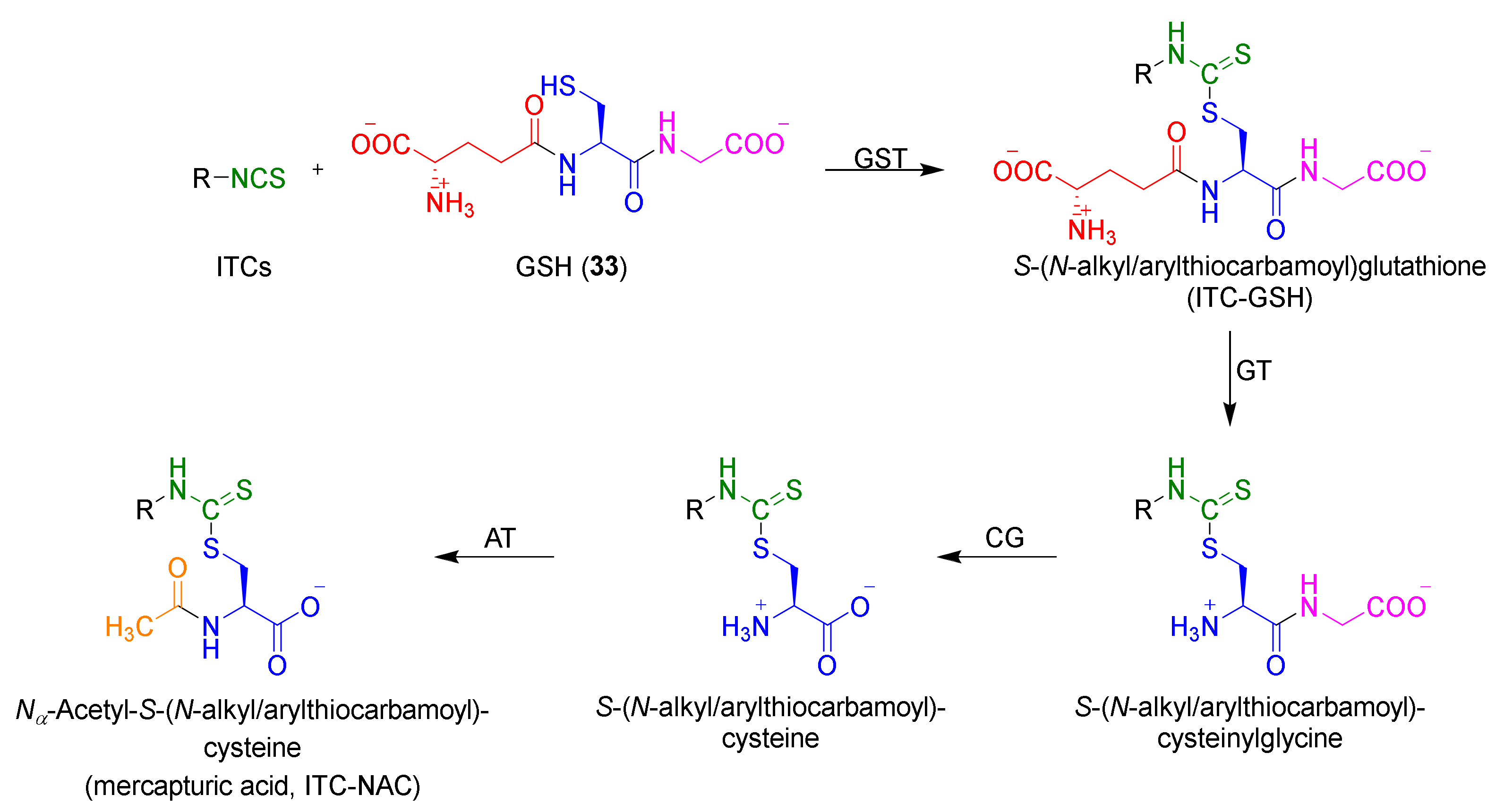{kind=link}
{kind=link}
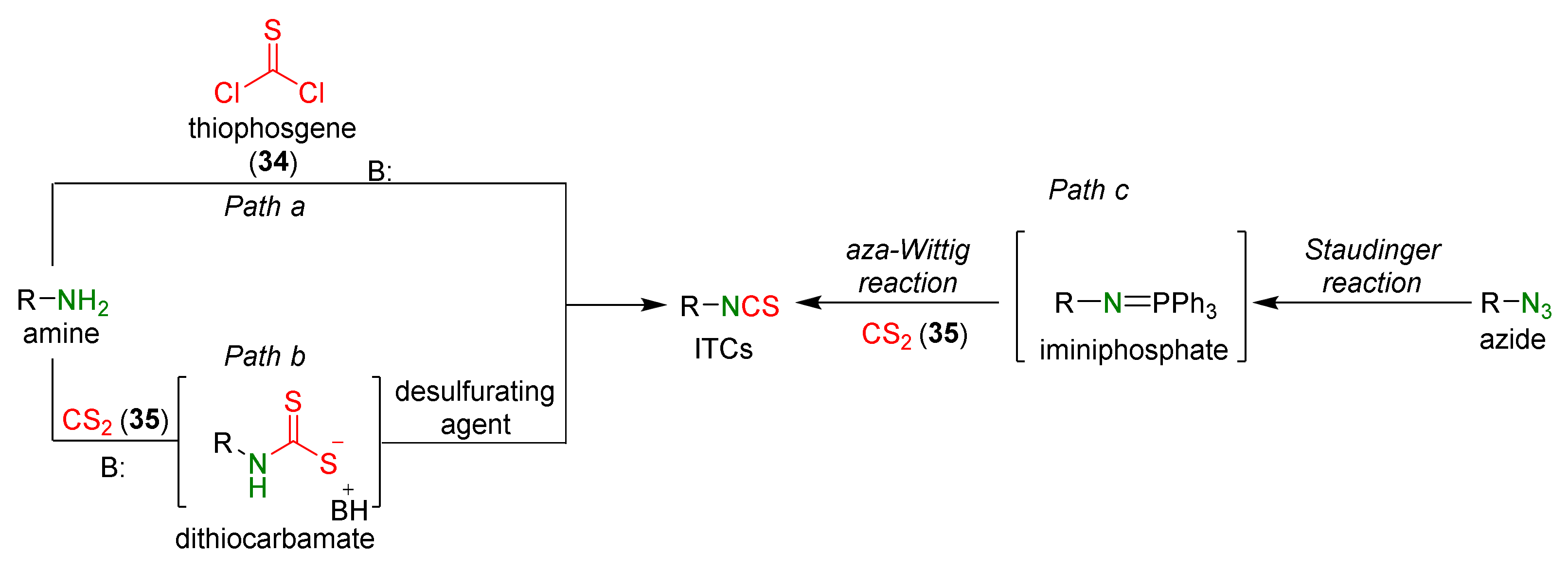{kind=link}
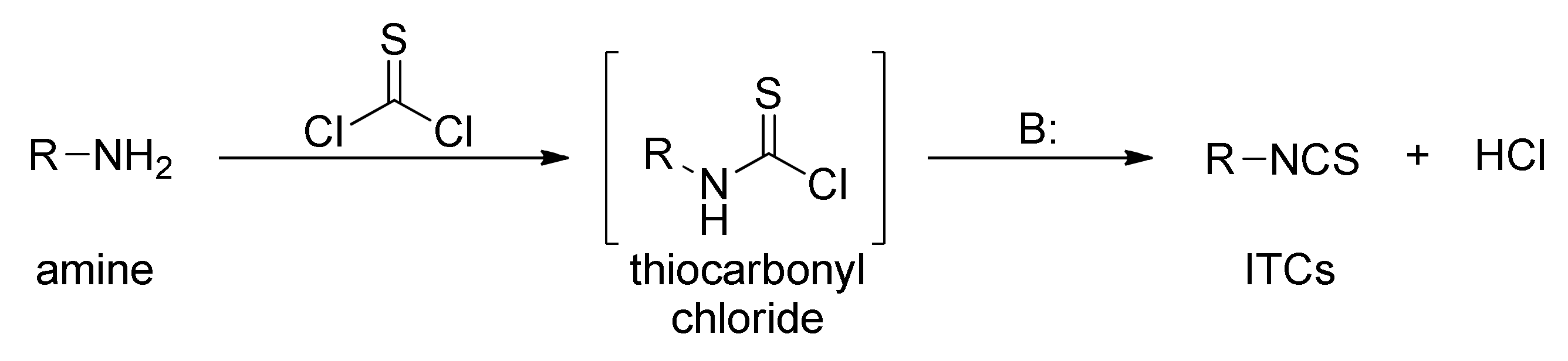{kind=link}
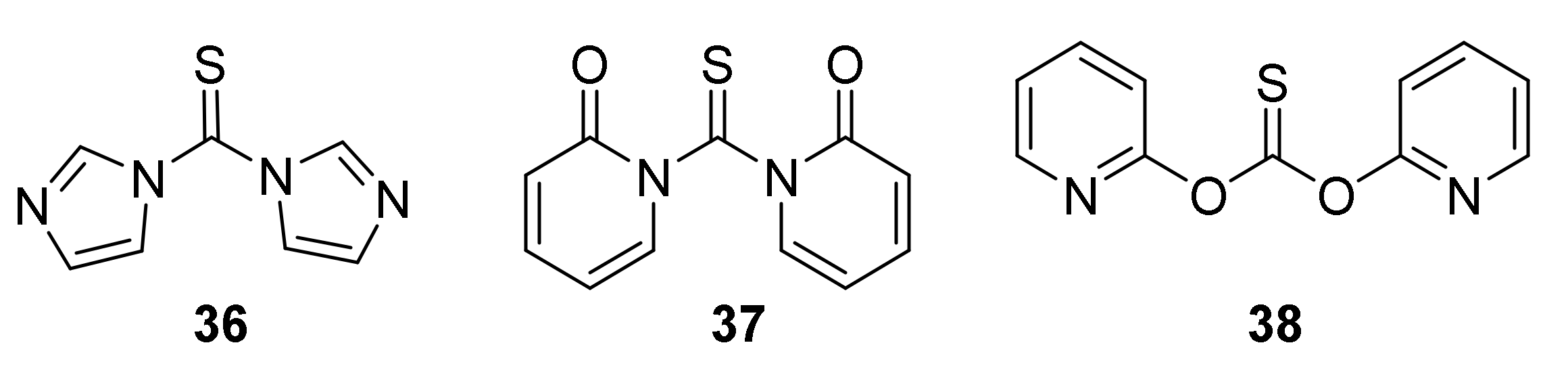{kind=link}
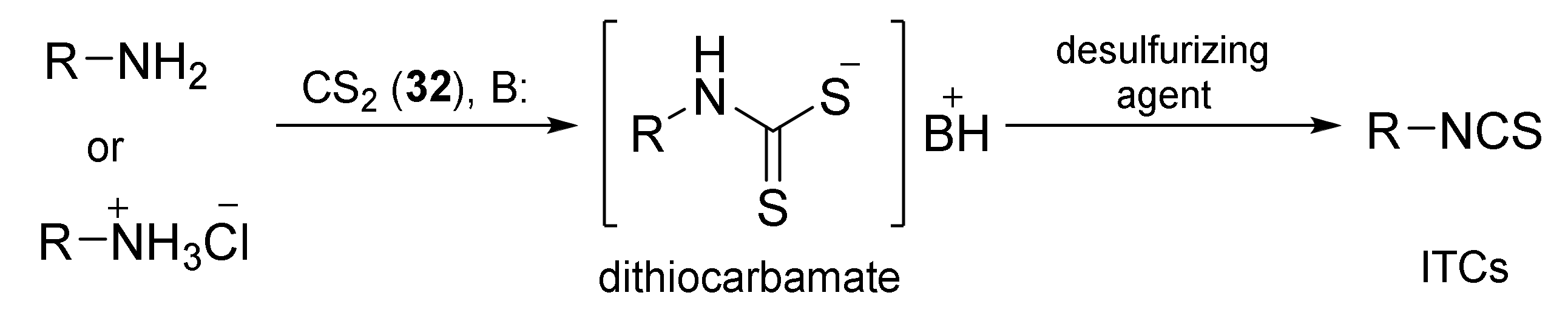{kind=link}
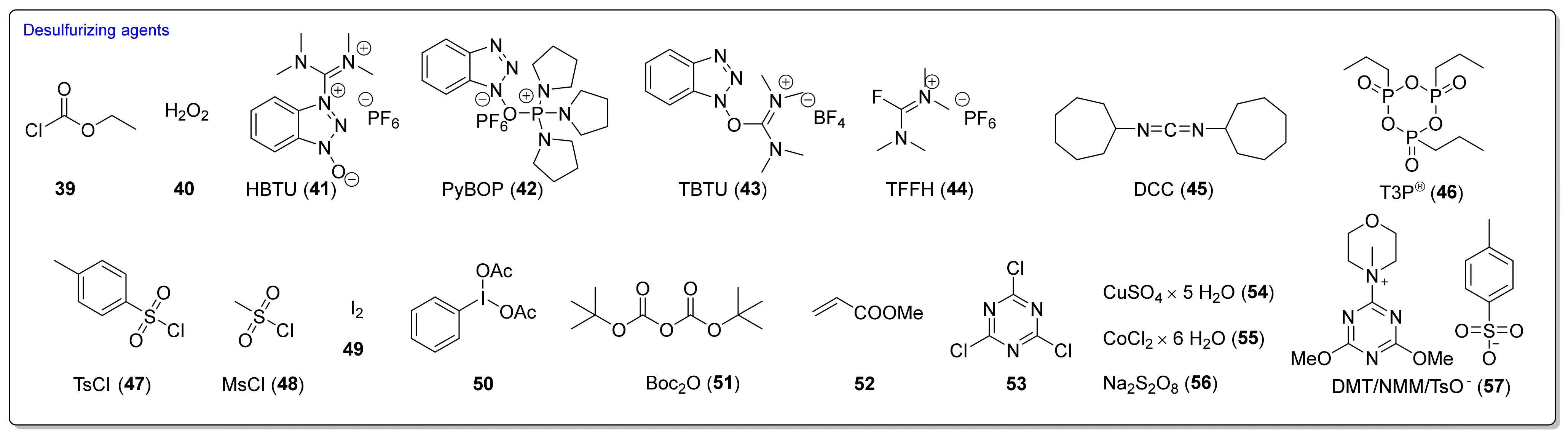{kind=link}
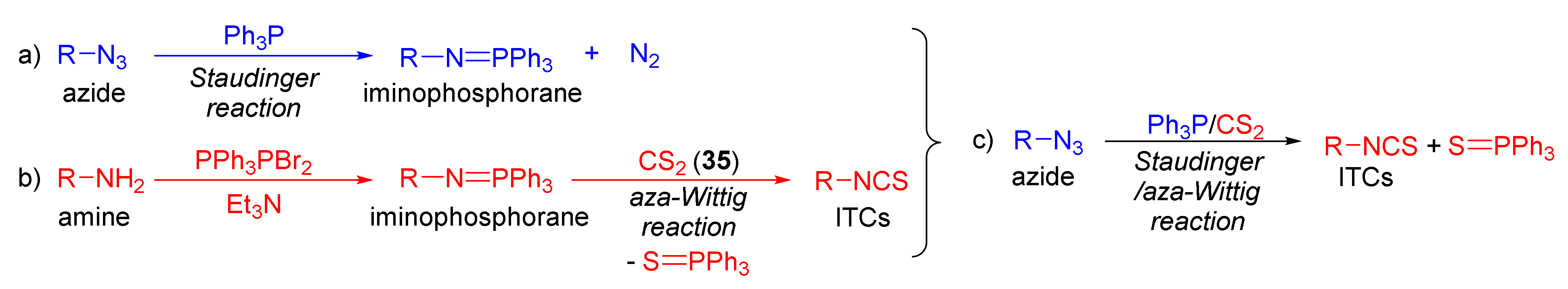{kind=link}
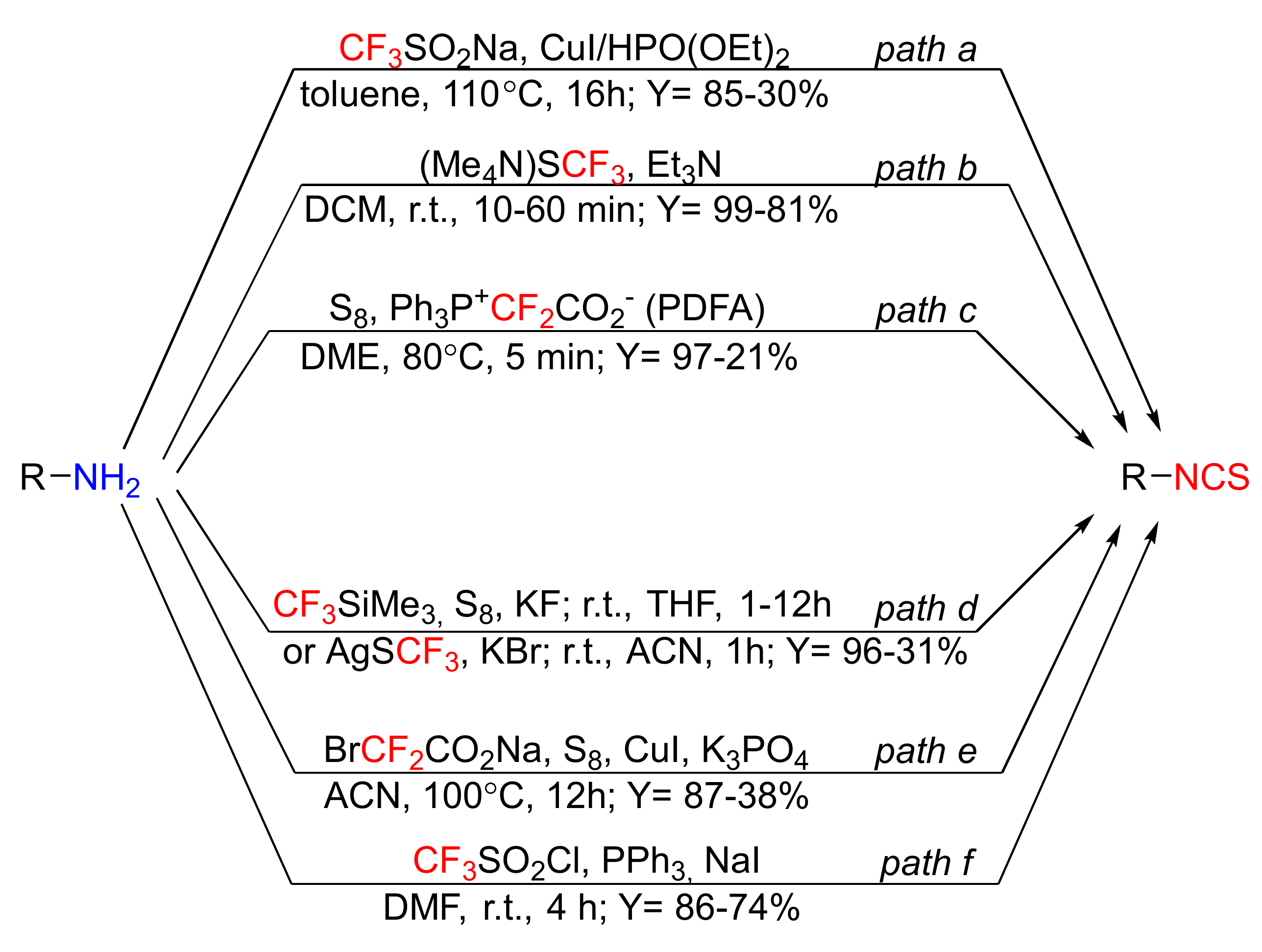{kind=link}
{kind=link}
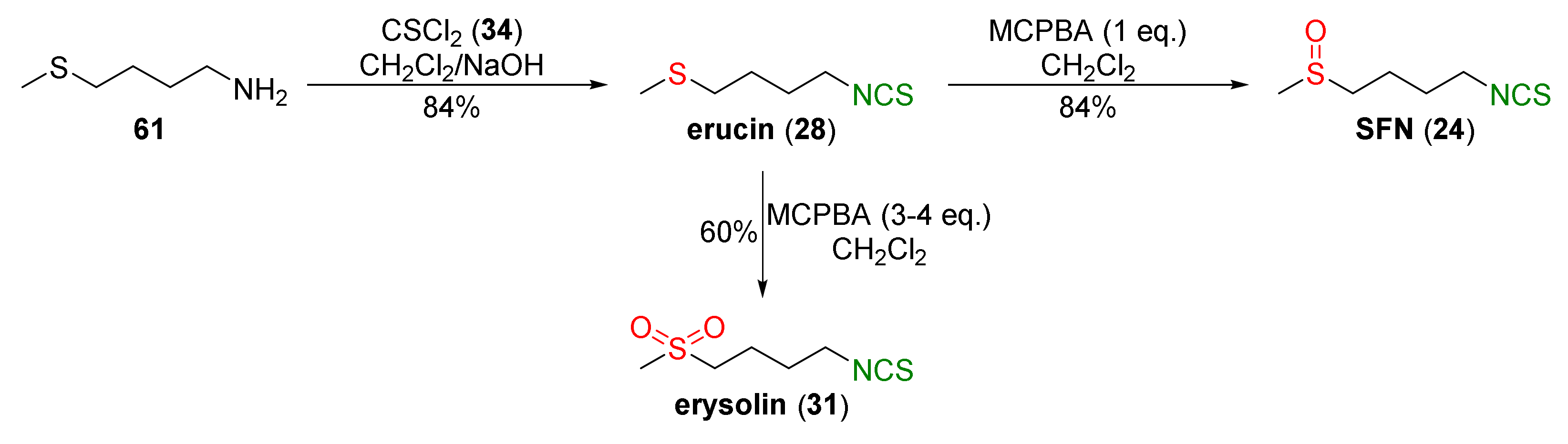{kind=link}
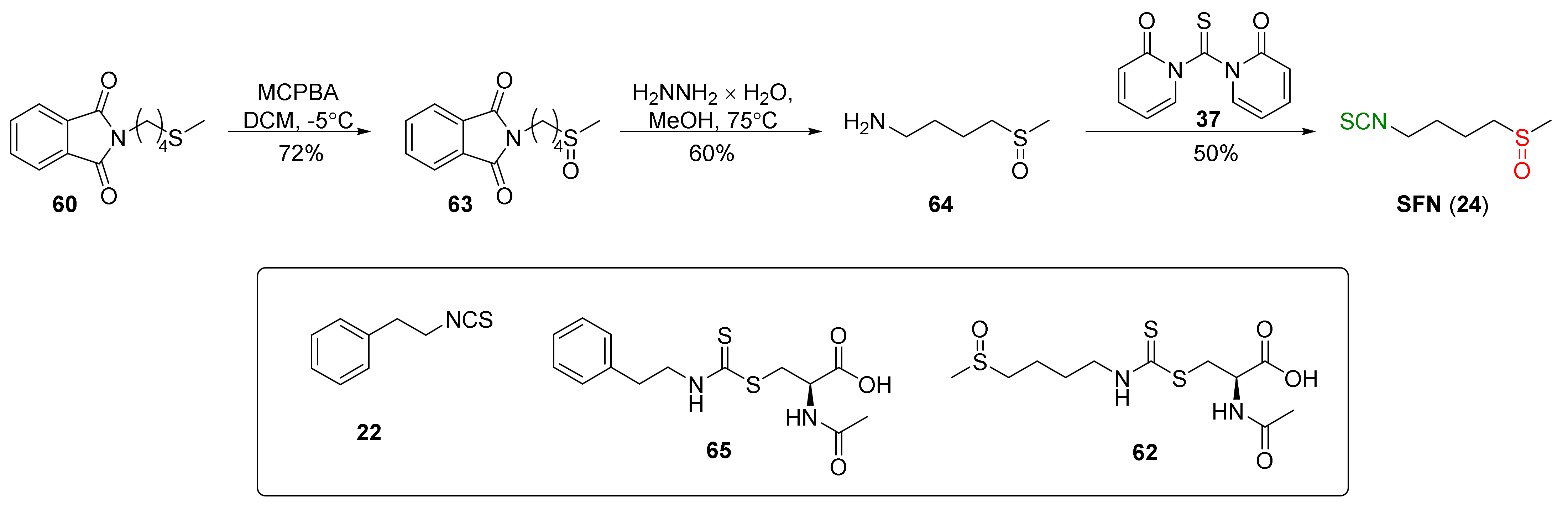{kind=link}
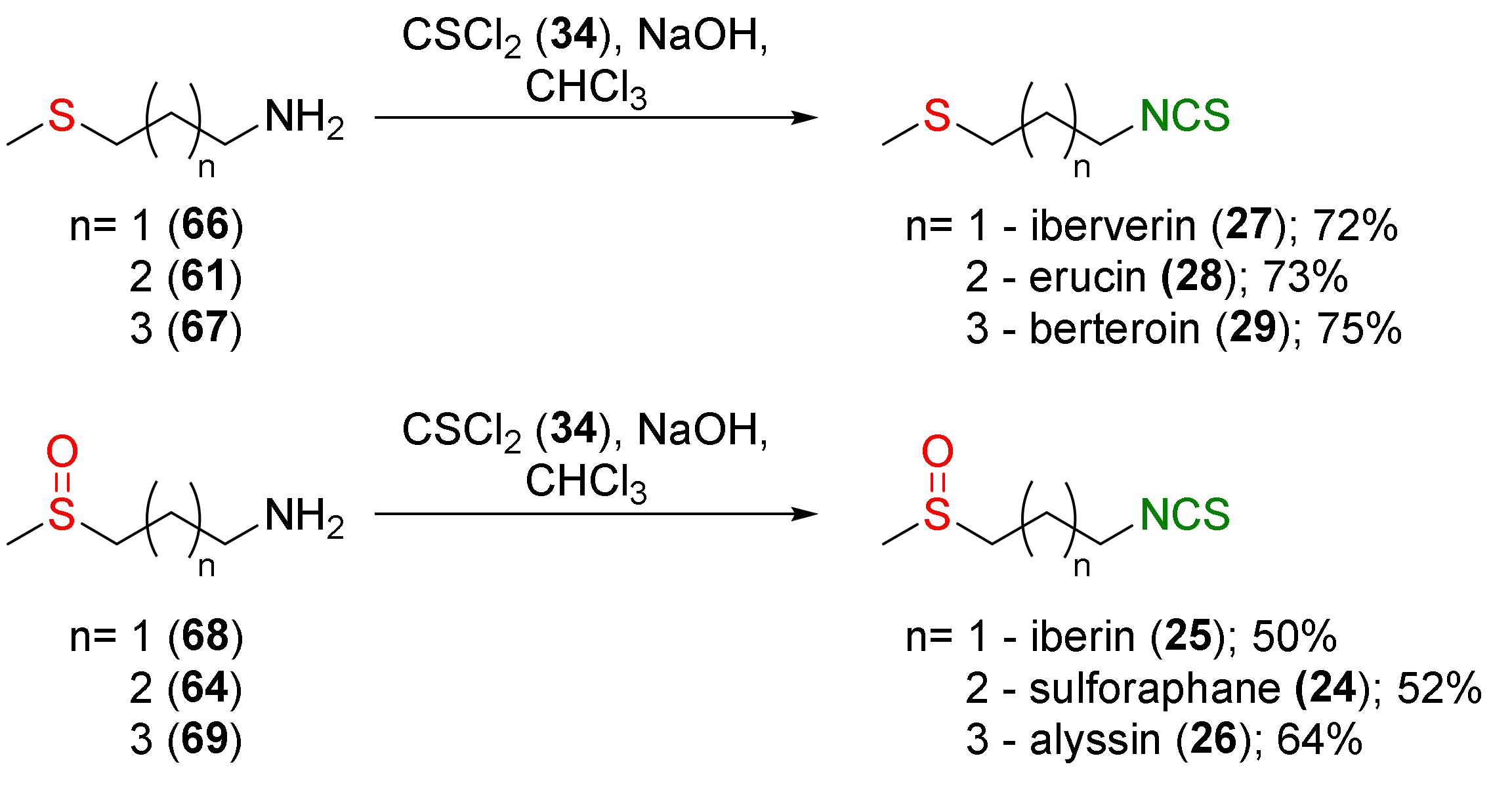{kind=link}
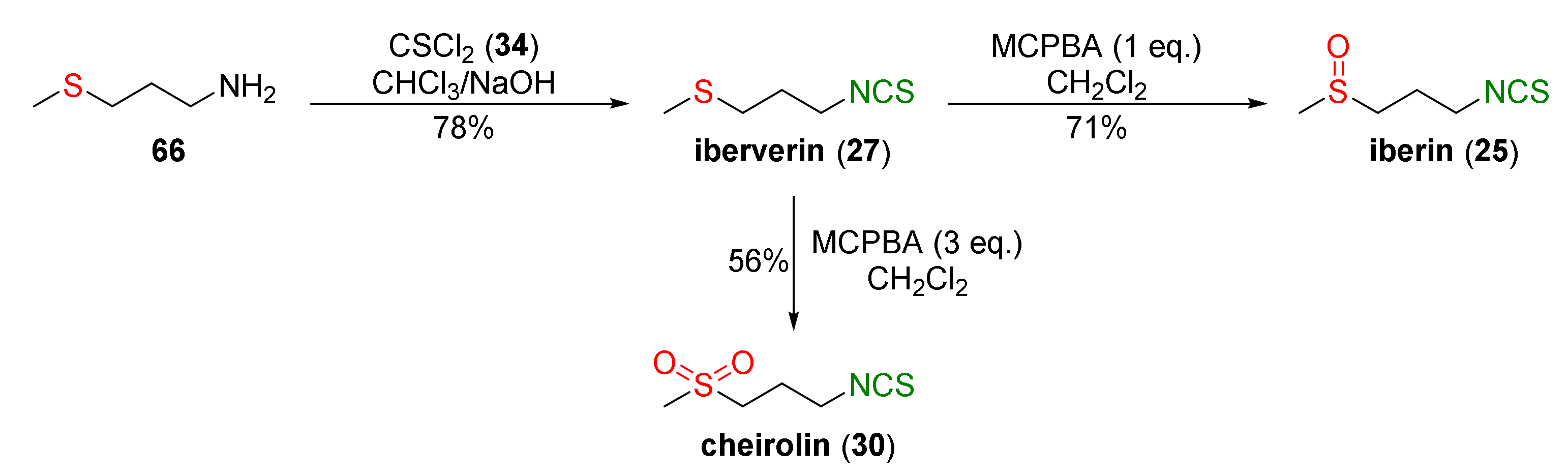{kind=link}
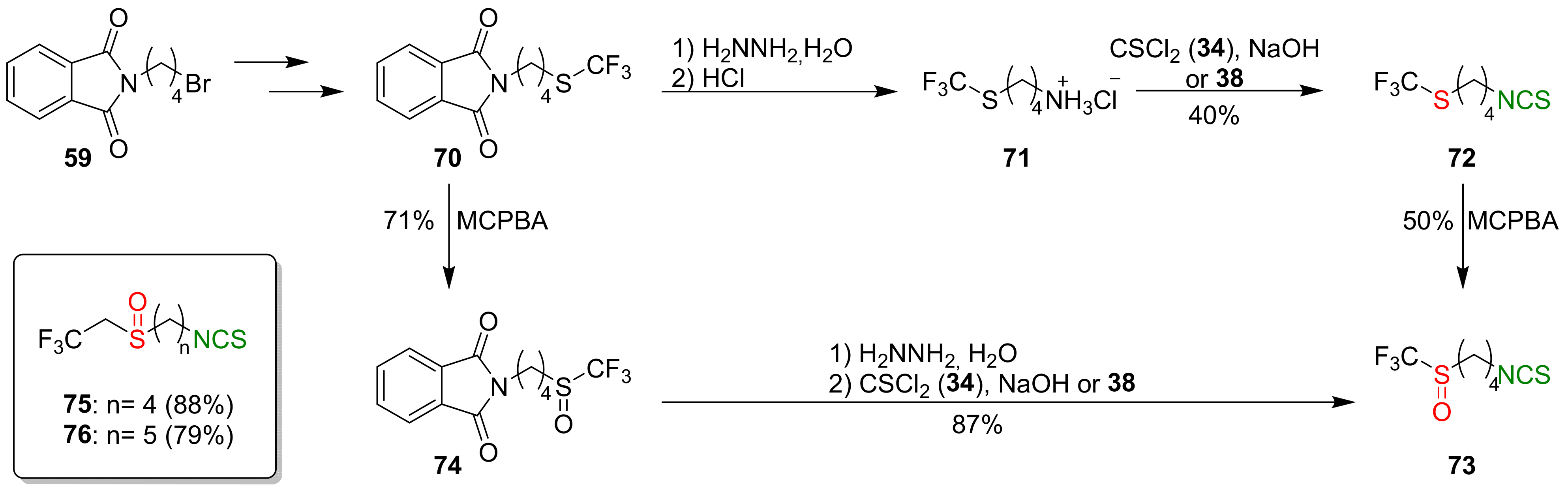{kind=link}
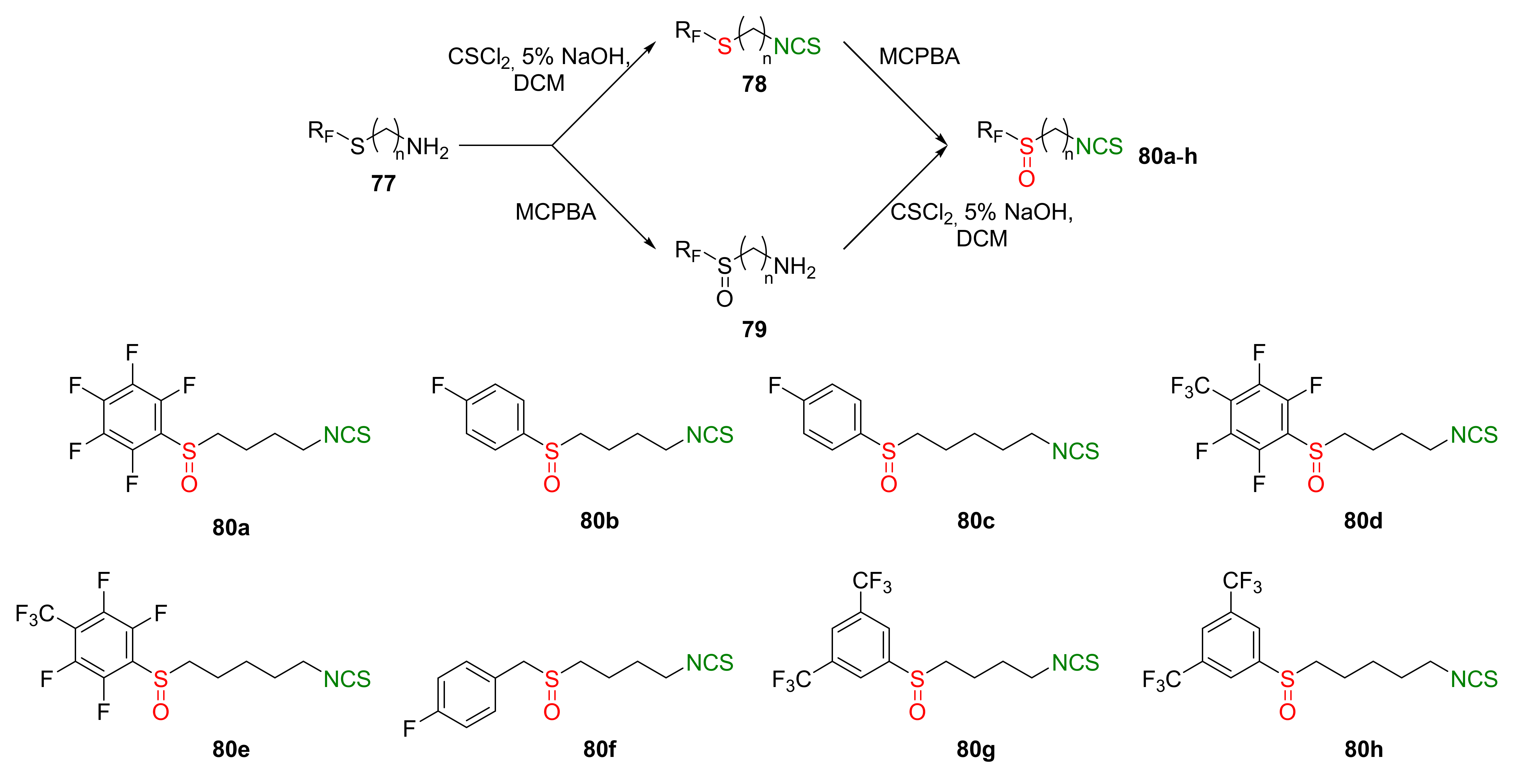{kind=link}
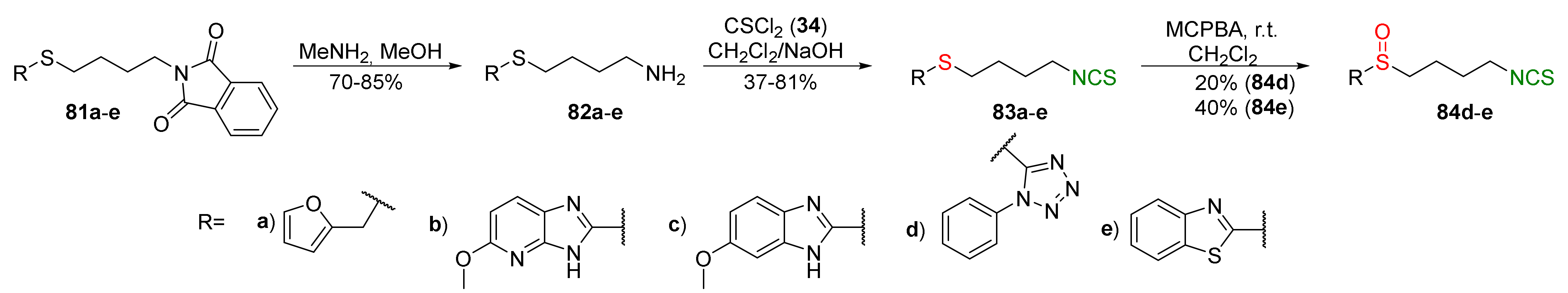{kind=link}
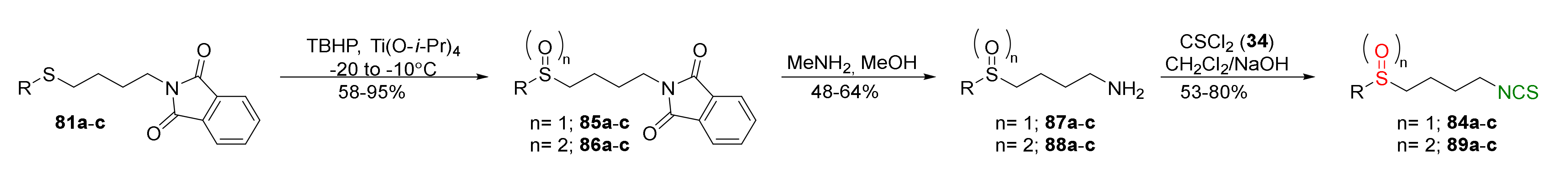{kind=link}
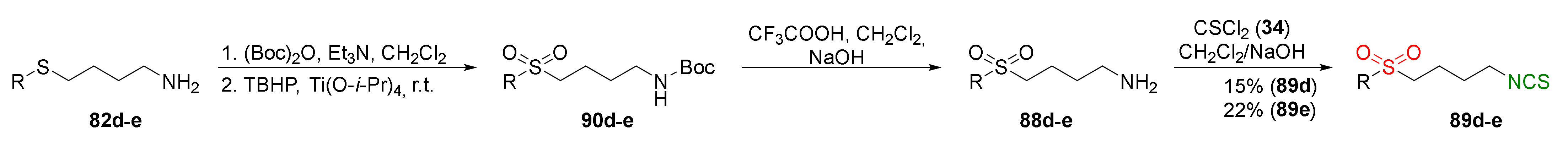{kind=link}
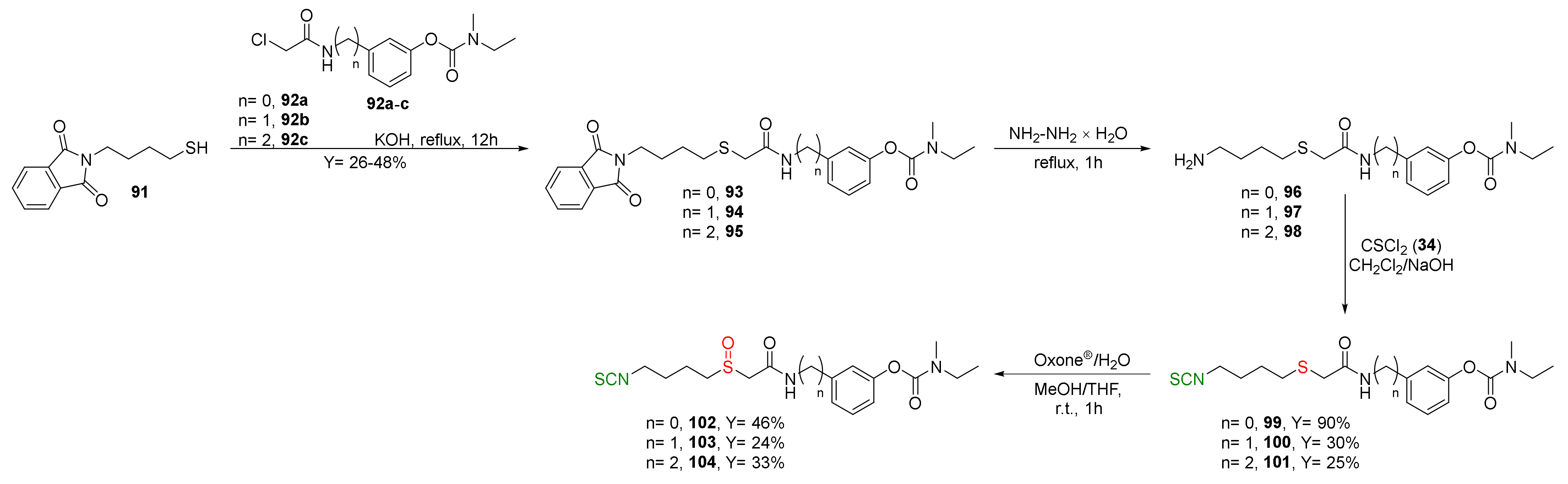{kind=link}
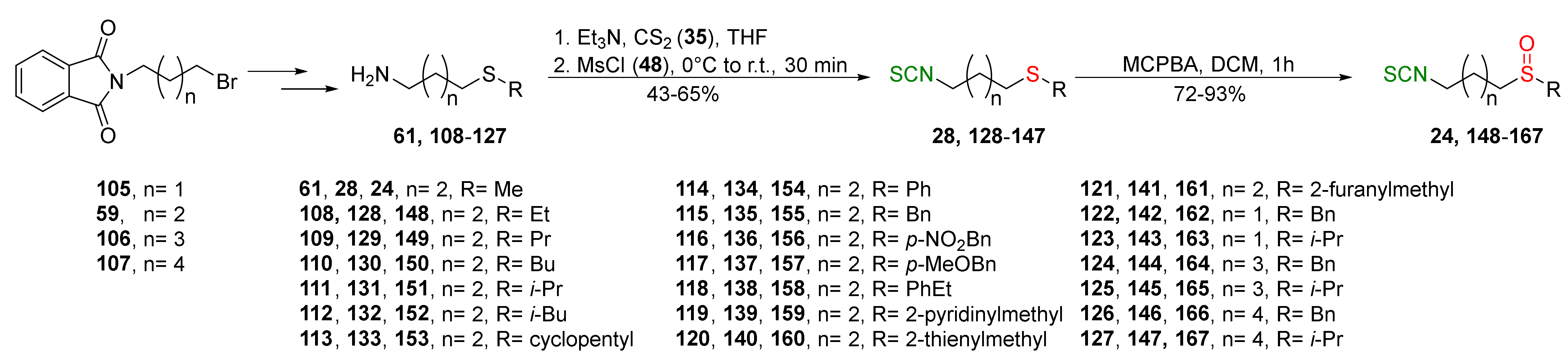{kind=link}
{kind=link}
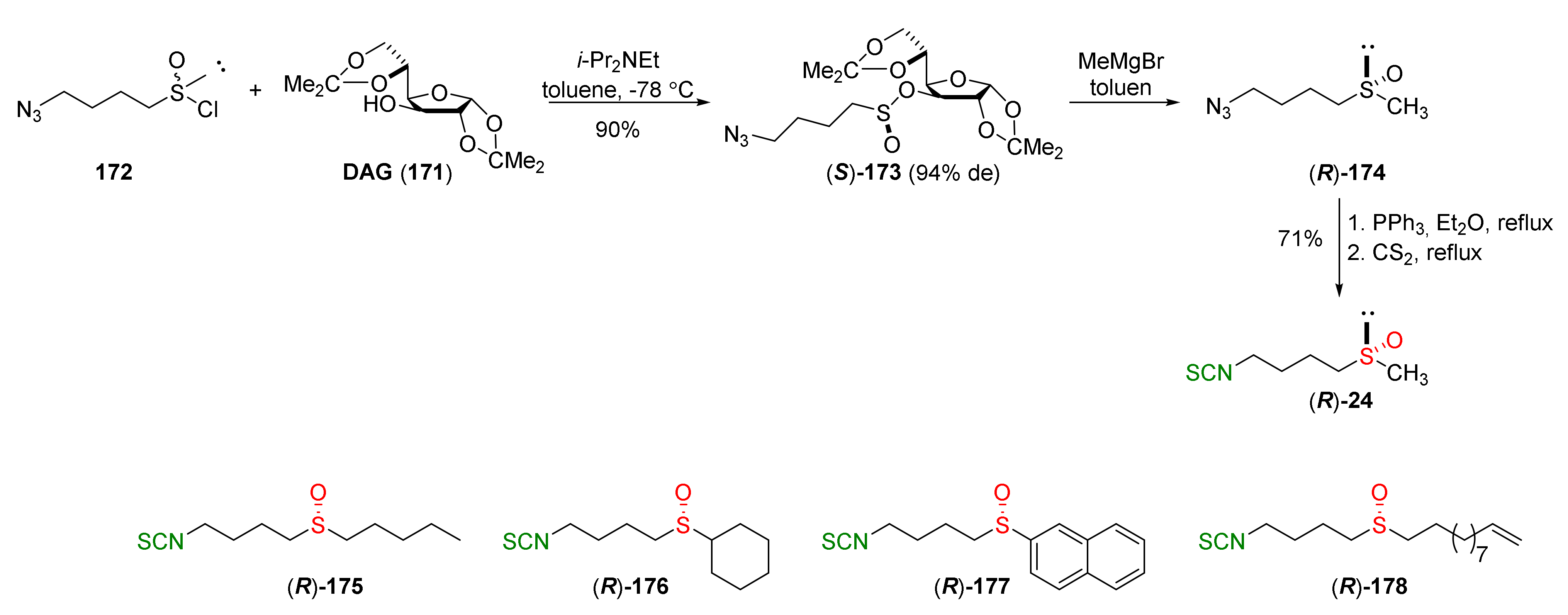{kind=link}
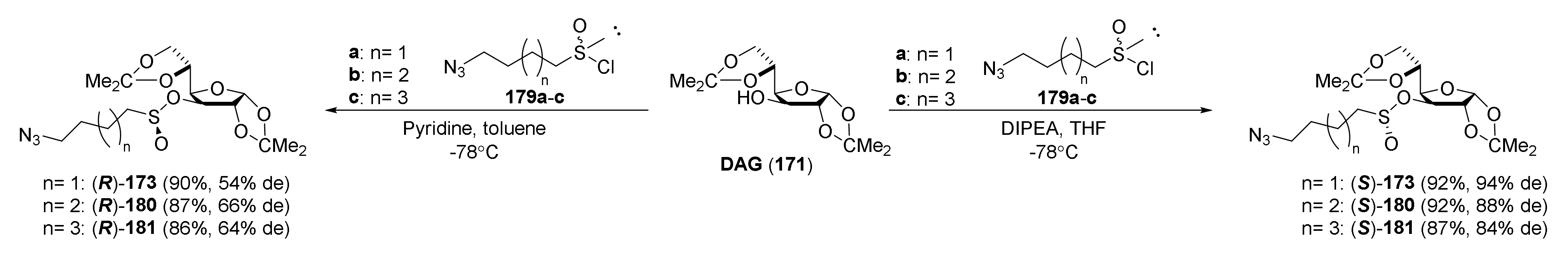{kind=link}
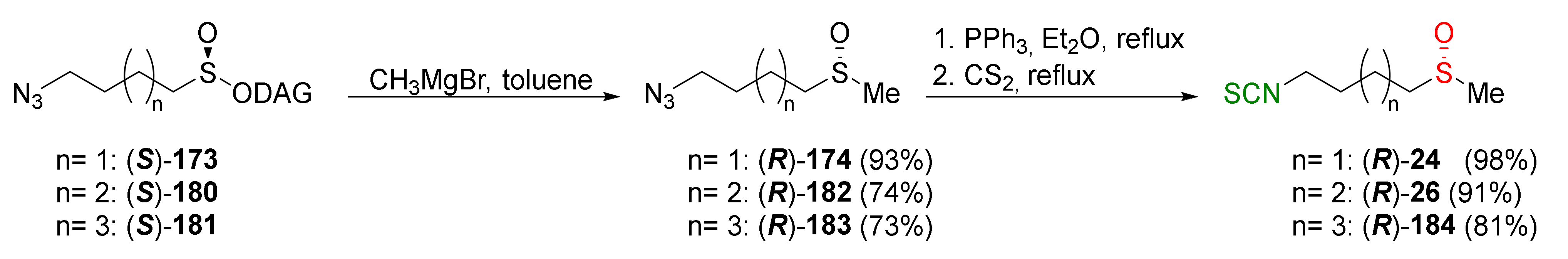{kind=link}
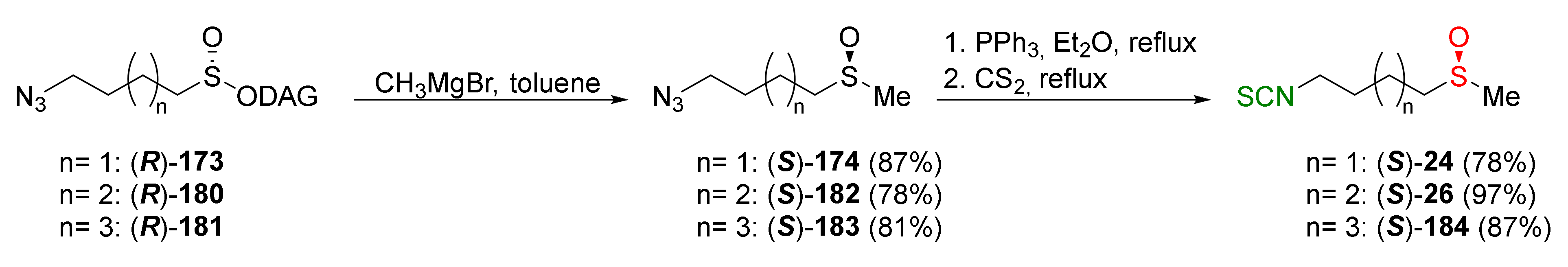{kind=link}
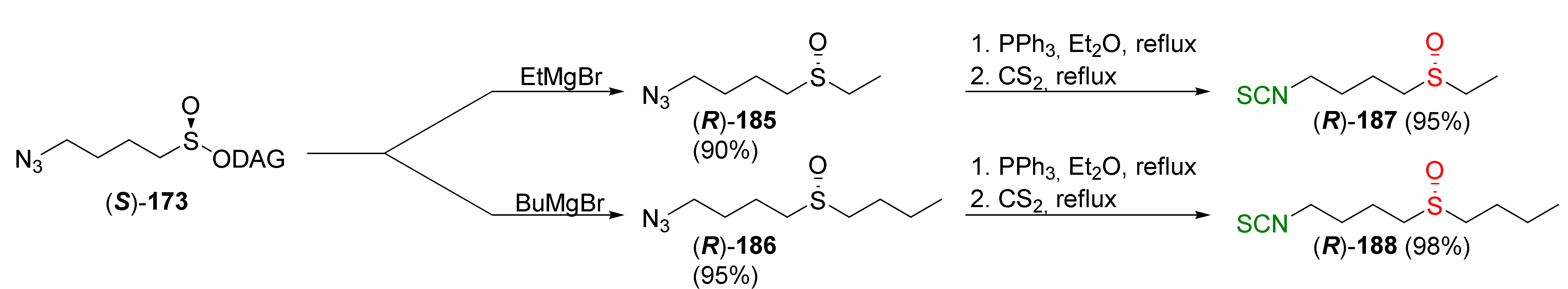{kind=link}
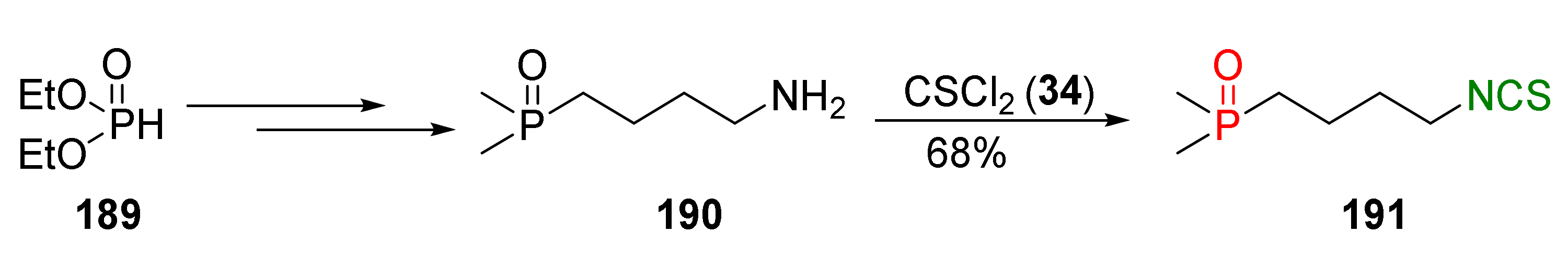{kind=link}
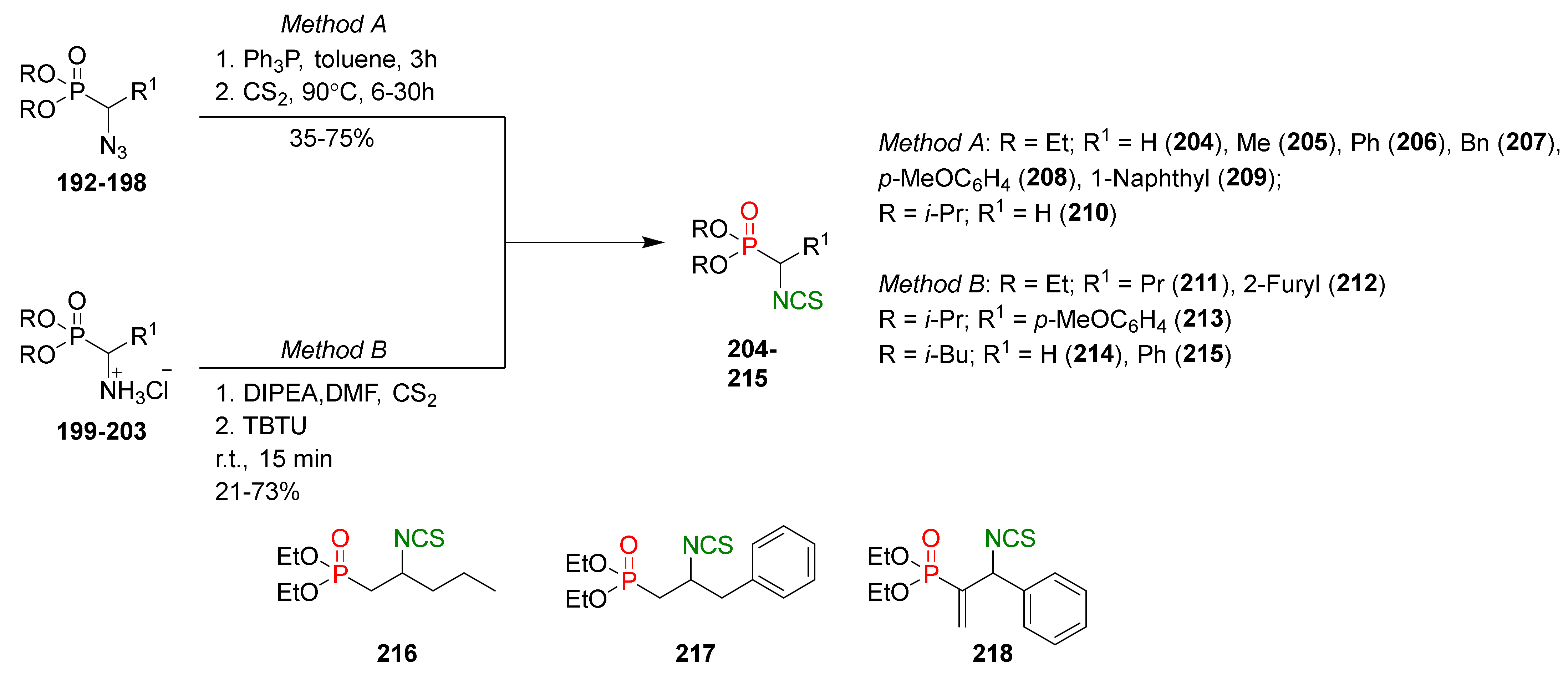{kind=link}
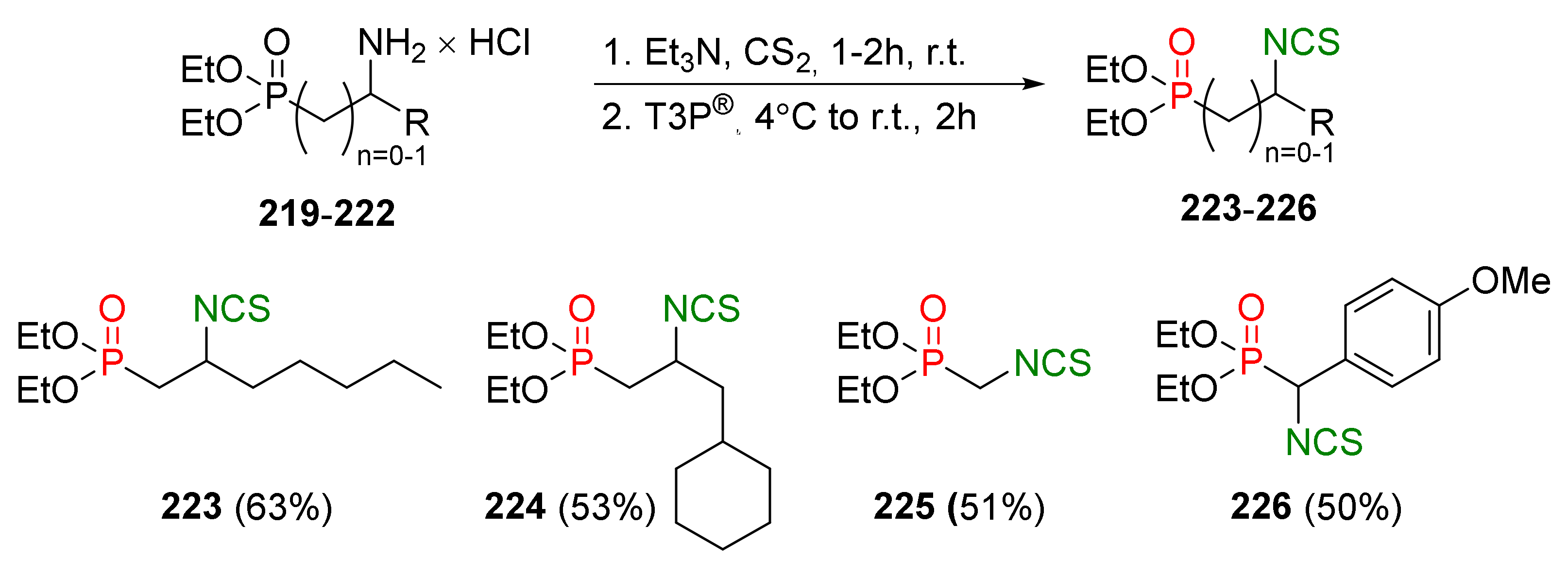{kind=link}
{kind=link}
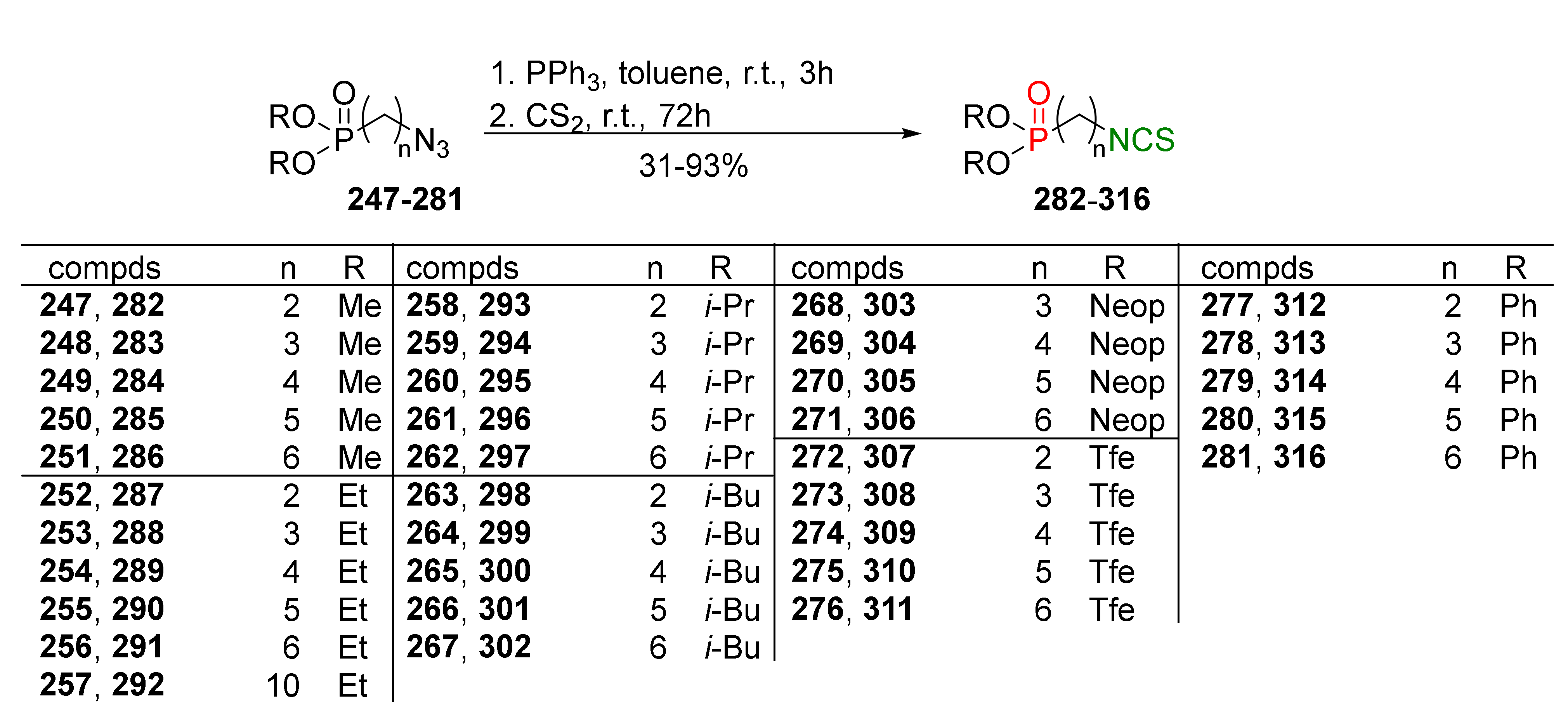{kind=link}
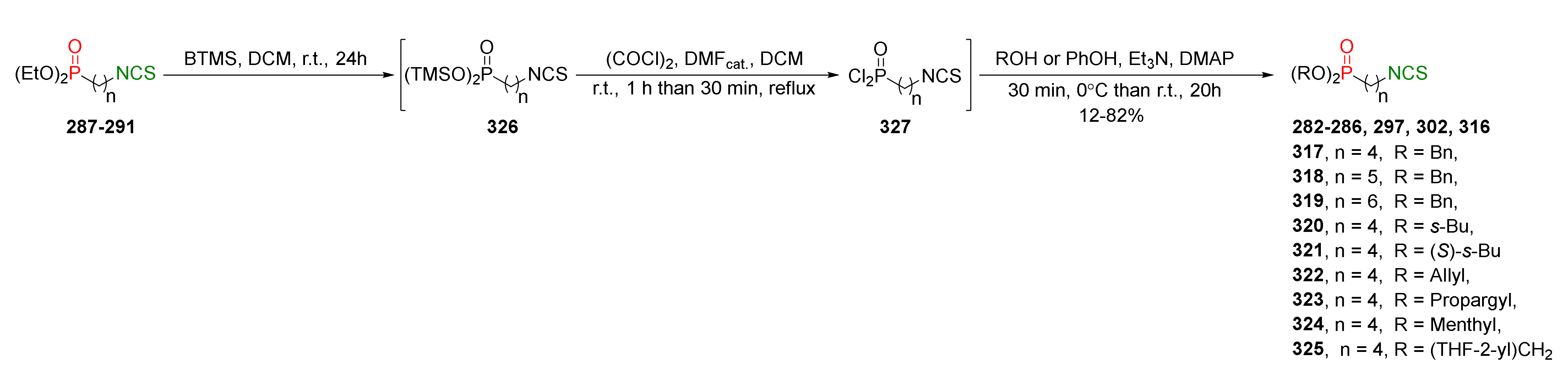{kind=link}
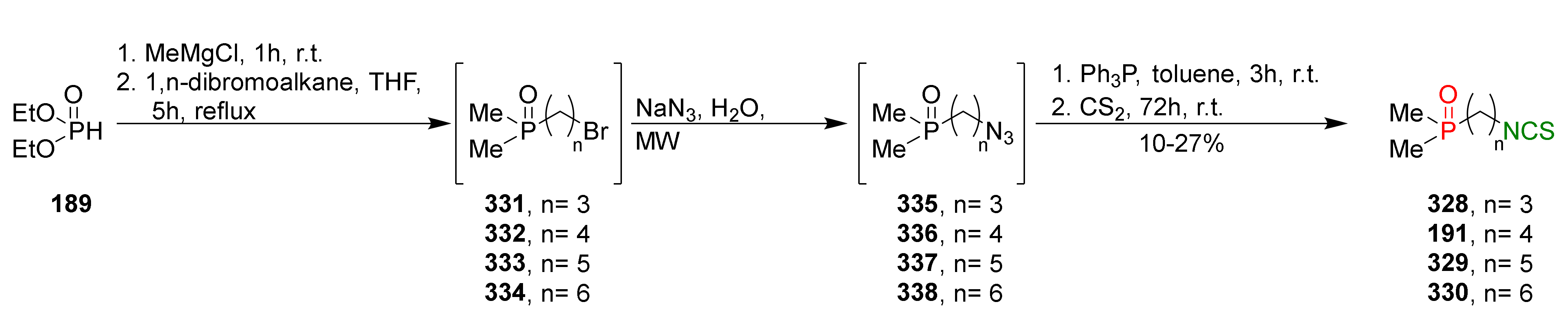{kind=link}
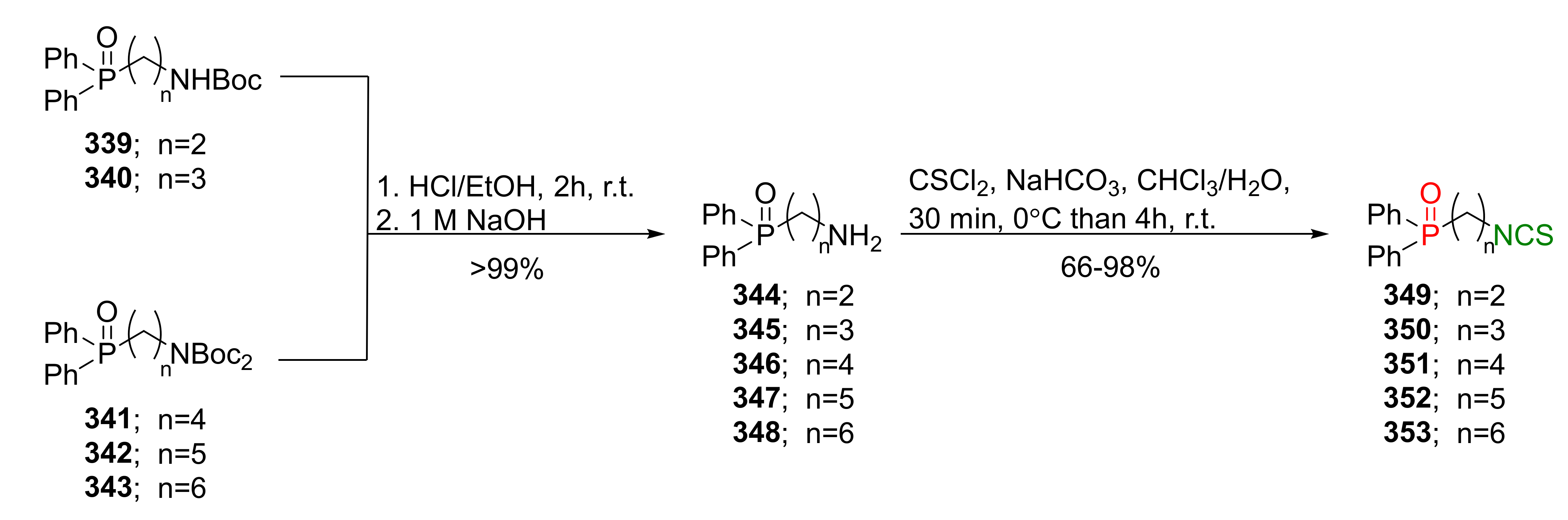{kind=link}
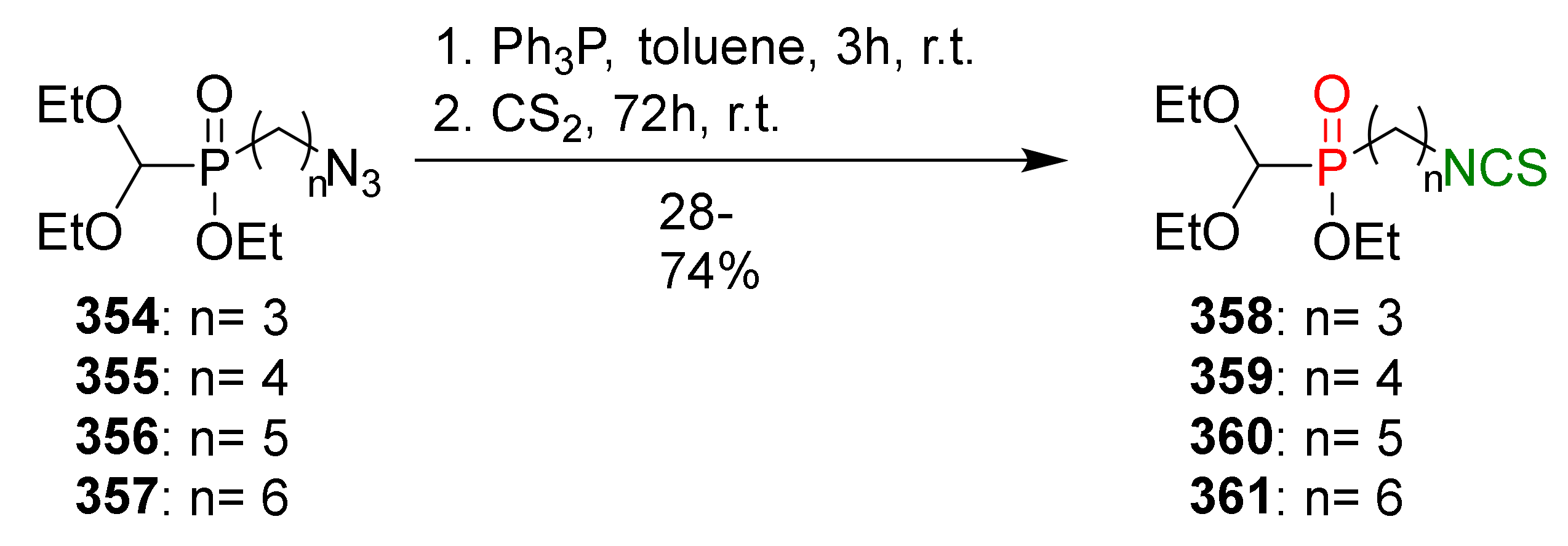{kind=link}
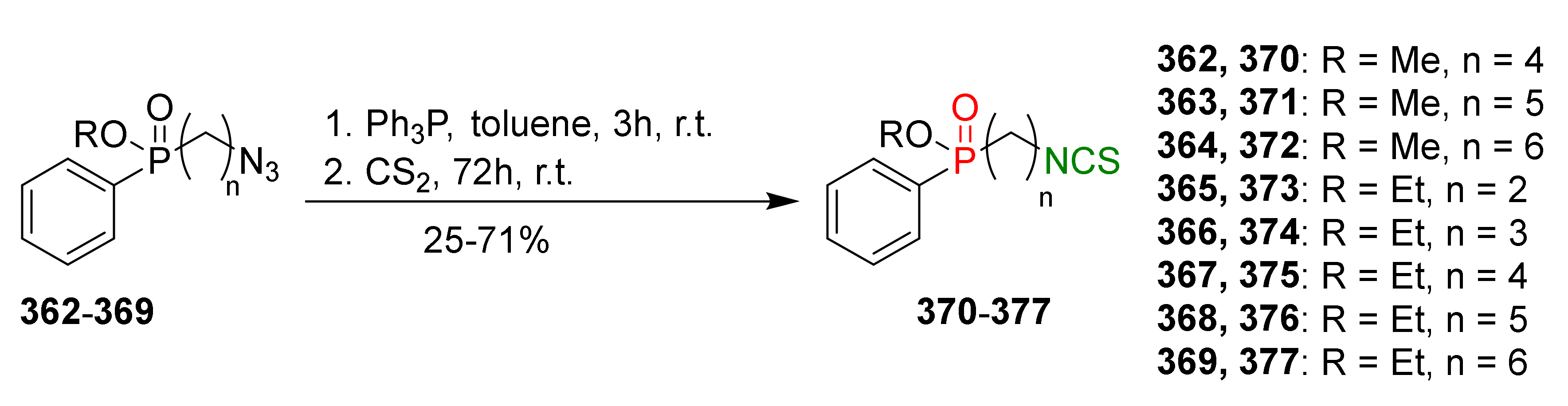{kind=link}
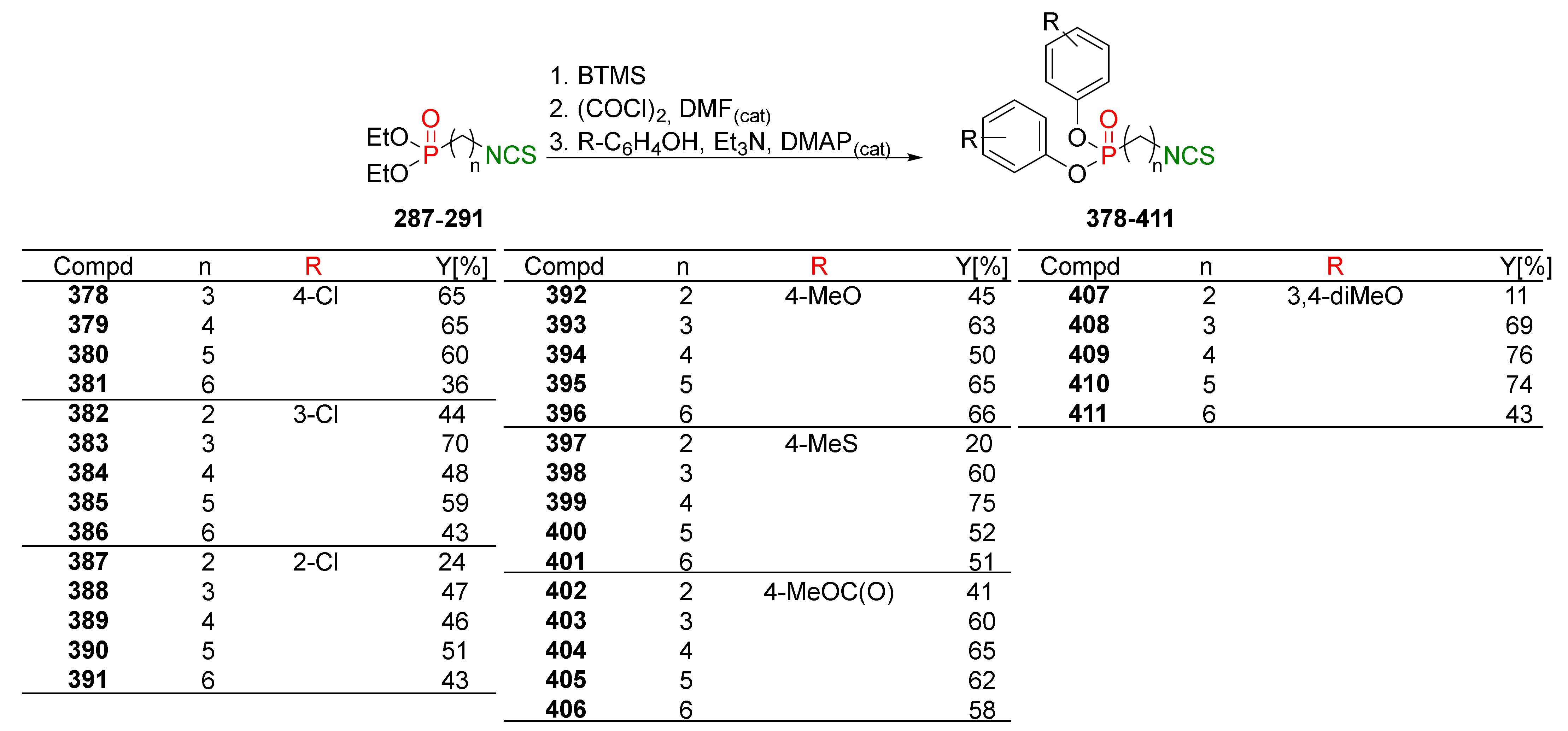{kind=link}
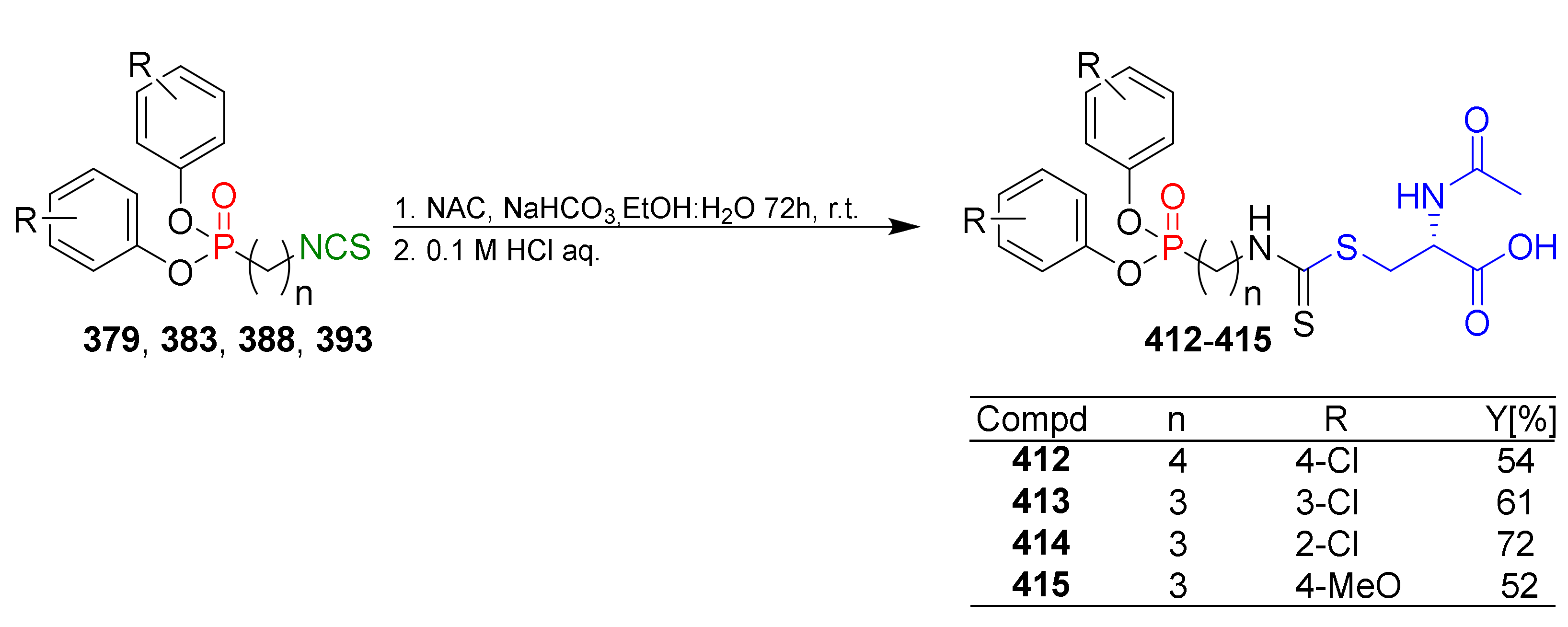{kind=link}
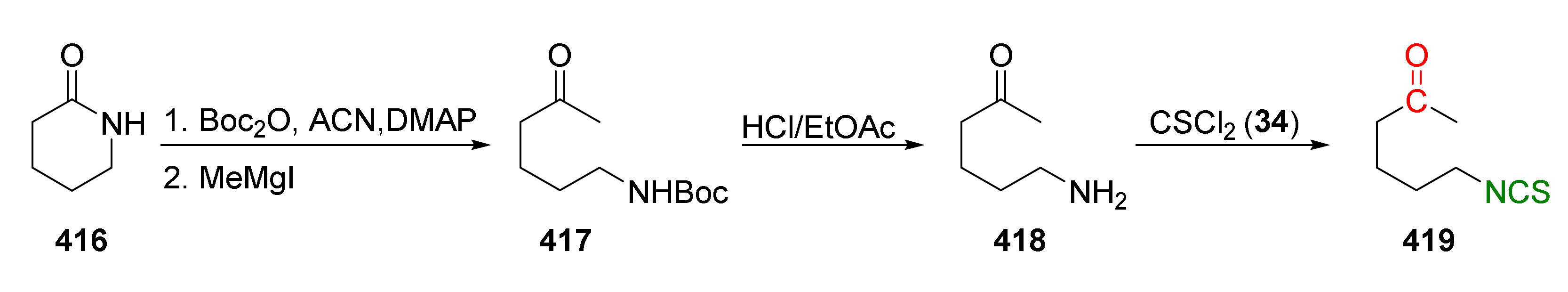{kind=link}
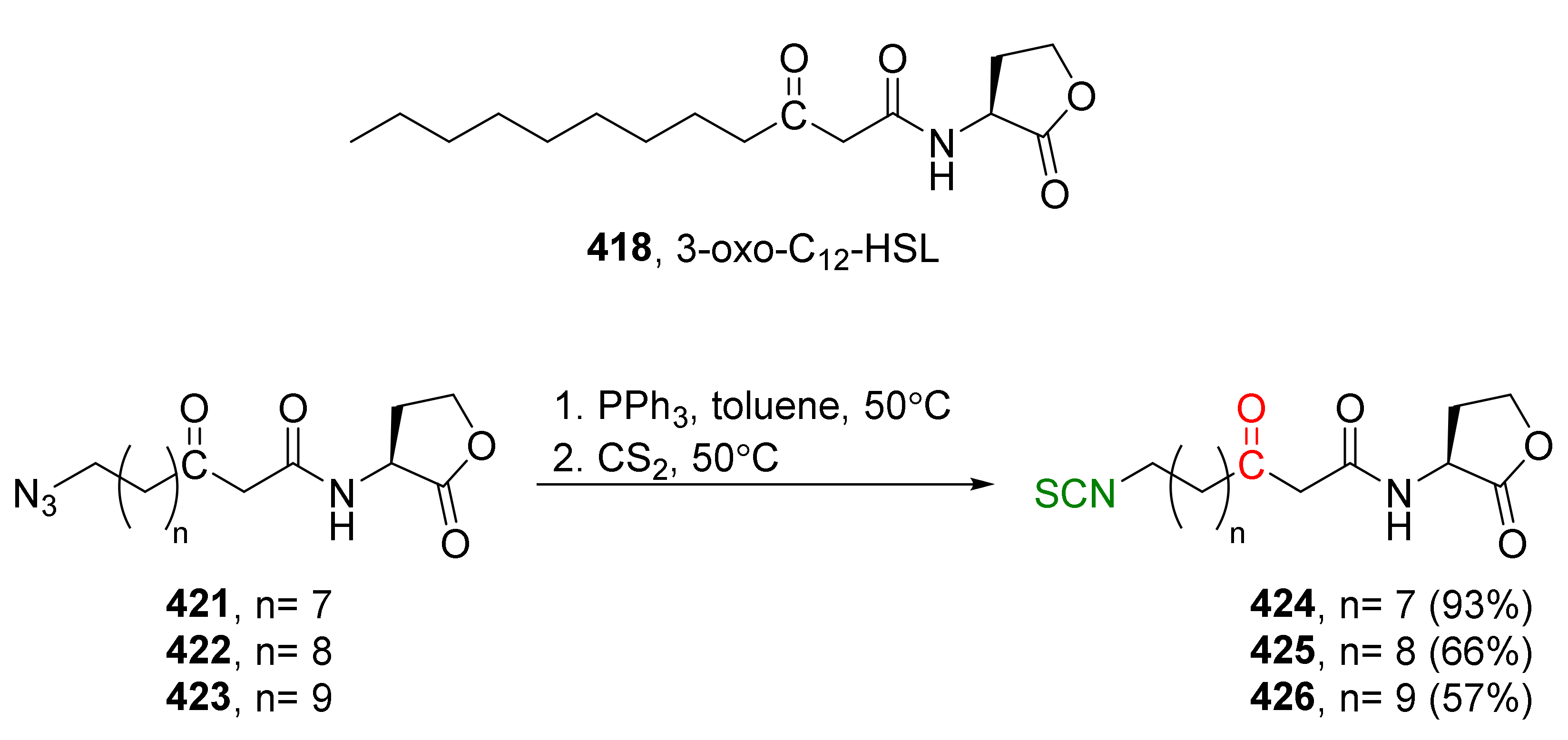{kind=link}
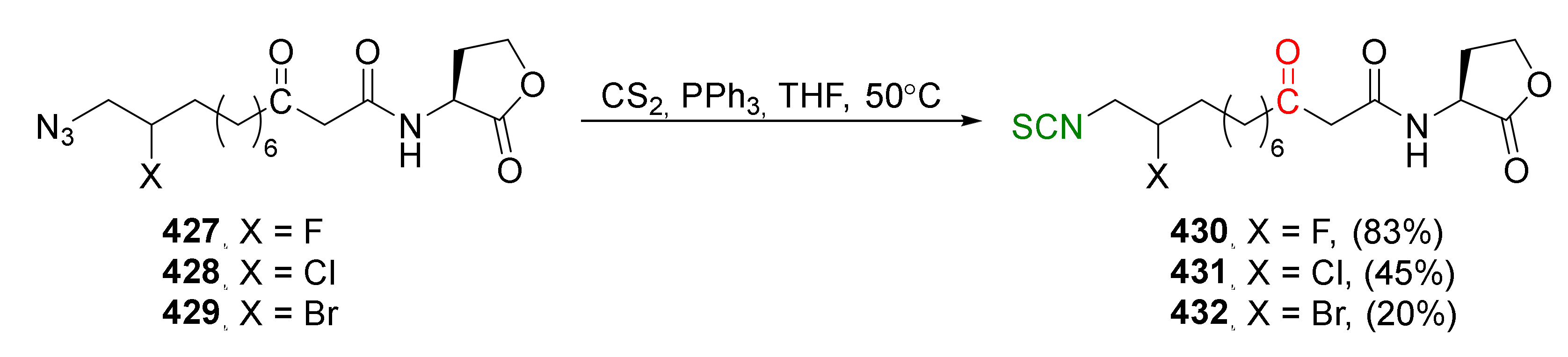{kind=link}
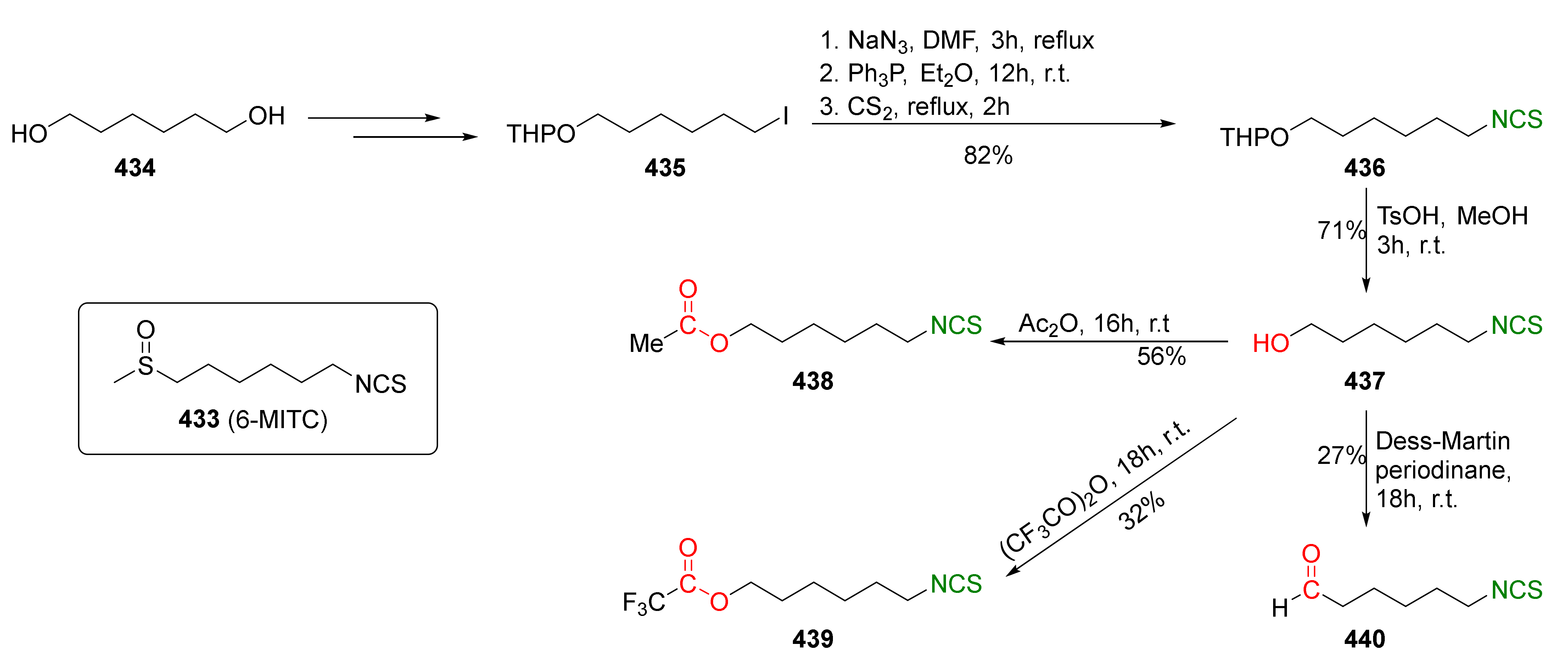{kind=link}
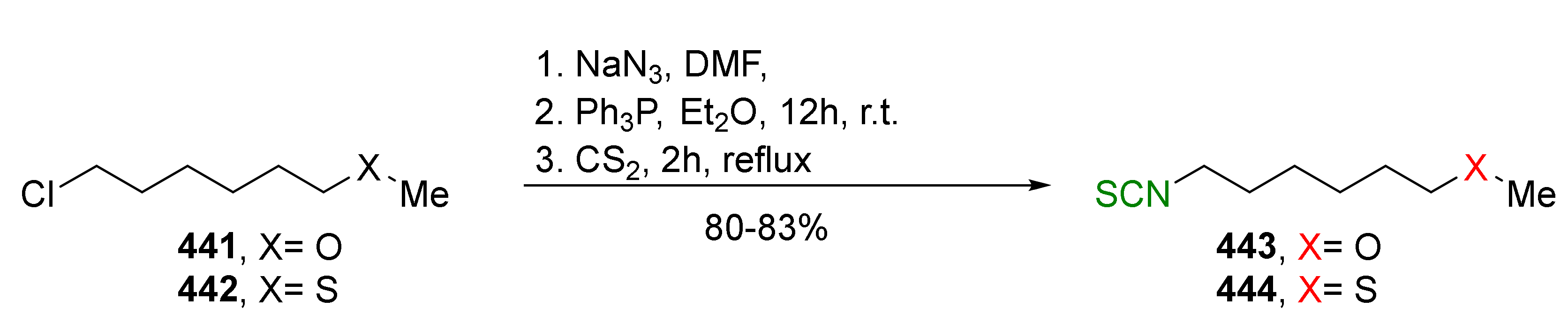{kind=link}
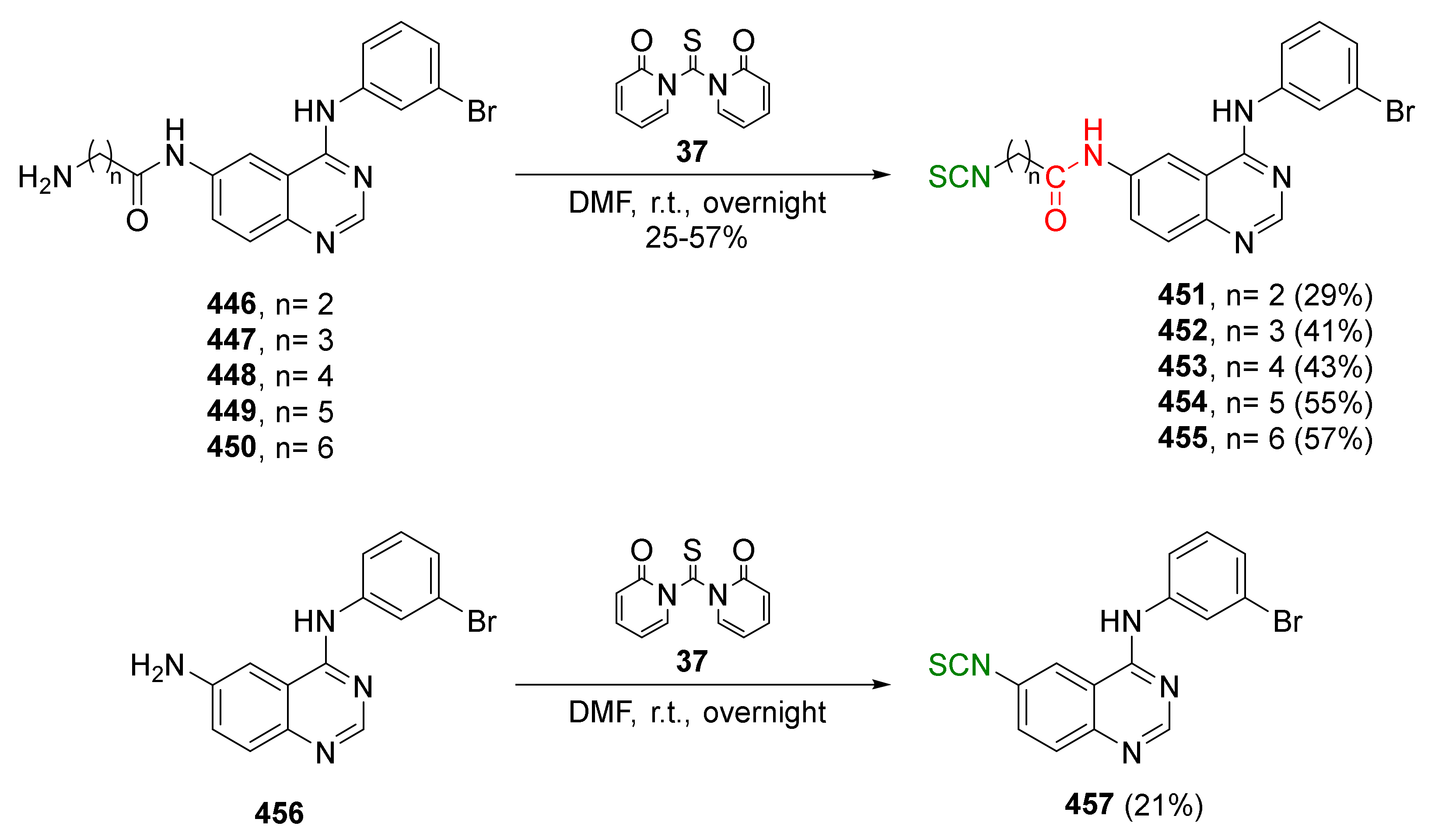{kind=link}
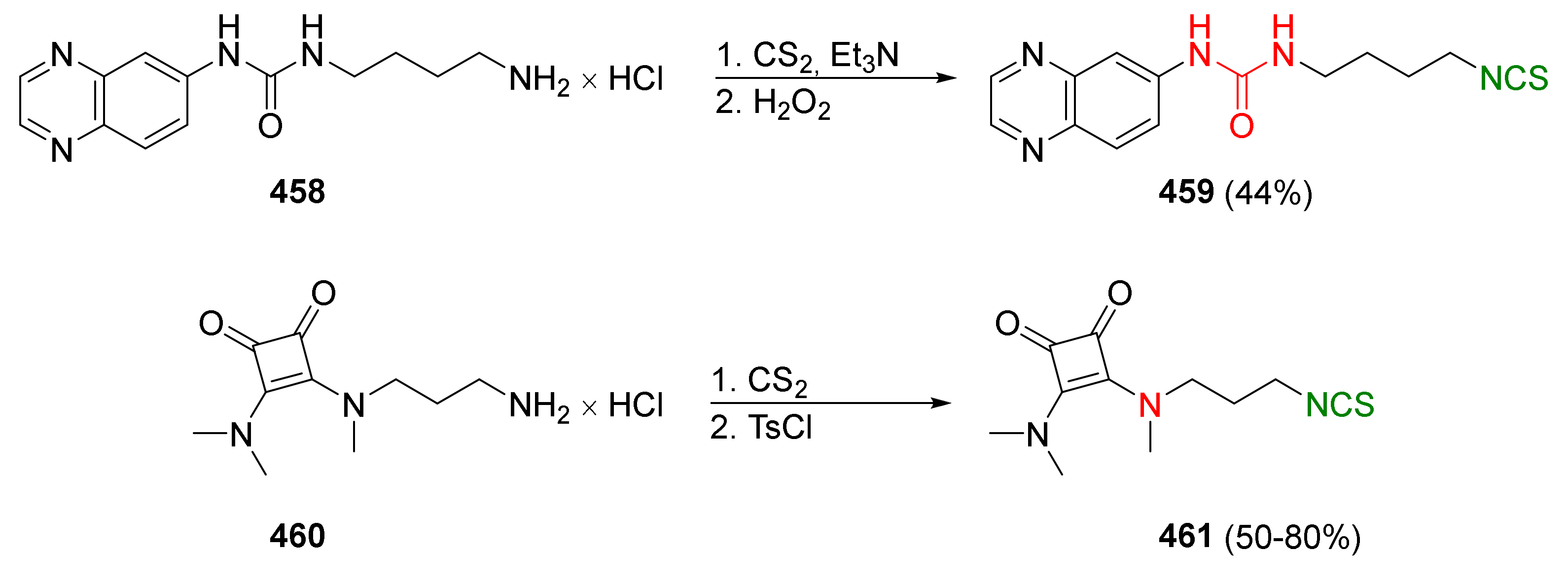{kind=link}
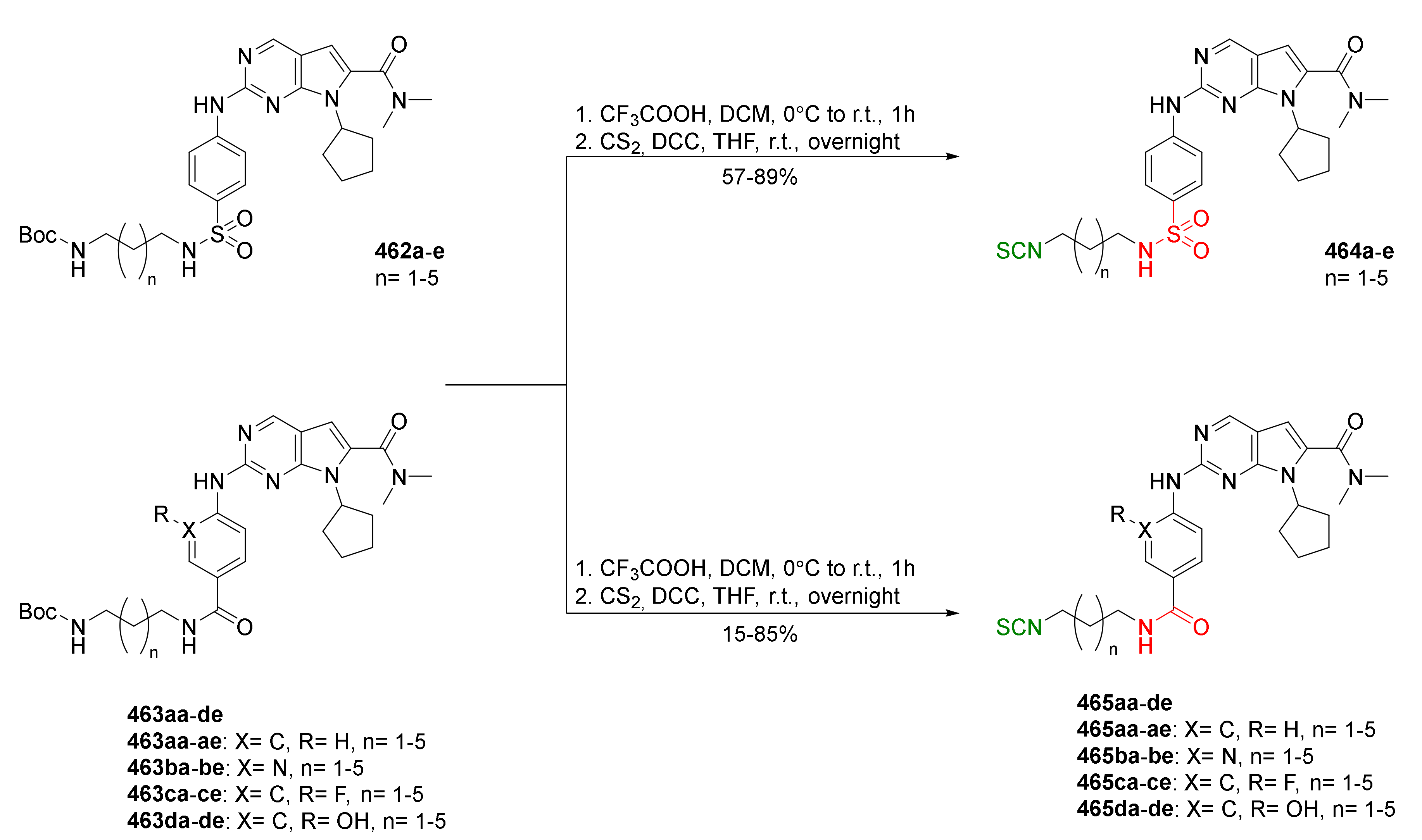{kind=link}
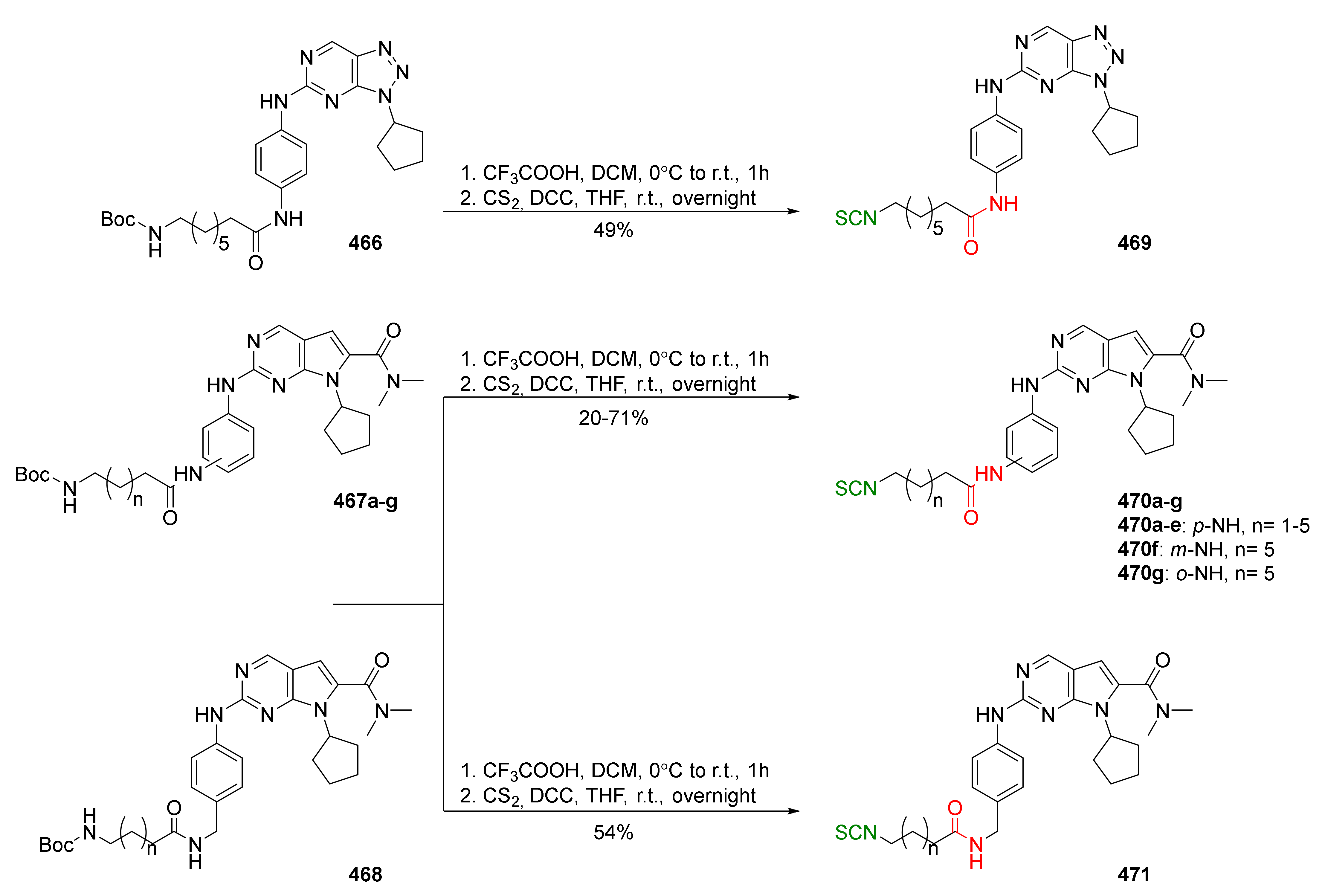{kind=link}
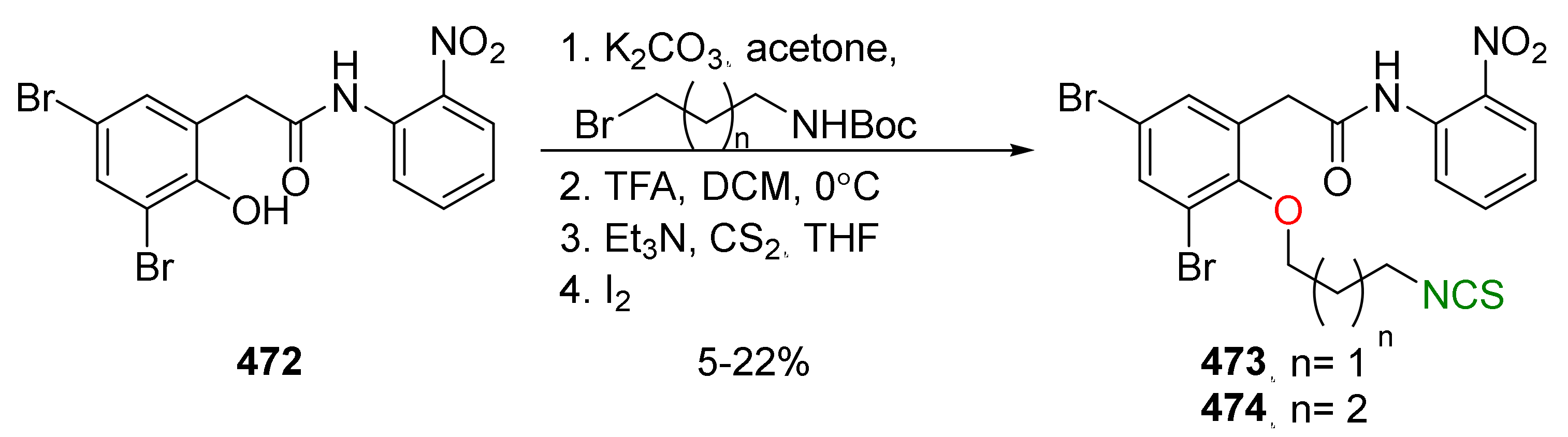{kind=link}
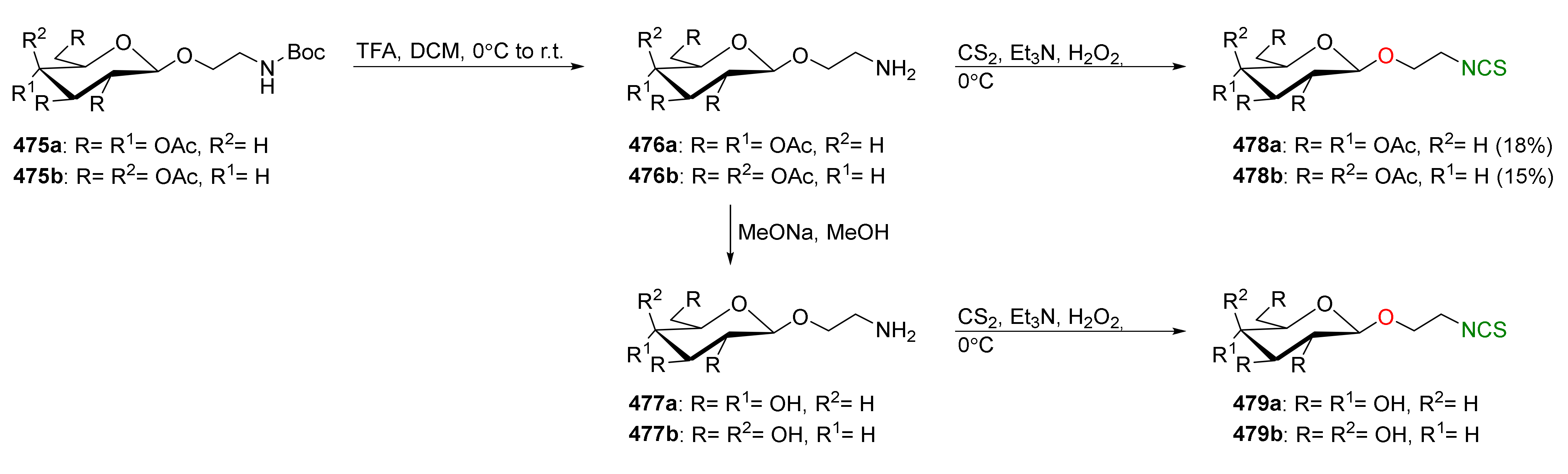{kind=link}
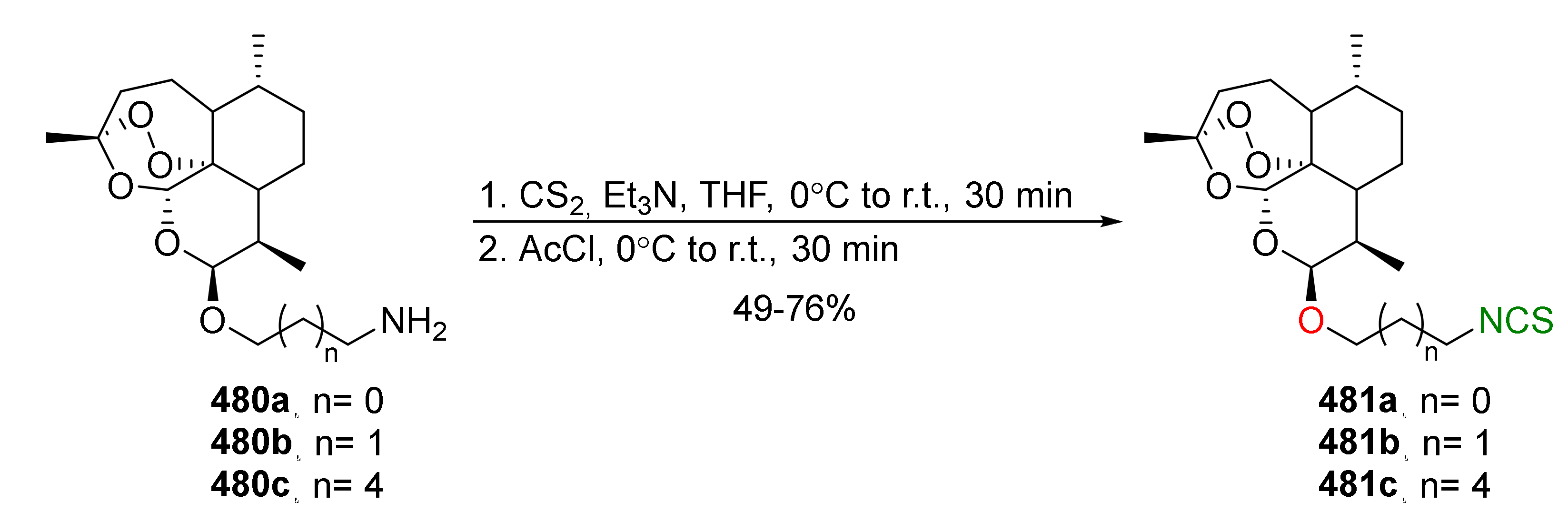{kind=link}
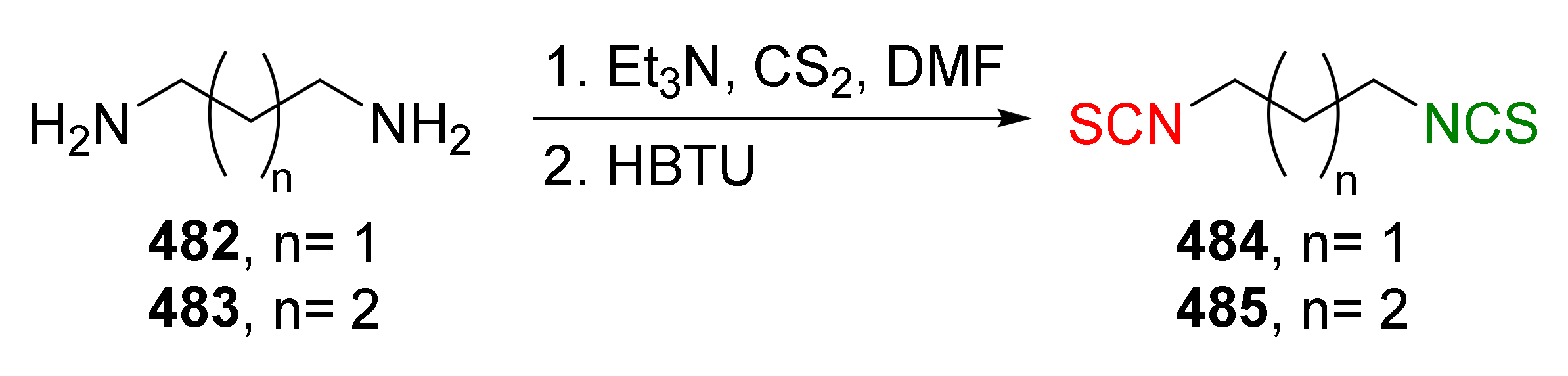{kind=link}
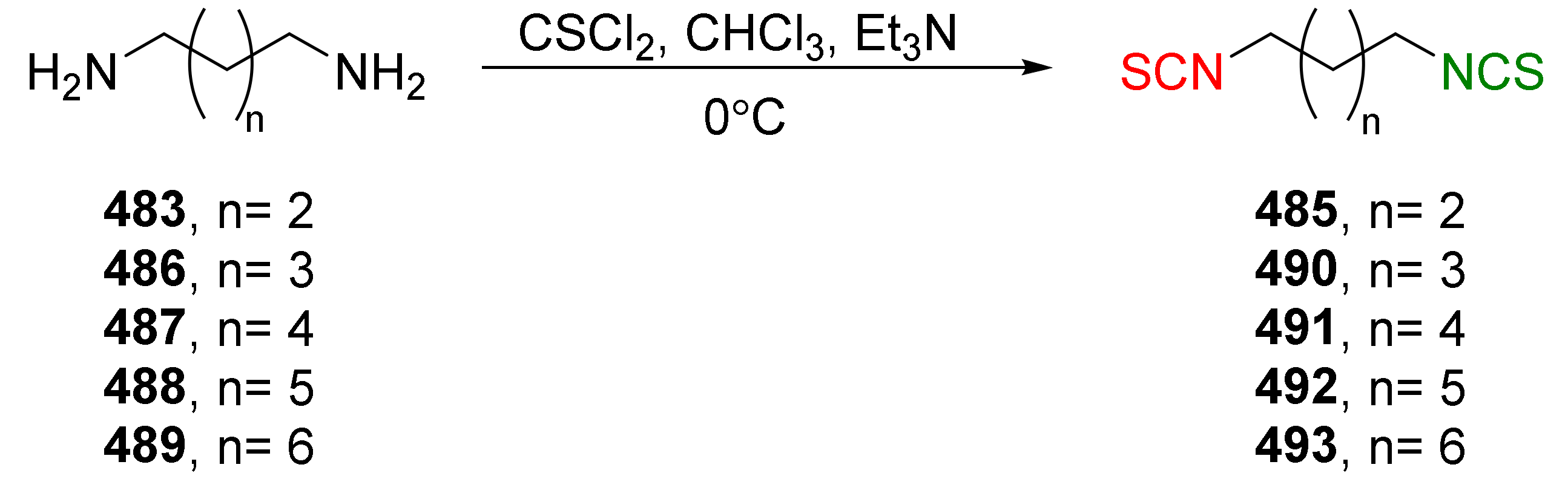{kind=link}
Table 1.
Cytotoxicity of SFN (24), erucin (28), and erysolin (31).
| Compound | Low IC50 (μM) | High IC50 (μM) | IC50 (NmuMG) (μM) |
|---|---|---|---|
| SFN (24) | 2.8 ± 0.1 (Hep3B) | 16.5 ± 1.4 (NCI/ADR RES) | 3.2 ± 0.2 |
| Erucin (28) | 8.9 ± 0.4 (SF-268) | 45.3 ± 4.9 (HCI-H460) | 23.5 |
| Erysolin (31) | 2.3 ± 0.8 (MCF-7) | 11.1 ± 0.6 (NCI/ADR RES) | 5.3 ± 0.4 |
Table 2.
Cytotoxicity of SFN and its fluorine-containing analogs 75.
| Compound | Malme-3M IC50 (μM) | Malme-3 IC50 (μM) |
|---|---|---|
| (R)-SFN | 25 | 38 |
| (S)-SFN | 30 | 26 |
| (R)-75 | 27 | 37 |
| (S)-75 | 25 | 35 |
Table 3.
The IC50 (μM) for 80d and 80e and SFN after 72 h of incubation (mean ± SD).
| IC50 (μM) ± SD | ||||
|---|---|---|---|---|
| Compound | MALME-3M | HT-29 | MCF-7 | MDA-MB-231 |
| 80d | 2.7 ± 0.7 | 1.2 ± 0.1 | 0.9 ± 0.1 | 0.5 ± 0.1 |
| 80e | 4.3 ± 0.7 | 1.4 ± 0.2 | 0.7 ± 0.1 | 1.2 ± 0.1 |
| SFN | 33.7 ± 0.6 | 11.4 ±0.1 | 11.9 ± 2.0 | 11.3 ± 0.7 |
Table 4.
Inhibitory effects of tetrazole analogues of SFN 83d, 84d, and 89d and SFN on MCF-7, SUM-159, and KG-1a.
Table 4.
Inhibitory effects of tetrazole analogues of SFN 83d, 84d, and 89d and SFN on MCF-7, SUM-159, and KG-1a.
| Compound | MCF-7 IC50 (μM) | SUM-159 IC50 (μM) | KG-1a IC50 (μM) |
|---|---|---|---|
| SFN | 24.11 ± 6.62 | 7.69 ± 0.92 | 8.24 ± 2.81 |
| 83d | 2.66 ± 0.25 | 1.46 ± 0.19 | 1.52 ± 0.38 |
| 84d | 4.11 ± 0.9 | 1.54 ± 0.29 | 0.51 ± 0.14 |
| 89d | 1.66 ± 0.23 | 2.08 ± 0.24 | 0.88 ± 0.28 |
Table 5.
The cytotoxicity of SFN and its derivatives.
| Compound | HepG2 IC50 (μM) | A549 IC50 (μM) | MCF-7 IC50 (μM) | HCT-116 IC50 (μM) | SH-SY5Y IC50 (μM) |
|---|---|---|---|---|---|
| SFN (24) | 14.05 | 21.99 | 17.66 | 11.59 | 13.72 |
| 2.05 (151) | 5.64 (161) | 3.3 (155) | 2.06 (152) | 2.79 (157) | |
| 135 | 12.56 | 51.34 | 38.28 | 41.45 | 20.71 |
 | 8.49 | 8.89 | 7.55 | 6.28 | 4.78 |
Table 6.
Selectively of (S)-SFN and (S)-184.
| Compound | A549 IC50 (μM) | MRC-5 IC50 (μM) |
|---|---|---|
| (S)-SFN ((S)-24) | 19.60 | 46.58 |
| (S)-184 | 7.54 | 17.58 |
Table 7.
Effects of SFN and its analogues 191 on inducer potency for QR in Hepa 1c1c7.
| Compound | CD (μM) |
|---|---|
| SFN (24) | 0.2 |
| 191 | 0.4 |
Table 8.
The antiproliferative activity of compound 246.
| IC50 (μM) ± SD | |||
|---|---|---|---|
| LoVo | LoVo/DX | A549 | MCF-7 |
| 7 ± 1 | 8 ± 1 | 31 ± 2 | 20 ± 1 |
Table 9.
Antiproliferative activity of isopropyl and isobutyl ω-(isothiocyanato)alkylphosphonates 293–302.
Table 9.
Antiproliferative activity of isopropyl and isobutyl ω-(isothiocyanato)alkylphosphonates 293–302.
| Compound | LoVo IC50 (μM) | LoVo/DX IC50 (μM) | Compound | LoVo IC50 (μM) | LoVo/DX IC50 (μM) |
|---|---|---|---|---|---|
| 293 | 2.7 ± 0.4 | 3.6 ± 0.9 | 298 | 1.9 ± 0.4 | 2.6 ± 0.4 |
| 294 | 2.6 ± 0.2 | 3.3 ± 0.9 | 299 | 2.4 ± 0.3 | 5.6 ± 3.2 |
| 295 | 2.5 ± 0.6 | 5.0 ± 3.7 | 300 | 3.3 ± 0.2 | 10.4 ± 1.4 |
| 296 | 2.6 ± 0.1 | 7.9 ± 1.6 | 301 | 2.4 ± 0.5 | 5.1 ± 4.4 |
| 297 | 2.7 ± 0.2 | 9.4 ± 1.6 | 302 | 2.7 ± 0.4 | 9.2 ± 1.4 |
| SFN | 22.9 ± 2.0 | 18.1 ± 3.0 |
Table 10.
Effect of SFN and its analogue 419 on the inducer potency for QR in Hepa 1c1c7.
| Compound | CD (μM) |
|---|---|
| SFN (24) | 0.2 |
| 419 | 0.2 |
Table 11.
In vitro NO production and tumor growth inhibitory activities of the most active ITCs 433, 437–439, 443, and thiocyanate 445.
Table 11.
In vitro NO production and tumor growth inhibitory activities of the most active ITCs 433, 437–439, 443, and thiocyanate 445.
| Compounds | Inhibition (IC50, μM) | |
|---|---|---|
| NO Production | Growth | |
| 433 (6-MITC) | 5.7 ± 0.5 | 8.0 ± 0.6 |
| 437 | 6.0 ± 1.2 | 4.4 ± 0.3 |
| 438 | 6.6 ± 1.2 | 4.1 ± 0.2 |
| 439 | 9.1 ± 1.0 | 5.6 ± 0.4 |
| 443 | 11.5 ± 5.9 | 8.1 ± 0.6 |
 | > 200 | > 200 |
Table 12.
Summary of the synthetic routes of SFN and its bifunctional analogs.
| Reaction | Reference |
|---|---|
| Synthesis of SFN and its sulfur analogues | |
 | [239,240,241,242,243,244,245,246,247] |
 | [249] |
 | [163,250,252] |
| Synthesis of phosphorous analogues of SFN | |
 | [188,257] |
 | [200,203,253] |
 | [200,254,257] |
| Synthesis of carbonyl and amide analogues of SFN | |
 | [188,263] |
 | [264,265] |
 | [260,261,262] |
| Synthesis of ether-linked analogues of SFN | |
 | [266,267,268] |
| Synthesis of diisothiocyanates | |
 | [269] |
 | [55] |
Table 13.
Summary of the relationship between the biological activities of the analogs of SFN and various functional groups.
Table 13.
Summary of the relationship between the biological activities of the analogs of SFN and various functional groups.
| Compound | Anticancer Activity | Reference | Compound | Anticancer Activity | Reference |
|---|---|---|---|---|---|
 | Malme-3M↑ Malme-3↓ | [244] |  | Malme-3M↑ HT-29↑ MCF-7↑ MDA-MB-231↑ | [245] |
 | MCF-7↑ SUM-159↑ KG-1a↑ | [246] |  | HepG2↑ A549↑ MCF-7↑ HCT-116↑ SH-SY5Y↑ | [249] |
 | CD↔ | [188] |  | LoVo↑ LoVo/DX↑ | [254] |
 | LoVo↑ LoVo/DX↑ | [257] |  | LoVo↑ LoVo/DX↑ | [258] |
 | CD↔ | [188] |  | A431↔ | [263] |
 | A549↑ H1299↑ MCF-7↑ MDA-MB-231↑ HepG2↑ Hela↑ | [264] |  | LoVo↑ LoVo/DX↑ | [269] |
↑—more active than SFN; ↓—less active than SFN; ↔—similar active to SFN.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Janczewski, Ł. Sulforaphane and Its Bifunctional Analogs: Synthesis and Biological Activity. Molecules 2022, 27, 1750. https://doi.org/10.3390/molecules27051750
AMA Style
Janczewski Ł. Sulforaphane and Its Bifunctional Analogs: Synthesis and Biological Activity. Molecules. 2022; 27(5):1750. https://doi.org/10.3390/molecules27051750
Chicago/Turabian StyleJanczewski, Łukasz. 2022. "Sulforaphane and Its Bifunctional Analogs: Synthesis and Biological Activity" Molecules 27, no. 5: 1750. https://doi.org/10.3390/molecules27051750