
An Orthogonal Synthetic Approach to Nonsymmetrical Bisazolyl 2,4,6-Trisubstituted Pyridines


 and
and
Abstract
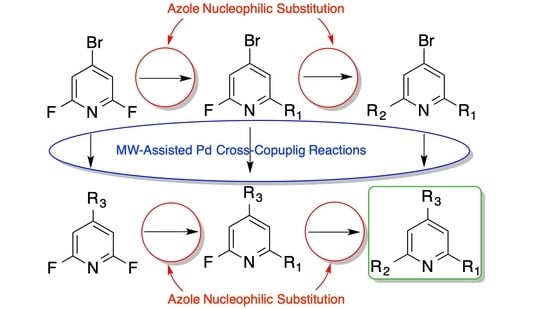
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Indazole Pyrazole Pyridine (ippy)
2.2. Bipyrazole Pyridine (bppy)
2.3. Biindazole Pyridine (bipy)
3. Conclusions and Outlook
4. Materials and Methods
4.1. General Methods
4.2. General Synthetic Procedures
4.2.1. Substitution of Fluorine Atoms by Pyrazolates or Indazolates: Method A: NaH in THF
4.2.2. Substitution of Fluorine Atoms by Pyrazolates or Indazolates: Method B: NaH in DMF
4.2.3. Substitution of Fluorine Atoms by Pyrazolates or Indazolates: Method C: K2CO3 in DMF and Microwave Radiation
4.2.4. Microwave-Assisted Suzuki-Miyaura Cross-Coupling Reaction with Boronic Acids
4.2.5. Microwave-Assisted Sonogashira Cross-Coupling Reaction with Phenylacetylene
4.3. Chemical Synthesis and Characterization
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Heller, M.; Schubert, U.S. Syntheses of Functionalized 2,2′:6′,2′′-Terpyridines. Eur. J. Org. Chem. 2003, 947–961. [Google Scholar]
- Campagna, S.; Puntoriero, F.; Nastasi, F.; Bergamini, G.; Balzani, V. Photochemistry and photophysics of coordination compounds: Ruthenium. Top. Curr. Chem. 2007, 280, 117–214. [Google Scholar]
- Pal, A.K. Hanan, Design, synthesis and excited-state properties of mononuclear Ru(ii) complexes of tridentate heterocyclic ligands. G.S. Chem. Soc. Rev. 2014, 43, 6184–6197. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.D.; Ortí, E.; Bolink, H.J.; Monti, F.; Accorsi, G.; Armaroli, N. Luminescent ionic transition-metal complexes for light-emitting electrochemical cells. Angew. Chem. Int. Ed. 2012, 51, 8178–8211. [Google Scholar] [CrossRef]
- Dreyse, P.; Loeb, B.; Soto-Arriaza, M.; Tordera, D.; Ortí, E.; Serrano-Pérez, J.J.; Bolink, H.J. Effect of free rotation in polypyridinic ligands of Ru (II) complexes applied in light-emitting electrochemical cells. Dalton Trans. 2013, 42, 15502–15513. [Google Scholar] [CrossRef]
- Gu, J.; Yan, Y.; Helbig, B.J.; Huang, Z.; Lian, T.; Schmehl, R.H. The influence of ligand localized excited states on the photophysics of second row and third row transition metal terpyridyl complexes: Recent examples and a case study. Coord. Chem. Rev. 2015, 100–109. [Google Scholar] [CrossRef] [Green Version]
- Schubert, U.S.; Winter, A.; Newkome, G.R. Terpyridine-Based Materials; Wiley-VCH: Weinheim, Germany, 2011; Chapter 4. [Google Scholar]
- Cargill-Thompson, A.M.W. The synthesis of 2, 2′:6′,2′′-terpyridine ligands—Versatile building blocks for supramolecular chemistry. Coord. Chem. Rev. 1997, 160, 1–52. [Google Scholar] [CrossRef]
- Byrne, J.P.; Kitchen, J.A.; Gunnaugsson, T. The btp [2, 6-bis (1, 2, 3-triazol-4-yl) pyridine] binding motif: A new versatile terdentate ligand for supramolecular and coordination chemistry. Chem. Soc. Rev. 2014, 43, 5302–5325. [Google Scholar] [CrossRef] [Green Version]
- de Bettencourt-Dias, A.; Barber, P.S.; Bauer, S. A water-soluble pybox derivative and its highly luminescent lanthanide ion complexes. J. Am. Chem. Soc. 2012, 134, 6987–6994. [Google Scholar] [CrossRef]
- Jameson, D.L.; Kruger, K.T.; Goldsby, K.A. Redox regulation in ruthenium(II) complexes of 2,6-bis(N-pyrazolyl)pyridine ligands: Synthetically versatile analogs of 2,2′:6′,2′′-terpyridine. Inorg. Chem. 1989, 4312–4314. [Google Scholar] [CrossRef]
- Jameson, D.L.; Goldsby, K.A. 2,6-bis(N-pyrazolyl)pyridines: The convenient synthesis of a family of planar tridentate N3 ligands that are terpyridine analogs. J. Org. Chem. 1990, 55, 4992–4994. [Google Scholar] [CrossRef]
- Starck, M.; Kadjane, P.; Bois, E.; Darbouret, B.; Incamps, A.; Ziessel, R.; Charbonniere, L.J. Towards libraries of luminescent lanthanide complexes and labels from generic synthons. Chem. Eur. J. 2011, 9164–9179. [Google Scholar] [CrossRef] [PubMed]
- Gamonal, A.; Brunet, E.; Juanes, O.; Rodriguez-Ubis, J.C. Pd cross-coupling reactions in the access to bis-pyrazole and bis-indazole pyridine-based nona-coordinated ligands. Luminescence properties of their lanthanide complexes. J. Photochem. Photobiol. A Chem. 2017, 342, 53–58. [Google Scholar] [CrossRef]
- Halcrow, M.A. Recent advances in the synthesis and applications of 2,6-dipyrazolylpyridine derivatives and their complexes. New J. Chem. 2014, 38, 1868–1882. [Google Scholar] [CrossRef]
- Zoppellaro, G.; Baumgarten, M. One-Step Synthesis of Symmetrically Substituted 2,6-Bis(pyrazol-1-yl)pyridine Systems. Eur. J. Org. Chem. 2005, 2888–2892. [Google Scholar] [CrossRef]
- Strohecker, D.J.; Lynch, V.M.; Holliday, B.J.; Jones, R.A. Synthesis and electronic investigation of mono-and di-substituted 4-nitro-and 4-amino-pyrazol-1-yl bis (pyrazol-1-yl) pyridine-type ligands and luminescent Eu (III) derivatives. Dalton Trans. 2017, 46, 7733–7742. [Google Scholar] [CrossRef]
- Halcrow, M.A. The synthesis and coordination chemistry of 2,6-bis(pyrazolyl)pyridines and related ligands—Versatile terpyridine analogues. Coord. Chem. Rev. 2005, 249, 2880–2908. [Google Scholar] [CrossRef]
- Brien, K.A.; Garner, C.M.; Pinney, K.G. Synthesis and characterization of 2, 6-bis-hydrazinopyridine, and its conversion to 2, 6-bis-pyrazolylpyridines. Tetrahedron 2006, 62, 3663–3666. [Google Scholar] [CrossRef]
- Basak, S.; Hui, P.; Chandrasekar, R. Regioselective, One-Pot Syntheses of Symmetrically and Unsymmetrically Halogenated 2′,6′-Bispyrazolylpyridines. Synthesis 2009, 23, 4042–4048. [Google Scholar]
- Vermonden, T.; Branowska, D.; Marcelis, A.T.M.; Sudhölter, E.J.R. Synthesis of 4-functionalized terdendate pyridine-based ligands. Tetrahedron 2003, 59, 5039–5045. [Google Scholar] [CrossRef]
- Santoro, A.; Cook, L.J.K.; Kulmaczewski, R.; Barret, S.A.; Cespedes, O.; Halcrow, M.A. Iron (II) complexes of tridentate indazolylpyridine ligands: Enhanced spin-crossover hysteresis and ligand-based fluorescence. Inorg. Chem. 2015, 54, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, R.; Kilner, C.; Halcrow, M.A. Unexpected product distributions in the synthesis of 2,6-bis-(indazolyl) pyridine and 2-(pyrazol-1-yl)-6-(indazolyl) pyridine. Tet. Lett. 2009, 50, 2484–2486. [Google Scholar] [CrossRef]
- Schlosser, M.; Bobbio, C.; Rauiss, T. Regiochemically Flexible Substitutions of Di-, Tri-, and Tetrahalopyridines: The Trialkylsilyl Trick. J. Org. Chem. 2005, 70, 2494–2502. [Google Scholar] [CrossRef] [PubMed]
- Schlosser, M.; Rauiss, T.; Bobbio, C. Rerouting Nucleophilic Substitution from the 4-Position to the 2- or 6-Position of 2,4-Dihalopyridines and 2,4,6-Trihalopyridines: The Solution to a Long-Standing Problem. Org. Lett. 2005, 7, 127–129. [Google Scholar] [CrossRef]
- Schlosser, M.; Rauiss, T. The Reactivity of 2-Fluoro- and 2-Chloropyridines toward Sodium Ethoxide: Factors Governing the Rates of Nucleophilic (Het)Aromatic Substitutions. Helv. Chim. Acta 2005, 88, 1240–1249. [Google Scholar] [CrossRef]
- Kadjane, P.; Starck, M.; Camerel, F.; Hill, D.; Hildebrandt, N.; Ziessel, R.; Charbonnière, L.J. Divergent Approach to a Large Variety of Versatile Luminescent Lanthanide Complexes. Inorg. Chem. 2009, 48, 4601–4603. [Google Scholar] [CrossRef]
- Brunet, E.; Juanes, O.; Sedano, R.; Rodriguez-Ubis, J.C. Synthesis of Novel Macrocyclic Lanthanide Chelates Derived from Bis-pyrazolylpyridine. Org. Lett. 2002, 4, 213–216. [Google Scholar] [CrossRef]
- Brunet, E.; Juanes, O.; Sedano, R.; Rodriguez-Ubis, J.C. Lanthanide complexes of new polyaminocarboxylate complexes with two chromophores derived from bispyrazolylpyridine and aceto or benzophenone: Synthesis, characterization and photophysical properties. Tetrahedron 2005, 61, 6757–6763. [Google Scholar] [CrossRef]
- Brunet, E.; Juanes, O.; Sedano, R.; Rodriguez-Ubis, J.C. Lanthanide complexes of polycarboxylate-bearing dipyrazolylpyridine ligands with near-unity luminescence quantum yields: The effect of pyridine substitution. Photochem. Photobiol. Sci. 2002, 1, 613–618. [Google Scholar] [CrossRef]
- Ye, Z.; Tan, M.; Wang, G.; Yuan, J. Preparation, characterization, and time-resolved fluorometric application of silica-coated terbium (III) fluorescent nanoparticles. Anal. Chem. 2004, 76, 513–518. [Google Scholar] [CrossRef]
- Yuan, J.; Tan, M.; Wang, G. Synthesis and luminescence properties of lanthanide (III) chelates with polyacid derivatives of thienyl-substituted terpyridine analogues. J. Lumin. 2004, 106, 91–101. [Google Scholar] [CrossRef]
- Karhunen, U.; Malmi, E.; Brunet, E.; Rodríguez-Ubis, J.C.; Soukka, T. Switchable lanthanide luminescent binary probes in efficient single nucleotide mismatch discrimination. Sens. Actuators B 2015, 211, 297–302. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gamonal Ruiz-Crespo, A.; Galán-Fernández, L.; Martínez-Martín, P.; Rodríguez-Ubis, J.C. An Orthogonal Synthetic Approach to Nonsymmetrical Bisazolyl 2,4,6-Trisubstituted Pyridines. Molecules 2022, 27, 1746. https://doi.org/10.3390/molecules27051746
Gamonal Ruiz-Crespo A, Galán-Fernández L, Martínez-Martín P, Rodríguez-Ubis JC. An Orthogonal Synthetic Approach to Nonsymmetrical Bisazolyl 2,4,6-Trisubstituted Pyridines. Molecules. 2022; 27(5):1746. https://doi.org/10.3390/molecules27051746
Chicago/Turabian StyleGamonal Ruiz-Crespo, Arturo, Laura Galán-Fernández, Paloma Martínez-Martín, and Juan Carlos Rodríguez-Ubis. 2022. "An Orthogonal Synthetic Approach to Nonsymmetrical Bisazolyl 2,4,6-Trisubstituted Pyridines" Molecules 27, no. 5: 1746. https://doi.org/10.3390/molecules27051746