
Designing Novel Compounds for the Treatment and Management of RET-Positive Non-Small Cell Lung Cancer—Fragment Based Drug Design Strategy

Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset Preparation
2.2. Fragment Tailoring Strategy
2.3. Hybrid Molecule Screening upon RET Target
2.4. Rescoring Validation of Hybrid Molecules
2.5. Binding Free Energy Analysis
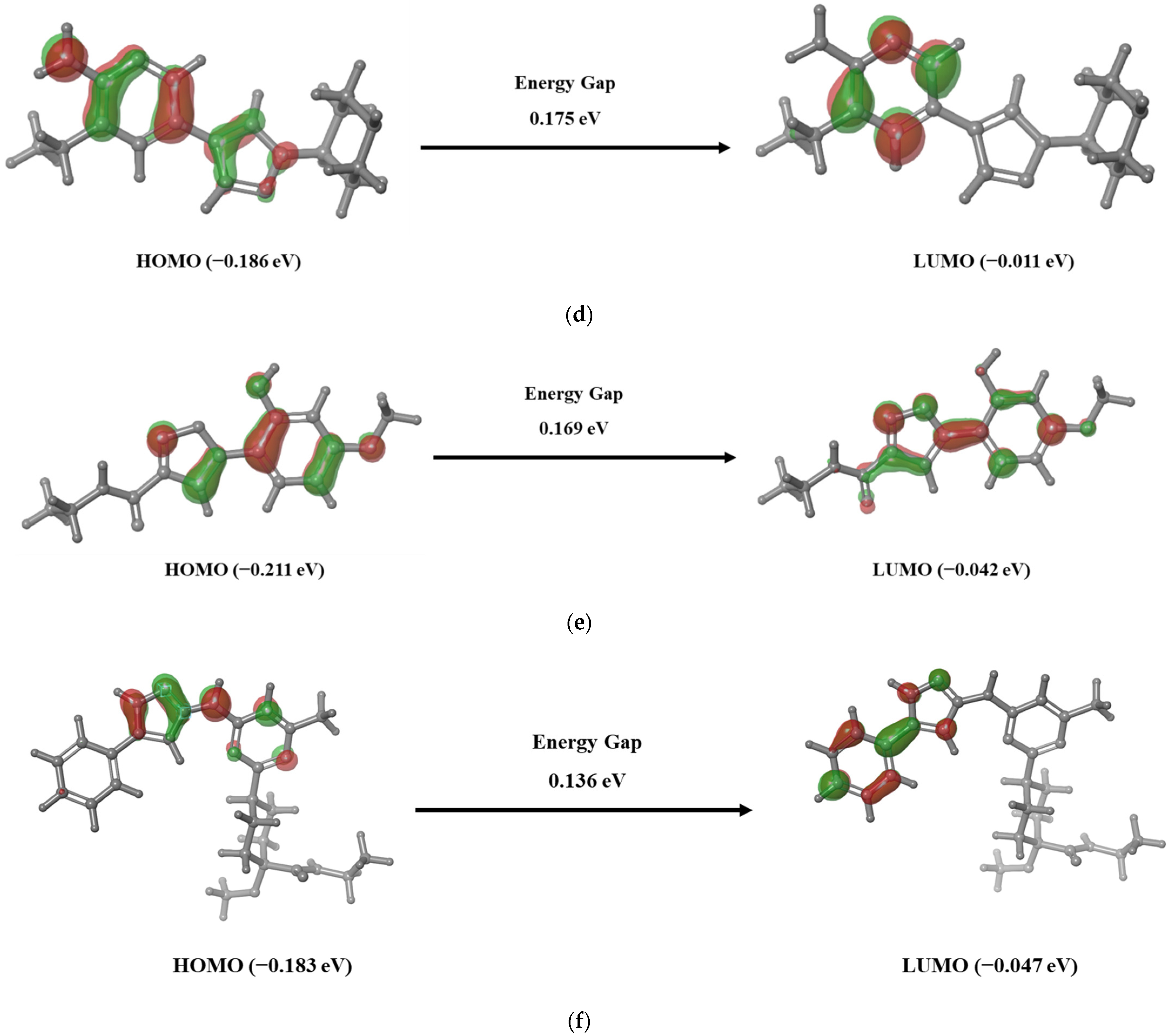
2.6. HOMO—LUMO Theory
2.7. Drug Likeness and Toxicity Analysis
2.8. Stability and Flexibility Assessment of Hybrid Molecules
2.9. Compound Reactivity Analysis Using PaccMann and MM-PBSA Analysis
2.10. Synergism Analysis of Parent Compounds
3. Results and Discussion
3.1. Fragments Tailoring and Docking Analysis
3.2. Rescoring with Machine Learning Algorithm
3.3. Binding Free Energy Calculations
3.4. Frontier Molecular Orbital Analysis
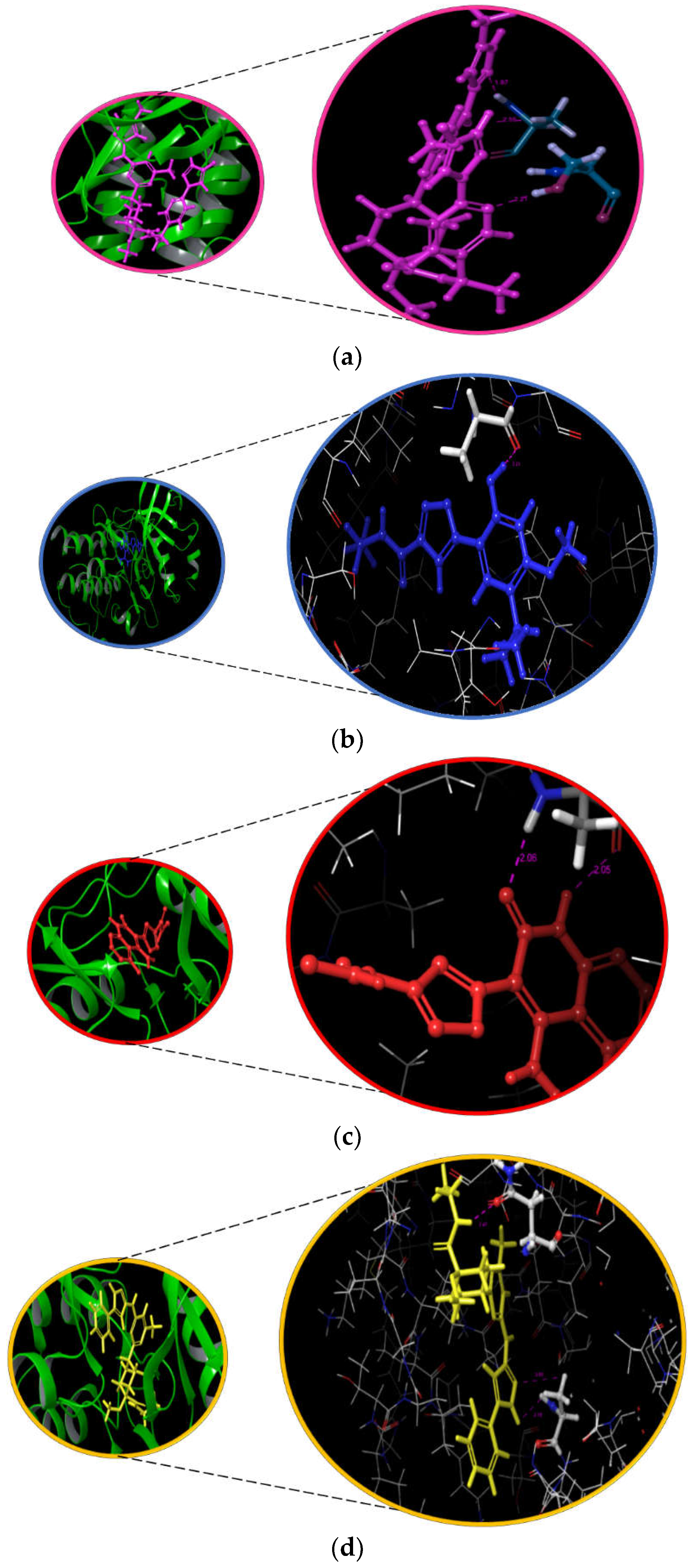
3.5. Interaction and ADMET Analysis
3.6. Molecular Dynamics Simulation
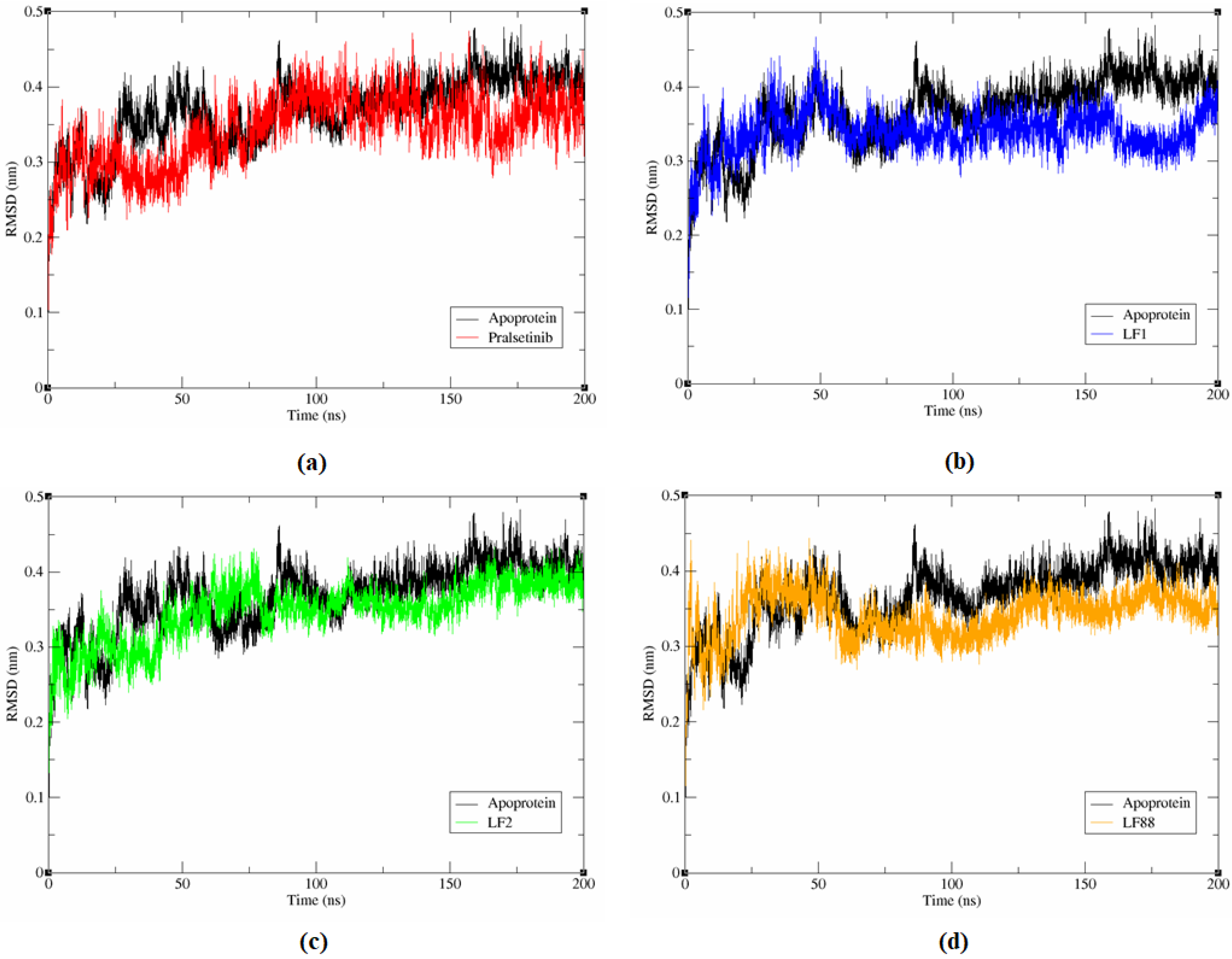
3.6.1. Stability Analysis of Complex System
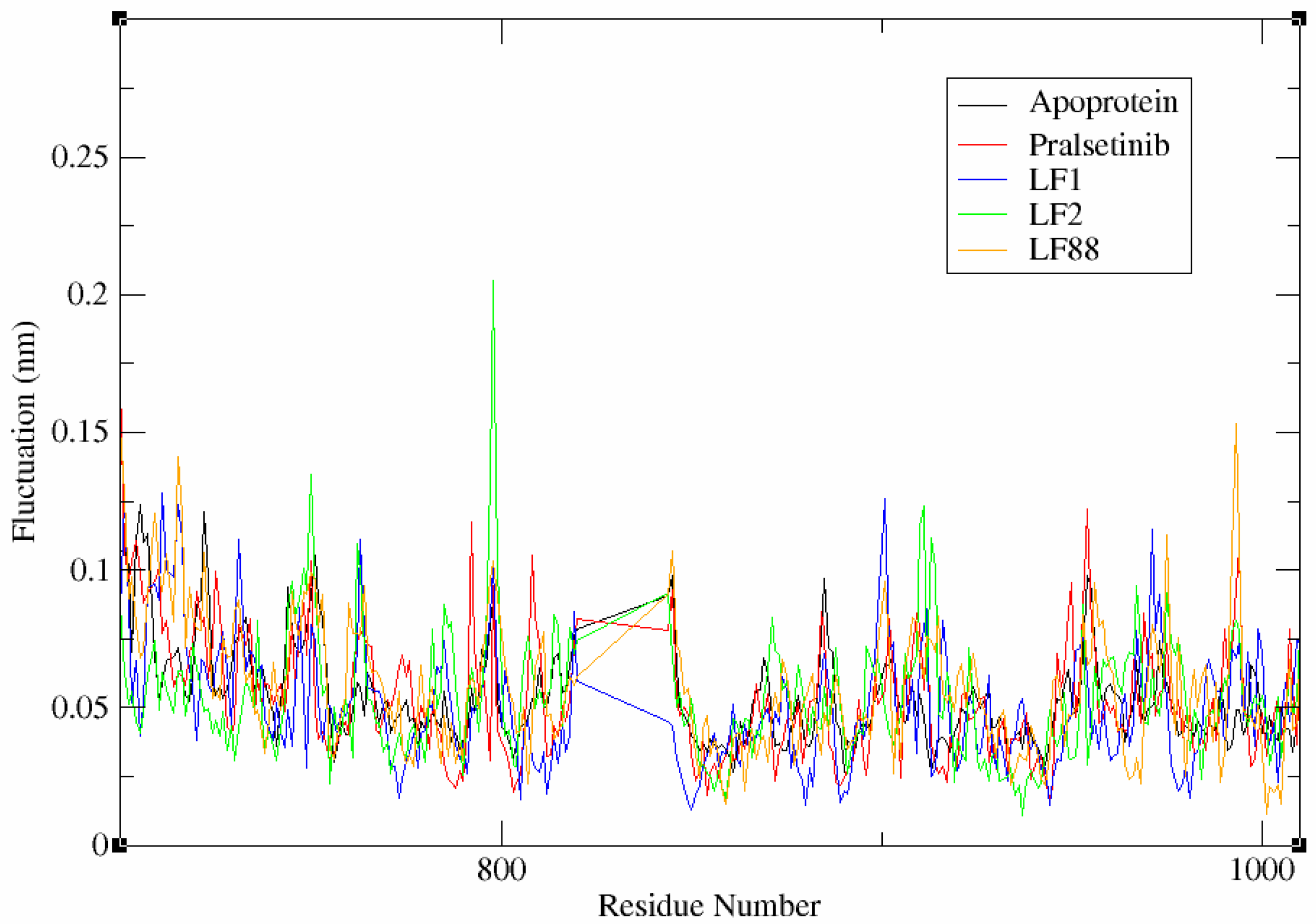
3.6.2. Flexibility Analysis of Complex System
3.6.3. Hydrogen Bond Interaction Analysis
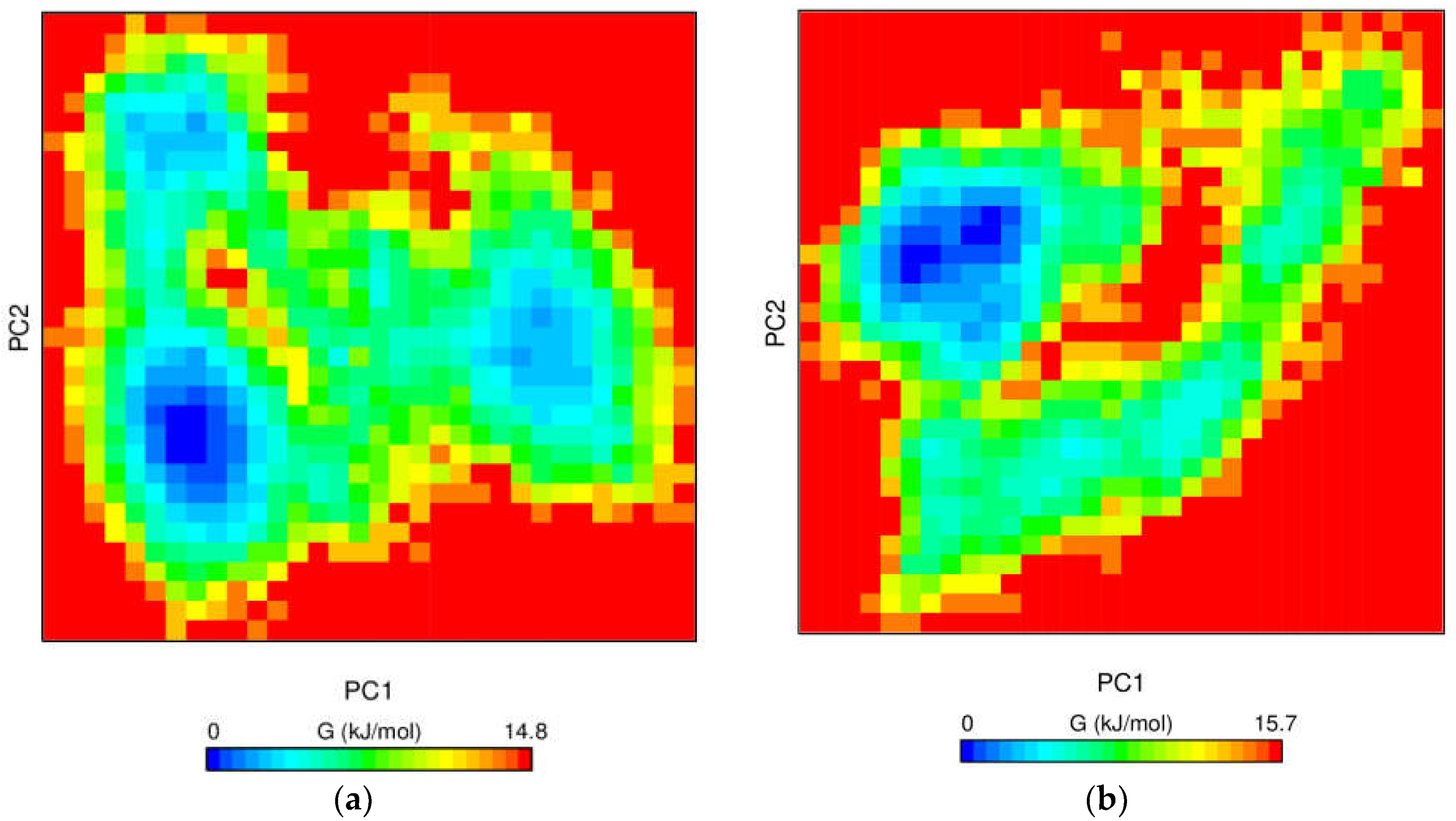
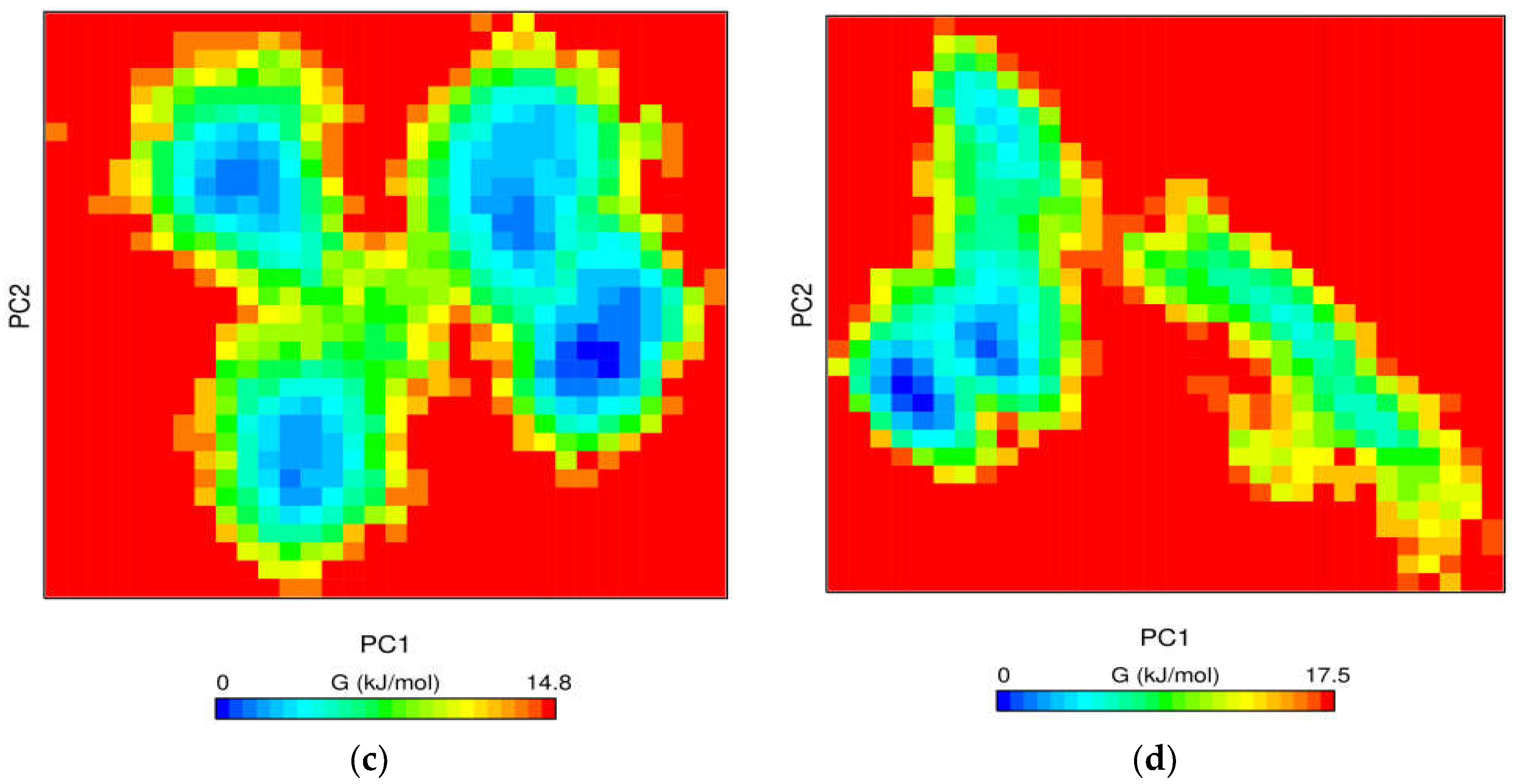
3.6.4. Free Energy Landscape
3.7. In Silico Compound Activity Analysis
3.8. Synergistic Effect Analysis
3.9. Evaluation of Hybrid Compounds against Mutant RET
4. Limitations and Future Prospective
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Woodman, C.; Vundu, G.; George, A.; Wilson, C. Applications and strategies in nanodiagnosis and nanotherapy in lung cancer. Semin. Cancer Biol. 2021, 69, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Cascetta, P.; Sforza, V.; Manzo, A.; Carillio, G.; Palumbo, G.; Esposito, G.; Montanino, A.; Costanzo, R.; Sandomenico, C.; De Cecio, R.; et al. RET Inhibitors in Non-Small-Cell Lung Cancer. Cancers 2021, 13, 4415. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Shen, T.; Terzyan, S.; Liu, X.; Hu, X.; Patel, K.; Hu, M.; Cabanillas, M.; Behrang, A.; Meric-Bernstam, F.; et al. Structural basis of acquired resistance to selpercatinib and pralsetinib mediated by non-gatekeeper RET mutations. Ann. Oncol. 2020, 32, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Fancelli, S.; Caliman, E.; Mazzoni, F.; Brugia, M.; Castiglione, F.; Voltolini, L.; Pillozzi, S.; Antonuzzo, L. Chasing the Target: New Phenomena of Resistance to Novel Selective RET Inhibitors in Lung Cancer. Updated Evidence and Future Perspectives. Cancers 2021, 13, 1091. [Google Scholar] [CrossRef]
- Drusbosky, L.M.; Rodriguez, E.; Dawar, R.; Ikpeazu, C.V. Therapeutic strategies in RET gene rearranged non-small cell lung cancer. J. Hematol. Oncol. 2021, 14, 1–8. [Google Scholar] [CrossRef]
- Drilon, A.; Hu, Z.I.; Lai, G.G.Y.; Tan, D.S.W. Targeting RET-driven cancers: Lessons from evolving preclinical and clinical landscapes. Nat. Rev. Clin. Oncol. 2018, 15, 151–167. [Google Scholar] [CrossRef]
- Gautschi, O.; Milia, J.; Filleron, T.; Wolf, J.; Carbone, D.P.; Owen, D.H.; Camidge, R.; Narayanan, V.; Doebele, R.C.; Besse, B.; et al. Targeting RET in Patients With RET-Rearranged Lung Cancers: Results From the Global, Multicenter RET Registry. J. Clin. Oncol. 2017, 35, 1403–1410. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Fu, S.; Patel, M.R.; Fakih, M.; Wang, D.; Olszanski, A.; Morgensztern, D.; Liu, S.V.; Cho, B.C.; Bazhenova, L.; et al. A Phase I/Ib Trial of the VEGFR-Sparing Multikinase RET Inhibitor RXDX-105. Cancer Discov. 2018, 9, 384–395. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Shen, T.; Mooers, B.H.M.; Hilberg, F.; Wu, J. Drug resistance profiles of mutations in the RET kinase domain. J. Cereb. Blood Flow Metab. 2018, 175, 3504–3515. [Google Scholar] [CrossRef] [Green Version]
- Bian, Y.; Xie, X.Q.S. Computational Fragment-Based Drug Design: Current Trends, Strategies, and Applications. AAPS J. 2018, 20, 59. [Google Scholar] [CrossRef] [PubMed]
- Parate, S.; Kumar, V.; Hong, J.C.; Lee, K.W. Investigating natural compounds against oncogenic RET tyrosine kinase using pharmacoinformatic approaches for cancer therapeutics. RSC Adv. 2021, 12, 1194–1207. [Google Scholar] [CrossRef]
- Pathak, P.; Naumovich, V.; Grishina, M.; Shukla, P.K.; Verma, A.; Potemkin, V. Quinazoline based 1,3,5-triazine derivatives as cancer inhibitors by impeding the phosphorylated RET tyrosine kinase pathway: Design, synthesis, docking, and QSAR study. Arch. Pharm. 2019, 352, 1900053. [Google Scholar] [CrossRef] [PubMed]
- El Hassab, M.A.; Shoun, A.A.; Al-Rashood, S.T.; Al-Warhi, T.; Eldehna, W.M. Identification of a New Potential SARS-CoV-2 RNA-Dependent RNA Polymerase Inhibitor via Combining Fragment-Based Drug Design, Docking, Molecular Dynamics, and MM-PBSA Calculations. Front. Chem. 2020, 8, 584894. [Google Scholar] [CrossRef]
- Li, Q. Application of Fragment-Based Drug Discovery to Versatile Targets. Front. Mol. Biosci. 2020, 7, 180. [Google Scholar] [CrossRef]
- Jain, A.; James, N.; Shanthi, V.; Ramanathan, K. Design of ALK Inhibitors for Non-Small Cell Lung Cancer–A Fragment Based Approach. Indian J. Pharm. Educ. Res. 2020, 54, 114–124. [Google Scholar] [CrossRef] [Green Version]
- Alkaff, A.H.; Saragih, M.; Imana, S.N.; Nasution, M.A.F.; Tambunan, U.S.F. Identification of DNA Methyltransferase-1 Inhibitor for Breast Cancer Therapy through Computational Fragment-Based Drug Design. Molecules 2021, 26, 375. [Google Scholar] [CrossRef]
- Mendoza, L. Clinical development of RET inhibitors in RET-rearranged non-small cell lung cancer: Update. Oncol. Rev. 2018, 12, 352. [Google Scholar] [CrossRef]
- Ferrara, R.; Auger, N.; Auclin, E.; Besse, B. Clinical and Translational Implications of RET Rearrangements in Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2018, 13, 27–45. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Knowles, P.P.; Murray-Rust, J.; Kjær, S.; Scott, R.; Hanrahan, S.; Santoro, M.; Ibáñez, C.F.; McDonald, N.Q. Structure and Chemical Inhibition of the RET Tyrosine Kinase Domain. J. Biol. Chem. 2006, 281, 33577–33587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thangapandian, S.; John, S.; Sakkiah, S.; Lee, K.W. Potential virtual lead identification in the discovery of renin inhibitors: Application of ligand and structure-based pharmacophore modeling approaches. Eur. J. Med. Chem. 2011, 46, 2469–2476. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, C. Fragment tailoring strategy to design novel chemical entities as potential binders of novel corona virus main protease. J. Biomol. Struct. Dyn. 2021, 39, 3733–3746. [Google Scholar] [CrossRef]
- Subbiah, V.; Gainor, J.F.; Rahal, R.; Brubaker, J.D.; Kim, J.L.; Maynard, M.; Hu, W.; Cao, Q.; Sheets, M.P.; Wilson, D.; et al. Precision Targeted Therapy with BLU-667 for RET-Driven Cancers. Cancer Discov. 2018, 8, 836–849. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Bradford, D.; Larkins, E.; Pai-Scherf, L.H.; Chatterjee, S.; Mishra-Kalyani, P.S.; Wearne, E.; Helms, W.S.; Ayyoub, A.; Bi, Y.; et al. FDA Approval Summary: Pralsetinib for the Treatment of Lung and Thyroid Cancers With RET Gene Mutations or Fusions. Clin. Cancer Res. 2021, 27, 5452–5456. [Google Scholar] [CrossRef]
- Wagner, J.R.; Churas, C.P.; Liu, S.; Swift, R.V.; Chiu, M.; Shao, C.; Feher, V.A.; Burley, S.K.; Gilson, M.K.; Amaro, R.E. Continuous Evaluation of Ligand Protein Predictions: A Weekly Community Challenge for Drug Docking. Structure 2019, 27, 1326–1335. [Google Scholar] [CrossRef]
- Zhang, L.; Ai, H.-X.; Li, S.-M.; Qi, M.-Y.; Zhao, J.; Zhao, Q.; Liu, H.-S. Virtual screening approach to identifying influenza virus neuraminidase inhibitors using molecular docking combined with machine-learning-based scoring function. Oncotarget 2017, 8, 83142–83154. [Google Scholar] [CrossRef] [Green Version]
- Madhavaram, M.; Nampally, V.; Gangadhari, S.; Palnati, M.K.; Tigulla, P. High-throughput virtual screening, ADME analysis, and estimation of MM/GBSA binding-free energies of azoles as potential inhibitors of Mycobacterium tuberculosis H37Rv. J. Recept. Signal Transduct. 2019, 39, 312–320. [Google Scholar] [CrossRef]
- Tripathy, S.; Sahu, S.K.; Azam, M.A.; Jupudi, S. Computer-aided identification of lead compounds as Staphylococcal epidermidis FtsZ inhibitors using molecular docking, virtual screening, DFT analysis, and molecular dynamic simulation. J. Mol. Model. 2019, 25, 360. [Google Scholar] [CrossRef]
- Obu, Q.S.; Louis, H.; Odey, J.O.; Eko, I.J.; Abdullahi, S.; Ntui, T.N.; Offiong, O.E. Synthesis, spectra (FT-IR, NMR) investigations, DFT study, in silico ADMET and Molecular docking analysis of 2-amino-4-(4-aminophenyl)thiophene-3-carbonitrile as a potential anti-tubercular agent. J. Mol. Struct. 2021, 1244, 130880. [Google Scholar] [CrossRef]
- Agwupuye, J.A.; Neji, P.A.; Louis, H.; Odey, J.O.; Unimuke, T.O.; Bisiong, E.A.; Eno, E.A.; Utsu, P.M.; Ntui, T.N. Investigation on electronic structure, vibrational spectra, NBO analysis, and molecular docking studies of aflatoxins and selected emerging mycotoxins against wild-type androgen receptor. Heliyon 2021, 7, 07544. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, F.; Siu, S.W.I. Identification of novel natural compound inhibitors for human complement component 5a receptor by homology modeling and virtual screening. Med. Chem. Res. 2016, 25, 1564–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, R.; Shukla, H.; Sonkar, A.; Pandey, T.; Tripathi, T. Structure-based screening and molecular dynamics simulations offer novel natural compounds as potential inhibitors of Mycobacterium tuberculosis isocitrate lyase. J. Biomol. Struct. Dyn. 2018, 36, 2045–2057. [Google Scholar] [CrossRef]
- Singh, N.; Dalal, V.; Kumar, P. Molecular docking and simulation analysis for elucidation of toxic effects of dicyclohexyl phthalate (DCHP) in glucocorticoid receptor-mediated adipogenesis. Mol. Simul. 2020, 46, 9–21. [Google Scholar] [CrossRef]
- Cadow, J.; Born, J.; Manica, M.; Oskooei, A.; Martínez, M.R. PaccMann: A web service for interpretable anticancer compound sensitivity prediction. Nucleic Acids Res. 2020, 48, W502–W508. [Google Scholar] [CrossRef]
- Singh, R.; Bhardwaj, V.; Purohit, R. Identification of a novel binding mechanism of Quinoline based molecules with lactate dehydrogenase of Plasmodium falciparum. J. Biomol. Struct. Dyn. 2021, 39, 348–356. [Google Scholar] [CrossRef]
- Muralidharan, N.; Sakthivel, R.; Velmurugan, D.; Gromiha, M.M. Computational studies of drug repurposing and synergism of lopinavir, oseltamivir and ritonavir binding with SARS-CoV-2 protease against COVID-19. J. Biomol. Struct. Dyn. 2021, 39, 2673–2678. [Google Scholar] [CrossRef]
- Yamaotsu, N.; Hirono, S. In silico fragment-mapping method: A new tool for fragment-based/structure-based drug discovery. J. Comput. Mol. Des. 2018, 32, 1229–1245. [Google Scholar] [CrossRef]
- Allen, W.J.; Rizzo, R.C. Implementation of the Hungarian Algorithm to Account for Ligand Symmetry and Similarity in Structure-Based Design. J. Chem. Inf. Model. 2014, 54, 518–529. [Google Scholar] [CrossRef]
- Singh, N.; Chaput, L.; Villoutreix, B.O. Fast Rescoring Protocols to Improve the Performance of Structure-Based Virtual Screening Performed on Protein–Protein Interfaces. J. Chem. Inf. Model. 2020, 60, 3910–3934. [Google Scholar] [CrossRef] [PubMed]
- Aaldering, L.J.; Vasanthanathan Poongavanam, D.; Langkjær, N.; Murugan, N.A.; Jørgensen, P.T.; Wengel, J.; Veedu, R.N. Development of an Efficient G-Quadruplex-Stabilised Thrombin-Binding Aptamer Containing a Three-Carbon Spacer Molecule. ChemBioChem 2017, 18, 755. [Google Scholar] [CrossRef] [Green Version]
- Palaka, B.K.; Venkatesan, R.; Ampasala, D.R.; Periyasamy, L. Identification of novel inhibitors of signal transducer and activator of transcription 3 over signal transducer and activator of transcription 1 for the treatment of breast cancer by in-silico and in-vitro approach. Process Biochem. 2019, 82, 153–166. [Google Scholar] [CrossRef]
- Jia, C.-C.; Chen, W.; Feng, Z.-L.; Liu, Z.-P. Recent developments of RET protein kinase inhibitors with diverse scaffolds as hinge binders. Future Med. Chem. 2021, 13, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Al-Majid, A.M.; Islam, M.S.; Atef, S.; El-Senduny, F.F.; Badria, F.A.; Elshaier, Y.A.M.M.; Ali, M.; Barakat, A.; Rahman, A.F.M.M. Synthesis of Pyridine-Dicarboxamide-Cyclohexanone Derivatives: Anticancer and α-Glucosidase Inhibitory Activities and In Silico Study. Molecules 2019, 24, 1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mani, G.S.; Shaik, S.P.; Tangella, Y.; Bale, S.; Godugu, C.; Kamal, A. A facile I2-catalyzed synthesis of imidazo[1,2-a]pyridines via sp3 C–H functionalization of azaarenes and evaluation of anticancer activity. Org. Biomol. Chem. 2017, 15, 6780–6791. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [Green Version]
- Guterres, H.; Im, W. Improving Protein-Ligand Docking Results with High-Throughput Molecular Dynamics Simulations. J. Chem. Inf. Model. 2020, 60, 2189–2198. [Google Scholar] [CrossRef]
- Kumar, R.; Jade, D.; Gupta, D. A novel identification approach for discovery of 5-HydroxyTriptamine 2A antagonists: Combination of 2D/3D similarity screening, molecular docking and molecular dynamics. J. Biomol. Struct. Dyn. 2018, 37, 931–943. [Google Scholar] [CrossRef]
- Londhe, A.M.; Gadhe, C.G.; Lim, S.M.; Pae, A.N. Investigation of Molecular Details of Keap1-Nrf2 Inhibitors Using Molecular Dynamics and Umbrella Sampling Techniques. Molecules 2019, 24, 4085. [Google Scholar] [CrossRef] [Green Version]
- Cloete, R.; Akurugu, W.A.; Werely, C.J.; van Helden, P.D.; Christoffels, A. Structural and functional effects of nucleotide variation on the human TB drug metabolizing enzyme arylamine N -acetyltransferase 1. J. Mol. Graph. Model. 2017, 75, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Aldeghi, M.; Bodkin, M.J.; Knapp, S.; Biggin, P.C. Statistical Analysis on the Performance of Molecular Mechanics Poisson–Boltzmann Surface Area versus Absolute Binding Free Energy Calculations: Bromodomains as a Case Study. J. Chem. Inf. Model. 2017, 57, 2203–2221. [Google Scholar] [CrossRef] [PubMed]
- Slynko, I.; Scharfe, M.; Rumpf, T.; Eib, J.; Metzger, E.; Schüle, R.; Jung, M.; Sippl, W. Virtual Screening of PRK1 Inhibitors: Ensemble Docking, Rescoring Using Binding Free Energy Calculation and QSAR Model Development. J. Chem. Inf. Model. 2014, 54, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Manica, M.; Oskooei, A.; Born, J.; Subramanian, V.; Saez-Rodriguez, J.; Martinez, M.R. Toward Explainable Anticancer Compound Sensitivity Prediction via Multimodal Attention-Based Convolutional Encoders. Mol. Pharm. 2019, 16, 4797–4806. [Google Scholar] [CrossRef]
- Tabata, M.; Tsubaki, M.; Takeda, T.; Tateishi, K.; Maekawa, S.; Tsurushima, K.; Imano, M.; Satou, T.; Ishizaka, T.; Nishida, S. Inhibition of HSP90 overcomes melphalan resistance through downregulation of Src in multiple myeloma cells. Clin. Exp. Med. 2019, 20, 63–71. [Google Scholar] [CrossRef]
- Solomon, B.J.; Tan, L.; Lin, J.J.; Wong, S.Q.; Hollizeck, S.; Ebata, K.; Tuch, B.B.; Yoda, S.; Gainor, J.F.; Sequist, L.V.; et al. RET Solvent Front Mutations Mediate Acquired Resistance to Selective RET Inhibition in RET-Driven Malignancies. J. Thorac. Oncol. 2020, 15, 541–549. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.-J.; Du, Y.; Wen, R.; Yang, M.; Xu, J. Drug resistance to targeted therapeutic strategies in non-small cell lung cancer. Pharmacol. Ther. 2020, 206, 107438. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Control and Linked Fragments | Stars | XP gScore (kcal/mol) | RMSD Å | RF-Score |
|---|---|---|---|---|---|
| 1 | Pralsetinib | 0 | −7.79 | 6.440 | 5.962 |
| 2 | LF1 | 0 | −8.2 | 0.257 | 6.334 |
| 3 | LF2 | 0 | −8.885 | 0.558 | 6.108 |
| 4 | LF3 | 0 | −11.039 | 0.535 | 6.397 |
| 5 | LF4 | 0 | −9.505 | 1.437 | 5.967 |
| 6 | LF5 | 0 | −7.817 | 0.717 | 5.968 |
| 7 | LF6 | 0 | −7.947 | 8.341 | 5.967 |
| 8 | LF7 | 0 | −8.134 | 0.304 | 6.135 |
| 9 | LF8 | 0 | −9.198 | 6.178 | 6.071 |
| 10 | LF9 | 0 | −9.19 | 1.144 | 5.973 |
| 11 | LF10 | 0 | −8.638 | 0.423 | 6.09 |
| 12 | LF11 | 0 | −8.25 | 0.798 | 5.976 |
| 13 | LF12 | 0 | −8.375 | 0.528 | 6.044 |
| 14 | LF13 | 0 | −8.257 | 0.733 | 5.967 |
| 15 | LF14 | 0 | −8.638 | 1.167 | 5.983 |
| 16 | LF15 | 0 | −9.439 | 7.719 | 5.976 |
| 17 | LF16 | 0 | −7.972 | 8.395 | 6.15 |
| 18 | LF17 | 0 | −9.577 | 1.175 | 5.98 |
| 19 | LF18 | 0 | −7.797 | 9.557 | 6.269 |
| 20 | LF19 | 0 | −7.985 | 7.017 | 6.154 |
| 21 | LF20 | 0 | −8.621 | 2.099 | 5.963 |
| 22 | LF21 | 0 | −8.828 | 0.597 | 5.961 |
| 23 | LF22 | 0 | −8.3 | 0.656 | 5.961 |
| 24 | LF23 | 0 | −8.17 | 0.228 | 5.958 |
| 25 | LF24 | 0 | −8.695 | 0.588 | 5.956 |
| 26 | LF25 | 0 | −9.611 | 1.041 | 5.955 |
| 27 | LF26 | 0 | −7.852 | 1.439 | 5.953 |
| 28 | LF27 | 0 | −7.913 | 0.224 | 5.951 |
| 29 | LF28 | 0 | −9.133 | 0.425 | 5.95 |
| 30 | LF29 | 0 | −9.835 | 4.270 | 5.948 |
| 31 | LF30 | 0 | −7.777 | 5.224 | 6 |
| 32 | LF31 | 0 | −7.746 | 0.218 | 6.2 |
| 33 | LF32 | 0 | −7.739 | 2.131 | 6.141 |
| 34 | LF33 | 0 | −7.696 | 5.693 | 6.116 |
| 35 | LF34 | 0 | −7.668 | 1.040 | 6.045 |
| 36 | LF35 | 0 | −7.619 | 0.605 | 5.983 |
| 37 | LF36 | 0 | −7.535 | 1.023 | 5.978 |
| 38 | LF37 | 0 | −7.526 | 0.564 | 5.983 |
| 39 | LF38 | 0 | −7.491 | 4.930 | 6.139 |
| 40 | LF39 | 0 | −7.49 | 0.329 | 6.085 |
| 41 | LF40 | 0 | −7.48 | 8.155 | 5.952 |
| 42 | LF41 | 0 | −7.468 | 1.141 | 5.973 |
| 43 | LF42 | 0 | −7.467 | 0.801 | 6.284 |
| 44 | LF43 | 0 | −7.458 | 0.789 | 5.965 |
| 45 | LF44 | 0 | −7.437 | 1.432 | 5.971 |
| 46 | LF45 | 0 | −7.421 | 3.320 | 5.955 |
| 47 | LF46 | 0 | −7.41 | 0.814 | 5.997 |
| 48 | LF47 | 0 | −7.389 | 1.163 | 6.102 |
| 49 | LF48 | 0 | −7.355 | 0.326 | 5.955 |
| 50 | LF49 | 0 | −7.339 | 2.510 | 5.976 |
| 51 | LF50 | 0 | −7.322 | 0.431 | 5.974 |
| 52 | LF51 | 0 | −7.289 | 6.173 | 5.97 |
| 53 | LF52 | 0 | −7.283 | 0.695 | 5.966 |
| 54 | LF53 | 0 | −7.207 | 3.886 | 6.215 |
| 55 | LF54 | 0 | −7.201 | 1.094 | 5.962 |
| 56 | LF55 | 0 | −7.199 | 1.717 | 6.123 |
| 57 | LF56 | 0 | −7.186 | 4.928 | 5.971 |
| 58 | LF57 | 0 | −7.139 | 1.687 | 6.114 |
| 59 | LF58 | 0 | −7.137 | 0.482 | 5.963 |
| 60 | LF59 | 0 | −7.087 | 0.569 | 6.087 |
| 61 | LF60 | 0 | −7.064 | 5.467 | 5.963 |
| 62 | LF61 | 0 | −7.014 | 0.255 | 5.963 |
| 63 | LF62 | 0 | −7.01 | 5.968 | 6.086 |
| 64 | LF63 | 0 | −6.992 | 0.257 | 5.957 |
| 65 | LF64 | 0 | −6.86 | 4.837 | 6.093 |
| 66 | LF65 | 0 | −6.835 | 4.914 | 5.974 |
| 67 | LF66 | 0 | −6.744 | 0.960 | 5.958 |
| 68 | LF67 | 0 | −6.72 | 0.984 | 5.97 |
| 69 | LF68 | 0 | −6.633 | 5.297 | 5.952 |
| 70 | LF69 | 0 | −6.603 | 0.990 | 5.985 |
| 71 | LF70 | 0 | −6.598 | 1.021 | 5.971 |
| 72 | LF71 | 0 | −6.535 | 1.020 | 5.99 |
| 73 | LF72 | 0 | −6.434 | 6.628 | 5.959 |
| 74 | LF73 | 0 | −6.424 | 6.309 | 6.117 |
| 75 | LF74 | 0 | −6.401 | 3.922 | 5.967 |
| 76 | LF75 | 0 | −6.375 | 0.630 | 6.011 |
| 77 | LF76 | 0 | −6.188 | 1.425 | 5.962 |
| 78 | LF77 | 0 | −6.094 | 0.317 | 6.184 |
| 79 | LF78 | 0 | −6.052 | 2.328 | 5.982 |
| 80 | LF79 | 0 | −5.865 | 3.356 | 6.172 |
| 81 | LF80 | 0 | −5.69 | 4.500 | 5.971 |
| 82 | LF81 | 0 | −5.583 | 3.759 | 6.036 |
| 83 | LF82 | 0 | −5.511 | 6.635 | 6.133 |
| 84 | LF83 | 0 | −5.344 | 2.498 | 5.95 |
| 85 | LF84 | 0 | −4.901 | 4.213 | 5.957 |
| 86 | LF85 | 0 | −4.717 | 2.020 | 6.083 |
| 87 | LF86 | 0 | −4.199 | 2.131 | 6.099 |
| 88 | LF87 | 0 | −3.846 | 0.665 | 5.964 |
| 89 | LF88 | 1 | −8.421 | 0.912 | 6.193 |
| 90 | LF89 | 1 | −8.639 | 0.666 | 6.01 |
| 91 | LF90 | 1 | −7.861 | 1.884 | 6.283 |
| 92 | LF91 | 1 | −9.161 | 9.304 | 5.957 |
| 93 | LF92 | 1 | −7.602 | 0.326 | 6.026 |
| 94 | LF93 | 1 | −6.7 | 7.458 | 6.044 |
| 95 | LF94 | 1 | −6.28 | 0.958 | 5.961 |
| 96 | LF95 | 1 | −6.021 | 3.630 | 7.361 |
| 97 | LF96 | 1 | −5.993 | 0.593 | 6.992 |
| 98 | LF97 | 1 | −5.952 | 10.779 | 5.962 |
| 99 | LF98 | 1 | −5.479 | 6.19 | 5.978 |
| 100 | LF99 | 2 | −7.692 | 6.267 | 5.984 |
| 101 | LF100 | 2 | −7.699 | 6.585 | 6.157 |
| 102 | LF101 | 2 | −8.121 | 0.669 | 6.045 |
| 103 | LF102 | 2 | −9.068 | 1.397 | 6.027 |
| 104 | LF103 | 2 | −8.167 | 0.789 | 5.952 |
| 105 | LF104 | 2 | −5.543 | 4.837 | 5.964 |
| 106 | LF105 | 4 | −6.998 | 3.025 | 6.035 |
| 107 | LF106 | 5 | −6.941 | 2.617 | 6.078 |
| 108 | LF107 | 5 | −3.98 | 2.962 | 5.957 |
| 109 | LF108 | 5 | −6.588 | 5.214 | 5.977 |
| 110 | LF109 | 6 | −4.871 | 1.902 | 5.971 |
| 111 | LF110 | 6 | −5.554 | 7.447 | 5.957 |
| 112 | LF111 | 7 | −6.089 | 0.562 | 5.96 |
| 113 | LF112 | 8 | −6.029 | 0.938 | 5.962 |
| 114 | LF113 | 8 | −5.723 | 0.491 | 6.178 |
| 115 | LF114 | 8 | −8.13 | 1.367 | 5.959 |
| 116 | LF115 | 8 | −6.545 | 1.339 | 5.96 |
| Control and Linked Fragments | dG Bind (kcal/mol) | Van der Waal’s Energy (kcal/mol) | Ligand Strain Energy (kcal/mol) | Electrostatic Potential (kcal/mol) | Covalent Interaction (kcal/mol) | Lipophilicity (kcal/mol) | Solvation Energy (kcal/mol) |
|---|---|---|---|---|---|---|---|
| Pralsetinib | −63.348 | −58.387 | 6.20432 | −12.472 | −0.4283 | −19.969 | 37.3355 |
| LF1 | −78.875 | −55.285 | 5.209 | −33.627 | 1.512 | −21.143 | 32.186 |
| LF27 | −73.11 | −60.237 | 2.678 | −21.049 | 2.615 | −25.583 | 32.147 |
| LF2 | −70.752 | −47.964 | 3.018 | −24.213 | 1.891 | −18.239 | 20.196 |
| LF21 | −63.654 | −54.224 | 2.992 | −19.372 | 1.127 | −24.461 | 34.469 |
| LF88 | −63.521 | −46.248 | 5.237 | −20.513 | 3.478 | −24.643 | 26.701 |
| LF29 | −62.965 | −45.4 | 2.562 | −25.554 | 2.113 | −22.992 | 30.221 |
| LF3 | −62.895 | −52.664 | 6.212 | −16.773 | −0.312 | −19.834 | 27.54 |
| LF4 | −60.017 | −39.524 | 2.804 | −21.897 | −0.561 | −21.251 | 25.189 |
| LF5 | −57.679 | −49.011 | 5.349 | −35.466 | 2.096 | −19.2 | 45.57 |
| LF89 | −55.77 | −44.968 | 6.72 | −19.658 | 5.17 | −18.602 | 23.929 |
| LF6 | −55.462 | −49.877 | 8.647 | −29.366 | 4.238 | −16.002 | 38.428 |
| LF25 | −55.094 | −50.213 | 8.401 | −10.118 | 2.115 | −19.658 | 24.019 |
| LF91 | −54.683 | −42.063 | 2.842 | −14.307 | 1.961 | −17.836 | 19.09 |
| LF23 | −53.86 | −44.928 | 4.357 | −13.386 | 1.727 | −20.908 | 25.182 |
| LF24 | −52.802 | −31.177 | 3.614 | −23.708 | 0.529 | −16.921 | 20.646 |
| LF7 | −52.465 | −51.814 | 4.835 | −35.392 | 3.19 | −15.905 | 49.912 |
| LF22 | −51.478 | −43.117 | 3.707 | −16.363 | 1.272 | −17.922 | 25.454 |
| LF8 | −51.337 | −44.893 | 5.922 | −14.472 | 5.267 | −15.919 | 20.795 |
| LF9 | −50.681 | −37.001 | 8.023 | −22.027 | 6.327 | −16.065 | 20.883 |
| LF10 | −50.255 | −43.526 | 4.463 | −13.965 | 4.363 | −14.628 | 19.622 |
| LF101 | −49.18 | −41.558 | 4.021 | −13.997 | 2.135 | −14.383 | 19.527 |
| LF102 | −48.459 | −33.805 | 4.821 | −18.293 | 3.508 | −16.351 | 18.643 |
| LF11 | −47.678 | −43.294 | 4.058 | −10.555 | 2.239 | −17.1 | 22.252 |
| LF103 | −47.375 | −50.103 | 8.598 | −28.738 | 3.098 | −17.355 | 48.194 |
| LF12 | −47.014 | −29.869 | 1.574 | −15.849 | 1.325 | −18.546 | 17.847 |
| LF20 | −46.6 | −32.975 | 1.8 | −12.629 | 1.051 | −20.039 | 18.639 |
| LF28 | −46.45 | −36.202 | 2.732 | −16.141 | 2.933 | −16.087 | 20.6 |
| LF114 | −46.195 | −42.487 | 7.755 | −12.376 | 4.829 | −20.4 | 25.46 |
| LF90 | −44.869 | −37.752 | 2.023 | −11.832 | 1.761 | −13.851 | 18.063 |
| LF13 | −44.188 | −34.743 | 1.058 | −10.255 | 0.955 | −15.561 | 16.05 |
| LF14 | −43.03 | −27.483 | 1.191 | −16.22 | 0.989 | −17.025 | 18.572 |
| LF15 | −41.74 | −42.888 | 15.734 | −25.959 | 9.447 | −18.956 | 38.089 |
| LF16 | −40.204 | −40.209 | 6.393 | −27.114 | 4.77 | −9.159 | 33.502 |
| LF17 | −39.071 | −42.268 | 22.468 | −27.647 | 12.815 | −25.637 | 45.318 |
| LF26 | −38.404 | −41.666 | 14.545 | −27.751 | 8.215 | −17.893 | 43.315 |
| LF18 | −31.506 | −35.323 | 16.143 | −0.157 | 0.576 | −16.869 | 20.436 |
| LF19 | −30.081 | −27.423 | 4.861 | −21.575 | 5.222 | −9.01 | 24.761 |
| Control and Linked Fragments | CNS | HOA | Hepatotoxicity | Carcinogenicity | Immunotoxicity | Mutagenicity | Cytotoxicity | Toxicity Class |
|---|---|---|---|---|---|---|---|---|
| Pralsetinib | −2 | 2 | Active | Inactive | Active | Inactive | Inactive | Class 4 |
| LF1 | −2 | 3 | Active | Inactive | Inactive | Inactive | Inactive | Class 5 |
| LF2 | −2 | 3 | Active | Active | Inactive | Active | Inactive | Class 5 |
| LF88 | −1 | 1 | Inactive | Active | Inactive | Inactive | Inactive | Class 5 |
| S. No. | Energy Terms (kJ/mol) | RET-Pralsetinib | RET-LF1 | RET-LF2 | RET-LF88 |
|---|---|---|---|---|---|
| 1 | Binding Energy | −9.445 ± 65.091 | −15 ± 22.651 | −13.158 ± 16.317 | −29.627 ± 27.501 |
| 2 | Van der Waal’s Energy | −23.022 ± 53.334 | −10.678 ± 11.115 | −8.669 ± 0.329 | −24.053 ± 31.102 |
| 3 | Electrostatic Energy | −0.074 ± 3.936 | −4.928 ± 25.728 | −7.359 ± 2.903 | −5.523 ± 13.183 |
| 4 | Polar Solvation Energy | 15.905 ± 55.514 | 3.008 ± 0.268 | 6.024 ± 1.240 | 4.021 ± 11.638 |
| 5 | SASA Energy | −2.254 ± 6.035 | −2.402 ± 0.107 | −3.154 ± 0.124 | −4.072 ± 8.184 |
| S. No. | Compound 1 | Compound 2 | Docking Score (kcal/mol) |
|---|---|---|---|
| 1 | Pralsetinib | Not applicable | −3.39 |
| 2 | Loxo-292 | NVP-AUY922 | −5.38 |
| 3 | Dovitinib | NVP-AUY922 | −4.06 |
| 4 | Pralsetinib | NVP-AUY922 | −3.84 |
| S. No | Native and Mutant RET Proteins | Glide XP Gscore (kcal/mol) | |||
|---|---|---|---|---|---|
| Pralsetinib | LF1 | LF2 | LF88 | ||
| 1 | Native | −7.79 | −8.2 | −8.885 | −8.421 |
| 2 | G810C | −3.69 | −4.035 | −4.404 | −3.655 |
| 3 | G810R | −3.356 | −5.741 | −3.845 | −4.842 |
| 4 | G810S | −6.69 | −4.403 | −4.404 | −3.655 |
| 5 | G810V | −5.684 | −3.97 | −4.547 | −4.86 |
| 6 | M918T | −4.85 | −6.736 | −8.235 | −7.647 |
| 7 | V738A | −4.555 | −5.715 | −6.457 | −5.282 |
| 8 | V804E | −6.682 | −4.114 | −4.533 | −5.61 |
| 9 | V804L | −8.067 | −5.186 | −8.445 | −8.412 |
| 10 | V804M | −7.625 | −5.46 | −8.337 | −7.754 |
| 11 | Y806C | −5.764 | −6.121 | −5.572 | −6.02 |
| 12 | Y806N | −4.810 | −6.590 | −5.685 | −6.297 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramesh, P.; Veerappapillai, S. Designing Novel Compounds for the Treatment and Management of RET-Positive Non-Small Cell Lung Cancer—Fragment Based Drug Design Strategy. Molecules 2022, 27, 1590. https://doi.org/10.3390/molecules27051590
Ramesh P, Veerappapillai S. Designing Novel Compounds for the Treatment and Management of RET-Positive Non-Small Cell Lung Cancer—Fragment Based Drug Design Strategy. Molecules. 2022; 27(5):1590. https://doi.org/10.3390/molecules27051590
Chicago/Turabian StyleRamesh, Priyanka, and Shanthi Veerappapillai. 2022. "Designing Novel Compounds for the Treatment and Management of RET-Positive Non-Small Cell Lung Cancer—Fragment Based Drug Design Strategy" Molecules 27, no. 5: 1590. https://doi.org/10.3390/molecules27051590