
Image-Based Annotation of Chemogenomic Libraries for Phenotypic Screening

 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
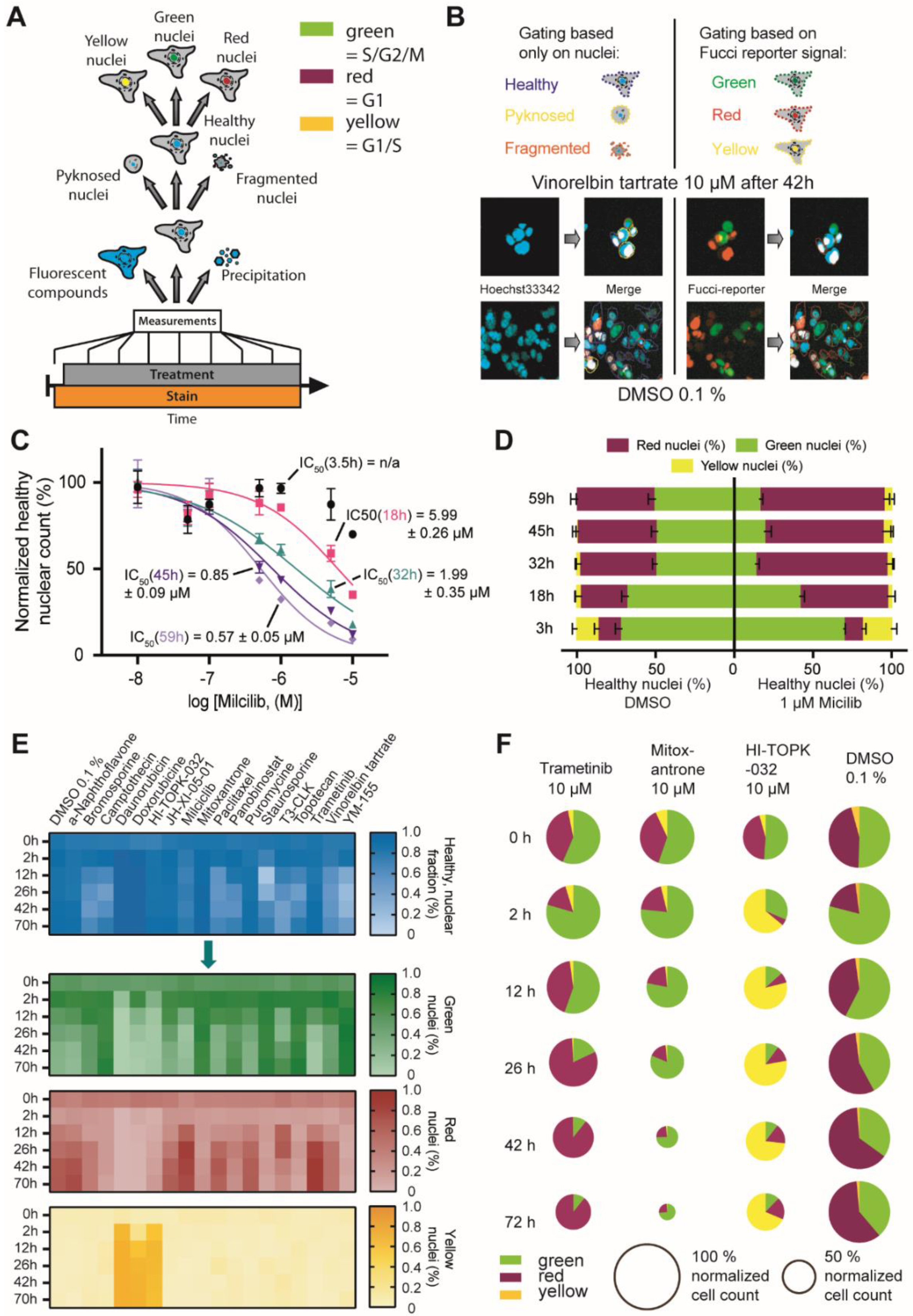
2.1. Optimization of HighVia Protocol and Validation of Cell Staining Dyes
2.2. Investigation of Nuclear Properties
2.3. FUCCI Cell Cycle Analysis
2.4. Multiplex Protocol
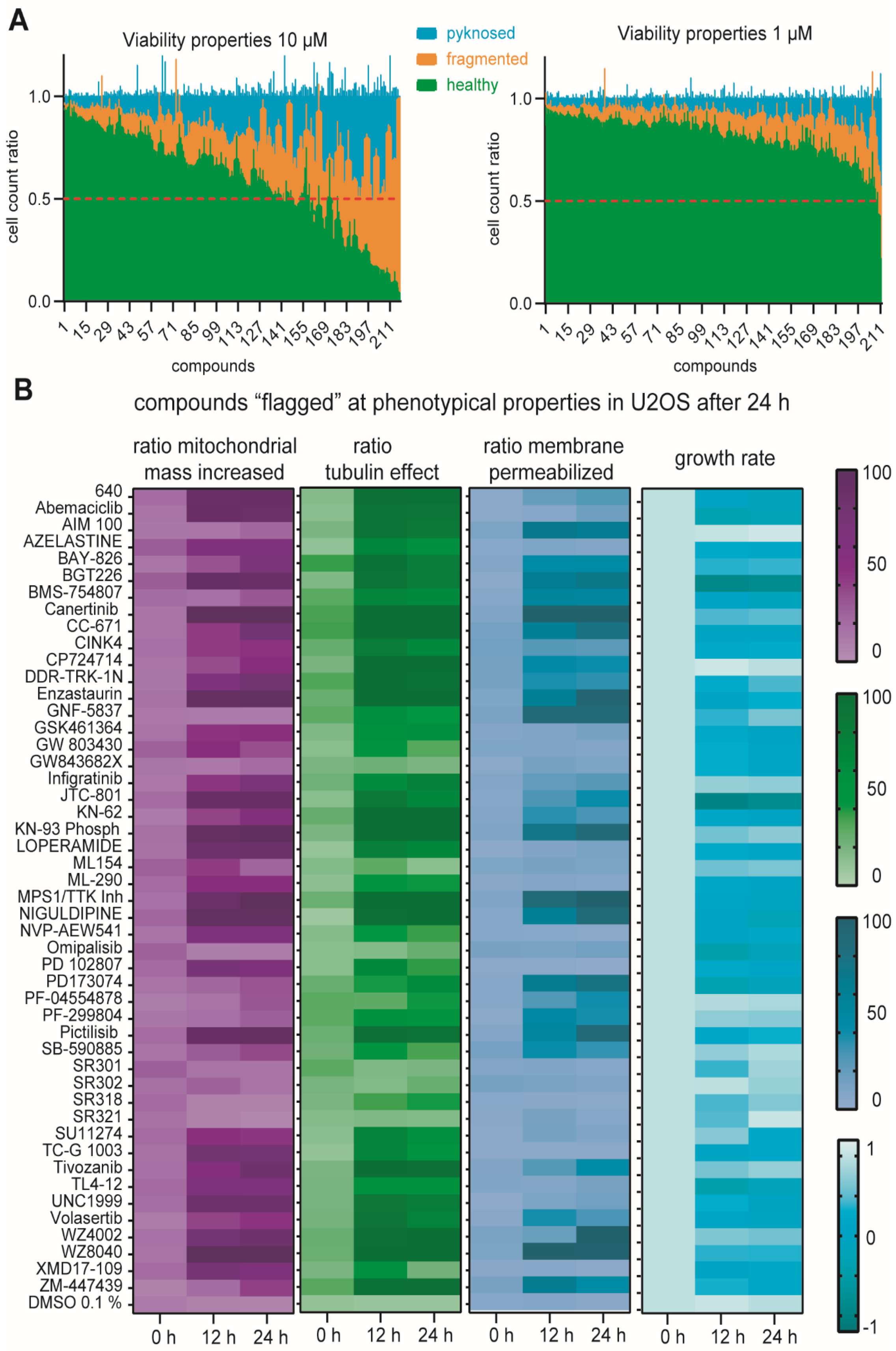
2.5. Multiplex Analysis of Chemogenomic Compounds
3. Discussion
3.1. Materials and Methods HighVia Extend Protocol
3.2. Multiplex Protocol
3.3. FUCCI Assay Protocol
3.4. Dye Titration CQ1 and Alamarblue Assay
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Haasen, D.; Schopfer, U.; Antczak, C.; Guy, C.; Fuchs, F.; Selzer, P. How Phenotypic Screening Influenced Drug Discovery: Lessons from Five Years of Practice. ASSAY Drug Dev. Technol. 2017, 15, 239–246. [Google Scholar] [CrossRef]
- Rietdijk, J.; Tampere, M.; Pettke, A.; Georgiev, P.; Lapins, M.; Warpman-Berglund, U.; Spjuth, O.; Puumalainen, M.-R.; Carreras-Puigvert, J. A phenomics approach for antiviral drug discovery. BMC Biol. 2021, 19, 156. [Google Scholar] [CrossRef]
- Bray, M.-A.; Singh, S.; Han, H.; Davis, C.T.; Borgeson, B.; Hartland, C.; Kost-Alimova, M.; Gustafsdottir, S.M.; Gibson, C.C.; Carpenter, A. Cell Painting, a high-content image-based assay for morphological profiling using multiplexed fluorescent dyes. Nat. Protoc. 2016, 11, 1757–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiff, L.; Migliori, B.; Chen, Y.; Carter, D. Deep learning and automated Cell Painting reveal Parkinson’s disease-specific signatures in primary patient fibroblasts. bioRxiv 2020. [Google Scholar] [CrossRef]
- Moffat, J.G.; Vincent, F.; Lee, J.A.; Eder, J.; Prunotto, M. Opportunities and challenges in phenotypic drug discovery: An industry perspective. Nat. Rev. Drug Discov. 2017, 16, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, C.; Audia, J.; Austin, C.; Baell, J.; Bennett, J.; Blagg, J.; Bountra, C.; Brennan, P.; Brown, P.; Bunnage, M.E.; et al. The promise and peril of chemical probes. Nat. Chem. Biol. 2015, 11, 536–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, P.J.; Müller, S. Open access chemical probes for epigenetic targets. Futur. Med. Chem. 2015, 7, 1901–1917. [Google Scholar] [CrossRef] [Green Version]
- Drewes, G.; Knapp, S. Chemoproteomics and Chemical Probes for Target Discovery. Trends Biotechnol. 2018, 36, 1275–1286. [Google Scholar] [CrossRef]
- Bunnage, M.E.; Chekler, E.L.P.; Jones, L. Target validation using chemical probes. Nat. Chem. Biol. 2013, 9, 195–199. [Google Scholar] [CrossRef]
- Wells, C.I.; Al-Ali, H.; Andrews, D.M.; Asquith, C.R.M.; Axtman, A.D.; Dikic, I.; Ebner, D.; Ettmayer, P.; Fischer, C.; Frederiksen, M.; et al. The Kinase Chemogenomic Set (KCGS): An Open Science Resource for Kinase Vulnerability Identification. Int. J. Mol. Sci. 2021, 22, 566. [Google Scholar] [CrossRef]
- Canham, S.M.; Wang, Y.; Cornett, A.; Auld, D.S.; Baeschlin, D.K.; Patoor, M.; Skaanderup, P.R.; Honda, A.; Llamas, L.; Wendel, G.; et al. Systematic Chemogenetic Library Assembly. Cell Chem. Biol. 2020, 27, 1124–1129. [Google Scholar] [CrossRef]
- Dafniet, B.; Cerisier, N.; Boezio, B.; Clary, A.; Ducrot, P.; Dorval, T.; Gohier, A.; Brown, D.; Audouze, K.; Taboureau, O. Development of a chemogenomics library for phenotypic screening. J. Chemin. 2021, 13, 91. [Google Scholar] [CrossRef]
- Müller, S.; Ackloo, S.; Arrowsmith, C.H.; Bauser, M.; Baryza, J.L.; Blagg, J.; Boettcher, J.; Bountra, C.; Brown, P.; Bunnage, M.; et al. Donated chemical probes for open science. eLife 2018, 7, 7. [Google Scholar] [CrossRef]
- Bredel, M.; Jacoby, E. Chemogenomics: An emerging strategy for rapid target and drug discovery. Nat. Rev. Genet. 2004, 5, 262–275. [Google Scholar] [CrossRef] [Green Version]
- Jones, L.; Bunnage, M.E. Applications of chemogenomic library screening in drug discovery. Nat. Rev. Drug Discov. 2017, 16, 285–296. [Google Scholar] [CrossRef]
- Caron, P.R.; Mullican, M.D.; Mashal, R.D.; Wilson, K.P.; Su, M.S.; Murcko, M. Chemogenomic approaches to drug discovery. Curr. Opin. Chem. Biol. 2001, 5, 464–470. [Google Scholar] [CrossRef]
- >EUbOPEN. Available online: https://www.eubopen.org/ (accessed on 5 January 2022).
- Carter, A.J.; Kraemer, O.; Zwick, M.; Mueller-Fahrnow, A.; Arrowsmith, C.H.; Edwards, A.M. Target 2035: Probing the human proteome. Drug Discov. Today 2019, 24, 2111–2115. [Google Scholar] [CrossRef]
- Kawamura, T.; Kawatani, M.; Muroi, M.; Kondoh, Y.; Futamura, Y.; Aono, H.; Tanaka, M.; Honda, K.; Osada, H. Proteomic profiling of small-molecule inhibitors reveals dispensability of MTH1 for cancer cell survival. Sci. Rep. 2016, 6, 26521. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Wang, Y.; Cheff, D.M.; Hall, M.D.; Shen, M. Predictive models for estimating cytotoxicity on the basis of chemical structures. Bioorg. Med. Chem. 2020, 28, 115422. [Google Scholar] [CrossRef]
- Tang, H.; Duggan, S.; Richardson, P.L.; Marin, V.; Warder, S.E.; McLoughlin, S.M. Target Identification of Compounds from a Cell Viability Phenotypic Screen Using a Bead/Lysate-Based Affinity Capture Platform. J. Biomol. Screen. 2015, 21, 201–211. [Google Scholar] [CrossRef] [Green Version]
- Howarth, A.; Schröder, M.; Montenegro, R.C.; Drewry, D.H.; Sailem, H.; Millar, V.; Müller, S.; Ebner, D.V. HighVia—A Flexible Live-Cell High-Content Screening Pipeline to Assess Cellular Toxicity. SLAS Discov. Adv. Sci. Drug Discov. 2020, 25, 801–811. [Google Scholar] [CrossRef]
- Chen, A.Y.; Yu, C.; Bodley, A.; Peng, L.F.; Liu, L. A new mammalian DNA topoisomerase I poison Hoechst 33342: Cytotoxicity and drug resistance in human cell cultures. Cancer Res. 1993, 53, 1332–1337. [Google Scholar] [PubMed]
- Durand, R.E.; Olive, P.L. Cytotoxicity, Mutagenicity and DNA damage by Hoechst 33342. J. Histochem. Cytochem. 1982, 30, 111–116. [Google Scholar] [CrossRef] [Green Version]
- Camilleri-Broët, S.; Vanderwerff, H.; Caldwell, E.; Hockenbery, D. Distinct Alterations in Mitochondrial Mass and Function Characterize Different Models of Apoptosis. Exp. Cell Res. 1998, 239, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Márquez-Jurado, S.; Díaz-Colunga, J.; das Neves, R.P.; Martinez-Lorente, A.; Almazan, F.; Guantes, R.; Iborra, F.J. Mitochondrial levels determine variability in cell death by modulating apoptotic gene expression. Nat. Commun. 2018, 9, 389. [Google Scholar] [CrossRef] [PubMed]
- Hsiang, Y.H.; Hertzberg, R.; Hecht, S.; Liu, L. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J. Biol. Chem. 1985, 260, 14873–14878. [Google Scholar] [CrossRef]
- Da Motta, L.L.; Ledaki, I.; Purshouse, K.; Haider, S.; De Bastiani, M.A.; Baban, D.; Morotti, M.; Steers, G.; Wigfield, S.; Bridges, E.; et al. The BET inhibitor JQ1 selectively impairs tumour response to hypoxia and downregulates CA9 and angiogenesis in triple negative breast cancer. Oncogene 2017, 36, 122–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francipane, M.G.; Lagasse, E. Selective targeting of human colon cancer stem-like cells by the mTOR inhibitor Torin-1. Oncotarget 2013, 4, 1948–1962. [Google Scholar] [CrossRef] [PubMed]
- Styrt, B.; Johnson, P.C.; Klempner, M.S. Differential lysis of plasma membranes and granules of human neutrophilis by digitonin. Tissue Cell 1985, 17, 793–800. [Google Scholar] [CrossRef]
- Wen, N.; Guo, B.; Zheng, H.; Xu, L.; Liang, H.; Wang, Q.; Wang, D.; Chen, X.; Zhang, S.; Li, Y.; et al. Bromodomain inhibitor jq1 induces cell cycle arrest and apoptosis of glioma stem cells through the VEGF/PI3K/AKT signaling pathway. Int. J. Oncol. 2019, 55, 879–895. [Google Scholar] [CrossRef] [PubMed]
- Vogl, D.T.; Raje, N.; Jagannath, S.; Richardson, P.; Hari, P.; Orlowski, R.; Supko, J.G.; Tamang, D.; Yang, M.; Jones, S.S.; et al. Ricolinostat, the First Selective Histone Deacetylase 6 Inhibitor, in Combination with Bortezomib and Dexamethasone for Relapsed or Refractory Multiple Myeloma. Clin. Cancer Res. 2017, 23, 3307–3315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.; Alvarado, A.S. Flow cytometry methods for the study of cell-cycle parameters of planarian stem cells. Dev. Dyn. 2009, 238, 1111–1117. [Google Scholar] [CrossRef] [PubMed]
- Sakaue-Sawano, A.; Kurokawa, H.; Morimura, T.; Hanyu, A.; Hama, H.; Osawa, H.; Kashiwagi, S.; Fukami, K.; Miyata, T.; Miyoshi, H.; et al. Visualizing Spatiotemporal Dynamics of Multicellular Cell-Cycle Progression. Cell 2008, 132, 487–498. [Google Scholar] [CrossRef] [Green Version]
- Yano, S.; Hoffman, R.M. Real-Time Determination of the Cell-Cycle Position of Individual Cells within Live Tumors Using FUCCI Cell-Cycle Imaging. Cells 2018, 7, 168. [Google Scholar] [CrossRef] [Green Version]
- Zielke, N.; Edgar, B.A. FUCCI sensors: Powerful new tools for analysis of cell proliferation. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 469–487. [Google Scholar] [CrossRef]
- Jorda, R.; Hendrychová, D.; Voller, J.; Řezníčková, E.; Gucký, T.; Kryštof, V. How Selective Are Pharmacological Inhibitors of Cell-Cycle-Regulating Cyclin-Dependent Kinases? J. Med. Chem. 2018, 61, 9105–9120. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martínez, C.; Gelbert, L.M.; Lallena, M.J.; de Dios, A. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs. Bioorg. Med. Chem. Lett. 2015, 25, 3420–3435. [Google Scholar] [CrossRef]
- Reiners, J.J.; Clift, R.; Mathieu, P. Suppression of cell cycle progression by flavonoids: Dependence on the aryl hydrocarbon receptor. Carcinogenesis 1999, 20, 1561–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demidenko, Z.N.; Kalurupalle, S.; Hanko, C.; Lim, C.-U.; Broude, E.; Blagosklonny, M.V. Mechanism of G1-like arrest by low concentrations of paclitaxel: Next cell cycle p53-dependent arrest with sub G1 DNA content mediated by prolonged mitosis. Oncogene 2008, 27, 4402–4410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.-H.; Wang, H.-S.; Soong, Y.-K. Paclitaxel-induced cell death. Cancer 2000, 88, 2619–2628. [Google Scholar] [CrossRef]
- Schick, U.; Kyula, J.; Barker, H.; Patel, R.; Zaidi, S.; Gregory, C.; Hafsi, H.; Roulstone, V.; Deutsch, E.; McLaughlin, M.; et al. Trametinib radiosensitises RAS- and BRAF-mutated melanoma by perturbing cell cycle and inducing senescence. Radiother. Oncol. 2015, 117, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Faulds, D.; Balfour, J.A.; Chrisp, P.; Langtry, H.D. Mitoxantrone. Drugs 1991, 41, 400–449. [Google Scholar] [CrossRef] [PubMed]
- Kluza, J.; Marchetti, P.; Gallego, M.-A.; Lancel, S. Mitochondrial proliferation during apoptosis induced by anticancer agents: Effects of doxorubicin and mitoxantrone on cancer and cardiac cells. Oncogene 2004, 23, 7018–7030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pommier, Y. Topoisomerase I inhibitors: Camptothecins and beyond. Nat. Cancer 2006, 6, 789–802. [Google Scholar] [CrossRef]
- Tsunetoh, S.; Terai, Y.; Sasaki, H.; Tanabe, A.; Tanaka, Y.; Sekijima, T.; Fujioka, S.; Kawaguchi, H.; Kanemura, M.; Yamashita, Y.; et al. Topotecan as a molecular targeting agent which blocks the Akt and VEGF cascade in platinum-resistant ovarian cancers. Cancer Biol. Ther. 2010, 10, 1137–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Aamri, H.M.; Ku, H.; Irving, H.R.; Tucci, J.; Meehan-Andrews, T.; Bradley, C. Time dependent response of daunorubicin on cytotoxicity, cell cycle and DNA repair in acute lymphoblastic leukaemia. BMC Cancer 2019, 19, 1–12. [Google Scholar] [CrossRef]
- Ishikawa, C.; Senba, M.; Mori, N. Mitotic kinase PBK/TOPK as a therapeutic target for adult T-cell leukemia/lymphoma. Int. J. Oncol. 2018, 53, 801–814. [Google Scholar] [CrossRef] [Green Version]
- Delaney, J.S. Predicting aqueous solubility from structure. Drug Discov. Today 2005, 10, 289–295. [Google Scholar] [CrossRef]
- Knick, V.C.; Eberwein, D.J.; Miller, C.G. Vinorelbine Tartrate and Paclitaxel Combinations: Enhanced Activity Against In Vivo P388 Murine Leukemia Cells. J. Natl. Cancer Inst. 1995, 87, 1072–1077. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.; Krona, C.; Kundu, S.; Doroszko, M.; Baskaran, S.; Schmidt, L.; Vinel, C.; Almstedt, E.; Elgendy, R.; Elfineh, L.; et al. A Patient-Derived Cell Atlas Informs Precision Targeting of Glioblastoma. Cell Rep. 2020, 32, 107897. [Google Scholar] [CrossRef] [PubMed]
- Röhm, S.; Berger, B.-T.; Schröder, M.; Chaikuad, A.; Winkel, R.; Hekking, K.F.W.; Benningshof, J.J.C.; Mueller, G.; Tesch, R.; Kudolo, M.; et al. Fast Iterative Synthetic Approach toward Identification of Novel Highly Selective p38 MAP Kinase Inhibitors. J. Med. Chem. 2019, 62, 10757–10782. [Google Scholar] [CrossRef] [PubMed]
- Pardo, O.; Latigo, J.; Jeffery, R.E.; Nye, E.; Poulsom, R.; Spencer-Dene, B.; Lemoine, N.; Stamp, G.W.; Aboagye, E.; Seckl, M.J. The Fibroblast Growth Factor Receptor Inhibitor PD173074 Blocks Small Cell Lung Cancer Growth In Vitro and In Vivo. Cancer Res. 2009, 69, 8645–8651. [Google Scholar] [CrossRef] [Green Version]
- Ippolito, T.; Tang, G.; Mavis, C.; Gu, J.J.; Hernandez-Ilizaliturri, F.J.; Barth, M.J. Omipalisib (GSK458), a Novel Pan-PI3K/mTOR Inhibitor, Exhibits In Vitro Anti-Lymphoma Activity in Chemotherapy-Sensitive and -Resistant Models of Burkitt Lymphoma. Blood 2016, 128, 5376. [Google Scholar] [CrossRef]
- Aveic, S.; Corallo, D.; Porcù, E.; Pantile, M.; Boso, D.; Zanon, C.; Viola, G.; Sidarovich, V.; Mariotto, E.; Quattrone, A.; et al. TP-0903 inhibits neuroblastoma cell growth and enhances the sensitivity to conventional chemotherapy. Eur. J. Pharmacol. 2018, 818, 435–448. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, H.; Chen, Y.; Wang, M.; Ding, G.; Li, T. Trk inhibitor GNF-5837 suppresses the tumor growth, survival and migration of renal cell carcinoma. Oncol. Rep. 2019, 42, 2039–2048. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.J.; Mittal, B.; Walsh, F.S.; Doherty, P. A Ca2+/Calmodulin Kinase Inhibitor, KN-62, Inhibits Neurite Outgrowth Stimulated by CAMs and FGF. Mol. Cell. Neurosci. 1995, 6, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.; Szabo, E.; Machado, R.A.C.; Broggini-Tenzer, A.; Walter, A.; Lobell, M.; Heldmann, D.; Süssmeier, F.; Grünewald, S.; Weller, M. Novel TIE-2 inhibitor BAY-826 displaysin vivoefficacy in experimental syngeneic murine glioma models. J. Neurochem. 2016, 140, 170–182. [Google Scholar] [CrossRef]
- Lino, M.; Wan, M.H.; Rocca, A.S.; Ngai, D.; Shobeiri, N.; Hou, G.; Ge, C.; Franceschi, R.T.; Bendeck, M.P. Diabetic Vascular Calcification Mediated by the Collagen Receptor Discoidin Domain Receptor 1 via the Phosphoinositide 3-Kinase/Akt/Runt-Related Transcription Factor 2 Signaling Axis. Arter. Thromb. Vasc. Biol. 2018, 38, 1878–1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reckamp, K.L.; Giaccone, G.; Camidge, D.R.; Gadgeel, S.M.; Khuri, F.R.; Engelman, J.A.; Koczywas, M.; Rajan, A.; Campbell, A.K.; Gernhardt, D.; et al. A phase 2 trial of dacomitinib (PF-00299804), an oral, irreversible pan-HER (human epidermal growth factor receptor) inhibitor, in patients with advanced non–small cell lung cancer after failure of prior chemotherapy and erlotinib. Cancer 2014, 120, 1145–1154. [Google Scholar] [CrossRef] [Green Version]
- Hafner, M.; Niepel, M.; Chung, M.; Sorger, P.K. Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nat. Methods 2016, 13, 521–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutros, M.; Heigwer, F.; Laufer, C. Microscopy-Based High-Content Screening. Cell 2015, 163, 1314–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrasekaran, S.N.; Ceulemans, H.; Boyd, J.D.; Carpenter, A.E. Image-based profiling for drug discovery: Due for a machine-learning upgrade? Nat. Rev. Drug Discov. 2021, 20, 145–159. [Google Scholar] [CrossRef]
- Cole, R. Live-cell imaging. Cell Adhes. Migr. 2014, 8, 452–459. [Google Scholar] [CrossRef] [Green Version]
- Neumann, B.; Held, M.; Liebel, U.; Erfle, H.; Rogers, P.; Pepperkok, R.; Ellenberg, J. High-throughput RNAi screening by time-lapse imaging of live human cells. Nat. Methods 2006, 3, 385–390. [Google Scholar] [CrossRef]
- Liu, Y.; Fares, M.; Dunham, N.P.; Gao, Z.; Miao, K.; Jiang, X.; Bollinger, S.S.; Boal, A.K.; Zhang, X. AgHalo: A Facile Fluorogenic Sensor to Detect Drug-Induced Proteome Stress. Angew. Chem. Int. Ed. 2017, 56, 8672–8676. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Nissink, J.W.M. Seven Year Itch: Pan-Assay Interference Compounds (PAINS) in 2017—Utility and Limitations. ACS Chem. Biol. 2018, 13, 36–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravorty, S.J.; Chan, J.; Greenwood, M.N.; Popa-Burke, I.; Remlinger, K.S.; Pickett, S.D.; Green, D.V.S.; Fillmore, M.C.; Dean, T.W.; Luengo, J.I.; et al. Nuisance Compounds, PAINS Filters, and Dark Chemical Matter in the GSK HTS Collection. SLAS Discov. Adv. Sci. Drug Discov. 2018, 23, 532–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jasial, S.; Hu, Y.; Bajorath, J. How Frequently Are Pan-Assay Interference Compounds Active? Large-Scale Analysis of Screening Data Reveals Diverse Activity Profiles, Low Global Hit Frequency, and Many Consistently Inactive Compounds. J. Med. Chem. 2017, 60, 3879–3886. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gul, N.; Karlsson, J.; Tängemo, C.; Linsefors, S.; Tuyizere, S.; Perkins, R.; Ala, C.; Zou, Z.; Larsson, E.; Bergö, M.O.; et al. The MTH1 inhibitor TH588 is a microtubule-modulating agent that eliminates cancer cells by activating the mitotic surveillance pathway. Sci. Rep. 2019, 9, 14667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowarz, E.; Löscher, D.; Marschalek, R. Optimized Sleeping Beauty transposons rapidly generate stable transgenic cell lines. Biotechnol. J. 2015, 10, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Izsvak, Z. Efficient stable gene transfer into human cells by the Sleeping Beauty transposon vectors. Methods 2009, 49, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Wiggers, C.R.M.; Govers, A.M.A.P.; Lelieveld, D.; Egan, D.A.; Zwaan, C.M.; Sonneveld, E.; Coffer, P.J.; Bartels, M. Epigenetic drug screen identifies the histone deacetylase inhibitor NSC3852 as a potential novel drug for the treatment of pediatric acute myeloid leukemia. Pediatric Blood Cancer 2019, 66, e27785. [Google Scholar] [CrossRef] [PubMed]
- Dilshara, M.G.; Jayasooriya, R.G.P.T.; Karunarathne, W.A.H.M.; Choi, Y.H.; Kim, G.-Y. Camptothecin induces mitotic arrest through Mad2-Cdc20 complex by activating the JNK-mediated Sp1 pathway. Food Chem. Toxicol. 2019, 127, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Lüpertz, R.; Wätjen, W.; Kahl, R.; Chovolou, Y. Dose- and time-dependent effects of doxorubicin on cytotoxicity, cell cycle and apoptotic cell death in human colon cancer cells. Toxicology 2010, 271, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, J.M.; Wu, G.; Zeng, C.; Zhu, J.; Meng, F.; Patel, S.; Wang, W.; Ficarro, S.B.; Leggett, A.L.; Powell, C.E.; et al. SRPKIN-1: A Covalent SRPK1/2 Inhibitor that Potently Converts VEGF from Pro-angiogenic to Anti-angiogenic Isoform. Cell Chem. Biol. 2018, 25, 460–470.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.N.; Lal, S.K.; Kumar, P.; Khan, A.U. Effect of mitoxantrone on proliferation dynamics and cell-cycle progression. Biosci. Rep. 2010, 30, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Bernhart, E.; Stuendl, N.; Kaltenegger, H.; Windpassinger, C.; Donohue, N.; Leithner, A.; Lohberger, B. Histone deacetylase inhibitors vorinostat and panobinostat induce G1 cell cycle arrest and apoptosis in multidrug resistant sarcoma cell lines. Oncotarget 2017, 8, 77254–77267. [Google Scholar] [CrossRef] [Green Version]
- Marshall, C.B.; Pippin, J.W.; Krofft, R.D.; Shankland, S.J. Puromycin aminonucleoside induces oxidant-dependent DNA damage in podocytes in vitro and in vivo. Kidney Int. 2006, 70, 1962–1973. [Google Scholar] [CrossRef] [Green Version]
- Bruno, S.; Ardelt, B.; Skierski, J.S.; Traganos, F.; Darzynkiewicz, Z. Different Effects of Staurosporine, an Inhibitor of Protein Kinases, on the Cell Cycle and Chromatin Structure of Normal and Leukemic Lymphocytes. Cancer Res. 1992, 52, 470–473. [Google Scholar]
- Murai, A.; Ebara, S.; Sasaki, S.; Ohashi, T.; Miyazaki, T.; Nomura, T.; Araki, S. Synergistic apoptotic effects in cancer cells by the combination of CLK and Bcl-2 family inhibitors. PLoS ONE 2020, 15, e0240718. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tjaden, A.; Chaikuad, A.; Kowarz, E.; Marschalek, R.; Knapp, S.; Schröder, M.; Müller, S. Image-Based Annotation of Chemogenomic Libraries for Phenotypic Screening. Molecules 2022, 27, 1439. https://doi.org/10.3390/molecules27041439
Tjaden A, Chaikuad A, Kowarz E, Marschalek R, Knapp S, Schröder M, Müller S. Image-Based Annotation of Chemogenomic Libraries for Phenotypic Screening. Molecules. 2022; 27(4):1439. https://doi.org/10.3390/molecules27041439
Chicago/Turabian StyleTjaden, Amelie, Apirat Chaikuad, Eric Kowarz, Rolf Marschalek, Stefan Knapp, Martin Schröder, and Susanne Müller. 2022. "Image-Based Annotation of Chemogenomic Libraries for Phenotypic Screening" Molecules 27, no. 4: 1439. https://doi.org/10.3390/molecules27041439