
Isolation and In Silico Anti-SARS-CoV-2 Papain-Like Protease Potentialities of Two Rare 2-Phenoxychromone Derivatives from Artemisia spp.

 , , , , , ,
, , , , , ,  and
and 
Abstract
:1. Introduction
2. Results
2.1. Isolation of Compounds
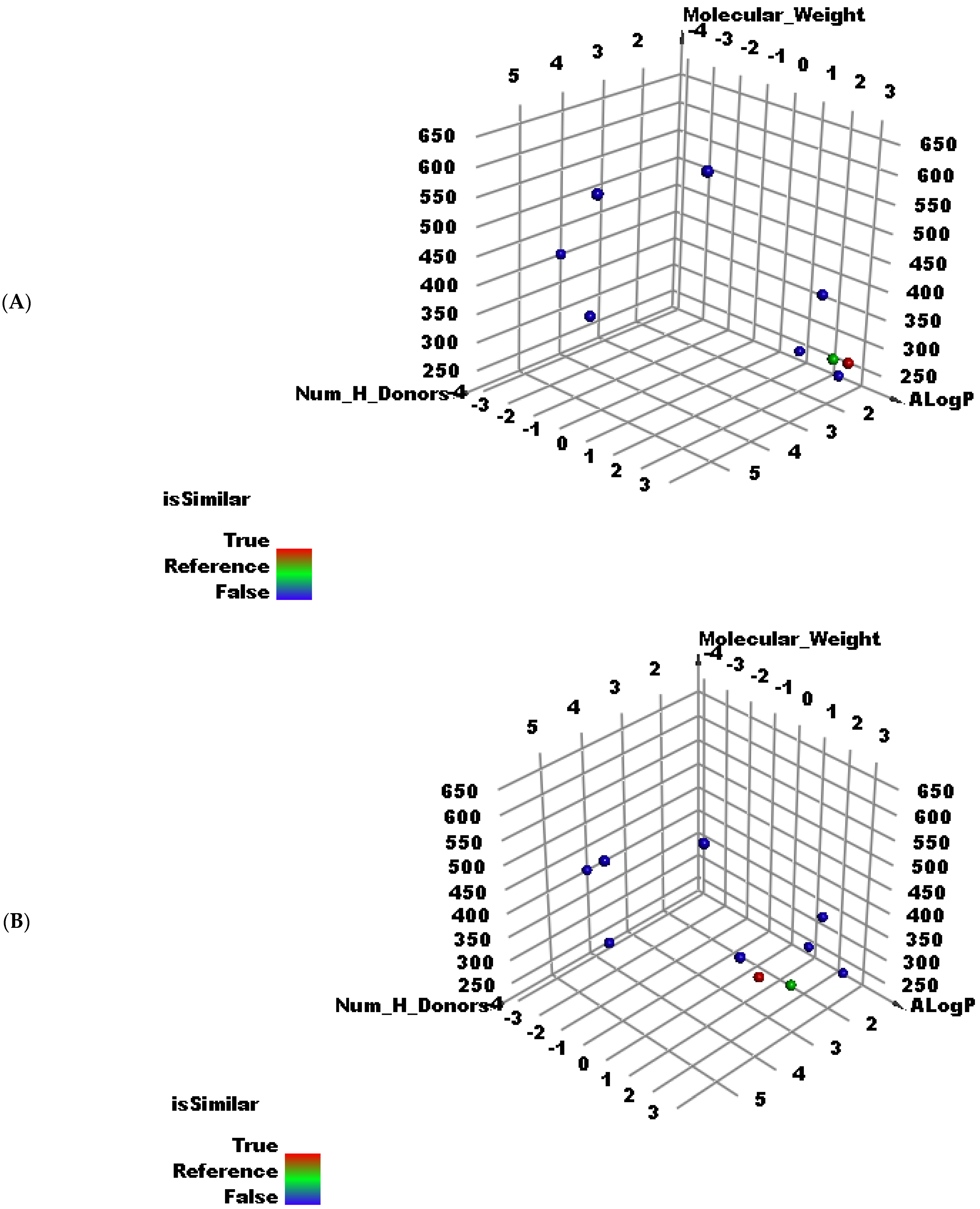
2.2. Molecular Similarity Studies
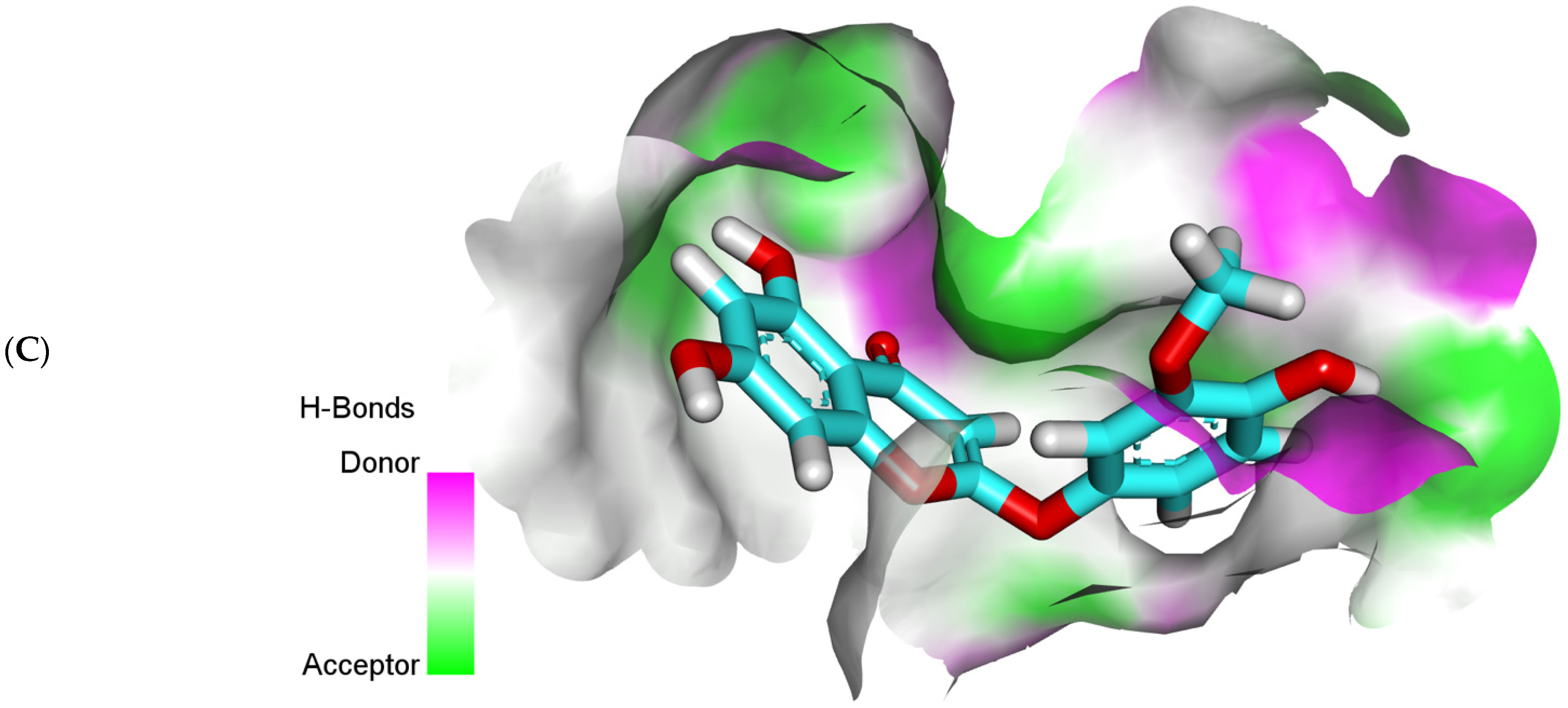
2.3. Fingerprints Studies
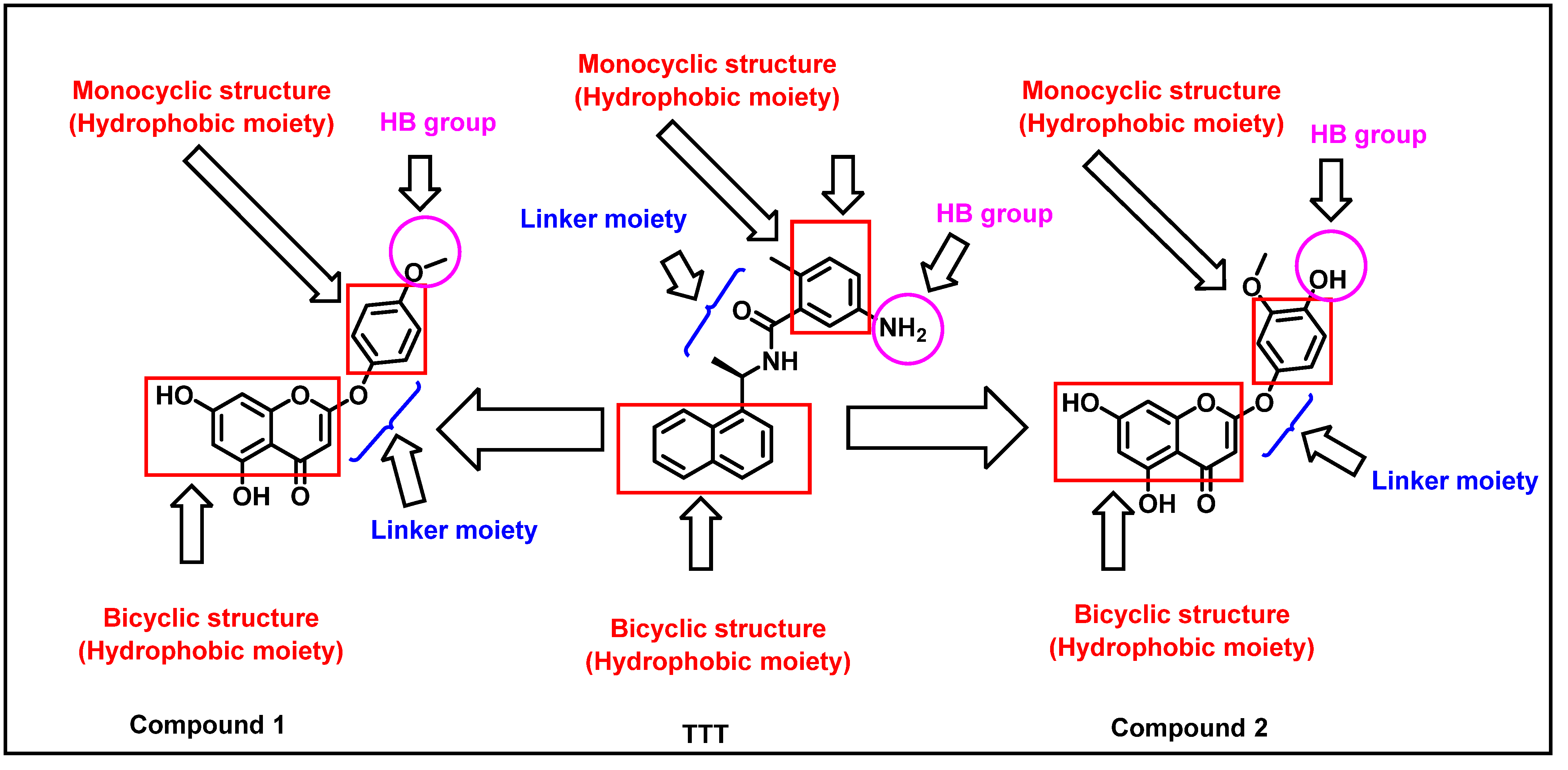
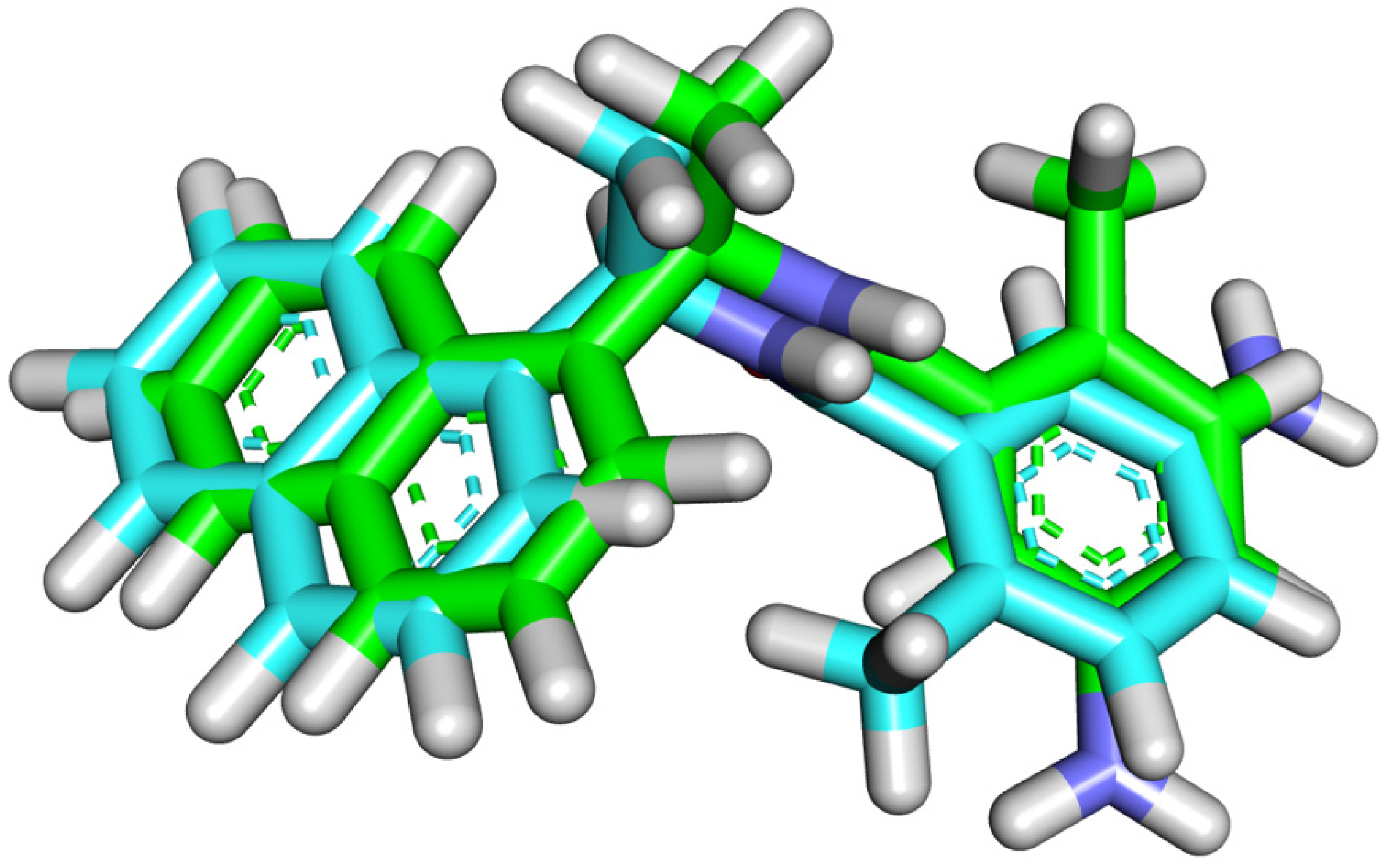
2.4. Pharmacophoric Features and Flexible Alignment Studies
2.5. Docking Studies
2.6. In Silico ADMET Study
2.7. In Silico Toxicity Studies
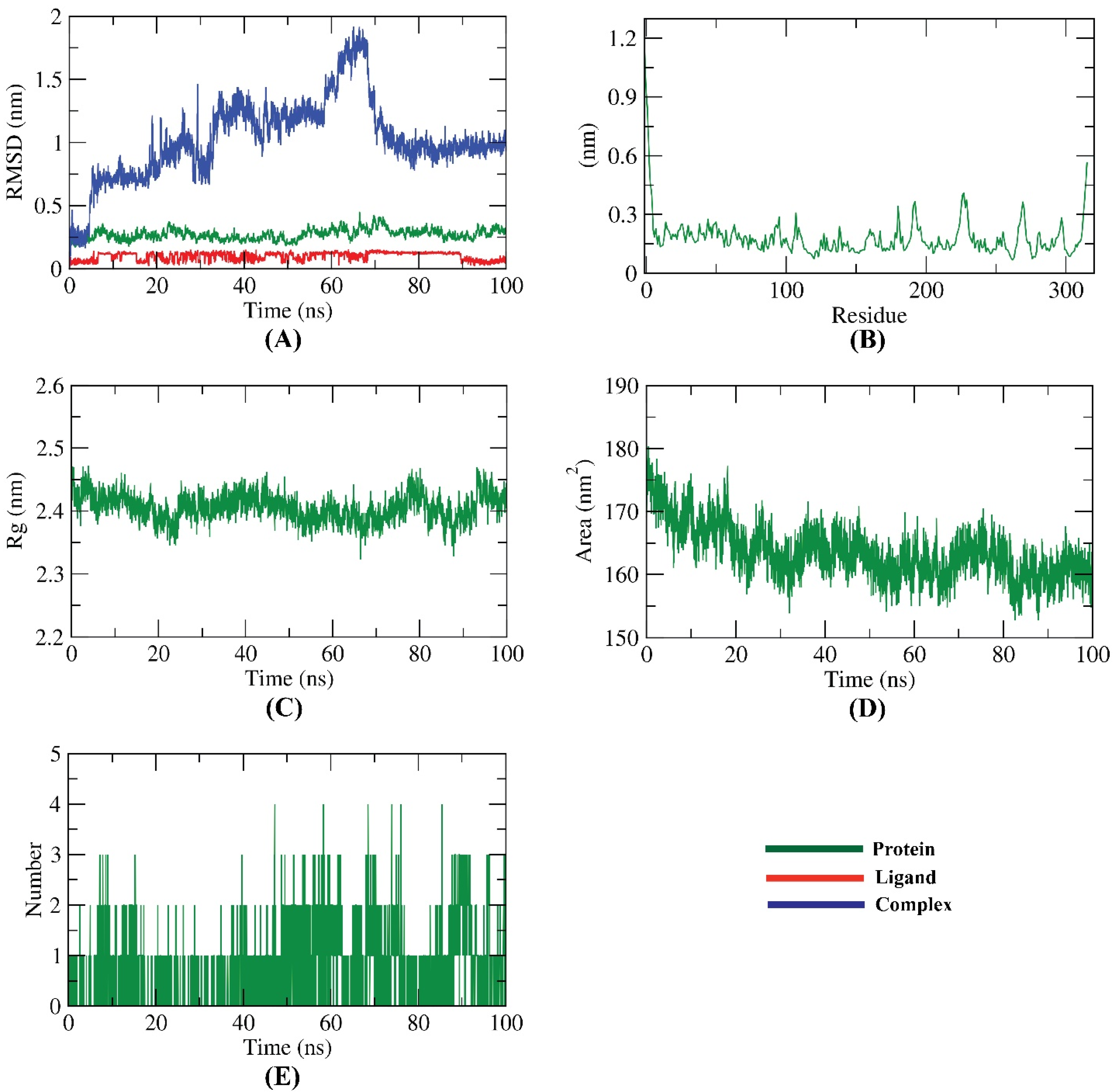
2.8. Molecular Dynamics (MD) Simulation Studies
3. Experimental
3.1. Isolation and Structure Elucidation of Compounds
3.2. Molecular Similarity
3.3. Fingerprint Study
3.4. DFT
3.5. Docking Studies
3.6. ADMET
3.7. Toxicity Studies
3.8. Molecular Dynamics Simulations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Metwaly, A.M.; Ghoneim, M.M.; Eissa, I.H.; Elsehemy, I.A.; Mostafa, A.E.; Hegazy, M.M.; Afifi, W.M.; Dou, D. Traditional ancient Egyptian medicine: A review. Saudi J. Biol. Sci. 2021, 28, 5823–5832. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yang, Y.; Metwaly, A.M.; Xue, Y.; Shi, Y.; Dou, D. The Chinese herbal formulae (Yitangkang) exerts an antidiabetic effect through the regulation of substance metabolism and energy metabolism in type 2 diabetic rats. J. Ethnopharmacol. 2019, 239, 111942. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Sun, B.; Huang, Q.; Peng, S.; Zhou, Y.; Zhang, Y. Quantitative knowledge presentation models of traditional Chinese medicine (TCM): A review. Artif. Intell. Med. 2020, 103, 101810. [Google Scholar] [CrossRef] [PubMed]
- Zhanzhaxina, A.; Suleimen, Y.; Metwaly, A.M.; Eissa, I.H.; Elkaeed, E.B.; Suleimen, R.; Ishmuratova, M.; Akatan, K.; Luyten, W. In Vitro and In Silico Cytotoxic and Antibacterial Activities of a Diterpene from Cousinia alata Schrenk. J. Chem. 2021, 2021, 5542455. [Google Scholar] [CrossRef]
- Hegazy, M.M.; Metwaly, A.M.; Mostafa, A.E.; Radwan, M.M.; Mehany, A.B.M.; Ahmed, E.; Enany, S.; Magdeldin, S.; Afifi, W.M.; ElSohly, M.A. Biological and chemical evaluation of some African plants belonging to Kalanchoe species: Antitrypanosomal, cytotoxic, antitopoisomerase I activities and chemical profiling using ultra-performance liquid chromatography/quadrupole-time-of-flight mass spectrometer. Pharmacogn. Mag. 2021, 17, 6. [Google Scholar]
- Imieje, V.O.; Zaki, A.A.; Metwaly, A.M.; Eissa, I.H.; Elkaeed, E.B.; Ali, Z.; Khan, I.A.; Falodun, A. Antileishmanial Derivatives of Humulene from Asteriscus hierochunticus with in silico Tubulin Inhibition Potential. Rec. Nat. Prod. 2021, 16, 150. [Google Scholar] [CrossRef]
- Jalmakhanbetova, R.; Elkaeed, E.B.; Eissa, I.H.; Metwaly, A.M.; Suleimen, Y.M. Synthesis and Molecular Docking of Some Grossgemin Amino Derivatives as Tubulin Inhibitors Targeting Colchicine Binding Site. J. Chem. 2021, 2021, 5586515. [Google Scholar] [CrossRef]
- Suleimen, Y.M.; Metwaly, A.M.; Mostafa, A.E.; Elkaeed, E.B.; Liu, H.-W.; Basnet, B.B.; Suleimen, R.N.; Ishmuratova, M.Y.; Turdybekov, K.M.; Van Hecke, K. Isolation, Crystal Structure, and In Silico Aromatase Inhibition Activity of Ergosta-5, 22-dien-3β-ol from the Fungus Gyromitra esculenta. J. Chem. 2021, 2021, 5529786. [Google Scholar] [CrossRef]
- Ghoneim, M.M.; Afifi, W.M.; Ibrahim, M.; Elagawany, M.; Khayat, M.T.; Aboutaleb, M.H.; Metwaly, A.M. Biological evaluation and molecular docking study of metabolites from Salvadora Persica L. Growing in Egypt. Pharmacogn. Mag. 2019, 15, 232. [Google Scholar]
- Liu, L.; Luo, S.; Yu, M.; Metwaly, A.M.; Ran, X.; Ma, C.; Dou, D.; Cai, D. Chemical Constituents of Tagetes patula and Their Neuroprotecting Action. Nat. Prod. Commun. 2020, 15. [Google Scholar] [CrossRef]
- Metwaly, A.M.; Ghoneim, M.M.; Musa, A. Two new antileishmanial diketopiperazine alkaloids from the endophytic fungus Trichosporum sp. Derpharmachemica 2015, 7, 322–327. [Google Scholar]
- Yassin, A.M.; El-Deeb, N.M.; Metwaly, A.M.; El Fawal, G.F.; Radwan, M.M.; Hafez, E.E. Induction of apoptosis in human cancer cells through extrinsic and intrinsic pathways by Balanites aegyptiaca furostanol saponins and saponin-coated silvernanoparticles. Appl. Biochem. Biotechnol. 2017, 182, 1675–1693. [Google Scholar] [CrossRef]
- Sharaf, M.H.; El-Sherbiny, G.M.; Moghannem, S.A.; Abdelmonem, M.; Elsehemy, I.A.; Metwaly, A.M.; Kalaba, M.H. New combination approaches to combat methicillin-resistant Staphylococcus aureus (MRSA). Sci. Rep. 2021, 11, 4240. [Google Scholar] [CrossRef]
- Metwaly, A.M.; Lianlian, Z.; Luqi, H.; Deqiang, D. Black ginseng and its saponins: Preparation, phytochemistry and pharmacological effects. Molecules 2019, 24, 1856. [Google Scholar] [CrossRef] [Green Version]
- Metwaly, A.M.; Kadry, H.A.; Atef, A.; Mohammad, A.-E.I.; Ma, G.; Cutler, S.J.; Ross, S.A. Nigrosphaerin A a new isochromene derivative from the endophytic fungus Nigrospora sphaerica. Phytochem. Lett. 2014, 7, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-M.; Ran, X.-K.; Riaz, M.; Yu, M.; Cai, Q.; Dou, D.-Q.; Metwaly, A.M.; Kang, T.-G.; Cai, D.-C. Chemical constituents of stems and leaves of Tagetespatula L. and its fingerprint. Molecules 2019, 24, 3911. [Google Scholar] [CrossRef] [Green Version]
- Metwaly, A.M.; Fronczek, F.R.; Ma, G.; Kadry, H.A.; Atef, A.; Mohammad, A.-E.I.; Cutler, S.J.; Ross, S.A. Antileukemic α-pyrone derivatives from the endophytic fungus Alternaria phragmospora. Tetrahedron Lett. 2014, 55, 3478–3481. [Google Scholar] [CrossRef] [Green Version]
- Metwaly, A.M.; Wanas, A.S.; Radwan, M.M.; Ross, S.A.; ElSohly, M.A. New α-Pyrone derivatives from the endophytic fungus Embellisia sp. Med. Chem. Res. 2017, 26, 1796–1800. [Google Scholar] [CrossRef]
- Suleimenov, E.; Raldugin, V.; Adekenov, S. Anhydroaustricin from Artemisia albida. Chem. Nat. Compd. 2008, 44, 541–542. [Google Scholar] [CrossRef]
- Suleimenov, E.; Smagulova, F.; Seidakhmetova, R.; Aksartov, R.; Raldugin, V.; Adekenov, S. 4-Epiashantin from Artemisia sieversiana. Chem. Nat. Compd. 2007, 43, 232–233. [Google Scholar] [CrossRef]
- Kikhanova, Z.S.; Iskakova, Z.B.; Dzhalmakhanbetova, R.; Seilkhanov, T.; Ross, S.; Suleimen, E. Constituents of Artemisia austriaca and their biological activity. Chem. Nat. Compd. 2013, 49, 967–968. [Google Scholar] [CrossRef]
- Tashenov, E.; Dzhalmakhanbetova, R.; Smagulova, F.; Dudkin, R.; Gorovoi, P.; Suleiman, E.; Ross, S. Cirsilineol and cubreuva lactone from Artemisia umbrosa and their biological activity. Chem. Nat. Compd. 2013, 49, 97–98. [Google Scholar] [CrossRef]
- Sisengalieva, G.; Suleimen, E.; Ishmuratova, M.Y.; Iskakova, Z.B.; Van Hecke, K. Constituents of Artemisia tschernieviana and their biological activity. Chem. Nat. Compd. 2015, 51, 544–547. [Google Scholar] [CrossRef]
- Suleimenov, E.; Smagulova, F.; Morozova, O.; Raldugin, V.; Bagryanskaya, I.Y.; Gatilov, Y.V.; Yamovoi, V.; Adekenov, S. Sesquiterpene lactones and flavonoids from Artemisia albida. Chem. Nat. Compd. 2005, 41, 689–691. [Google Scholar] [CrossRef]
- Suleimen, E.; Dzhalmakhanbetova, R.; Ishmuratova, M.Y. Flavonoids from Artemisia santolinifolia. Chem. Nat. Compd. 2014, 50, 918–919. [Google Scholar] [CrossRef]
- Suleimenov, E.; Ozek, T.; Demirci, F.; Demirci, B.; Baser, K.; Adekenov, S. Component composition and antimicrobial activity of essential oil from Artemisia kasakorum. Chem. Nat. Compd. 2008, 44, 263–265. [Google Scholar] [CrossRef]
- Suleimenov, E.; Ozek, T.; Demirci, F.; Demirci, B.; Baser, K.; Adekenov, S. Component composition of essential oils of Artemisia lercheana and A. sieversiana of the flora of Kazakhstan. Antimicrobial activity of A. sieversiana essential oil. Chem. Nat. Compd. 2009, 45, 120–123. [Google Scholar] [CrossRef]
- Suleimen, E.M.; Dudkin, R.V.; Gorovoi, P.G.; Wang, M.; Khan, I.; Ross, S.A. Composition and Bioactivity of Artemisia umbrosa Essential Oil. Chem. Nat. Compd. 2014, 50, 545–546. [Google Scholar] [CrossRef]
- Suleimen, E.M.; Ibataev, Z.A.; Iskakova, Z.B.; Ishmuratova, M.Y. Constituent Composition and Biological Activity of Essential Oil from Artemisia gurganica. Chem. Nat. Compd. 2015, 51, 1184–1185. [Google Scholar] [CrossRef]
- Sampietro, D.A.; Lizarraga, E.F.; Ibatayev, Z.A.; Omarova, A.B.; Suleimen, Y.M.; Catalán, C.A.N. Chemical composition and antimicrobial activity of essential oils from Acantholippia deserticola, Artemisia proceriformis, Achillea micrantha and Libanotis buchtormensis against phytopathogenic bacteria and fungi. Nat. Prod. Res. 2016, 30, 1950–1955. [Google Scholar] [CrossRef]
- Suleimen, E.M.; Ibataev, Z.A.; Iskakova, Z.B.; Ishmuratova, M.Y.; Ross, S.A.; Martins, C.H.G. Constituent Composition and Biological Activity of Essential Oil from Artemisia terrae-albae. Chem. Nat. Compd. 2016, 52, 173–175. [Google Scholar] [CrossRef]
- Suleimen, E.M.; Sisengalieva, G.G.; Adilkhanova, A.A.; Dudkin, R.V.; Gorovoi, P.G.; Iskakova, Z.B. Composition and Biological Activity of Essential Oil from Artemisia keiskeana. Chem. Nat. Compd. 2019, 55, 154–156. [Google Scholar] [CrossRef]
- Suleimen, E.M.; Dudkin, R.V.; Gorovoi, P.G.; Wang, M.; Khan, I.; Ross, S.A. Constituent Compositions of Essential Oils from Artemisia littoricola and A. mandshurica. Chem. Nat. Compd. 2015, 51, 790–792. [Google Scholar] [CrossRef]
- Shatar, S.; Altantsetseg, S.; Darijimaa, S. The essential oil composition of six Artemisia species from Mongolia. J. Essent. Oil Bear. Plants 1999, 2, 56–67. [Google Scholar]
- Ozek, G.; Suleimen, Y.; Tabanca, N.; Doudkin, R.; Gorovoy, P.G.; Goger, F.; Wedge, D.E.; Ali, A.; Khan, I.A.; Baser, K.H. Chemical Diversity and Biological Activity of the Volatiles of Five Artemisia Species from Far East Russia; Agricultural Research Service University, Natural Products Utilization: Oxford, MS, USA, 2014. [Google Scholar]
- Vasilyeva, A.P.G.; Goloskokov, V.P.; Zaitseva, L.G. Flora of Kazakhstan. Alma-Ata 1966, 9, 106–107. [Google Scholar]
- Plantarium. Artemisia glauca, Taxon Details. Available online: https://www.plantarium.ru/page/view/item/3891.html (accessed on 23 November 2021).
- Vasilyeva, A.P.G.; Goloskokov, V.P.; Zaitseva, L.G. Flora of Kazakhstan, Science; N. V. Pavlova, Alma-Ata, 1966; Volume V. [Google Scholar]
- WHO. WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 25 December 2021).
- Bharatam, P.V. Computer-Aided Drug Design; Springer: Singapore, 2021; pp. 137–210. [Google Scholar] [CrossRef]
- Sabe, V.T.; Ntombela, T.; Jhamba, L.A.; Maguire, G.E.; Govender, T.; Naicker, T.; Kruger, H.G. Current trends in computer aided drug design and a highlight of drugs discovered via computational techniques: A review. Eur. J. Med. Chem. 2021, 224, 113705. [Google Scholar] [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [Green Version]
- Jalmakhanbetova, R.I.; Suleimen, Y.M.; Oyama, M.; Elkaeed, E.B.; Eissa, I.; Suleimen, R.N.; Metwaly, A.M.; Ishmuratova, M.Y. Isolation and In Silico Anti-COVID-19 Main Protease (Mpro) Activities of Flavonoids and a Sesquiterpene Lactone from Artemisia sublessingiana. J. Chem. 2021, 2021, 5547013. [Google Scholar] [CrossRef]
- Al-Karmalawy, A.A.; Dahab, M.A.; Metwaly, A.M.; Elhady, S.S.; Elkaeed, E.B.; Eissa, I.H.; Darwish, K.M. Molecular Docking and Dynamics Simulation Revealed the Potential Inhibitory Activity of ACEIs Against SARS-CoV-2 Targeting the hACE2 Receptor. Front. Chem. 2021, 9, 661230. [Google Scholar] [CrossRef]
- Alesawy, M.S.; Abdallah, A.E.; Taghour, M.S.; Elkaeed, E.B.; H Eissa, I.; Metwaly, A.M. In Silico Studies of Some Isoflavonoids as Potential Candidates against COVID-19 Targeting Human ACE2 (hACE2) and Viral Main Protease (Mpro). Molecules 2021, 26, 2806. [Google Scholar] [CrossRef]
- El-Demerdash, A.; Metwaly, A.M.; Hassan, A.; El-Aziz, A.; Mohamed, T.; Elkaeed, E.B.; Eissa, I.H.; Arafa, R.K.; Stockand, J.D. Comprehensive virtual screening of the antiviral potentialities of marine polycyclic guanidine alkaloids against SARS-CoV-2 (COVID-19). Biomolecules 2021, 11, 460. [Google Scholar] [CrossRef]
- Eissa, I.H.; Khalifa, M.M.; Elkaeed, E.B.; Hafez, E.E.; Alsfouk, A.A.; Metwaly, A.M. In Silico Exploration of Potential Natural Inhibitors against SARS-CoV-2 nsp10. Molecules 2021, 26, 6151. [Google Scholar] [CrossRef]
- León, L.; Maldonado, E.; Cruz, A.; Ortega, A. Tenuiflorins A–C: New 2-Phenoxychromones from the Leaves of Mimosa tenuiflora. Planta Med. 2004, 70, 536–539. [Google Scholar] [CrossRef]
- Hashidoko, Y.; Tahara, S.; Mizutani, J. 2-Phenoxychromones and a structurally related flavone from leaves ofRosa rugosa. Phytochemistry 1991, 30, 3837–3838. [Google Scholar] [CrossRef]
- Aisa, H.A.; Zhao, Y.; He, C. A 2-phenoxychromone from Artemisia rupestris. Chem. Nat. Compd. 2006, 42, 16–18. [Google Scholar] [CrossRef]
- Nasser, M.; Salim, N.; Hamza, H.; Saeed, F.; Rabiu, I. Improved deep learning based method for molecular similarity searching using stack of deep belief networks. Molecules 2021, 26, 128. [Google Scholar] [CrossRef]
- Turchi, M.; Cai, Q.; Lian, G. An evaluation of in-silico methods for predicting solute partition in multiphase complex fluids–A case study of octanol/water partition coefficient. Chem. Eng. Sci. 2019, 197, 150–158. [Google Scholar] [CrossRef]
- Sullivan, K.M.; Enoch, S.J.; Ezendam, J.; Sewald, K.; Roggen, E.L.; Cochrane, S. An adverse outcome pathway for sensitization of the respiratory tract by low-molecular-weight chemicals: Building evidence to support the utility of in vitro and in silico methods in a regulatory context. Appl. In Vitro Toxicol. 2017, 3, 213–226. [Google Scholar] [CrossRef] [Green Version]
- Altamash, T.; Amhamed, A.; Aparicio, S.; Atilhan, M. Effect of hydrogen bond donors and acceptors on CO2 absorption by deep eutectic solvents. Processes 2020, 8, 1533. [Google Scholar] [CrossRef]
- Wan, Y.; Tian, Y.; Wang, W.; Gu, S.; Ju, X.; Liu, G. In silico studies of diarylpyridine derivatives as novel HIV-1 NNRTIs using docking-based 3D-QSAR, molecular dynamics, and pharmacophore modeling approaches. RSC Adv. 2018, 8, 40529–40543. [Google Scholar] [CrossRef] [Green Version]
- Escamilla-Gutiérrez, A.; Ribas-Aparicio, R.M.; Córdova-Espinoza, M.G.; Castelán-Vega, J.A. In silico strategies for modeling RNA aptamers and predicting binding sites of their molecular targets. Nucleosides Nucleotides Nucleic Acids 2021, 40, 798–807. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, A.C.; Kumar, A.; Bharadwaj, S.; Chaudhary, R.; Sahi, S. Ligand-Based Approach for In-silico Drug Designing. In Bioinformatics Techniques for Drug Discovery; Springer: Singapore, 2018; pp. 11–19. [Google Scholar]
- Rarey, M.; Dixon, J.S. Feature trees: A new molecular similarity measure based on tree matching. J. Comput.-Aided Mol. Des. 1998, 12, 471–490. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ren, J.-X.; Ma, J.-X.; Ding, L. Development of an in silico prediction model for chemical-induced urinary tract toxicity by using naïve Bayes classifier. Mol. Divers. 2019, 23, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Ieritano, C.; Campbell, J.L.; Hopkins, W.S. Predicting differential ion mobility behaviour in silico using machine learning. Analyst 2021, 146, 4737–4743. [Google Scholar] [CrossRef]
- Taha, M.; Ismail, N.H.; Ali, M.; Rashid, U.; Imran, S.; Uddin, N.; Khan, K.M. Molecular hybridization conceded exceptionally potent quinolinyl-oxadiazole hybrids through phenyl linked thiosemicarbazide antileishmanial scaffolds: In silico validation and SAR studies. Bioorganic Chem. 2017, 71, 192–200. [Google Scholar] [CrossRef]
- Chu, H.; He, Q.-X.; Wang, J.; Hu, Y.; Wang, Y.-Q.; Lin, Z.-H. In silico design of novel benzohydroxamate-based compounds as inhibitors of histone deacetylase 6 based on 3D-QSAR, molecular docking, and molecular dynamics simulations. New J. Chem. 2020, 44, 21201–21210. [Google Scholar] [CrossRef]
- Opo, F.A.D.M.; Rahman, M.M.; Ahammad, F.; Ahmed, I.; Bhuiyan, M.A.; Asiri, A.M. Structure based pharmacophore modeling, virtual screening, molecular docking and ADMET approaches for identification of natural anti-cancer agents targeting XIAP protein. Sci. Rep. 2021, 11, 4049. [Google Scholar] [CrossRef]
- Heidrich, J.; Sperl, L.E.; Boeckler, F.M. Embracing the diversity of halogen bonding motifs in fragment-based drug discovery—construction of a diversity-optimized halogen-enriched fragment library. Front. Chem. 2019, 7, 9. [Google Scholar] [CrossRef]
- Szatylowicz, H.; Stasyuk, O.A.; Krygowski, T.M. Calculating the aromaticity of heterocycles. Adv. Heterocycl. Chem. 2016, 120, 301–327. [Google Scholar]
- Andreeva, E.; Raevsky, O. Lipophilicity of organic compounds calculated using structural similarity and molecular physicochemical descriptors. Pharm. Chem. J. 2009, 43, 258. [Google Scholar] [CrossRef]
- Alesawy, M.S.; Elkaeed, E.B.; Alsfouk, A.A.; Metwaly, A.M.; Eissa, I.J.M. In Silico Screening of Semi-Synthesized Compounds as Potential Inhibitors for SARS-CoV-2 Papain-Like Protease: Pharmacophoric Features, Molecular Docking, ADMET, Toxicity and DFT Studies. Molecules 2021, 26, 6593. [Google Scholar] [CrossRef]
- Norinder, U.; Bergström, C.A. Prediction of ADMET properties. ChemMedChem Chem. Enabling Drug Discov. 2006, 1, 920–937. [Google Scholar]
- Hansson, T.; Oostenbrink, C.; van Gunsteren, W. Molecular dynamics simulations. Curr. Opin. Struct. Biol. 2002, 12, 190–196. [Google Scholar] [CrossRef]
- Liu, X.; Shi, D.; Zhou, S.; Liu, H.; Liu, H.; Yao, X. Molecular dynamics simulations and novel drug discovery. Expert Opin. Drug Discov. 2018, 13, 23–37. [Google Scholar] [CrossRef]
- Parmar, D.R.; Soni, J.Y.; Guduru, R.; Rayani, R.H.; Kusurkar, R.V.; Vala, A.G.; Talukdar, S.N.; Eissa, I.H.; Metwaly, A.M.; Khalil, A. Discovery of new anticancer thiourea-azetidine hybrids: Design, synthesis, in vitro antiproliferative, SAR, in silico molecular docking against VEGFR-2, ADMET, toxicity, and DFT studies. Bioorganic Chem. 2021, 115, 105206. [Google Scholar] [CrossRef]
- Amer, H.H.; Alotaibi, S.H.; Trawneh, A.H.; Metwaly, A.M.; Eissa, I.H. Anticancer activity, spectroscopic and molecular docking of some new synthesized sugar hydrazones, Arylidene and α-Aminophosphonate derivatives. Arab. J. Chem. 2021, 14, 103348. [Google Scholar] [CrossRef]
- El-Adl, K.; Sakr, H.M.; Yousef, R.G.; Mehany, A.B.; Metwaly, A.M.; Elhendawy, M.A.; Radwan, M.M.; ElSohly, M.A.; Abulkhair, H.S.; Eissa, I.H. Discovery of new quinoxaline-2 (1H)-one-based anticancer agents targeting VEGFR-2 as inhibitors: Design, synthesis, and anti-proliferative evaluation. Bioorganic Chem. 2021, 114, 105105. [Google Scholar] [CrossRef]
- Eissa, I.H.; Ibrahim, M.K.; Metwaly, A.M.; Belal, A.; Mehany, A.B.; Abdelhady, A.A.; Elhendawy, M.A.; Radwan, M.M.; ElSohly, M.A.; Mahdy, H.A. Design, molecular docking, in vitro, and in vivo studies of new quinazolin-4 (3H)-ones as VEGFR-2 inhibitors with potential activity against hepatocellular carcinoma. Bioorganic Chem. 2021, 107, 104532. [Google Scholar] [CrossRef]
- Yousef, R.G.; Sakr, H.M.; Eissa, I.H.; Mehany, A.B.; Metwaly, A.M.; Elhendawy, M.A.; Radwan, M.M.; ElSohly, M.A.; Abulkhair, H.S.; El-Adl, K. New quinoxaline-2 (1 H)-ones as potential VEGFR-2 inhibitors: Design, synthesis, molecular docking, ADMET profile and anti-proliferative evaluations. New J. Chem. 2021, 45, 16949–16964. [Google Scholar] [CrossRef]
- Eissa, I.H.; El-Helby, A.-G.A.; Mahdy, H.A.; Khalifa, M.M.; Elnagar, H.A.; Mehany, A.B.; Metwaly, A.M.; Elhendawy, M.A.; Radwan, M.M.; ElSohly, M.A. Discovery of new quinazolin-4 (3H)-ones as VEGFR-2 inhibitors: Design, synthesis, and anti-proliferative evaluation. Bioorganic Chem. 2020, 105, 104380. [Google Scholar] [CrossRef]
- El-Adl, K.; El-Helby, A.-G.A.; Ayyad, R.R.; Mahdy, H.A.; Khalifa, M.M.; Elnagar, H.A.; Mehany, A.B.; Metwaly, A.M.; Elhendawy, M.A.; Radwan, M.M. Design, synthesis, and anti-proliferative evaluation of new quinazolin-4 (3H)-ones as potential VEGFR-2 inhibitors. Bioorganic Med. Chem. 2021, 29, 115872. [Google Scholar] [CrossRef]
- El-Helby, A.-G.A.; Sakr, H.; Ayyad, R.R.; Mahdy, H.A.; Khalifa, M.M.; Belal, A.; Rashed, M.; El-Sharkawy, A.; Metwaly, A.M.; Elhendawy, M.A. Design, synthesis, molecular modeling, in vivo studies and anticancer activity evaluation of new phthalazine derivatives as potential DNA intercalators and topoisomerase II inhibitors. Bioorganic Chem. 2020, 103, 104233. [Google Scholar] [CrossRef]
- Eissa, I.H.; Metwaly, A.M.; Belal, A.; Mehany, A.B.; Ayyad, R.R.; El-Adl, K.; Mahdy, H.A.; Taghour, M.S.; El-Gamal, K.M.; El-Sawah, M.E. Discovery and antiproliferative evaluation of new quinoxalines as potential DNA intercalators and topoisomerase II inhibitors. Arch. Pharm. 2019, 352, 1900123. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; Mackerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | ALog p | M. W | HBA | HBD | Rotatable Bonds | Rings | Aromatic Rings | MFPSA | Minimum Distance |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 3.129 | 300.263 | 6 | 2 | 3 | 3 | 2 | 0.302 | 0 |
| 2 | 2.887 | 316.262 | 7 | 3 | 3 | 3 | 2 | 0.359 | 0 |
| TTT | 3.647 | 304.386 | 2 | 2 | 3 | 3 | 3 | 0.171 | 0.723 |
| Compound | Similarity | SA | SB | SC |
|---|---|---|---|---|
| 1 | 1 | 299 | 0 | 0 |
| 2 | 1 | 289 | 0 | 0 |
| TTT | 0.579 | 276 | 178 | 23 |
| Compound | ∆G [Kcal/mol] |
|---|---|
| 1 | −18.86 |
| 2 | −18.37 |
| TTT | −20.32 |
| Compound | FDA Rodent Carcinogenicity | Carcinogenicity TD50 a | Rat MTD b | Rat Oral LD50 b | LOAEL b | Ocular Irritancy c | Skin Irritancy c |
|---|---|---|---|---|---|---|---|
| 1 | Not a carcinogen | 144.939 | 0.289735 | 0.363122 | 0.0221904 | + | - |
| 2 | Not a carcinogen | 113.277 | 0.44318 | 0.549081 | 0.0362493 | + | - |
| remdesivir | Not a carcinogen | 9.2458 | 0.234965 | 0.308859 | 0.0037911 | + | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suleimen, Y.M.; Jose, R.A.; Suleimen, R.N.; Arenz, C.; Ishmuratova, M.; Toppet, S.; Dehaen, W.; Alsfouk, A.A.; Elkaeed, E.B.; Eissa, I.H.; et al. Isolation and In Silico Anti-SARS-CoV-2 Papain-Like Protease Potentialities of Two Rare 2-Phenoxychromone Derivatives from Artemisia spp. Molecules 2022, 27, 1216. https://doi.org/10.3390/molecules27041216
Suleimen YM, Jose RA, Suleimen RN, Arenz C, Ishmuratova M, Toppet S, Dehaen W, Alsfouk AA, Elkaeed EB, Eissa IH, et al. Isolation and In Silico Anti-SARS-CoV-2 Papain-Like Protease Potentialities of Two Rare 2-Phenoxychromone Derivatives from Artemisia spp. Molecules. 2022; 27(4):1216. https://doi.org/10.3390/molecules27041216
Chicago/Turabian StyleSuleimen, Yerlan M., Rani A. Jose, Raigul N. Suleimen, Christoph Arenz, Margarita Ishmuratova, Suzanne Toppet, Wim Dehaen, Aisha A. Alsfouk, Eslam B. Elkaeed, Ibrahim H. Eissa, and et al. 2022. "Isolation and In Silico Anti-SARS-CoV-2 Papain-Like Protease Potentialities of Two Rare 2-Phenoxychromone Derivatives from Artemisia spp." Molecules 27, no. 4: 1216. https://doi.org/10.3390/molecules27041216