
A New Series of Aryloxyacetic Acids Endowed with Multi-Target Activity towards Peroxisome Proliferator-Activated Receptors (PPARs), Fatty Acid Amide Hydrolase (FAAH), and Acetylcholinesterase (AChE)

 , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
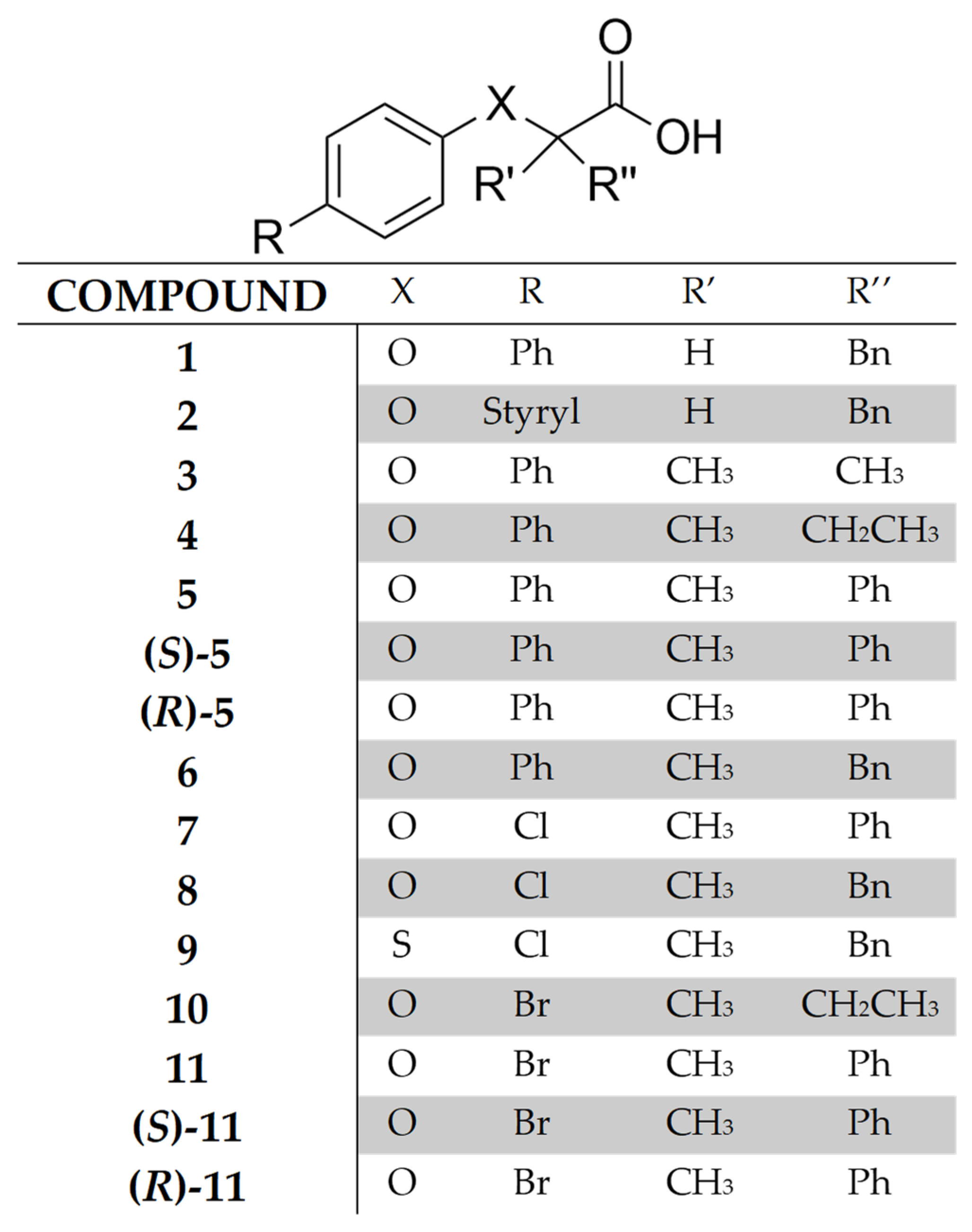
2. Results and Discussion
2.1. Dockings Studies
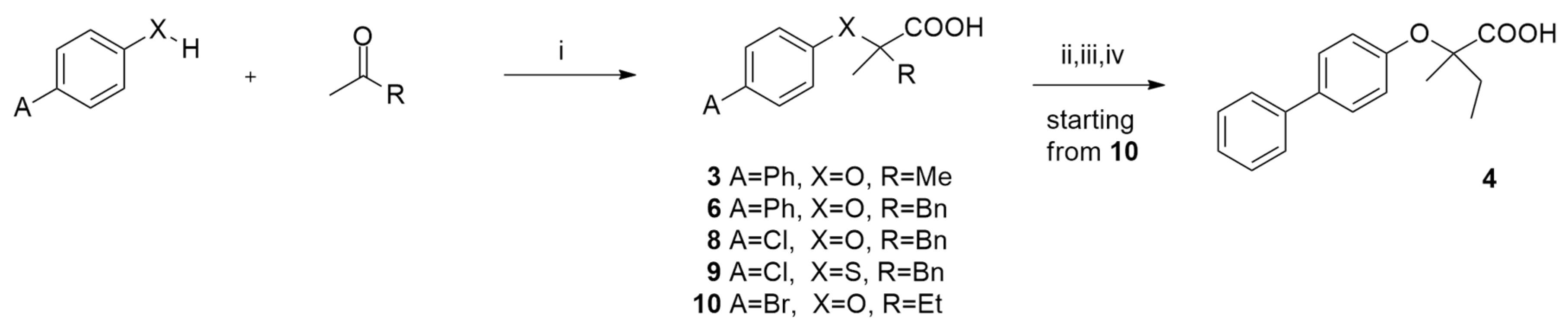
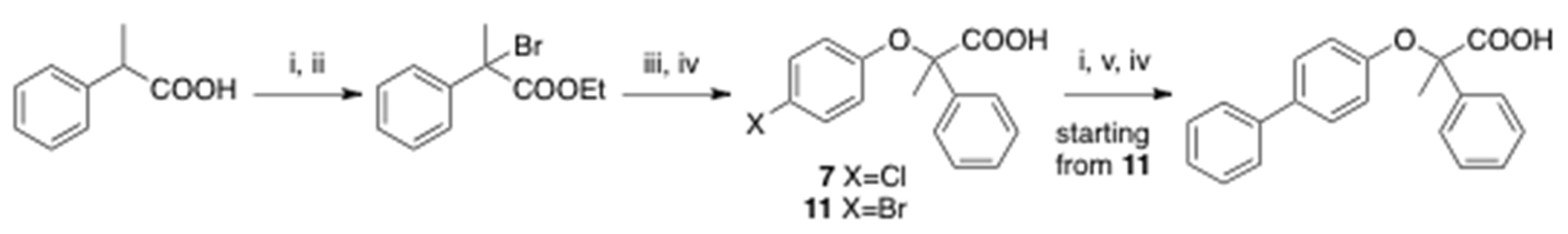
2.2. Synthesis
2.3. Biological Activity
2.3.1. PPARα Activity
2.3.2. PPARγ Activity
2.3.3. FAAH Inhibition Assay
2.3.4. Inhibition of Cholinesterases and Aβ Peptide Aggregation
2.3.5. ADME Properties
3. Materials and Methods
3.1. Chemical Methods
3.1.1. Preparation of (4-Phenylphenoxy)-2-methylpropanoic Acid (3)
3.1.2. Preparation of (4-Phenylphenoxy)-2-methyl-3-phenylpropanoic Acid (6)
3.1.3. Preparation of (4-Chlorophenoxy)-2-methyl-3-phenylpropanoic Acid (8)
3.1.4. Preparation of (4-Chloro-phenylsulfanyl)-2-methyl-3-phenylpropanoic Acid (9)
3.1.5. Preparation of (4-Bromophenoxy)-2-methylbutanoic Acid (10)
3.1.6. Preparation of Methyl (4-Bromophenoxy)-2-methylbutanoate
3.1.7. Synthesis of Methyl (4-Phenylphenoxy)-2-methylbutanoate
3.1.8. Synthesis of Methyl (4-Phenylphenoxy)-2-methylbutanoic Acid (4)
3.1.9. Preparation of Ethyl 2-Phenylpropanoate
3.1.10. Preparation of Ethyl 2-Bromo-2-phenylpropanoate
3.1.11. Preparation of Ethyl 2-(4-Bromophenoxy)-2-phenylpropanoate
3.1.12. Preparation of Ethyl 2-(4-Chlorophenoxy)-2-phenylpropanoate
3.1.13. Synthesis of Ethyl (4-Phenylphenoxy)-2-phenylpropanoate
3.1.14. Preparation of (4-Phenylphenoxy)-, (4-Chlorophenoxy)- and (4-Bromophenoxy)-2 phenylpropanoic Acids (5, 7 and 11)
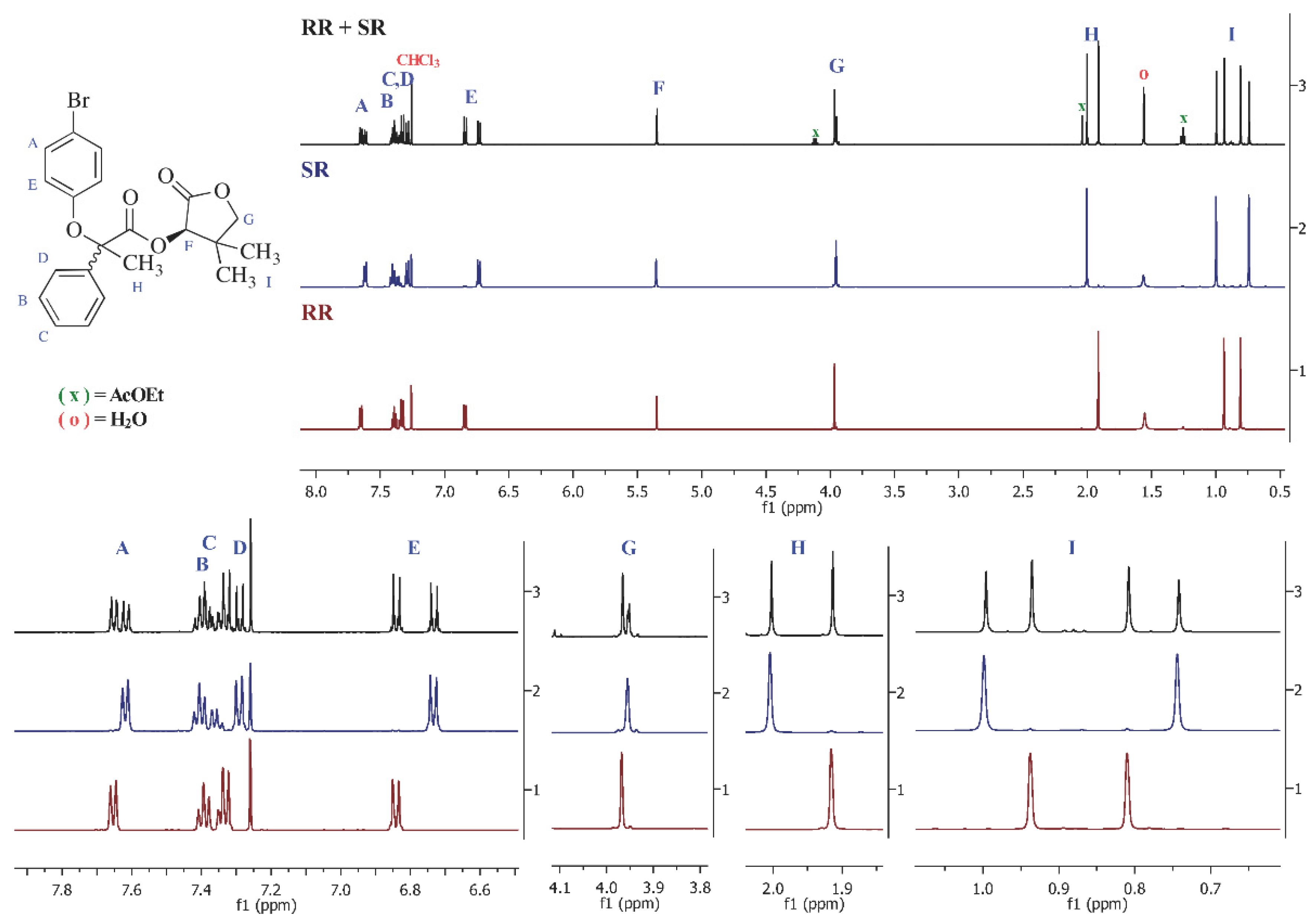
3.1.15. Synthesis of (R,R)- and (S,R)-Tetrahydro-4,4-dimethyl-2-oxofuran-3-yl-2-(4-bromophenoxy)-2-phenylpropanoates (12a and 12b)
3.1.16. Synthesis of (R,R)- and (S,R)-Tetrahydro-4,4-dimethyl-2-oxofuran-3-yl 2-(4-phenylphenoxy)-2-phenylpropanoate (13a and 13b)
3.1.17. Preparation of (R)- and (S)-(4-Phenylphenoxy)-, (4-bromophenoxy)-2 phenylpropanoic acids (R-5, S-5 and R-11, S-11)
3.2. PPAR Assay
3.3. FAAH Inhibition Assay
3.4. AChE and BuChE Inhibition Assay
3.5. Inhibition of Aβ40 Aggregation
3.6. Molecular Dockings
3.7. Statistical Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laganà, A.S.; Vitale, S.G.; Nigro, A.; Sofo, V.; Salmeri, F.M.; Rossetti, P.; Rapisarda, A.M.C.; La Vignera, S.; Condorelli, R.A.; Rizzo, G.; et al. Pleiotropic Actions of Peroxisome Proliferator-Activated Receptors (PPARs) in Dysregulated Metabolic Homeostasis, Inflammation and Cancer: Current Evidence and Future Perspectives. Int. J. Mol. Sci. 2016, 17, 999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansour, M. The roles of peroxisome proliferator-activated receptors in the metabolic syndrome. Prog. Mol. Biol. Transl. Sci. 2014, 121, 217–266. [Google Scholar]
- Hong, F.; Xu, P.; Zhai, Y. The Opportunities and Challenges of Peroxisome Proliferator-Activated Receptors Ligands in Clinical Drug Discovery and Development. Int. J. Mol. Sci. 2018, 19, 2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadayat, T.M.; Shrestha, A.; Jeon, Y.H.; An, H.; Kim, J.; Cho, S.J.; Chin, J. Targeting Peroxisome Proliferator-Activated Receptor Delta (PPARδ): A Medicinal Chemistry Perspective. J. Med. Chem. 2020, 63, 10109–10134. [Google Scholar] [CrossRef] [PubMed]
- Bortolini, M.; Wright, M.B.; Bopst, M.; Balas, B. Examining the Safety of PPAR Agonists—Current Trends and Future Prospects. Expert Opin. Drug Saf. 2013, 12, 65–79. [Google Scholar] [CrossRef]
- Pirat, C.; Farce, A.; Lebègue, N.; Renault, N.; Furman, C.; Millet, R.; Yous, S.; Speca, S.; Berthelot, P.; Desreumaux, P.; et al. Targeting Peroxisome Proliferator-Activated Receptors (PPARs): Development of Modulators. J. Med. Chem. 2012, 55, 4027–4061. [Google Scholar] [CrossRef]
- Lavecchia, A.; Cerchia, C. Selective PPARγ Modulators for Type 2 Diabetes Treatment: How Far Have We Come and What Does the Future Hold? Future Med. Chem. 2018, 10, 703–705. [Google Scholar] [CrossRef]
- Iannotti, F.A.; Vitale, R.M. The Endocannabinoid System and PPARs: Focus on Their Signalling Crosstalk, Action and Transcriptional Regulation. Cells 2021, 10, 586. [Google Scholar] [CrossRef]
- O’Sullivan, S.E. An update on PPAR activation by cannabinoids. Br. J. Pharmacol. 2016, 173, 1899–1910. [Google Scholar] [CrossRef] [Green Version]
- Lago-Fernandez, A.; Zarzo-Arias, S.; Jagerovic, N.; Morales, P. Relevance of Peroxisome Proliferator Activated Receptors in Multitarget Paradigm Associated with the Endocannabinoid System. Int. J. Mol. Sci. 2021, 22, 1001. [Google Scholar] [CrossRef] [PubMed]
- Toczek, M.; Malinowska, B. Enhanced Endocannabinoid Tone as a Potential Target of Pharmacotherapy. Life Sci. 2018, 204, 20–45. [Google Scholar] [CrossRef] [PubMed]
- Bottemanne, P.; Muccioli, G.G.; Alhouayek, M. N-Acylethanolamine Hydrolyzing Acid Amidase Inhibition: Tools and Potential Therapeutic Opportunities. Drug Discov. Today 2018, 23, 1520–1529. [Google Scholar] [CrossRef] [PubMed]
- Petrosino, S.; Di Marzo, V. FAAH and MAGL Inhibitors: Therapeutic Opportunities from Regulating Endocannabinoid Levels. Curr. Opin. Investig. Drugs 2010, 11, 51–62. [Google Scholar]
- Brunetti, L.; Loiodice, F.; Piemontese, L.; Tortorella, P.; Laghezza, A. New Approaches to Cancer Therapy: Combining Fatty Acid Amide Hydrolase (FAAH) Inhibition with Peroxisome Proliferator-Activated Receptors (PPARs) Activation. J. Med. Chem. 2019, 62, 10995–11003. [Google Scholar] [CrossRef]
- Brunetti, L.; Laghezza, A.; Loiodice, F.; Tortorella, P.; Piemontese, L. Combining fatty acid amide hydrolase (FAAH) inhibition with peroxisome proliferator-activated receptor (PPAR) activation: A new potential multi-target therapeutic strategy for the treatment of Alzheimer’s disease. Neural Regen. Res. 2020, 15, 67–68. [Google Scholar]
- Panlilio, L.V.; Justinova, Z.; Goldberg, S.R. Inhibition of FAAH and activation of PPAR: New approaches to the treatment of cognitive dysfunction and drug addiction. Pharmacol. Ther. 2013, 138, 84–102. [Google Scholar] [CrossRef] [Green Version]
- Fruchart, J.-C. Selective peroxisome proliferator-activated receptorα modulators (SPPARMα): The next generation of peroxisome proliferator-activated receptor α-agonists. Cardiovasc. Diabetol. 2013, 12, 82. [Google Scholar] [CrossRef] [Green Version]
- Cariou, B.; Zair, Y.; Staels, B.; Bruckert, E. Effects of the new dual PPAR alpha/delta agonist GFT505 on lipid and glucose homeostasis in abdominally obese patients with combined dyslipidemia or impaired glucose metabolism. Diabetes Care 2011, 34, 2008–2014. [Google Scholar] [CrossRef] [Green Version]
- Hughes, T.S.; Giri, P.K.; de Vera, I.M.; Marciano, D.P.; Kuruvilla, D.S.; Shin, Y.; Blayo, A.L.; Kamenecka, T.M.; Burris, T.P.; Griffin, P.R.; et al. An alternate binding site for PPARgamma ligands. Nat. Commun. 2014, 5, 3571. [Google Scholar] [CrossRef] [Green Version]
- Laghezza, A.; Piemontese, L.; Cerchia, C.; Montanari, R.; Capelli, D.; Giudici, M.; Crestani, M.; Tortorella, P.; Peiretti, F.; Pochetti, G.; et al. Identification of the First PPARα/γ Dual Agonist Able To Bind to Canonical and Alternative Sites of PPARγ and To Inhibit Its Cdk5-Mediated Phosphorylation. J. Med. Chem. 2018, 61, 8282–8298. [Google Scholar] [CrossRef] [PubMed]
- De Vivo, M.; Scarpelli, R.; Cavalli, A.; Migliore, M.; Piomelli, D.; Habrant, D.; Favia, A. Multi-target FAAH and Cox Inhibitors and Therapeutical Uses Thereof. U.S. Patent WO2014023643A1, 13 February 2014. [Google Scholar]
- Favia, A.D.; Habrant, D.; Scarpelli, R.; Migliore, M.; Albani, C.; Bertozzi, S.M.; Dionisi, M.; Tarozzo, G.; Piomelli, D.; Cavalli, A.; et al. Identification and Characterization of Carprofen as a Multi-target Fatty Acid Amide Hydrolase/Cyclooxygenase Inhibitor. J. Med. Chem. 2012, 55, 8807–8826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolacci, L.; Romeo, E.; Veronesi, M.; Magotti, P.; Albani, C.; Dionisi, M.; Lambruschini, C.; Scarpelli, R.; Cavalli, A.; De Vivo, M.; et al. A Binding Site for Nonsteroidal Anti-inflammatory Drugs in Fatty Acid Amide Hydrolase. J. Am. Chem. Soc. 2013, 135, 22–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunetti, L.; Carrieri, A.; Piemontese, L.; Tortorella, P.; Loiodice, F.; Laghezza, A. Beyond the Canonical Endocannabinoid System. A Screening of PPAR Ligands as FAAH Inhibitors. Int. J. Mol. Sci. 2020, 21, 7026. [Google Scholar] [CrossRef]
- Gilardi, F.; Giudici, M.; Mitro, N.; Maschi, O.; Guerrini, U.; Rando, G.; Maggi, A.; Cermenati, G.; Laghezza, A.; Loiodice, F.; et al. LT175 is a novel PPARα/γ ligand with potent insulin-sensitizing effects and reduced adipogenic properties. J. Biol. Chem. 2014, 289, 6908–6920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talele, T.T. Opportunities for Tapping into Three-Dimensional Chemical Space through a Quaternary Carbon. J. Med. Chem. 2020, 63, 13291–13315. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2019 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Blennow, K.; Zetterberg, H. Biomarkers for Alzheimer’s disease: Current status and prospects for the future. J. Intern. Med. 2018, 284, 643–663. [Google Scholar] [CrossRef] [Green Version]
- Soto-Rojas, L.O.; de la Cruz-López, F.; Torres, M.A.O.; Viramontes-Pintos, A.; Cárdenas-Aguayo, M.; Meraz-Ríos, M.A.; Salinas-Lara, C.; Florán-Garduño, B.; Luna-Muñoz, J. Neuro-inflammation and alteration of the blood-brain barrier in Alzheimers disease. In Alzheimer’s Disease—Challenges for the Future; InTechOpen: London, UK, 2015. [Google Scholar]
- Masters, C.L.; Selkoe, D.J. Biochemistry of amyloid beta-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006262. [Google Scholar] [CrossRef]
- Van der Velpen, V.; Teav, T.; Gallart-Ayala, H.; Mehl, F.; Konz, I.; Clark, C.; Oikonomidi, A.; Peyratout, G.; Henry, H.; Delorenzi, M.; et al. Systemic and central nervous system metabolic alterations in Alzheimer’s disease. Alzheimer’s Res. Ther. 2019, 11, 93. [Google Scholar] [CrossRef] [Green Version]
- Contestabile, A. The history of the cholinergic hypothesis. Behav. Brain Res. 2011, 221, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Orhan, I.E.; Senol, F.S. Designing multi-targeted therapeutics for the treatment of Alzheimer’s disease. Curr. Top. Med. Chem. 2016, 16, 1889–1896. [Google Scholar] [CrossRef]
- Daoud, I.; Melkemi, N.; Salah, T.; Ghalem, S. Combined QSAR, molecular docking and molecular dynamics study on new Acetylcholinesterase and Butyrylcholinesterase inhibitors. Comput. Biol. Chem. 2018, 74, 304–326. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Aisen, P.S.; DuBois, B.; Frölich, L.; Jack, C.R., Jr.; Jones, R.W.; Morris, J.C.; Raskin, J.; Dowsett, S.A.; Scheltens, P. Drug development in Alzheimer’s disease: The path to 2025. Alzheimers Res. Ther. 2016, 8, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzior, N.; Wieckowska, A.; Panek, D.; Malawska, B. Recent development of multifunctional agents as potential drug candidates for the treatment of Alzheimer’s disease. Curr. Med. Chem. 2015, 22, 373–404. [Google Scholar] [CrossRef]
- Piemontese, L.; Loiodice, F.; Chaves, S.; Santos, M.A. The Therapy of Alzheimer’s Disease: Towards a New Generation of Drugs. In Frontiers in Clinical Drug Research—Alzheimer Disorders; Bentham Science Publishers: Al Sharjah, United Arab Emirates, 2019; Volume 8, pp. 33–80. [Google Scholar]
- Bedse, G.; Romano, A.; Lavecchia, A.M.; Cassano, T.; Gaetani, S. The role of endocannabinoid signaling in the molecular mechanisms of neurodegeneration in Alzheimer’s disease. J. Alzheimers Dis. 2015, 43, 1115–1136. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Ruiz, J.; Romero, J.; Ramos, J.A. Endocannabinoids and neurodegenerative disorders: Parkinson’s disease, Huntington’s chorea, Alzheimer’s disease, and others. Handb. Exp. Pharmacol. 2015, 231, 233–259. [Google Scholar]
- Ikegai, K.; Fukumoto, K.; Mukaiyama, T. Copper(II)-catalyzed O-Phenylation of Tertiary Alcohols with Organobismuth(V) Reagents. Chem. Lett. 2006, 35, 612–613. [Google Scholar] [CrossRef]
- Pinelli, A.; Godio, C.; Laghezza, A.; Mitro, N.; Fracchiolla, G.; Tortorella, V.; Lavecchia, A.; Novellino, E.; Fruchart, J.-C.; Staels, B.; et al. Synthesis, biological evaluation, and molecular modeling investigation of new chiral fibrates with PPARalpha and PPARgamma agonist activity. J. Med. Chem. 2005, 48, 5509–5519. [Google Scholar] [CrossRef]
- Fracchiolla, G.; Laghezza, A.; Piemontese, L.; Carbonara, G.; Lavecchia, A.; Tortorella, P.; Crestani, M.; Novellino, E.; Loiodice, F. Synthesis, biological evaluation and molecular modeling investigation of chiral phenoxyacetic acid analogues with PPARalpha and PPARgamma agonist activity. ChemMedChem 2007, 2, 641–654. [Google Scholar] [CrossRef]
- Pisani, L.; Catto, M.; De Palma, A.; Farina, R.; Cellamare, S.; Altomare, C.D. Discovery of Potent Dual Binding Site Acetylcholinesterase Inhibitors via Homo- and Heterodimerization of Coumarin-based Moieties. ChemMedChem 2017, 12, 1349–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campora, M.; Canale, C.; Gatta, E.; Tasso, B.; Laurini, E.; Relini, A.; Pricl, S.; Catto, M.; Tonelli, M. Multi-target Biological Profiling of New Naphthoquinone and Anthraquinone-Based Derivatives for the Treatment of Alzheimer’s Disease. ACS Chem. Neurosci. 2021, 12, 447–461. [Google Scholar] [CrossRef] [PubMed]
- Szałaj, N.; Bajda, M.; Dudek, K.; Brus, B.; Gobec, S.; Malawska, B. Multiple Ligands Targeting Cholinesterases and β-Amyloid: Synthesis, Biological Evaluation of Heterodimeric Compounds with Benzylamine Pharmacophore. Arch. Pharm. 2015, 348, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Pisani, L.; De Palma, A.; Giangregorio, N.; Miniero, D.V.; Pesce, P.; Nicolotti, O.; Campagna, F.; Altomare, C.D.; Catto, M. Mannich base approach to 5-methoxyisatin 3-(4-isopropylphenyl)hydrazone: A water-soluble prodrug for a multi-target inhibition of cholinesterases, beta-amyloid fibrillization and oligomer-induced cytotoxicity. Eur. J. Pharm. Sci. 2017, 109, 381–388. [Google Scholar] [CrossRef]
- Convertino, M.; Pellarin, R.; Catto, M.; Carotti, A.; Caflisch, A. 9,10-Anthraquinone hinders beta-aggregation: How does a small molecule interfere with Abeta-peptide amyloid fibrillation? Protein Sci. 2009, 18, 792–800. [Google Scholar]
- Daina, A.; Zoete, V. A Boiled-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- Hollon, T.; Yoshimura, F.K. Variation in enzymatic transient gene expression assays. Anal. Biochem. 1989, 182, 411–418. [Google Scholar] [CrossRef]
- Schrödinger Release, version 2021-4. Desmond Molecular Dynamics System. Maestro-Desmond Interoperability Tools, Schrödinger: New York, NY, USA, 2021.
- QUACPAC, version 2.1.0.4; OpenEye Scientific Software: Santa Fe, NM, USA, 2020.
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminf. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Sierra, M.L.; Beneton, V.; Boullay, A.B.; Boyer, T.; Brewster, A.G.; Donche, F.; Forest, M.C.; Fouchet, M.H.; Gellibert, F.J.; Grillot, D.A.; et al. Substituted 2-[(4-aminomethyl)phenoxy]-2-methylpropionic acid PPARalpha agonists. 1. Discovery of a novel series of potent HDLc raising agents. J. Med. Chem. 2007, 50, 685–695. [Google Scholar] [CrossRef]
- Montanari, R.; Saccoccia, F.; Scotti, E.; Crestani, M.; Godio, C.; Gilardi, F.; Loiodice, F.; Fracchiolla, G.; Laghezza, A.; Tortorella, P.; et al. Crystal structure of the peroxisome proliferator-activated receptor gamma (PPARgamma) ligand binding domain complexed with a novel partial agonist: A new region of the hydrophobic pocket could be exploited for drug design. J. Med. Chem. 2008, 51, 7768–7776. [Google Scholar] [CrossRef]
- Gerlits, O.; Ho, K.Y.; Cheng, X.; Blumenthal, D.; Taylor, P.; Kovalevsky, A.; Radić, Z. A new crystal form of human acetylcholinesterase for exploratory room-temperature crystallography studies. Chem. Biol. Interact. 2019, 309, 108698. [Google Scholar] [CrossRef] [PubMed]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995, 117, 5179–5193. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- El Khoury, L.; Santos-Martins, D.; Sasmal, S.; Eberhardt, J.; Bianco, G.; Ambrosio, F.A.; Solis-Vasquez, L.; Koch, A.; Forli, S.; Mobley, D.L. Comparison of affinity ranking using AutoDock-GPU and MM-GBSA scores for BACE-1 inhibitors in the D3R Grand Challenge 4. J. Comput. Aided Mol. Des. 2019, 33, 1011–1020. [Google Scholar] [CrossRef]
- Forli, S.; Olson, A.J. A force field with discrete displaceable waters and desolvation entropy for hydrated ligand docking. J. Med. Chem. 2012, 55, 623–638. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Target | FEB (a) | ΔE (b) | EFF (c) | POP (d) |
|---|---|---|---|---|---|
| (S)-5 | PPARα | −8.52 | 0.00 | −0.355 | 66/1000 |
| (R)-5 | −9.56 | 0.00 | −0.370 | 16/1000 | |
| (S)-11 | −7.44 | 0.65 | −0.392 | 28/1000 | |
| (R)-11 | −9.21 | 0.00 | −0.485 | 310/1000 | |
| Wy-14,643 | −10.08 | 0.00 | −0.480 | 194/1000 | |
| (S)-5 | PPARγ | −10.67 | 0.00 | −0.445 | 643/1000 |
| (R)-5 | −10.03 | 0.00 | −0.418 | 96/1000 | |
| (S)-11 | −10.37 | 0.00 | −0.546 | 715/1000 | |
| (R)-11 | −9.21 | 0.00 | −0.485 | 231/1000 | |
| rosiglitazone | −9.41 | 0.81 | −0.376 | 125/1000 | |
| (S)-5 | FAAH | −8.30 | 0.41 | −0.346 | 62/1000 |
| (R)-5 | −9.08 | 0.00 | −0.378 | 590/1000 | |
| (S)-11 | −7.41 | 0.42 | −0.390 | 167/1000 | |
| (R)-11 | −8.06 | 0.48 | −0.424 | 134/1000 | |
| JZL195 | −11.24 | 0.00 | −0.351 | 44/1000 | |
| (S)-5 | AChE | −9.54 | 0.00 | −0.498 | 350/1000 |
| (R)-5 | −9.58 | 0.00 | −0.399 | 540/1000 | |
| (S)-11 | −8.47 | 0.00 | −0.446 | 206/1000 | |
| (R)-11 | −8.54 | 0.00 | −0.449 | 612/1000 | |
| donepezil | −10.30 | 0.10 | −0.368 | 508/1000 |
| Compound | PPARα | PPARγ | FAAH | ||
|---|---|---|---|---|---|
| EC50 (µM) | Emax a (%) | EC50 (µM) | Emax a (%) | IC50 (µM) | |
| 1 | 0.19 ± 0.04 | 116 ± 4 | 0.55 ± 0.12 | 62 ± 7 | >50 |
| 2 | 1.75 ± 0.12 | 57 ± 4 | 0.72 ± 0.27 | 50 ± 1 | 24 ± 2.5 |
| 3 | 2.4 ± 0.4 | 69 ± 5 | 14.6 ± 3.5 | 9.6 ± 1.7 | 7.6 ± 0.9 |
| 4 | 4.7 ± 2.7 | 108 ± 12 | 13 ± 1.1 | 23 ± 9 | 6.9 ± 0.5 |
| 5 | 0.46 ± 0.04 | 105 ± 10 | 2.5 ± 0.6 | 39 ± 7 | n.t. |
| (S)-5 | 0.126 ± 0.011 | 86 ± 4 | 1.54 ± 0.24 | 38.7 ± 3.4 | 5.3 ± 2.0 |
| (R)-5 | - | i | 7.9 ± 2.5 | 14 ± 3 | 6.0 ± 0.8 |
| 6 | 0.20 ± 0.03 | 129 ± 12 | 0.88 ± 0.11 | 91 ± 12 | 14.8 ± 0.4 |
| 7 | 1.99 ± 0.23 | 63 ± 1 | 20.2 ± 1.2 | 22.5 ± 0.4 | n.t. |
| 8 | 1.57 ± 0.42 | 92 ± 6 | 5.06 ± 1.12 | 64 ± 1 | n.t. |
| 9 | 0.86 ± 0.03 | 86 ± 6 | 11.4 ± 1.8 | 18 ± 1 | n.t. |
| 10 | 7.9 ± 2.1 | 92 ± 22 | - | 8.1 ± 1.6 | 8.8 ± 0.5 |
| 11 | 0.73 ± 0.12 | 97 ± 1 | 13.8 ± 3.8 | 21 ± 2 | n.t. |
| (S)-11 | 0.233 ± 0.034 | 77 ± 4 | 3.9 ± 0.5 | 40 ± 1.3 | 10.0 ± 1.8 |
| (R)-11 | 22 ± 4 | 15.1 ± 2.5 | 6.8 ± 1.9 | ||
| Wy-14,643 | 1.56 ± 0.30 | 100 ± 10 | |||
| Rosiglitazone | 0.039 ± 0.003 | 100 ± 9 | |||
| JZL195 | 0.019 ± 0.003 | ||||
| Compound | AChE | Aβ40 Aggr. |
|---|---|---|
| i% @10 µM a | i% @100 µM a | |
| 3 | 39 ± 5 | 29 ± 4 |
| 4 | 49 ± 1 | 25 ± 5 |
| 5 | n.t. | n.t. |
| (S)-5 | 37 ± 5 | 37 ± 5 |
| (R)-5 | 37 ± 3 | 39 ± 3 |
| 6 | 44 ± 4 | 17 ± 2 |
| 7 | n.t. | n.t. |
| 8 | n.t. | n.t. |
| 9 | n.t. | n.t. |
| 10 | 35 ± 1 | 25 ± 6 |
| 11 | n.t. | n.t. |
| (S)-11 | 47 ± 3 | 35 ± 4 |
| (R)-11 | 38 ± 4 | 44 ± 4 |
| Donepezil | 0.017 ± 0.002 b | 14 ± 7 c |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leuci, R.; Brunetti, L.; Laghezza, A.; Piemontese, L.; Carrieri, A.; Pisani, L.; Tortorella, P.; Catto, M.; Loiodice, F. A New Series of Aryloxyacetic Acids Endowed with Multi-Target Activity towards Peroxisome Proliferator-Activated Receptors (PPARs), Fatty Acid Amide Hydrolase (FAAH), and Acetylcholinesterase (AChE). Molecules 2022, 27, 958. https://doi.org/10.3390/molecules27030958
Leuci R, Brunetti L, Laghezza A, Piemontese L, Carrieri A, Pisani L, Tortorella P, Catto M, Loiodice F. A New Series of Aryloxyacetic Acids Endowed with Multi-Target Activity towards Peroxisome Proliferator-Activated Receptors (PPARs), Fatty Acid Amide Hydrolase (FAAH), and Acetylcholinesterase (AChE). Molecules. 2022; 27(3):958. https://doi.org/10.3390/molecules27030958
Chicago/Turabian StyleLeuci, Rosalba, Leonardo Brunetti, Antonio Laghezza, Luca Piemontese, Antonio Carrieri, Leonardo Pisani, Paolo Tortorella, Marco Catto, and Fulvio Loiodice. 2022. "A New Series of Aryloxyacetic Acids Endowed with Multi-Target Activity towards Peroxisome Proliferator-Activated Receptors (PPARs), Fatty Acid Amide Hydrolase (FAAH), and Acetylcholinesterase (AChE)" Molecules 27, no. 3: 958. https://doi.org/10.3390/molecules27030958