
Discovery and Characterization of a Cryptic Secondary Binding Site in the Molecular Chaperone HSP70

 , , , ,
, , , ,  , and
, and
Abstract
:1. Introduction
2. Results
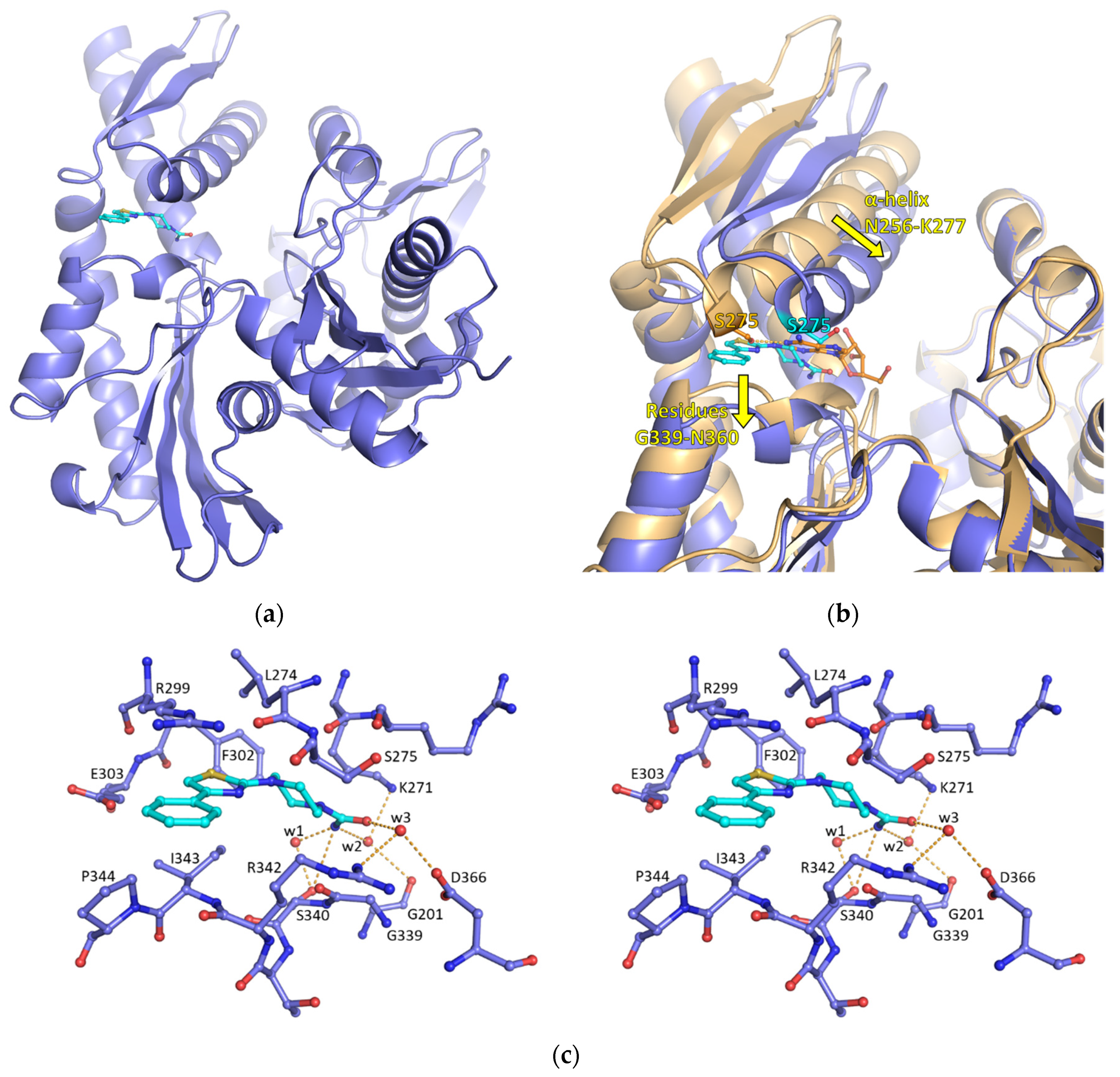
2.1. Discovery of a New Cryptic Binding Site in HSP70
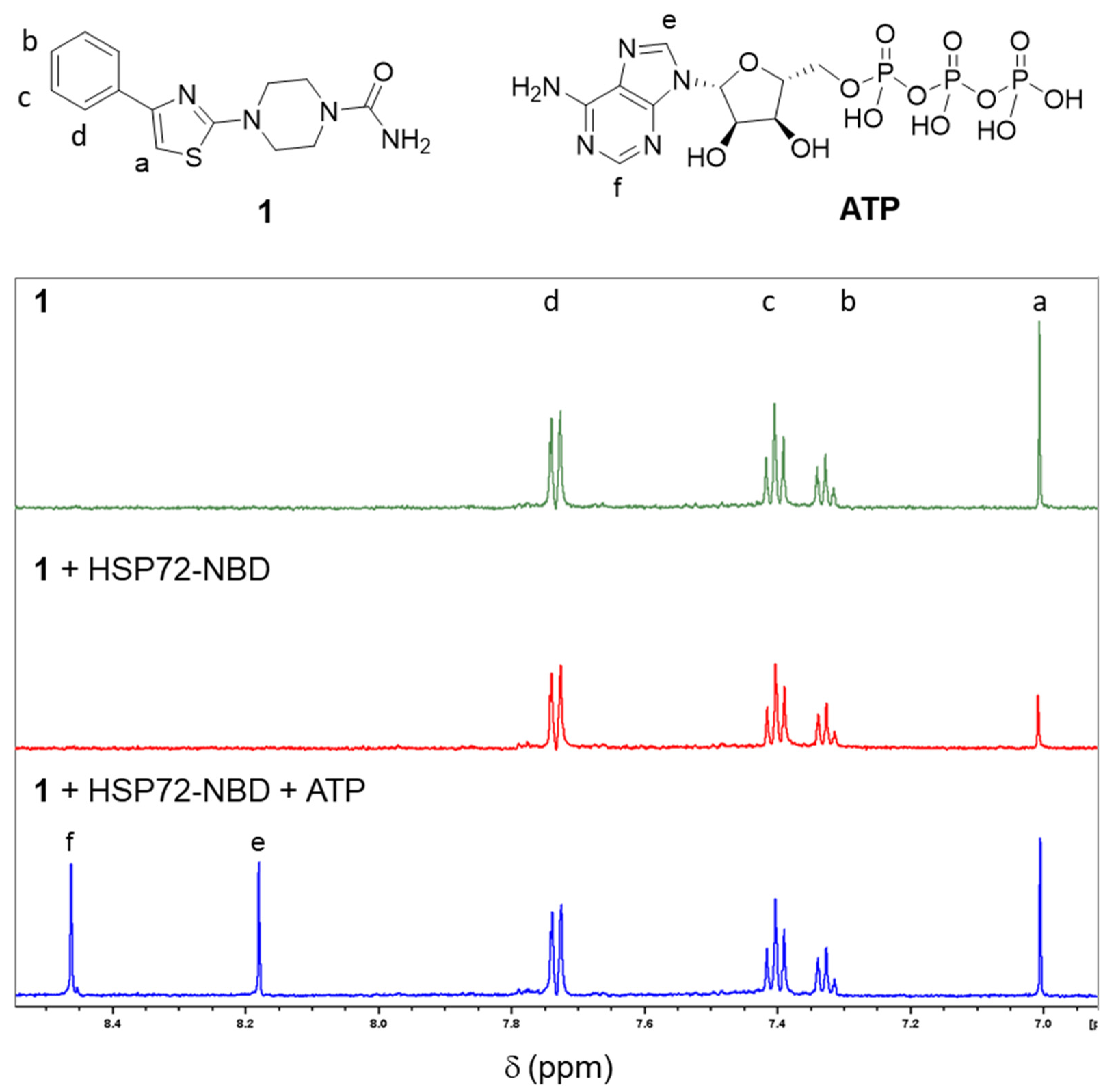
2.2. Validation of Fragment Binding through Characterization of Solubilized Analogs in Orthogonal Biophysical Assays
2.3. Exploring the Interactions of 1 with the Secondary Binding Site by Molecular Dynamics
2.4. Virtual High-Thoughput Screening to Identify Alternative Compounds Targeting the Secondary Binding Site
3. Discussion
4. Materials and Methods
4.1. Synthetic Chemistry
4.1.1. General Synthetic Method
4.1.2. 4-(4-Phenylthiazol-2-yl)piperazine-1-carboxamide (1)
4.1.3. 4-[4-[4-(3-Morpholinoprop-1-ynyl)phenyl]thiazol-2-yl]piperazine-1-carboxamide (3)
4.2. Biophysical Evaluation of Compounds
4.2.1. SPR Experiments
4.2.2. Ligand Observed NMR Experiments
4.2.3. Determination of the Solubility of 1 by Quantitative NMR
4.3. Protein Crystallography
4.3.1. HSP72-NBD Expression
4.3.2. HSP72-NBD Purification
4.3.3. HSP72-NBD Crystallization
4.3.4. Crystallographic Data Collection, Processing and Refinement
4.4. Molecular Dynamics Simulations
4.5. Docking and Virtual Screening Experiments
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Powers, M.V.; Workman, P. Inhibitors of the Heat Shock Response: Biology and Pharmacology. FEBS Lett. 2007, 581, 3758–3769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, M.P.; Lila, X.; Gierasch, M. Recent Advances in the Structural and Mechanistic Aspects of Hsp70 Molecular Chaperones. J. Biol. Chem. 2019, 294, 2085–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrose, A.J.; Chapman, E. Function, Therapeutic Potential, and Inhibition of Hsp70 Chaperones. J. Med. Chem. 2021, 64, 7060–7082. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.G.; Chang, L.; Gestwicki, J.E. Heat Shock Protein 70 (Hsp70) as an Emerging Drug Target. J. Med. Chem. 2010, 53, 4585–4602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, M.Y.; Gabai, V.L. Hsp70 in Cancer: Back to the Future. Oncogene 2015, 34, 4153–4161. [Google Scholar] [CrossRef] [Green Version]
- Powers, M.V.; Jones, K.; Barillari, C.; Westwood, I.; Van Montfort, R.L.M.; Workman, P. Targeting HSP70: The Second Potentially Druggable Heat Shock Protein and Molecular Chaperone? Cell Cycle 2010, 9, 1542–1550. [Google Scholar] [CrossRef]
- Powers, M.V.; Clarke, P.A.; Workman, P. Dual Targeting of HSC70 and HSP72 Inhibits HSP90 Function and Induces Tumor-Specific Apoptosis. Cancer Cell 2008, 14, 250–262. [Google Scholar] [CrossRef] [Green Version]
- Jones, A.M.; Westwood, I.M.; Osborne, J.D.; Matthews, T.P.; Cheeseman, M.D.; Rowlands, M.G.; Jeganathan, F.; Burke, R.; Lee, D.; Kadi, N.; et al. A Fragment-Based Approach Applied to a Highly Flexible Target: Insights and Challenges towards the Inhibition of HSP70 Isoforms. Sci. Rep. 2016, 6, 34701. [Google Scholar] [CrossRef]
- Williamson, D.S.; Borgognoni, J.; Clay, A.; Daniels, Z.; Dokurno, P.; Drysdale, M.J.; Foloppe, N.; Francis, G.L.; Graham, C.J.; Howes, R.; et al. Novel Adenosine-Derived Inhibitors of 70 KDa Heat Shock Protein, Discovered Through Structure-Based Design. J. Med. Chem. 2009, 52, 1510–1513. [Google Scholar] [CrossRef]
- Keseru, G.M.; Erlanson, D.A.; Ferenczy, G.G.; Hann, M.M.; Murray, C.W.; Pickett, S.D. Design Principles for Fragment Libraries: Maximizing the Value of Learnings from Pharma Fragment-Based Drug Discovery (FBDD) Programs for Use in Academia. J. Med. Chem. 2016, 59, 8189–8206. [Google Scholar] [CrossRef] [Green Version]
- Massey, A.J. ATPases as Drug Targets: Insights from Heat Shock Proteins 70 and 90. J. Med. Chem. 2010, 53, 7280–7286. [Google Scholar] [CrossRef] [PubMed]
- Ludlow, R.F.; Verdonk, M.L.; Saini, H.K.; Tickle, I.J.; Jhoti, H. Detection of Secondary Binding Sites in Proteins Using Fragment Screening. Proc. Natl. Acad. Sci. USA 2015, 112, 15910–15915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, R.A.E.; Congreve, M.; Murray, C.W.; Rees, D.C. Fragment-based lead discovery: Leads by design. Drug Discov. Today 2005, 10, 987–992. [Google Scholar] [CrossRef]
- Ayotte, Y.; Marando, V.M.; Vaillancourt, L.; Bouchard, P.; Heffron, G.; Coote, P.W.; Larda, S.T.; La Plante, S.R. Exposing Small-Molecule Nanoentities by a Nuclear Magnetic Resonance Relaxation Assay. J. Med. Chem. 2019, 62, 7885–7896. [Google Scholar] [CrossRef]
- Eurtivong, C.; Reynisson, J. The Development of a Weighted Index to Optimise Compound Libraries for High Throughput Screening. Mol. Inf. 2019, 38, 1800068. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef]
- Marcel, L.; Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Oliver Korb, O.; Stützle, T.; Exner, T.E. Empirical scoring functions for advanced protein-ligand docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef]
- Mooij, W.T.M.; Verdonk, M.L. General and targeted statistical potentials for protein–ligand interactions. Proteins 2005, 61, 272–287. [Google Scholar] [CrossRef]
- Mak, O.W.; Sharma, N.; Reynisson, J.; Leung, I.K.H. Discovery of novel Hsp90 C-terminal domain inhibitors that disrupt co-chaperone binding. Bioorg. Med. Chem. Lett. 2021, 38, 127857. [Google Scholar] [CrossRef]
- Leung, E.; Ayine-Tora, D.M.; Santos-Ledo, A.; Korolchuk, V.I.; Reynisson, J. Identification of novel Atg3-Atg8 inhibitors using virtual screening for autophagy modulation. Bioorg. Chem. 2021, 114, 105092. [Google Scholar] [CrossRef] [PubMed]
- Khomenko, T.; Zakharenko, A.; Odarchenko, O.; Arabshahi, H.J.; Sannikova, V.; Zakharova, O.; Korchagina, D.; Reynisson, J.; Volcho, K.; Salakhutdinov, N.; et al. New inhibitors of tyrosyl-DNA phosphodiesterase I (Tdp 1) combining 7-hydroxycoumarin and monoterpenoid moieties. Bioorg. Med. Chem. 2016, 24, 5573–5581. [Google Scholar] [CrossRef] [PubMed]
- Bhusal, R.P.; Patel, K.; Kwai, B.X.C.; Swartjes, A.; Bashiri, G.; Reynisson, J.; Sperry, J.; Leung, I.K.H. Development of NMR and thermal shift assays for the evaluation of Mycobacterium tuberculosis isocitrate lyase inhibitor. Med. Chem. Commun. 2017, 8, 255–2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axerio-Cilies, P.; Castañeda, I.P.; Mirza, A.; Reynisson, J. Investigation of the incidence of “undesirable” molecular moieties for high-throughput screening compound libraries in marketed drug compounds. Eur. J. Med. Chem. 2009, 44, 1128–1134. [Google Scholar] [CrossRef]
- Chavanieu, A.; Pugniere, M. Developments in SPR Fragment Screening. Expert Opin. Drug Discov. 2016, 11, 489–499. [Google Scholar] [CrossRef]
- Siegal, G.; Hollander, J.G. Target immobilization and NMR screening of fragments in early drug discovery. Curr. Topics Med. Chem. 2009, 9, 1736–1745. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H. Biological assay challenges from compound solubility: Strategies for bioassay optimization. Drug Discov. Today 2006, 11, 446–451. [Google Scholar] [CrossRef]
- Vajda, S.; Beglov, D.; Egbert, M.; Wakefield, A.E.; Whitty, A. Cryptic binding sites on proteins: Definition, detection, and druggability. Curr. Opin. Chem. Biol. 2018, 44, 1–8. [Google Scholar] [CrossRef]
- Mizukoshi, Y.; Takeuchi, K.; Tokunaga, Y.; Matsuo, H.; Imai, M.; Fujisaki, M.; Kamoshida, H.; Takizawa, T.; Hanzawa, H.; Shimada, I. Targeting the cryptic sites: NMR-based strategy to improve protein druggability by controlling the conformational equilibrium. Sci. Adv. 2020, 6, eabd0480. [Google Scholar] [CrossRef]
- Pettinger, J.; Le Bihan, Y.-V.; Widya, M.; van Montfort, R.L.M.; Jones, K.; Cheeseman, M.D. An irreversible inhibitor of HSP72 that unexpectedly targets lysine-56. Angew. Chem. Int. Ed. Engl. 2017, 56, 3536–3540. [Google Scholar] [CrossRef] [PubMed]
- Pettinger, J.; Carter, M.; Jones, K.; Cheeseman, M.D. Kinetic Optimization of Lysine-Targeting Covalent Inhibitors of HSP72. J. Med. Chem. 2019, 62, 11383–11398. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Murkin, A.S. New electrophiles and strategies for mechanism-based and targeted covalent inhibitor design. Biochemistry 2019, 58, 5234–5244. [Google Scholar] [CrossRef] [PubMed]
- Miyata, Y.; Rauch, J.N.; Jinwal, U.K.; Thompson, A.D.; Srinivasan, S.; Dickey, C.A.; Gestwicki, J.E. Cysteine Reactivity Distinguishes Redox Sensing by the Heat-Inducible and Constitutive Forms of Heat Shock Protein 70. Chem. Biol. 2012, 19, 1391–1399. [Google Scholar] [CrossRef] [Green Version]
- Sondermann, H.; Scheufler, C.; Schneider, C.; Hohfeld, J.; Hartl, F.U.; Moarefi, I. Structure of a Bag/Hsc70 complex: Convergent functional evolution of Hsp70 nucleotide exchange factors. Science 2001, 291, 1553–1557. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Evans, P. Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 72–82. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [Green Version]
- Bricogne, G.; Blanc, E.; Brandl, M.; Flensburg, C.; Keller, P.; Paciorek, W.; Roversi, P.; Sharff, A.; Smart, O.S.; Vonrhein, C.; et al. BUSTER, Version 2.10.4; Global Phasing Ltd.: Cambridge, UK, 2021. [Google Scholar]
- Smart, O.S.; Holstein, J.; Womack, T.O.; Sharff, A.; Flensburg, C.; Keller, P.; Paciorek, W.; Vonrhein, C.; Bricogne, G. Grade, Version 1.2.20; Global Phasing Ltd.: Cambridge, UK, 2020. [Google Scholar]
- Bruno, I.J.; Cole, J.C.; Lommerse, J.P.M.; Rowland, R.S.; Taylor, R.; Verdonk, M.L. IsoStar: A library of information about nonbonded interactions. J. Comput. Aided Mol. Des. 1997, 11, 525–537. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B., III; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, I.W.; Leaver-Fay, A.; Chen, V.B.; Block, J.N.; Kapral, G.J.; Wang, X.; Murray, L.W.; Arendall, W.B., III; Snoeyink, J.; Richardson, J.S.; et al. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007, 35, W375–W383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Wu, Y.; Tepper, H.L.; Voth, G.A. Flexible simple point-charge water model with improved liquid-state properties. J. Chem. Phys. 2006, 124, 024503. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Batcho, P.F.; Case, D.A.; Schlick, T. Optimized particle-mesh Ewald/multiple-time step integration for molecular dynamics simulations. J. Chem. Phys. 2001, 115, 4003–4018. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Malde, A.K.; Zuo, L.; Breeze, M.; Stroet, M.; Poger, D.; Nair, P.C.; Oostenbrink, C.; Mark, A.E. An Automated Force Field Topology Builder (ATB) and Repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. Multidimensional free-energy calculations using the weighted histogram analysis method. J. Comput. Chem. 1995, 16, 1339–1350. [Google Scholar] [CrossRef]
- Chembridge DIVERSet™ Diverse Screening Libraries. Available online: https://www.chembridge.com/screening_libraries/diversity_libraries/ (accessed on 2 December 2021).
- Ioakimidis, L.; Thoukydidis, L.; Mirza, A.; Naeem, S.; Reynisson, J. Benchmarking the Reliability of QikProp. Correlation between Experimental and Predicted Values. QSAR. Comb. Sci. 2008, 27, 445–456. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | KD (μM) 1 HSC70-NBD | Ligand Efficiency (kJ mol−1 HA−1) 2 | KD (μM) 1 S275W HSC70 |
|---|---|---|---|
| 3 | 388 | 0.16 | 352 |
| Reduction in 1H NMR Peak Intensity Relative to 1 Alone 1 | ||||
|---|---|---|---|---|
| Conditions | Peak a | Peak b | Peak c | Peak d |
| 1 + HSP72-NBD | 58% | 7% | 14% | 13% |
| 1 + HSP72-NBD + ATP | 8% | 12% | 2% | 2% |
| vHTS Hit | HSC70-NBD SPR KD (μM) 1 | Ligand Efficiency (kJ mol−1 HA−1) | HSC70-NBD S275W SPR KD (μM) 2 |
|---|---|---|---|
 4 | 406 (±110) | 0.18 | 385 (±97) |
 5 | 295 (±59) 2 | 0.19 | 176 (±88) |
 6 | 13 (±1) 2 | 0.24 | 12 3 |
 7 | 134 (±69) | 0.21 | 159 (±55) |
 8 | 99 (±14) | 0.20 | 86 (±12) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Connor, S.; Le Bihan, Y.-V.; Westwood, I.M.; Liu, M.; Mak, O.W.; Zazeri, G.; Povinelli, A.P.R.; Jones, A.M.; van Montfort, R.; Reynisson, J.; et al. Discovery and Characterization of a Cryptic Secondary Binding Site in the Molecular Chaperone HSP70. Molecules 2022, 27, 817. https://doi.org/10.3390/molecules27030817
O’Connor S, Le Bihan Y-V, Westwood IM, Liu M, Mak OW, Zazeri G, Povinelli APR, Jones AM, van Montfort R, Reynisson J, et al. Discovery and Characterization of a Cryptic Secondary Binding Site in the Molecular Chaperone HSP70. Molecules. 2022; 27(3):817. https://doi.org/10.3390/molecules27030817
Chicago/Turabian StyleO’Connor, Suzanne, Yann-Vaï Le Bihan, Isaac M. Westwood, Manjuan Liu, Oi Wei Mak, Gabriel Zazeri, Ana P. R. Povinelli, Alan M. Jones, Rob van Montfort, Jóhannes Reynisson, and et al. 2022. "Discovery and Characterization of a Cryptic Secondary Binding Site in the Molecular Chaperone HSP70" Molecules 27, no. 3: 817. https://doi.org/10.3390/molecules27030817