

Development and Validation of the LC–MS/MS Method for Determination of 130 Natural and Synthetic Cannabinoids in Cannabis Oil

Abstract
:1. Introduction
2. Results and Discussion
2.1. LC–MS/MS Method
2.2. Extraction
2.3. Method Validation
2.3.1. Selectivity and Sensitivity
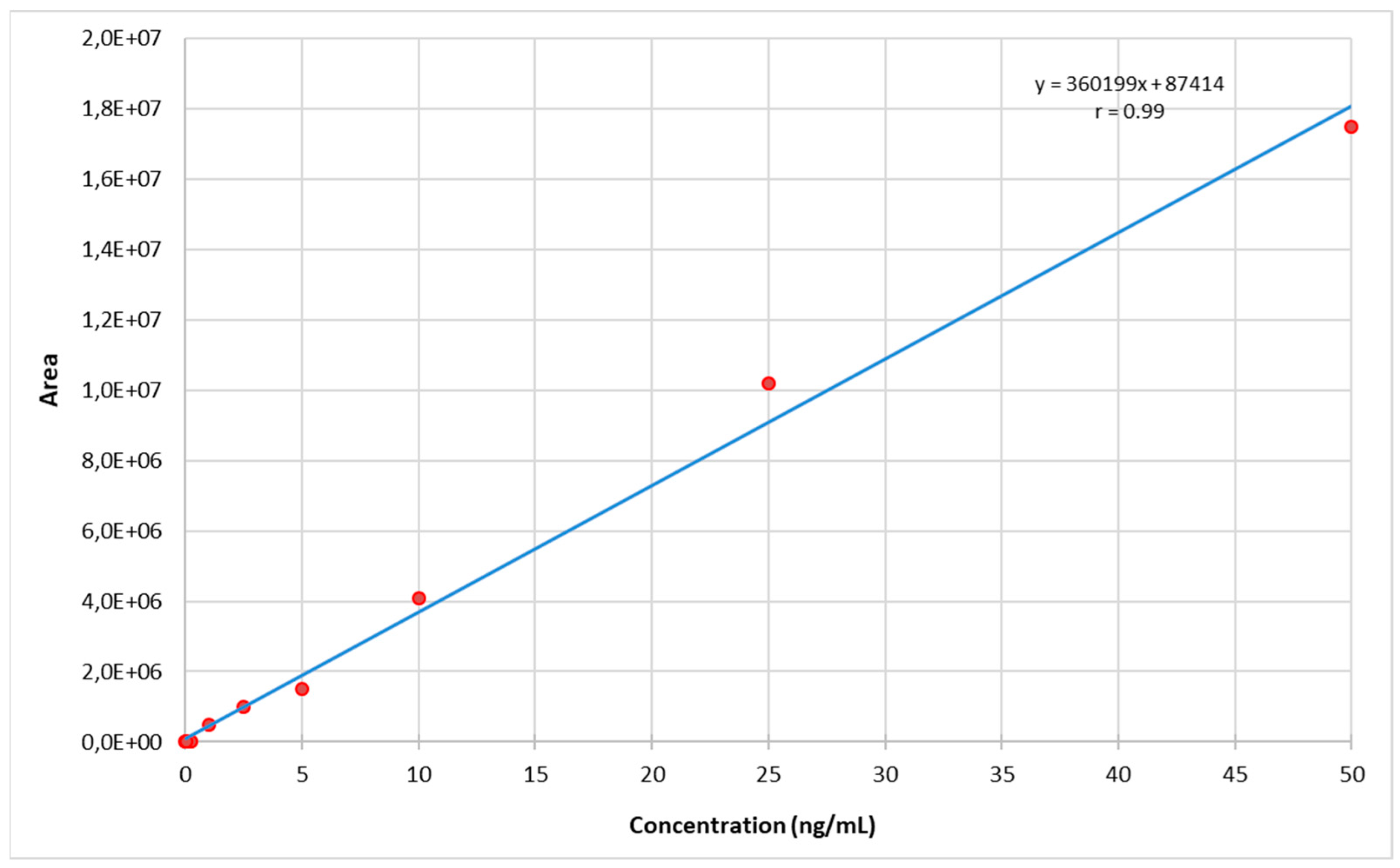
2.3.2. Calibration Model
2.3.3. Precision and Bias
2.3.4. Carryover
2.3.5. Matrix Effect
2.3.6. Dilution Integrity
2.4. Application to Dietary Supplements and Food Samples
3. Materials and Methods
3.1. Preparation of Standards and Solutions
3.2. Sample Preparation
3.3. HPLC
3.4. MS/MS
3.5. Validation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- White, C.M. Dietary Supplements Pose Real Dangers to Patients. Ann. Pharmacother. 2020, 54, 815–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernieri, C.; Nichetti, F.; Raimondi, A.; Pusceddu, S.; Platania, M.; Berrino, F.; de Braud, F. Diet and supplements in cancer prevention and treatment: Clinical evidences and future perspectives. Crit. Rev. Oncol. Hematol. 2018, 123, 57–73. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, W.T.; Gao, Y.; Xue, X.; Pang, H.; Hao, W.; Xia, Y.; Wang, S.; Su, X.; Zhao, L.; et al. Analysis of Dietary Supplement Use and Influencing Factors in the Mongolian Population. Biomed. Res. Int. 2022, 22, 4064588. [Google Scholar] [CrossRef] [PubMed]
- Adegboye, A.R.A.; Ojo, O.; Begum, G. The use of dietary supplements among African and Caribbean women living in the UK: A cross-sectional study. Nutrients 2020, 12, 847. [Google Scholar] [CrossRef] [Green Version]
- LeDoux, M.A.; Appelhans, K.R.; Braun, L.A.; Dziedziczak, D.; Jennings, S.; Liu, L.; Osiecki, H.; Wyszumiala, E.; Griffiths, J.C. A quality dietary supplement: Before you start and after it’s marketed—A conference report. Eur. J. Nutr. 2015, 54, S1–S8. [Google Scholar] [CrossRef] [Green Version]
- Marcus, D.M. Dietary supplements: What’s in a name? What’s in the bottle? Drug Test. Anal. 2016, 8, 410–412. [Google Scholar] [CrossRef]
- Ratajczak, M.; Kaminska, D.; Światły-Błaszkiewicz, A.; Matysiak, J. Quality of Dietary Supplements Containing Plant-Derived Ingredients Reconsidered by Microbiological Approach. Int. J. Environ. Res. Public Health 2020, 17, 6837. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, J.Y.; Cho, S.H.; Jeong, J.H.; Cho, S.; Park, H.J.; Baek, S.Y. Determination of Miroestrol and Isomiroestrol from Pueraria mirifica (White Kwao Krua) in Dietary Supplements by LC-MS-MS and LC-Q-Orbitrap/MS. J. Chromatogr. Sci. 2017, 55, 214–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fibigr, J.; Šatínský, D.; Solich, P. Current trends in the analysis and quality control of food supplements based on plant extracts. Anal. Chim. Acta 2018, 1036, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Muschietti, L.; Redko, F.; Ulloa, J. Adulterants in selected dietary supplements and their detection methods. Drug Test. Anal. 2020, 12, 861–886. [Google Scholar] [CrossRef] [PubMed]
- Pawar, R.S.; Sagi, S.; Leontyev, D. Analysis of bitter orange dietary supplements for natural and synthetic phenethylamines by LC-MS/MS. Drug Test. Anal. 2020, 12, 1241–1251. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Kang, H.S.; Kim, H.; Moon, G. Multi-Class Determination of 64 Illicit Compounds in Dietary Supplements Using Liquid Chromatography-Tandem Mass Spectrometry. Molecules 2020, 25, 4399. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.S.; Lim, N.Y.; Choi, H.S.; Lee, J.H.; Moon, S.H.; Kim, H.; Baek, S.Y. Simultaneous screening of dietary supplements for 25 anti-hyperlipidemic substances using ultra-performance liquid chromatography and liquid chromatography/electrospray ionization tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2021, 35, e8989. [Google Scholar] [CrossRef] [PubMed]
- Musiał, J.; Powierska-Czarny, J.; Czarny, J.; Raczkowski, M.; Galant, N.; Buszewski, B.; Gadzała-Kopciuch, R. One-step extraction and determination of 513 psychoactive substances, drugs, and their metabolites from hair by LC-MS/MS. Arch. Toxicol. 2022, 96, 2927–2933. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.A.; Travis, J.C.; Venhuis, B.J. A synthetic stimulant never tested in humans, 1,3-dimethylbutylamine (DMBA), is identified in multiple dietary supplements. Drug Test. Anal. 2015, 7, 83–87. [Google Scholar] [CrossRef]
- Liang, Q.; Qu, J.; Luo, G.; Wang, Y. Rapid and reliable determination of illegal adulterant in herbal medicines and dietary supplements by LC/MS/MS. J. Pharm. Biomed. Anal. 2006, 40, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Lee, J.H.; Park, H.J.; Cho, S.H.; Cho, S.; Kim, W.S. Monitoring of 29 weight loss compounds in foods and dietary supplements by LC-MS/MS. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2014, 31, 777–783. [Google Scholar] [CrossRef]
- Choi, J.Y.; Heo, S.; Yoo, G.J.; Park, S.K.; Yoon, C.Y.; Baek, S.Y. Development and validation of an LC-MS/MS method for the simultaneous analysis of 28 specific narcotic adulterants used in dietary supplements. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2015, 32, 1029–1039. [Google Scholar] [CrossRef] [PubMed]
- Bogusz, M.J.; Hassan, H.; Al-Enazi, E.; Ibrahim, Z.; Al-Tufail, M. Application of LC-ESI-MS-MS for detection of synthetic adulterants in herbal remedies. J. Pharm. Biomed. Anal. 2006, 41, 554–564. [Google Scholar] [CrossRef]
- Lu, Y.; Anderson, H.D. Cannabinoid signaling in health and disease. Can. J. Physiol. Pharmacol. 2017, 95, 311–327. [Google Scholar] [CrossRef]
- Starowicz, K.; Finn, D.P. Cannabinoids and Pain: Sites and Mechanisms of Action. Adv. Pharmacol. 2017, 80, 437–475. [Google Scholar] [PubMed] [Green Version]
- Fraguas-Sánchez, A.I.; Torres-Suárez, A.I. Medical Use of Cannabinoids. Drugs 2018, 78, 1665–1703. [Google Scholar] [CrossRef] [PubMed]
- Jones, É.; Vlachou, S.A. Critical Review of the Role of the Cannabinoid Compounds Δ9-Tetrahydrocannabinol (Δ9-THC) and Cannabidiol (CBD) and their Combination in Multiple Sclerosis Treatment. Molecules 2020, 25, 4930. [Google Scholar] [CrossRef]
- Dos Reis Rosa Franco, G.; Smid, S.; Viegas, C. Phytocannabinoids: General Aspects and Pharmacological Potential in Neurodegenerative Diseases. Curr. Neuropharmacol. 2021, 19, 449–464. [Google Scholar] [CrossRef]
- Lafaye, G.; Karila, L.; Blecha, L.; Benyamina, A. Cannabis, cannabinoids, and health. Dialogues Clin. Neurosci. 2017, 19, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Burstein, S. Cannabidiol (CBD) and its analogs: A review of their effects on inflammation. Bioorg. Med. Chem. 2015, 23, 1377–1385. [Google Scholar] [CrossRef]
- Christinat, N.; Savoy, M.C.; Mottier, P. Development, validation and application of a LC-MS/MS method for quantification of 15 cannabinoids in food. Food Chem. 2020, 318, 126469. [Google Scholar] [CrossRef]
- Cerino, P.; Buonerba, C.; Cannazza, G.; D’Auria, J.; Ottoni, E.; Fulgione, A.; Di Stasio, A.; Pierri, B.; Gallo, A. A Review of Hemp as Food and Nutritional Supplement. Cannabis Cannabinoid Res. 2021, 6, 19–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanabus, J.; Bryła, M.; Roszko, M.; Modrzewska, M.; Pierzgalski, A. Cannabinoids-Characteristics and Potential for Use in Food Production. Molecules 2021, 26, 6723. [Google Scholar] [CrossRef] [PubMed]
- Zivovinovic, S.; Alder, R.; Allenspach, M.D.; Steuer, C. Determination of cannabinoids in Cannabis sativa L. samples for recreational, medical, and forensic purposes by reversed-phase liquid chromatography-ultraviolet detection. J. Anal. Sci. Technol. 2018, 9, 27. [Google Scholar] [CrossRef]
- Kim, J.Y.; Park, H.J.; Kim, J.W.; Lee, J.H.; Heo, S.; Yoon, C.Y.; Cho, S. Development and validation of UPLC and LC-MS/MS methods for the simultaneous determination of anti-obesity drugs in foods and dietary supplements. Arch. Pharm. Res. 2016, 39, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Le Boisselier, R.; Alexandre, J.; Lelong-Boulouard, V.; Debruyne, D. Focus on cannabinoids and synthetic cannabinoids. Clin. Pharmacol. Ther. 2017, 101, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Heo, S.; Yoo, G.J.; Choi, J.Y.; Park, H.J.; Do, J.A.; Cho, S.; Baek, S.Y.; Park, S.K. Simultaneous Analysis of Cannabinoid and Synthetic Cannabinoids in Dietary Supplements Using UPLC with UV and UPLC-MS-MS. J. Anal. Toxicol. 2016, 40, 350–359. [Google Scholar] [CrossRef] [Green Version]
- Meng, Q.; Buchanan, B.; Zuccolo, J.; Poulin, M.M.; Gabriele, J.; Baranowski, D.C. A reliable and validated LC-MS/MS method for the simultaneous quantification of 4 cannabinoids in 40 consumer products. PLoS ONE 2018, 13, e0196396. [Google Scholar] [CrossRef]
- Nie, B.; Henion, J.; Ryona, I. The Role of Mass Spectrometry in the Cannabis Industry. J. Am. Soc. Mass Spectrom. 2019, 30, 719–730. [Google Scholar] [CrossRef]
- Lee, J.H.; Min, A.Y.; Han, J.H.; Yang, Y.J.; Kim, H.; Shin, D. Development and validation of LC-MS/MS method with QuEChERS clean-up for detecting cannabinoids in foods and dietary supplements. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2020, 37, 1413–1424. [Google Scholar] [CrossRef]
- McRae, G.; Melanson, J.E. Quantitative determination and validation of 17 cannabinoids in cannabis and hemp using liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2020, 412, 7381–7393. [Google Scholar] [CrossRef]
- Takashina, S.; Takahashi, M.; Morimoto, K.; Inoue, K. LC-MS/MS Assay for the Measurement of Cannabidiol Profiling in CBD Oil from Japanese Market and Application for Convertible Tetrahydrocannabinol in Acetic Acid Condition. Chem. Pharm. Bull. 2022, 70, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Stefkov, G.; Cvetkovikj Karanfilova, I.; Stoilkovska Gjorgievska, V.; Trajkovska, A.; Geskovski, N.; Karapandzova, M.; Kulevanova, S. Analytical Techniques for Phytocannabinoid Profiling of Cannabis and Cannabis-Based Products-A Comprehensive Review. Molecules 2022, 27, 975. [Google Scholar] [CrossRef] [PubMed]
- Al Ubeed, H.M.S.; Bhuyan, D.J.; Alsherbiny, M.A.; Basu, A.; Vuong, Q.V. A Comprehensive Review on the Techniques for Extraction of Bioactive Compounds from Medicinal Cannabis. Molecules 2022, 27, 604. [Google Scholar] [CrossRef]
- Kim, S.H.; Lee, J.; Yoon, T.; Choi, J.; Choi, D.; Kim, D.; Kwon, S.W. Simultaneous determination of anti-diabetes/anti-obesity drugs by LC/PDA, and targeted analysis of sibutramine analog in dietary supplements by LC/MS/MS. Biomed. Chromatogr. 2009, 23, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Y.; Wang, Y. Validated quantification method for five ephedrines in dietary supplements using LC-MS/MS: Application to 503 cases. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1039, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.; Kim, J.W.; Han, K.M.; Lee, J.H.; Hwang, I.S.; Lee, J.H.; Kim, J.; Kweon, S.J.; Cho, S.; Chae, K.R.; et al. Simultaneous analysis of 17 diuretics in dietary supplements by HPLC and LC-MS/MS. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2013, 30, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Van Thuyne, W.; Delbeke, F.T. Validation of a GC-MS screening method for anabolizing agents in solid nutritional supplements. Biomed. Chromatogr. 2004, 18, 155–159. [Google Scholar] [CrossRef]
- Uekusa, K.; Ono, T.; Hayashida, M.; Nihira, M.; Ohno, Y. GC/MS analysis of an herbal dietary supplement containing ephedrine. Leg. Med. 2009, 1, S573–S575. [Google Scholar] [CrossRef] [PubMed]
- Jang, E.; Kim, H.; Jang, S.; Lee, J.; Baeck, S.; In, S.; Kim, E.; Kim, Y.U.; Han, E. Concentrations of THC, CBD, and CBN in commercial hemp seeds and hempseed oil sold in Korea. Forensic. Sci. Int. 2020, 306, 110064. [Google Scholar] [CrossRef]
- Di Marco Pisciottano, I.; Guadagnuolo, G.; Soprano, V.; Esposito, M.; Gallo, P. A survey of Δ9-THC and relevant cannabinoids in products from the Italian market: A study by LC-MS/MS of food, beverages and feed. Food Chem. 2021, 346, 128898. [Google Scholar] [CrossRef] [PubMed]
- Scientific Working Group for Forensic Toxicology. Scientific Working Group for Forensic Toxicology (SWGTOX) standard practices for method validation in forensic toxicology. J. Anal. Toxicol. 2013, 452–474. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Cannabis Oil Samples | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | |
| DELTA-9-TETRAHYDROCANNABINOL (THC) | <0.01 | 0.03 | 0.07 | 0.07 | 0.09 | 0.09 | <0.01 | 0.02 | 0.02 | <0.01 | <0.01 | 0.06 |
| DELTA-9-TETRAHYDROCANNABINOL ACID (THCA-A) | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | 0.01 |
| CANNABIDIOL (CBD) | 28.38 | 3.85 | 15.71 | 10.75 | 7.32 | 4.74 | 1.54 | 2.04 | 0.84 | <0.01 | <0.01 | 6.09 |
| CANNABIDIOLIC ACID (CBDA) | 0.01 | 1.63 | 0.26 | <0.01 | 0.66 | 0.01 | <0.01 | <0.01 | 0.33 | <0.01 | <0.01 | 2.41 |
| CANNABIGEROL (CBG) | 0.02 | 0.01 | <0.01 | 0.32 | 1.12 | 5.06 | 0.16 | 0.05 | 0.01 | <0.01 | <0.01 | 0.23 |
| CANNABIGEROLIC ACID (CBGA) | <0.01 | <0.01 | <0.01 | <0.01 | 0.06 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | 0.02 |
| CANNABICHROMENE (CBC) | <0.01 | 0.05 | 0.13 | 0.12 | 0.19 | 0.21 | <0.01 | 0.08 | 0.03 | <0.01 | <0.01 | 0.11 |
| CANNABIVARIN (CBV) | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 |
| CANNABIDIVARINIC ACID (CBDVA) | <0.01 | 0.02 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | 0.03 |
| CANNABIGEROVARINIC ACID (CBGVA) | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 |
| CANNABINOL (CBN) | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | 0.04 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 |
| TETRAHYDROCANNABIVARINIC ACID (THCVA) | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 |
| TETRAHYDROCANNABIVARIN (THCV) | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 |
| SYNTHETIC CANNABINOIDS (117 COMPOUNDS) | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galant, N.; Czarny, J.; Powierska-Czarny, J.; Piotrowska-Cyplik, A. Development and Validation of the LC–MS/MS Method for Determination of 130 Natural and Synthetic Cannabinoids in Cannabis Oil. Molecules 2022, 27, 8601. https://doi.org/10.3390/molecules27238601
Galant N, Czarny J, Powierska-Czarny J, Piotrowska-Cyplik A. Development and Validation of the LC–MS/MS Method for Determination of 130 Natural and Synthetic Cannabinoids in Cannabis Oil. Molecules. 2022; 27(23):8601. https://doi.org/10.3390/molecules27238601
Chicago/Turabian StyleGalant, Natalia, Jakub Czarny, Jolanta Powierska-Czarny, and Agnieszka Piotrowska-Cyplik. 2022. "Development and Validation of the LC–MS/MS Method for Determination of 130 Natural and Synthetic Cannabinoids in Cannabis Oil" Molecules 27, no. 23: 8601. https://doi.org/10.3390/molecules27238601