
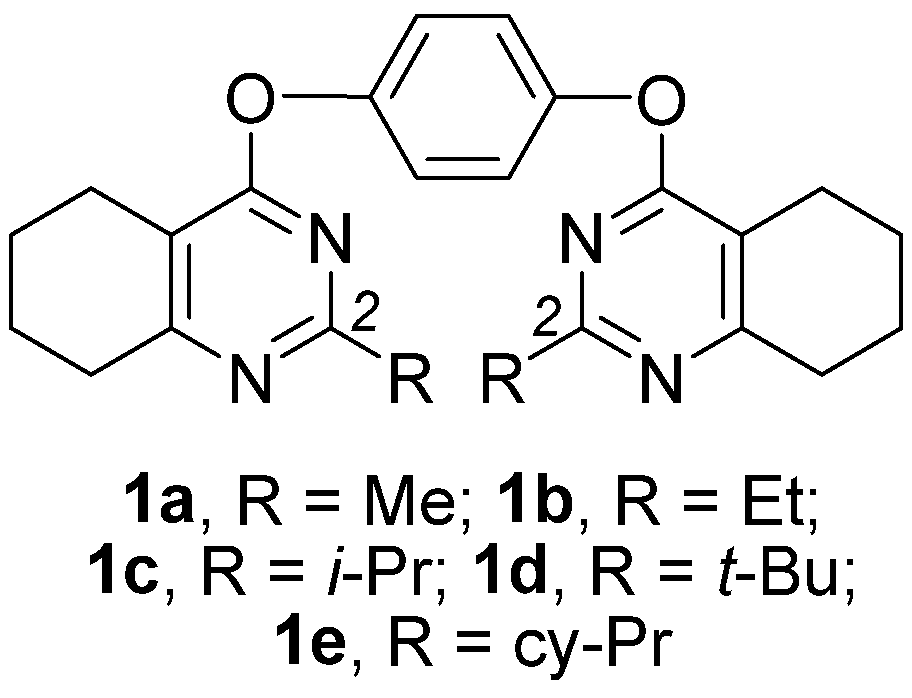
Novel Nanomolar Allosteric Modulators of AMPA Receptor of Bis(pyrimidine) Series: Synthesis, Biotesting and SAR Analysis
 , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Electrophysiological Evaluation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Number of Neurons n | Currents (%) for Various Concentrations of Compounds (M, Control = 100%) | ||||||
|---|---|---|---|---|---|---|---|---|
| 10−12 | 10−11 | 10−10 | 10−9 | 10−8 | 10−7 | 10−6 | ||
| 1a [30] | 7 | 108 ± 5 | 132 ± 5 | 143 ± 9 | 170 ± 11 | 123 ± 8 | 85 ± 6 | 78 ± 4 |
| 1b [30] | 5 | 100 ± 2 | 117 ± 6 | 126 ± 8 | 155 ± 5 | 128 ± 7 | 100 ± 8 | – |
| 1c [30] | 4 | 100 ± 2 | 84 ± 5 | 72 ± 6 | 82 ± 7 | 92 ± 4 | 98 ± 5 | – |
| 1d [30] | 5 | – | 100 ± 2 | 108 ± 4 | 120 ± 4 | 125 ± 5 | 133 ± 6 | 145 ± 7 |
| 1e [30] | 5 | – | 100 ± 2 | 100 ± 2 | 95 ± 4 | 96 ± 3 | 97 ± 2 | 96 ± 5 |
| 1f | 4 | 101 ± 9 | 118 ± 10 | 147 ± 12 | 166 ± 12 | 157 ± 11 | 144 ± 12 | 122 ± 8 |
| 1g | 3 | 100 ± 3 | 100 ± 3 | 108 ± 3 | 118 ± 4 | 129 ± 4 | 115 ± 4 | 101 ± 3 |
| 1h | 5 | 100 ± 4 | 141 ± 9 | 149 ± 10 | 153 ± 9 | 129 ± 6 | 110 ± 5 | 100 ± 4 |
| 1i | 5 | 103 ± 3 | 129 ± 9 | 141 ± 9 | 151 ± 12 | 134 ± 9 | 109 ± 4 | 102 ± 3 |
| 1j | 6 | 94 ± 2 | 82 ± 3 | 76 ± 4 | 71 ± 4 | 64 ± 6 | 57 ± 6 | 52 ± 7 |
| 1k | 5 | 105 ± 2 | 138 ± 5 | 149 ± 6 | 177 ± 6 | 163 ± 6 | 155 ± 7 | 117 ± 4 |
| 1l | 4 | 111 ± 9 | 117 ± 9 | 124 ± 7 | 129 ± 6 | 138 ± 8 | 132 ± 8 | 124 ± 7 |
| 1m | 4 | 120 ± 9 | 133 ± 9 | 129 ± 9 | 119 ± 9 | 117 ± 9 | 115 ± 9 | 102 ± 9 |
| 1n | 3 | 100 ± 3 | 121 ± 4 | 129 ± 5 | 138 ± 6 | 136 ± 5 | 136 ± 4 | 112 ± 3 |
| 1o | 5 | 115 ± 3 | 138 ± 9 | 147 ± 10 | 161 ± 9 | 160 ± 10 | 158 ± 11 | 116 ± 4 |
| 1p | 4 | 117 ± 9 | 126 ± 11 | 128 ± 10 | 133 ± 11 | 125 ± 10 | 120 ± 11 | 102 ± 3 |
| CTZ | 8 | - | - | - | - | - | 100 ± 3 | 145 ± 11 |
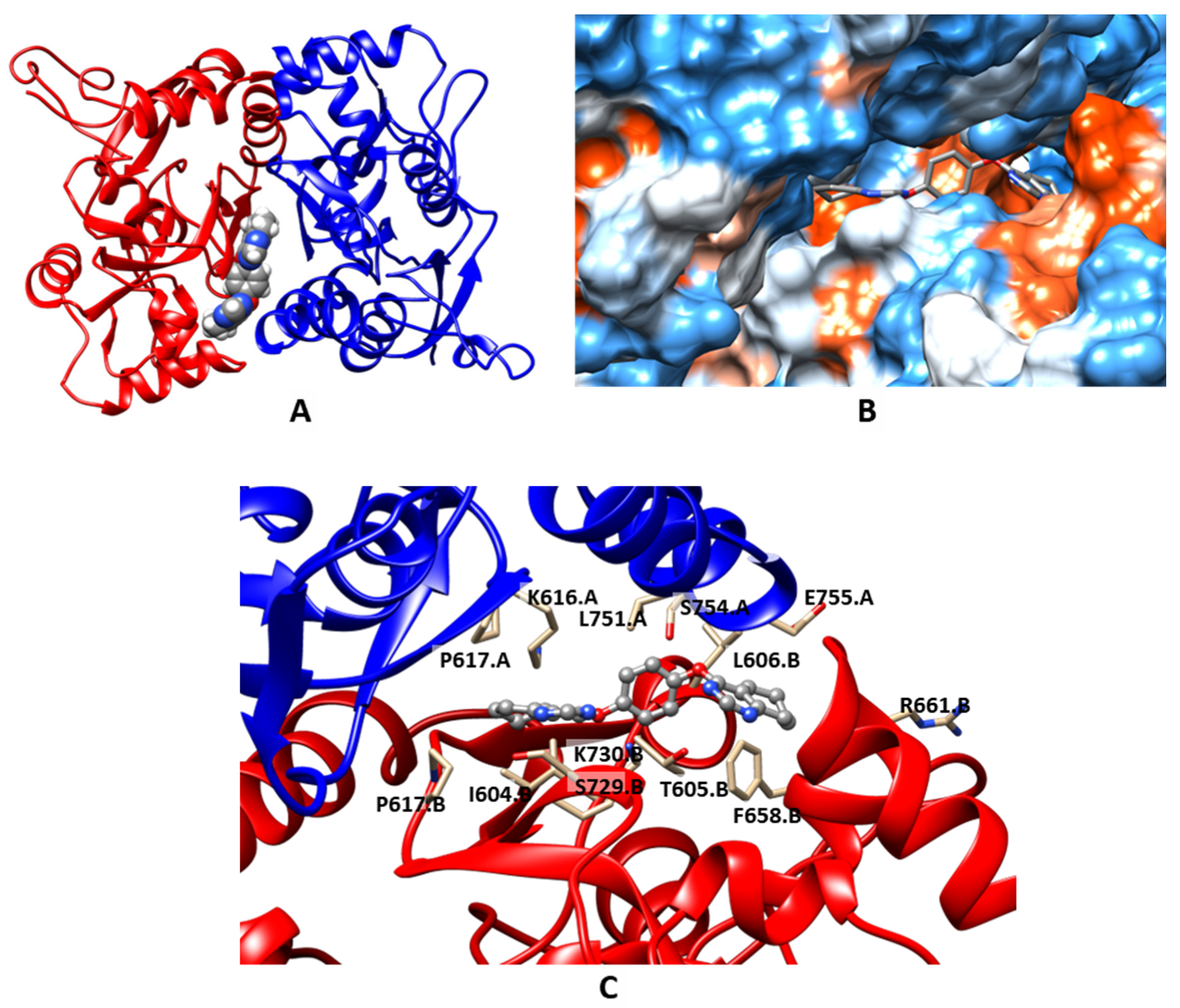
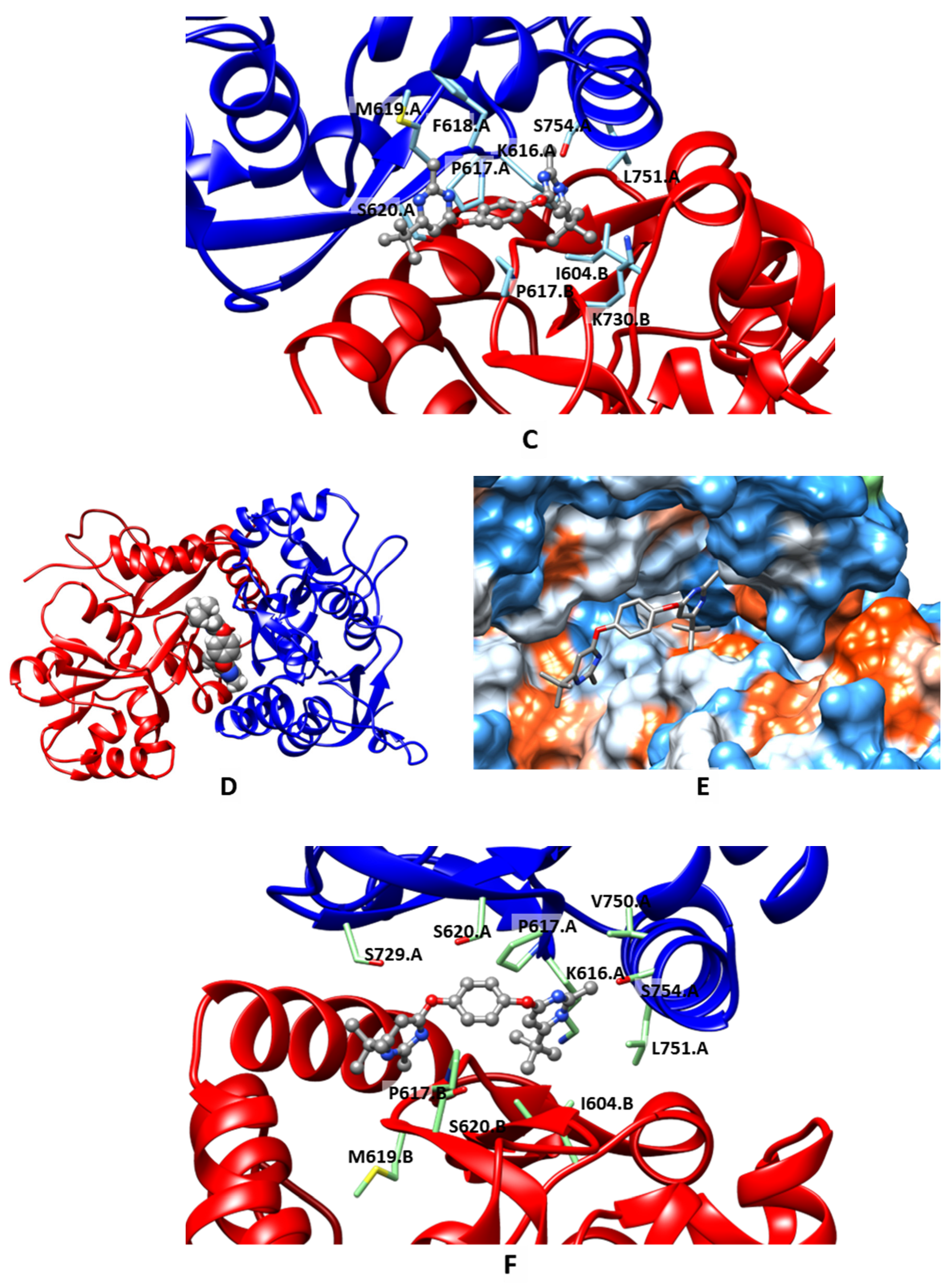
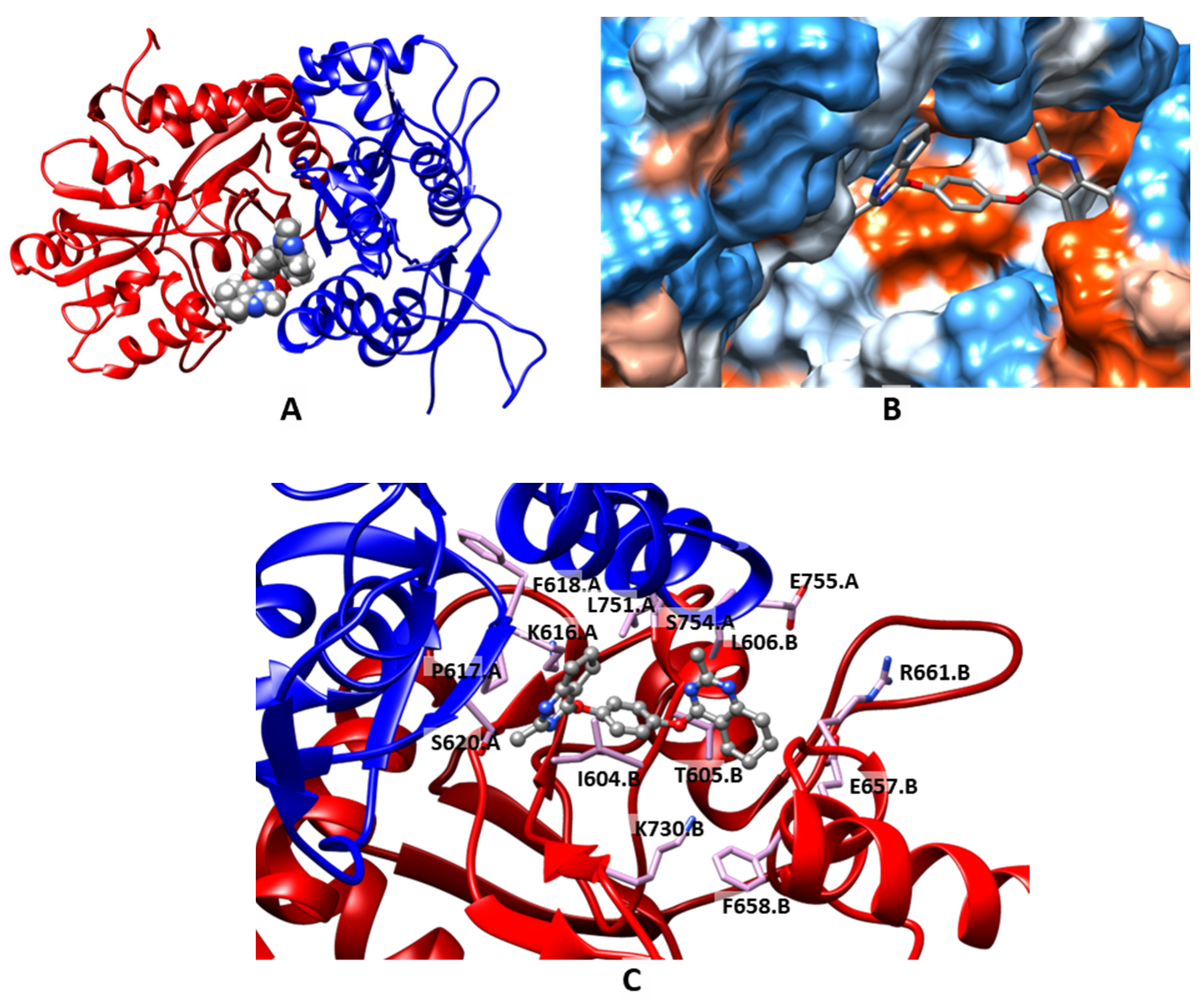
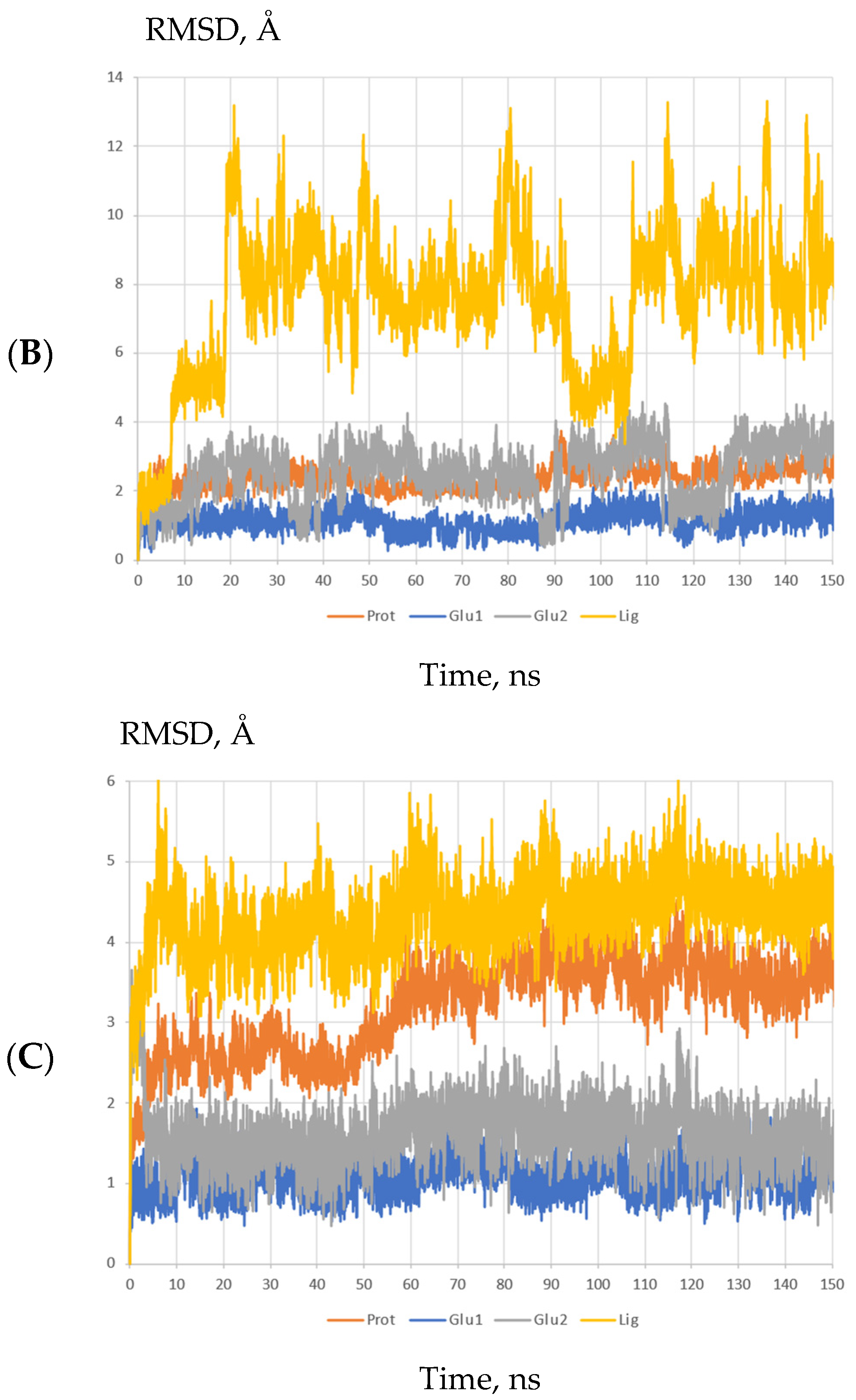
2.3. Molecular Modeling
2.4. Prediction of ADMET, Physicochemical, and PAINS Profiles
3. Materials and Methods
3.1. Chemistry
3.1.1. General Remarks
3.1.2. Synthesis of 4-Chloro-2-methyl-6,7,8,9-tetrahydro-5H-cyclohepta[d]pyrimidine (2c)
3.1.3. Synthesis of 4-(4-(Benzyloxy)phenoxy)pyrimidines 4a–e,7 (General Method)
4-(4-(Benzyloxy)phenoxy)-2-methyl-5,6,7,8-tetrahydroquinazoline (4a)
4-(4-(Benzyloxy)phenoxy)-2-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidine (4b)
4-(4-(Benzyloxy)phenoxy)-2-methyl-6,7,8,9-tetrahydro-5H-cyclohepta[d]pyrimidine (4c)
4-(4-(Benzyloxy)phenoxy)-6-(tert-butyl)-2-methylpyrimidine (4d)
4-(4-(Benzyloxy)phenoxy)-5,6,7,8-tetrahydroquinazoline (4e)
4-(4-(Benzyloxy)phenoxy)-2-methyl-5,6,7,8-tetrahydroquinazoline 1-Oxide (7)
3.1.4. Synthesis of 4-((Pyrimidin-4-yl)oxy)phenols 5a–e,8 (General Method)
4-((2-Methyl-5,6,7,8-tetrahydroquinazolin-4-yl)oxy)phenol (5a)
4-((2-Methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)oxy)phenol (5b)
4-((2-Methyl-6,7,8,9-tetrahydro-5H-cyclohepta[d]pyrimidin-4-yl)oxy)phenol (5c)
4-((6-(tert-Butyl)-2-methylpyrimidin-4-yl)oxy)phenol (5d)
4-((5,6,7,8-Tetrahydroquinazolin-4-yl)oxy)phenol (5e)
4-[(2-Methyl-1-oxido-5,6,7,8-tetrahydroquinazolin-4-yl)oxy]phenol (8) [30]
3.1.5. Synthesis of Bis(pyrimidines) 1a–p (General Method)
1,4-Bis((2-methyl-5,6,7,8-tetrahydroquinazolin-4-yl)oxy)benzene (1a) [30]
1,4-Bis((5,6,7,8-tetrahydroquinazolin-4-yl)oxy)benzene (1f)
1,4-Bis((2-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)oxy)benzene (1g)
1,4-Bis((2-methyl-6,7,8,9-tetrahydro-5H-cyclohepta[d]pyrimidin-4-yl)oxy)benzene (1h)
1,4-Bis((6-(tert-butyl)-2-methylpyrimidin-4-yl)oxy)benzene (1i)
2-Methyl-4-(4-((2-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)oxy)phenoxy)-5,6,7,8-tetrahydroquinazoline (1j)
2-Methyl-4-(4-((2-methyl-5,6,7,8-tetrahydroquinazolin-4-yl)oxy)phenoxy)-6,7,8,9-tetrahydro-5H-cyclohepta[d]pyrimidine (1k)
4-(4-((6-(tert-Butyl)-2-methylpyrimidin-4-yl)oxy)phenoxy)-2-methyl-5,6,7,8-tetrahydroquinazoline (1l)
4-(4-((6-(tert-Butyl)-2-methylpyrimidin-4-yl)oxy)phenoxy)-2-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidine (1m)
4-(4-((6-(tert-Butyl)-2-methylpyrimidin-4-yl)oxy)phenoxy)-2-methyl-6,7,8,9-tetrahydro-5H-cyclohepta[d]pyrimidine (1n)
2-Methyl-4-(4-((2-methyl-5,6,7,8-tetrahydroquinazolin-4-yl)oxy)phenoxy)-5,6,7,8-tetrahydroquinazoline 1-Oxide (1o)
4,4′-[1,4-Phenylenebis(oxy)]bis(2-methyl-5,6,7,8-tetrahydroquinazoline) 1,1′-Dioxide (1p) [30]
3.2. Electrophysiological Evaluation
3.3. Molecular Modeling
3.4. Prediction of ADMET, Physicochemical, and PAINS Profiles
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Reiner, A.; Levitz, J. Glutamatergic signaling in the central nervous system: Ionotropic and metabotropic receptors in concert. Neuron 2018, 98, 1080–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brogi, S.; Campiani, G.; Brindisi, M.; Butini, S. Allosteric modulation of ionotropic glutamate receptors: An outlook on new therapeutic approaches to treat central nervous system disorders. ACS Med. Chem. Lett. 2019, 10, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radchenko, E.V.; Tarakanova, A.S.; Karlov, D.S.; Lavrov, M.I.; Palyulin, V.A. Ligands of the AMPA-subtype glutamate receptors: Mechanisms of action and novel chemotypes. Biomed. Khim. 2021, 67, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Partin, K.M. AMPA receptor potentiators: From drug design to cognitive enhancement. Curr. Opin. Pharmacol. 2015, 20, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Lauterborn, J.C.; Palmer, L.C.; Jia, Y.; Pham, D.T.; Hou, B.; Wang, W.; Trieu, B.H.; Cox, C.D.; Kantorovich, S.; Gall, C.M.; et al. Chronic ampakine treatments stimulate dendritic growth and promote learning in middle-aged rats. J. Neurosci. 2016, 36, 1636–1646. [Google Scholar] [CrossRef] [Green Version]
- Arai, A.C.; Xia, Y.-F.; Rogers, G.; Lynch, G.; Kessler, M. Benzamide-type AMPA receptor modulators form two subfamilies with distinct modes of action. J. Pharmacol. Exp. Ther. 2002, 303, 1075–1085. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Goodman, L.; Fourie, C.; Schenk, S.; Leitch, B.; Montgomery, J.M. AMPA receptors as therapeutic targets for neurological disorders. In Ion Channels as Therapeutic Targets, Part A; Advances in Protein Chemistry and Structural Biology; Donev, R., Ed.; Academic Press: Cambridge, MA, USA, 2016; Volume 103, pp. 203–261. [Google Scholar] [CrossRef]
- Twomey, E.C.; Sobolevsky, A.I. Structural mechanisms of gating in ionotropic glutamate receptors. Biochemistry 2018, 57, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Lynch, G. Glutamate-based therapeutic approaches: Ampakines. Curr. Opin. Pharmacol. 2006, 6, 82–88. [Google Scholar] [CrossRef]
- Chen, S.; Gouaux, E. Structure and mechanism of AMPA receptor—auxiliary protein complexes. Curr. Opin. Struct. Biol. 2019, 54, 104–111. [Google Scholar] [CrossRef]
- Ren, J.; Lenal, F.; Yang, M.; Ding, X.; Greer, J.J. Coadministration of the AMPAKINE CX717 with propofol reduces respiratory depression and fatal apneas. Anesthesiology 2013, 118, 1437–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Beugen, B.J.; Qiao, X.; Simmons, D.H.; De Zeeuw, C.I.; Hansel, C. Enhanced AMPA receptor function promotes cerebellar long-term depression rather than potentiation. Learn. Mem. 2014, 21, 662–667. [Google Scholar] [CrossRef] [Green Version]
- Lauterborn, J.C.; Truong, G.S.; Baudry, M.; Bi, X.; Lynch, G.; Gall, C.M. Chronic elevation of brain-derived neurotrophic factor by ampakines. J. Pharmacol. Exp. Ther. 2003, 307, 297–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmons, D.A.; Rex, C.S.; Palmer, L.; Pandyarajan, V.; Fedulov, V.; Gall, C.M.; Lynch, G. Up-regulating BDNF with an ampakine rescues synaptic plasticity and memory in Huntington’s disease knockin mice. Proc. Natl. Acad. Sci. USA 2009, 106, 4906–4911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radin, D.P.; Johnson, S.; Purcell, R.; Lippa, A.S. Effects of chronic systemic low-impact ampakine treatment on neurotrophin expression in rat brain. Biomed. Pharmacother. 2018, 105, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Freudenberg, F.; Celikel, T.; Reif, A. The role of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors in depression: Central mediators of pathophysiology and antidepressant activity? Neurosci. Biobehav. Rev. 2015, 52, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Murakami, K.; Tajima, Y.; Hara, H.; Kunugi, A.; Kimura, H. TAK-137, an AMPA receptor potentiator with little agonistic effect, produces antidepressant-like effect without causing psychotomimetic effects in rats. Pharmacol. Biochem. Behav. 2019, 183, 80–86. [Google Scholar] [CrossRef]
- Gordillo-Salas, M.; Pascual-Antón, R.; Ren, J.; Greer, J.; Adell, A. Antidepressant-like effects of CX717, a positive allosteric modulator of AMPA receptors. Mol. Neurobiol. 2020, 57, 3498–3507. [Google Scholar] [CrossRef]
- Suzuki, A.; Tajima, Y.; Kunugi, A.; Kimura, H. Electrophysiological characterization of a novel AMPA receptor potentiator, TAK-137, in rat hippocampal neurons. Neurosci. Lett. 2019, 712, 134488. [Google Scholar] [CrossRef]
- Ward, S.E.; Harries, M.H.; Aldegheri, L.; Bradford, A.M.; Ballini, E.; Dawson, L.; Lacroix, L.; Pardoe, J.; Starr, K.; Weil, A.; et al. Pharmacological characterisation of MDI-222, a novel AMPA receptor positive allosteric modulator with an improved safety profile. J. Psychopharmacol. 2020, 34, 93–102. [Google Scholar] [CrossRef]
- Goffin, E.; Drapier, T.; Larsen, A.P.; Geubelle, P.; Ptak, C.P.; Laulumaa, S.; Rovinskaja, K.; Gilissen, J.; de Tullio, P.; Olsen, L.; et al. 7-Phenoxy-substituted 3,4-dihydro-2H-1,2,4-benzothiadiazine 1,1-dioxides as positive allosteric modulators of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors with nanomolar potency. J. Med. Chem. 2018, 61, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Hanada, T. The discovery and development of perampanel for the treatment of epilepsy. Expert Opin. Drug Discov. 2014, 9, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Potschka, H.; Trinka, E. Perampanel: Does it have broad-spectrum potential? Epilepsia 2019, 60, 22–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenum-Berg, C.; Musgaard, M.; Chavez-Abiega, S.; Thisted, C.L.; Barrella, L.; Biggin, P.C.; Kristensen, A.S. Mutational analysis and modeling of negative allosteric modulator binding sites in AMPA receptors. Mol. Pharmacol. 2019, 96, 835–850. [Google Scholar] [CrossRef]
- Narangoda, C.; Sakipov, S.N.; Kurnikova, M.G. AMPA receptor noncompetitive inhibitors occupy a promiscuous binding site. ACS Chem. Neurosci. 2019, 10, 4511–4521. [Google Scholar] [CrossRef]
- Drapier, T.; Geubelle, P.; Bouckaert, C.; Nielsen, L.; Laulumaa, S.; Goffin, E.; Dilly, S.; Francotte, P.; Hanson, J.; Pochet, L.; et al. Enhancing action of positive allosteric modulators through the design of dimeric compounds. J. Med. Chem. 2018, 61, 5279–5291. [Google Scholar] [CrossRef]
- Lavrov, M.I.; Karlov, D.S.; Voronina, T.A.; Grigoriev, V.V.; Ustyugov, A.A.; Bachurin, S.O.; Palyulin, V.A. Novel positive allosteric modulators of AMPA receptors based on 3,7-diazabicyclo[3.3.1]nonane scaffold. Mol. Neurobiol. 2020, 57, 191–199. [Google Scholar] [CrossRef]
- Temnyakova, N.S.; Vasilenko, D.A.; Lavrov, M.I.; Karlov, D.S.; Grishin, Y.K.; Zamoyski, V.L.; Grigoriev, V.V.; Averina, E.B.; Palyulin, V.A. Novel bivalent positive allosteric AMPA receptor modulator of bis-amide series. Mend. Comm. 2021, 31, 216–218. [Google Scholar] [CrossRef]
- Nazarova, A.A.; Sedenkova, K.N.; Karlov, D.S.; Lavrov, M.I.; Grishin, Y.K.; Kuznetsova, T.S.; Zamoyski, V.L.; Grigoriev, V.V.; Averina, E.B.; Palyulin, V.A. Bivalent AMPA receptor positive allosteric modulators of the bis(pyrimidine) series. Med. Chem. Commun. 2019, 10, 1615–1619. [Google Scholar] [CrossRef]
- Lavrov, M.I.; Karlov, D.S.; Palyulin, V.A.; Grigoriev, V.V.; Zamoyski, V.L.; Brkich, G.E.; Pyatigorskaya, N.V.; Zapolskiy, M.E. Novel positive allosteric modulator of AMPA-receptors based on tricyclic scaffold. Mend. Comm. 2018, 28, 311–313. [Google Scholar] [CrossRef]
- Karlov, D.S.; Lavrov, M.I.; Palyulin, V.A.; Zefirov, N.S. Pharmacophore analysis of positive allosteric modulators of AMPA receptors. Russ. Chem. Bull. 2016, 65, 581–587. [Google Scholar] [CrossRef]
- Radchenko, E.V.; Karlov, D.S.; Lavrov, M.I.; Palyulin, V.A. Structural requirements for molecular design of positive allosteric modulators of AMPA receptor. Mend. Comm. 2017, 27, 623–625. [Google Scholar] [CrossRef]
- Karlov, D.S.; Lavrov, M.I.; Palyulin, V.A.; Zefirov, N.S. MM-GBSA and MM-PBSA performance in activity evaluation of AMPA receptor positive allosteric modulators. J. Biomol. Struct. Dyn. 2018, 36, 2508–2516. [Google Scholar] [CrossRef] [PubMed]
- Lavrov, M.I.; Grigor’ev, V.V.; Bachurin, S.O.; Palyulin, V.A.; Zefirov, N.S. Novel bivalent positive allosteric modulators of AMPA receptor. Dokl. Biochem. Biophys. 2015, 464, 322–324. [Google Scholar] [CrossRef]
- Lavrov, M.I.; Veremeeva, P.N.; Karlov, D.S.; Zamoyski, V.L.; Grigoriev, V.V.; Palyulin, V.A. Tricyclic derivatives of bispidine as AMPA receptor allosteric modulators. Mend. Comm. 2019, 29, 619–621. [Google Scholar] [CrossRef]
- Vasilenko, D.A.; Sadovnikov, K.S.; Sedenkova, K.N.; Karlov, D.S.; Radchenko, E.V.; Grishin, Y.K.; Rybakov, V.B.; Kuznetsova, T.S.; Zamoyski, V.L.; Grigoriev, V.V.; et al. A facile approach to bis(isoxazoles), promising ligands of the AMPA receptor. Molecules 2021, 26, 6411. [Google Scholar] [CrossRef] [PubMed]
- Lavrov, M.I.; Veremeeva, P.N.; Golubeva, E.A.; Radchenko, E.V.; Zamoyski, V.L.; Grigoriev, V.V.; Palyulin, V.A. Positive and negative AMPA receptor modulators based on tricyclic bispidine derivative: Minor structural change inverts the type of activity. Mend. Comm. 2022, 32, 360–363. [Google Scholar] [CrossRef]
- Das, P.; Takada, M.; Matsuzaki, K.; Saito, N.; Shibata, N. SF5-Pyridylaryl-λ3-iodonium salts and their utility as electrophilic reagents to access SF5-pyridine derivatives in the late-stage of synthesis. Chem. Commun. 2017, 53, 3850–3853. [Google Scholar] [CrossRef]
- Wang, Y.; Wan, S.; Li, Z.; Fu, Y.; Wang, G.; Zhang, J.; Wu, X. Design, synthesis, biological evaluation and molecular modeling of novel 1H-pyrazolo[3,4-d]pyrimidine derivatives as BRAFV600E and VEGFR-2 dual inhibitors. Eur. J. Med. Chem. 2018, 155, 210–228. [Google Scholar] [CrossRef]
- Potashman, M.H.; Bready, J.; Coxon, A.; DeMelfi, T.M.; DiPietro, L.; Doerr, N.; Elbaum, D.; Estrada, J.; Gallant, P.; Germain, J.; et al. Design, synthesis, and evaluation of orally active benzimidazoles and benzoxazoles as vascular endothelial growth factor-2 receptor tyrosine kinase inhibitors. J. Med. Chem. 2007, 50, 4351–4373. [Google Scholar] [CrossRef]
- Sedenkova, K.N.; Averina, E.B.; Grishin, Y.K.; Kutateladze, A.G.; Rybakov, V.B.; Kuznetsova, T.S.; Zefirov, N.S. Three-component heterocyclization of gem-bromofluorocyclopropanes with NOBF4: Access to 4-fluoropyrimidine N-oxides. J. Org. Chem. 2012, 77, 9893–9899. [Google Scholar] [CrossRef]
- Sedenkova, K.N.; Averina, E.B.; Grishin, Y.K.; Bacunov, A.B.; Troyanov, S.I.; Morozov, I.V.; Deeva, E.B.; Merkulova, A.V.; Kuznetsova, T.S.; Zefirov, N.S. Nitronium salts as novel reagents for the heterocyclization of gem-bromofluorocyclopropanes into pyrimidine derivatives. Tetrahedron Lett. 2015, 56, 4927–4930. [Google Scholar] [CrossRef]
- Miller, G.W.; Rose, F.L. S-Triazolopyrimidines. Part I. Synthesis as potential therapeutic agents. J. Chem. Soc. 1963, 5642–5659. [Google Scholar] [CrossRef]
- Gangjee, A.; Zhao, Y.; Raghavan, S.; Rohena, C.C.; Mooberry, S.L.; Hamel, E. Structure-activity relationship and in vitro and in vivo evaluation of the potent cytotoxic anti-microtubule agent N-(4-methoxyphenyl)-N,2,6-trimethyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-aminium chloride and its analogues as antitumor agents. J. Med. Chem. 2013, 56, 6829–6844. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Kim, D.; Kang, S.Y.; Park, W.-K.; Kim, H.J.; Jung, M.E.; Son, E.-J.; Pae, A.N.; Kim, J.; Lee, J. Arylpiperazine-containing pyrimidine 4-carboxamide derivatives targeting serotonin 5-HT2A, 5-HT2C, and the serotonin transporter as a potential antidepressant. Bioorg. Med. Chem. Lett. 2010, 20, 6439–6442. [Google Scholar] [CrossRef]
- Buděšínský, Z.; Roubínek, F. In Stellung 4 und 2,4 substituierte 5,6-Tetramethylenpyrimidine. Collect. Czech. Chem. Commun. 1964, 29, 2341–2350. [Google Scholar] [CrossRef]
- Bernath, G.; Lazar, J.; Gera, L.; Goendoes, G.; Ecsery, Z. Saturated heterocycles. XLVI. Synthesis of 2-substituted-5,6- pentamethylene-, 5,6-hexamethylene- and 5,6-decamethylenepyrimidin-4(3H)-ones. Acta Chim. Hung. 1984, 115, 231–235. [Google Scholar] [CrossRef]
- Harms, J.E.; Benveniste, M.; Maclean, J.K.F.; Partin, K.M.; Jamieson, C. Functional analysis of a novel positive allosteric modulator of AMPA receptors derived from a structure-based drug design strategy. Neuropharmacology 2013, 64, 45–52. [Google Scholar] [CrossRef] [Green Version]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; MacKerell, A.D. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. WIREs Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A new tool to perform end-state free energy calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Sushko, I.; Novotarskyi, S.; Körner, R.; Pandey, A.K.; Rupp, M.; Teetz, W.; Brandmaier, S.; Abdelaziz, A.; Prokopenko, V.V.; Tanchuk, V.Y.; et al. Online chemical modeling environment (OCHEM): Web platform for data storage, model development and publishing of chemical information. J. Comput.-Aided Mol. Des. 2011, 25, 533–554. [Google Scholar] [CrossRef] [Green Version]
- Radchenko, E.V.; Dyabina, A.S.; Palyulin, V.A.; Zefirov, N.S. Prediction of human intestinal absorption of drug compounds. Russ. Chem. Bull. 2016, 65, 576–580. [Google Scholar] [CrossRef]
- Dyabina, A.S.; Radchenko, E.V.; Palyulin, V.A.; Zefirov, N.S. Prediction of blood-brain barrier permeability of organic compounds. Dokl. Biochem. Biophys. 2016, 470, 371–374. [Google Scholar] [CrossRef]
- Radchenko, E.V.; Dyabina, A.S.; Palyulin, V.A. Towards deep neural network models for the prediction of the blood-brain barrier permeability for diverse organic compounds. Molecules 2020, 25, 5901. [Google Scholar] [CrossRef]
- Radchenko, E.V.; Rulev, Y.A.; Safanyaev, A.Y.; Palyulin, V.A.; Zefirov, N.S. Computer-aided estimation of the hERG-mediated cardiotoxicity risk of potential drug components. Dokl. Biochem. Biophys. 2017, 473, 128–131. [Google Scholar] [CrossRef]
- ADMET Prediction Service. Available online: http://qsar.chem.msu.ru/admet/ (accessed on 10 October 2022).
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef]
- RDKit: Open-Source Cheminformatics Software. Available online: https://www.rdkit.org/ (accessed on 10 October 2022).











| Compound | MW | LogPow | pSaq | LogBB | HIA | hERG pKi | hERG pIC50 | QED |
|---|---|---|---|---|---|---|---|---|
| 1a | 402.50 | 5.11 | 6.63 | −0.34 | 84 | 5.37 | 4.33 | 0.60 |
| 1b | 430.55 | 5.75 | 6.91 | −1.43 | 84 | 5.64 | 4.36 | 0.50 |
| 1c | 458.61 | 5.93 | 7.13 | −0.29 | 93 | 5.37 | 4.63 | 0.40 |
| 1d | 486.66 | 6.09 | 7.89 | −0.27 | 100 | 6.35 | 4.41 | 0.39 |
| 1e | 454.57 | 5.48 | 7.08 | −0.23 | 93 | 5.78 | 4.56 | 0.43 |
| 1f | 374.44 | 4.36 | 5.49 | −1.60 | 84 | 5.37 | 4.59 | 0.66 |
| 1g | 374.44 | 4.36 | 5.77 | −0.40 | 84 | 5.13 | 4.57 | 0.67 |
| 1h | 430.55 | 5.64 | 7.23 | −0.28 | 84 | 5.39 | 4.64 | 0.47 |
| 1i | 406.53 | 5.01 | 6.59 | 0.22 | 100 | 7.37 | 4.69 | 0.52 |
| 1j | 388.47 | 4.75 | 6.21 | −0.37 | 84 | 5.25 | 4.45 | 0.64 |
| 1k | 416.53 | 5.38 | 6.92 | −0.31 | 84 | 5.38 | 4.48 | 0.51 |
| 1l | 404.51 | 5.15 | 6.60 | −0.05 | 97 | 6.39 | 4.51 | 0.56 |
| 1m | 390.49 | 4.80 | 6.24 | −0.08 | 97 | 6.26 | 4.63 | 0.61 |
| 1n | 418.54 | 5.42 | 6.88 | −0.02 | 97 | 6.39 | 4.66 | 0.48 |
| 1o | 418.50 | 3.52 | 4.03 | −0.44 | 84 | 5.23 | 4.40 | 0.46 |
| 1p | 434.50 | 2.37 | 2.96 | −0.53 | 84 | 5.09 | 4.48 | 0.46 |
| Energy Terms, kcal/mol | Compound 1f | Compound 1i | Compound 1j |
|---|---|---|---|
| ΔEint | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| ΔEele | −0.76 ± 0.08 | −2.4 ± 0.1 | −2.0 ± 0.1 |
| ΔEvdw | −43.2 ± 0.3 | −29.65 ± 0.3 | −37.2 ± 0.2 |
| ΔEMM = ΔEint + ΔEele + ΔEvdw | −44.0 ± 0.3 | −32.1 ± 0.4 | −39.2 ± 0.2 |
| ΔGGB | 5.6 ± 0.1 | 7.2 ± 0.1 | 8.8 ± 0.1 |
| ΔGSA | −5.79 ± 0.02 | −4.51 ± 0.05 | −4.71 ± 0.02 |
| ΔGsol = ΔGGB + ΔGSA | −0.19 ± 0.06 | 2.6 ± 0.1 | 4.1 ± 0.1 |
| ΔGMMGBSA = ΔEMM + ΔGsol | −44.2 ± 0.3 | −29.4 ± 0.4 | −35.1 ± 0.2 |
| −TΔS | 2.6 ± 0.1 | 3.8 ± 0.2 | 2.2 ± 0.2 |
| ΔGb = ΔGMMGBSA − TΔS | −41.6 ± 0.3 | −26.2 ± 0.4 | −30.9 ± 0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sedenkova, K.N.; Zverev, D.V.; Nazarova, A.A.; Lavrov, M.I.; Radchenko, E.V.; Grishin, Y.K.; Gabrel’yan, A.V.; Zamoyski, V.L.; Grigoriev, V.V.; Averina, E.B.; et al. Novel Nanomolar Allosteric Modulators of AMPA Receptor of Bis(pyrimidine) Series: Synthesis, Biotesting and SAR Analysis. Molecules 2022, 27, 8252. https://doi.org/10.3390/molecules27238252
Sedenkova KN, Zverev DV, Nazarova AA, Lavrov MI, Radchenko EV, Grishin YK, Gabrel’yan AV, Zamoyski VL, Grigoriev VV, Averina EB, et al. Novel Nanomolar Allosteric Modulators of AMPA Receptor of Bis(pyrimidine) Series: Synthesis, Biotesting and SAR Analysis. Molecules. 2022; 27(23):8252. https://doi.org/10.3390/molecules27238252
Chicago/Turabian StyleSedenkova, Kseniya N., Denis V. Zverev, Anna A. Nazarova, Mstislav I. Lavrov, Eugene V. Radchenko, Yuri K. Grishin, Alexey V. Gabrel’yan, Vladimir L. Zamoyski, Vladimir V. Grigoriev, Elena B. Averina, and et al. 2022. "Novel Nanomolar Allosteric Modulators of AMPA Receptor of Bis(pyrimidine) Series: Synthesis, Biotesting and SAR Analysis" Molecules 27, no. 23: 8252. https://doi.org/10.3390/molecules27238252