2.1. Dip-Coating (DC) Experiments on the Bare Nitinol Wire and Pre-Plated Nitinol Wire—Evaluation of Chemistry MPS and SPS under Non-Electrochemical Condition
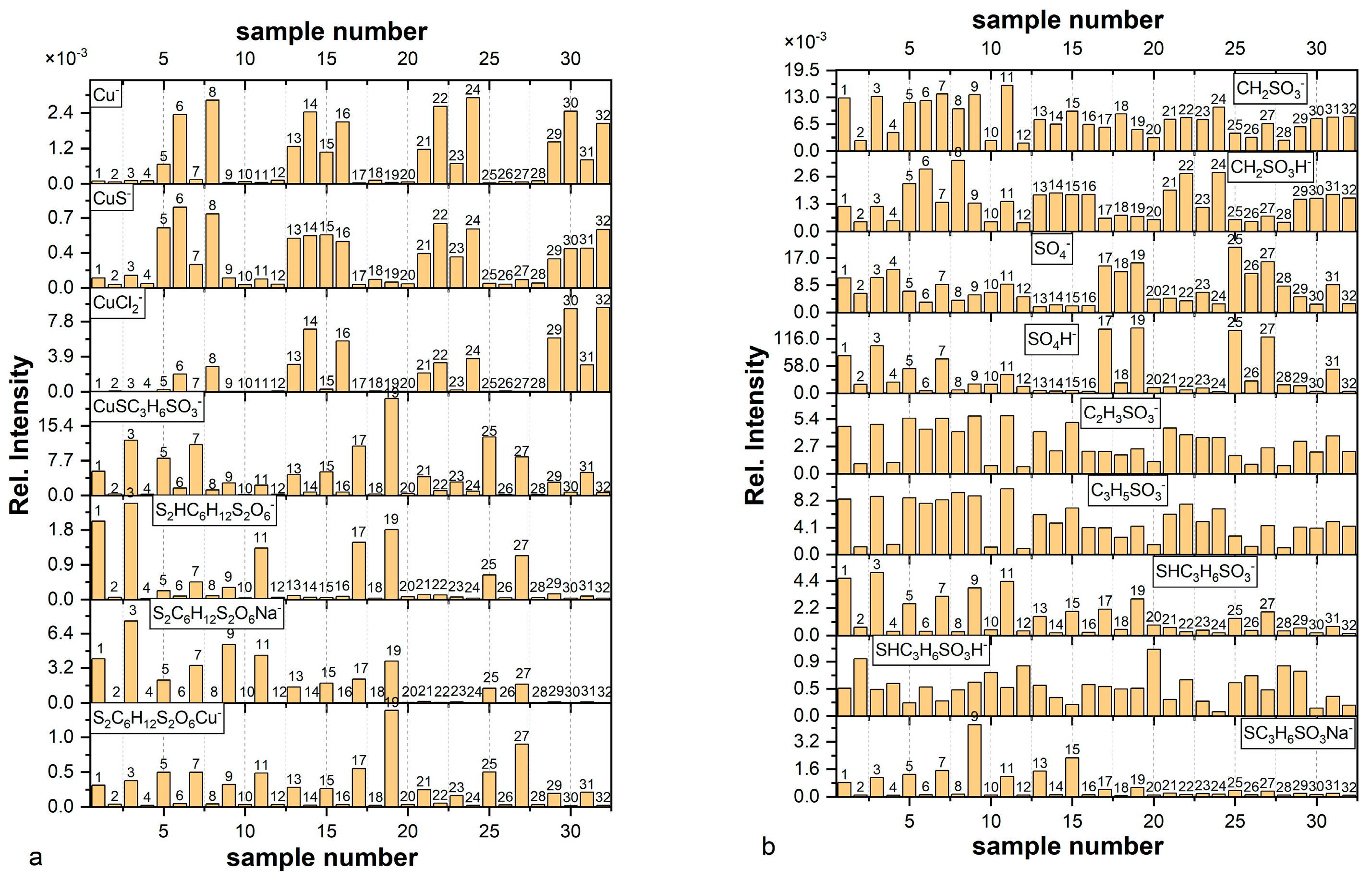
Figure 1a,b shows distribution of the most prominent fragments identified in the TOF-SIMS spectra. The
m/z ratios and assignments of selected fragments are included in
Table 1.
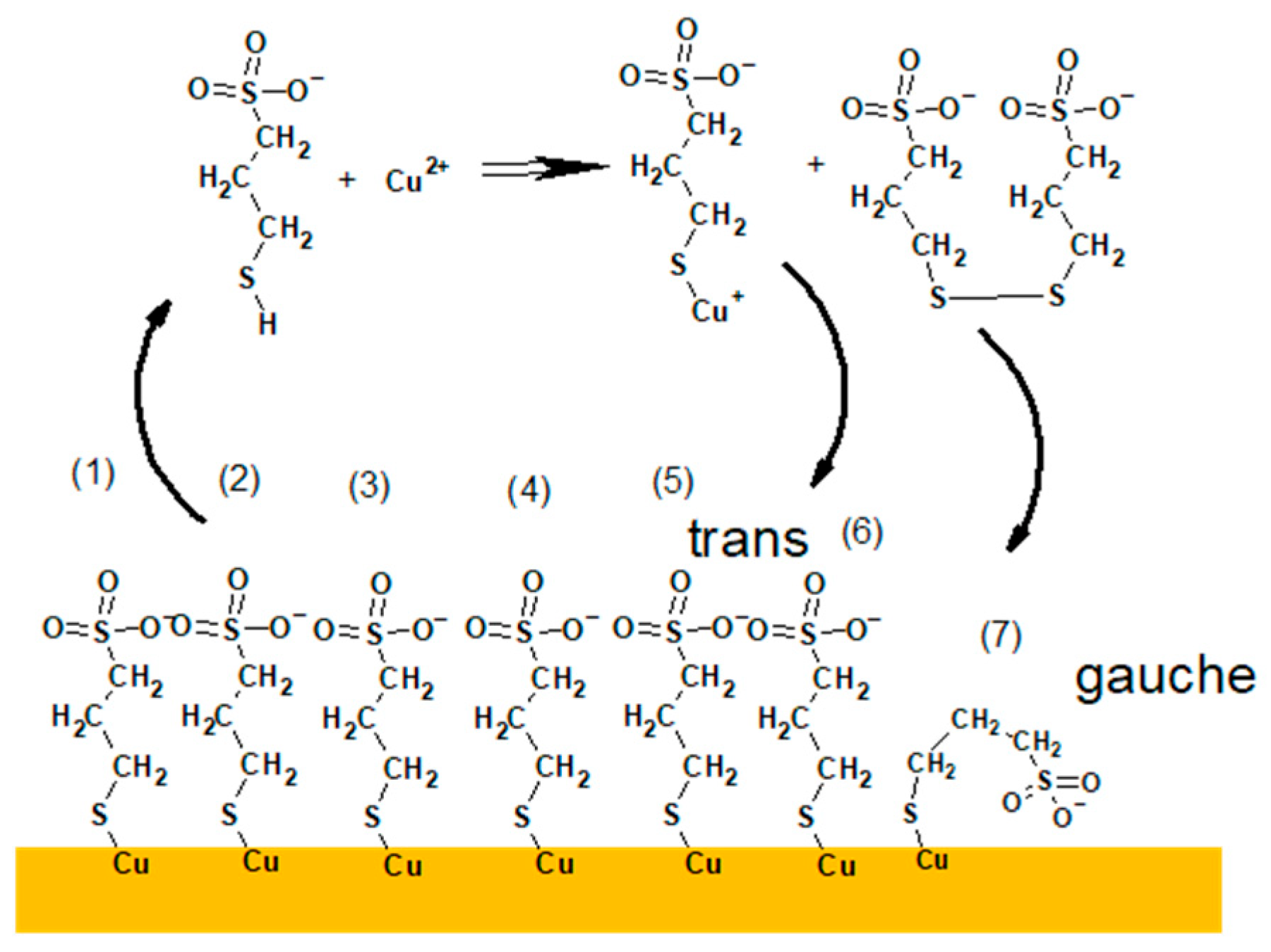
The SO3− fragment can be yielded from sulfonate ends of thiolate molecules as well as from sulphate ions. SO4− and HSO4− correspond to sulphate ions. CH2SO3−, C2H3SO3−, C3H5SO3− are yielded from complexated and uncomplexated forms of thiolate.
The fragment CH
2SO
3− is yielded through homolytic cleavage C–C bond of terminal sulfonate end whilst the possible fragmentation pathways that lead to fragments: C
2H
3SO
3−, C
3H
5SO
3− are as follows:
For all nitinol substrates the intensity of Cu
− fragment is negligible in comparison to the copper pre-plated nitinol wires (
Figure 1a,b). It unambiguously means that the Cu
− fragment is mainly yielded from the copper substrate. Low intensity of Cu
− for samples with copper substrate is determined by the thicker thiolate or thiolate/Cl layer. For example, the thickest layer among layers deposited on the copper substrate is observed for Sample 7 (copper substrate immersed in MPS solution for 60 s). On the other hand, Sample 8 prepared in the same way but demonstrates high intensity of Cu
− after rinsing because only a thin layer, very likely monolayer of MPS, remains on the substrate. We can observe similar behaviour for all unrinsed/rinsed copper samples: 5/6, 7/8, 13/14, 15/16, 21/22, 23/24, 29/30, and 31/32.
Similarly, a high intensity of CuS
− fragment was identified for the samples containing thiolate molecules adsorbed on the copper substrate (Samples 5–8, 13–16, 21–24, 29–32). It can strongly suggest that the CuS
− fragment can be assigned to Cu–S and proves the existence of the bond between S from thiolate group of MPS and metallic copper of the substrate. On the other hand, low intensity CuS
− fragment was also identified for layers deposited on the nitinol wire (Samples 1–4, 9–12, 17–20, 25–28). As for the TOF-SIMS spectra, it is not possible directly to distinguish the oxidation state of copper atom; this finding is helpful to distinguish Cu(0)-SC
3H
6SO
3 and Cu(I)-SC
3H
6SO
3−. For Samples 1–4, 9–12, 17–20, and 25–28 the CuS
− fragment can be unambiguously assigned to Cu(I)-S bond yielded from the Cu(I)-SC
3H
6SO
3− complex. The latter complex is considered as the main product of reaction Cu
2+ with MPS and it is assumed as one of the crucial steps in the close loop cycle in the near electrode surface during copper electrodeposition [
25] as it is shown in Reaction (3).
Interaction of sulfonate group through oxygen localized in S–O with the copper substrate is determined by CuSO− and CuSO3− fragments. Distributions of CuSO− and CuSO3− are very similar to the Cu−. If a sulfonate group is in close proximity to the copper substrate, sputtered copper ions react easily with sulfonate yielding CuSO− and CuSO3−. We can assume that a higher yield of CuSO3− may be favoured for higher symmetry of SO3−, whilst for asymmetric structure in the case of greater stretch of S=O, a higher yield of CuSO− may be generated. For example, the rinsing of Samples 29–31 provides a rise in intensity of CuSO− and reduces the intensity of CuSO3− simultaneously. It may suggest that after rinsing at lower thiolate coverage the lower asymmetry of the sulfonate group is determined by greater interaction of oxygen in S–O with copper. It arises from the fact that oxygen in S–O is in closer proximity to the copper surface due to higher availability of copper substrate.
The next important fragment is CuCl
2−. In the first assumption it is rational to assign this fragment to the Cl layer adsorbed on the copper substrate. However, surprisingly, CuCl
2 is detected not only for the layers deposited on copper from solution containing Cl
− ions (Samples 21, 22, 24, 29, 30, 31, 32), but also from solution without Cl
− ions (Samples 6, 8, 13, 14, 16). It is known that the TOF-SIMS technique is very sensitive to detection of chloride ions in the negative mode as demonstrated by high secondary ion fields [
34,
35,
42]. In practice, the detection limit of elemental analysis reaches the level of 10 ppb. Due to this reason, even a trace amount of chloride impurities from the air or residual gases inside the TOF-SIMS instrument can be adsorbed on bare copper and can be easily detected in the form of CuCl
2− and Cl
− (not shown in
Figure 1). We detected CuCl
2− fragments in the TOF-SIMS spectra even for extremely pure copper layer deposited by physical vapor deposition (magnetron sputtering) on silicon wafer (see
Supplement Figure S1) due to contamination of copper surface by chloride impurities.
Figure S1 shows distribution of Cu
−- and CuCl
2−-negative ions. It is clearly seen that the intensity of CuCl
2− is nearly ten times higher than the Cu
− fragment.
This means that the CuCl2− fragment can be a good indicator for detection of not only copper substrate covered by chloride ions from the solution, but also for identification of the bare copper substrate. For example, we did not detect CuCl2− for Samples 5 and 7 because the layer containing Cu(I)-MPS is sufficiently thick to cover copper substrate, although after rinsing (Sample 6, 8) a significant area of bare copper can easily adsorb Cl− contamination from the air-yielding CuCl2− fragments. A similar situation occurred for rinsed layers deposited from SPS solution (Samples 14, 16). On the other hand, the unrinsed layer for Sample 13 is too thin to protect the bare copper from contamination by chloride ions.
Identification of SPS molecules was determined by distribution of S
2HC
6H
12S
2O
6 (
m/z = 309.10), SPS–H fragment, the protonated form of SPS that unambiguously proves existence of a protonated form of SPS in an acidic environment. The significant number of SPS–H fragments is clearly visible only for unrinsed layers deposited on the nitinol substrate. The highest intensity of SPS was detected for layers obtained from the MPS-containing solutions (Samples 1, 3, 17, and 19) as an effect of Reaction (3) and for layers deposited from solutions containing SPS (11, 25, 27). For the layers received from solution containing SPS and Cl (25, 27), a significant quantity of SPS–H fragment was observed. Accordingly, the relative intensity for the fragment SPS–H for Sample 27 is about two times higher than for Sample 25. It shows that a 30-time increase in immersion time gives rise to a 2-time increase in the intensity (amount) of SPS–H. It may suggest that the amount of SPS–H is not linearly proportional to the immersion time, while increasing amount of SPS–H in a function of immersion time is obvious. On the other hand, for samples obtained from SPS-exclusive solution (without Cl) with a shorter immersion time (Sample 9) the amount of SPS–H was significantly reduced. In the case of the copper substrates long immersion time (Sample 7) the MPS-containing solution allowed for the acquisition of a noticeable amount of SPS, because at that time (60 s) a thick layer was deposited that prevented some parts of SPS molecules from direct contact with the copper substrate. Unlike Sample 5, SPS–H amount is significantly reduced due to a shorter immersion time. The lack of SPS–H for unrinsed samples received from SPS solutions on the copper substrates (Samples 13, 15, 29, 31) indicates that during contact with copper substrate disulphide bonds of SPS molecules are dissociatively retained. This observation is in full agreement with previous findings [
12,
43].
We also identified an unprotonated form of SPS in the layers. Assuming the number of SPS moles formed in Reaction (3) is equal to ½ moles of MPS for Samples 1–8 and 17–24 the solution can receive 0.1 M of SPS and 0.1 M of Cu(I)-SC3H6SO3− in. Then, if [H+] is equal to 0.05 M, the number of moles in the protonated form of SPS (SPS–H) is equal to 0.05 M (if we assume that all SPS molecules can be protonated), whilst the remaining 0.05 moles of SPS remain unprotonated. The unprotonated forms of SPS were identified as the sodium and copper adducts: SPS–Na (m/z = 331) and SPS–Cu (m/z = 371) occurring in the unrinsed layers deposited on nitinol as well as copper substrate (samples with odd numbers).
The existence of the fragment with mass 371 (Cu–SPS) was previously reported by Frank et el. [
19] in the solution phase identified by means of the LC-MS technique as an effect of reaction:
Reaction (4) suggests that Cu(I) is bonded to the S-thiolate group.
The occurrence of a sodium adduct of SPS (SPS–Na) is natural because a commercially available SPS is manufactured in that form. On the other hand, complex Cu
2+(SPS) was also crystallized and identified in solid state by Pasquale [
22] as a product of reaction MPS with Cu
2+ (Reaction 3). Pasquale reported that Cu
2+ ions are formed during air oxidation and are bound to the sulfonate group. Our results obtained for the MPS solution (Samples 1, 3, 5, 7) show that the amounts of SPS–Na are proportional to the immersion times of both nitinols as well as copper substrates. The lower amount of SPS–Na for copper substrate may suggests that additional amounts of Cu
+ allow formation of SPS–Cu that has been confirmed by distribution of SPS–Cu for Samples 5 and 7. In reality, fragment 371 assigned as Cu(II)-SPS can also correspond to Cu(I)[MPS]
2 complex [
22] as a product of reaction:
For Samples 9–16 we should expect a reaction between SPS and Cu
2+ in the solution phase as it was reported (UV-VIS, NMR) [
21,
24,
25].
For Samples 9, 11, 13, and 15 we identified significant amounts of SHC
3H
6SO
3− fragments (
Figure 1b) assigned to the MPS molecule. For Samples 13 and 15 on the copper surface, MPS is yielded from Cu(I)MPS (a product of Reaction (3)). For Samples 9 and 11, prepared on the nitinol substrate, the fragment SHC
3H
6SO
3− is yielded by homolytical cleavage of S-S disulphide bonds in the SPS molecules.
Moreover, for Samples 9, 11, 13, and 15, the CuSC3H6SO3− (m/z = 217) fragment was also identified and can be assigned to the complex Cu(I)SC3H6SO3−, the product of the reaction of SC3H6SO3−- (released from SPS during sputtering) with Cu2+ ions.
After rinsing (Samples 12, 14, 16, 18), there is a lack of Cu(I)SC3H6SO3 that is soluble in the water.
Moreover, the SPS molecules (Samples 16 and 18) after adsorption on copper immediately dissociate releasing two MPS molecules that immediately adsorb on the copper, remaining after rinsing as it was identified in the form CuS−. A similar behavioural pathway occurs for the unrinsed MPS/Cl and SPS/Cl samples.
Overall, for solutions consisting of SPS, two possible pathways of formation and decomposition of fragment 371 should be considered. The first pathway, known from the literature [
12,
44,
45,
46], relies on two the following stages: (1) disulphide bond in SPS molecule is cleaved into two MPS units, and (2) a reaction between MPS molecules and Cu
2+ occurs according to Reaction (3). In the excess of MPS, a Cu(I)[SC
3H
6SO
3]
2− complex (Reaction (5)) is formed. In the second pathway, the Cu
2+(SPS) complex (identified previously by Pasquale [
22]) in the layer is simply cleaved into CuSC
3H
6 SO
3 and MPS. Considering that the last proposed scheme is only a single step we can claim that the existence of the Cu
2+(SPS) complex is more favourable than Cu(I)[SC
3H
6SO
3]
2−.
Very low amounts of the SPS–Na (
m/z = 331) were detected for unrinsed layers (Samples 21, 23, 29, 31) deposited on the copper surface from solutions containing Cl ions. In turn, the SPS–H fragment (
m/z = 309) was not identified for Samples 21, 23, 29, 31. It suggests that the Cu–SPS complex is preferentially created over the sodium and protonated form of SPS in the chloride-consisting environment in near the copper surface in the crystallized phase. The affinity of Na
+, H
+, and Cu
2+ ions for the sulfonate groups of SPS molecules can be estimated from the following reactions:
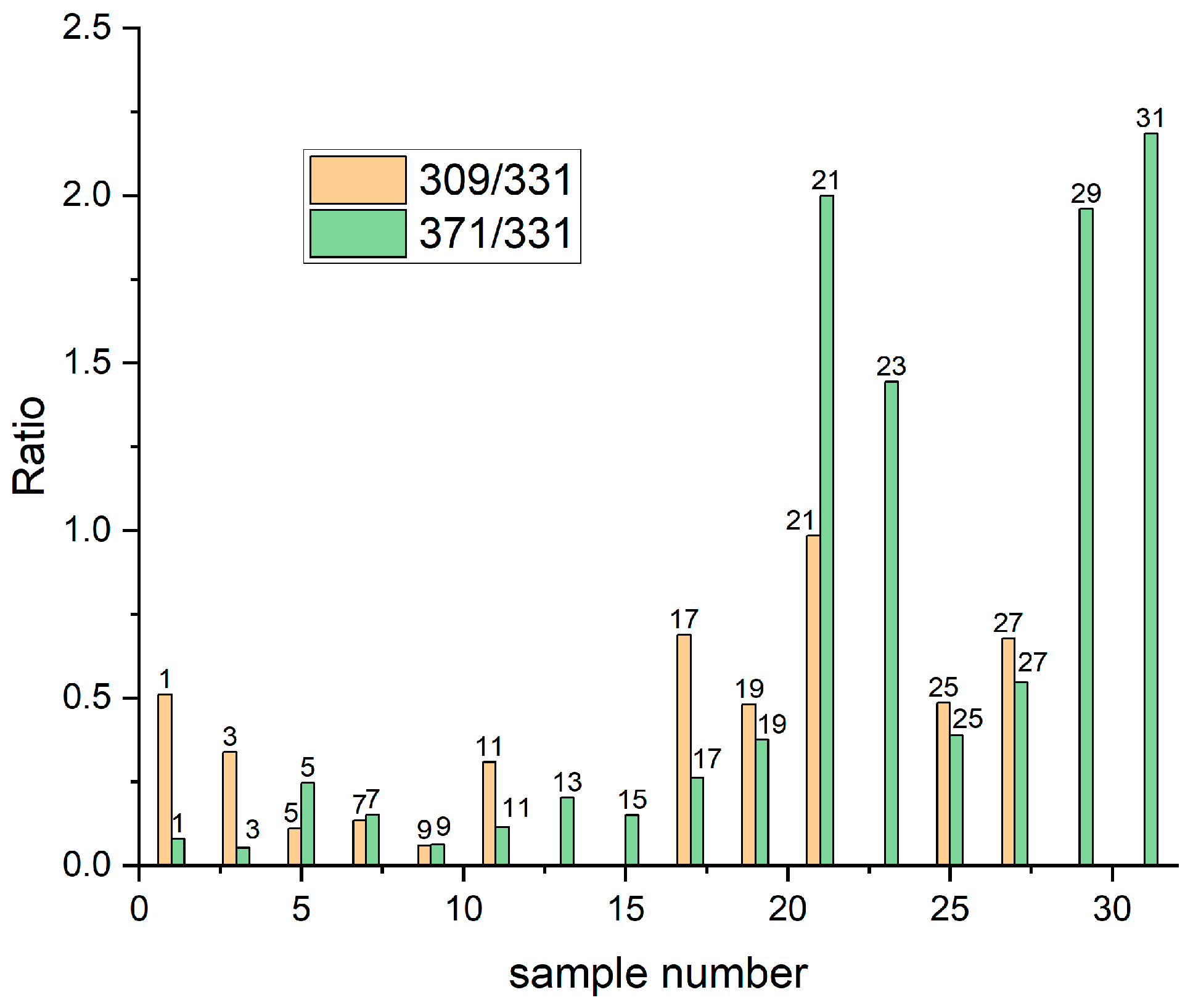
To estimate the shift of Reactions (6) and (7) the intensity ratio of fragments 309(SPSH)/331(SPSNa) and 371(SPSCu)/331(SPSNa) can be used. Assuming that C
SPSNa = 0.1 M = [H
+] from sulphuric acid, we suppose that the shift of Reactions (6) and (7) to the right side occurred when the ratios 309/331 and 371/331 were higher than one, and to the left side for ratios lower than one. In
Figure 2 the distributions of both 309/331 and 317/331 ratios for all unrinsed samples are shown. For Samples 25 and 27 we observed a significant shift in Reactions (6) and (7) to the right side after addition of chloride ions in comparison to Samples 9 and 11 containing only SPS. It is possible that chloride ions can form a pair Cl—Na
+ that prevents the shift of Reactions (5) and (6) to the left side during crystallization. For Sample 25, based on Reactions (6) and (7), one mole of SPS–Na
+ should react with 0.025 moles of Cu
2+ and 0.025 moles of H
+, providing 0.025 moles of SPS–H and 0.025 moles of Cu–SPS. Basically, two sulfonate groups of SPS can be protonated and cationizated by Cu
2+, but as TOF-SIMS spectrometry only yielded single charged fragments, we only consider reactions with one sulfonate group.
In Samples 1 and 3 containing Cu(I)C
3H
6SO
3 and SPS, all copper ions are complexed in the form MPS-Cu. The fragment Cu–SPS has the same mass as Cu[MPS]
2, so we conclude that the 371 fragment forms in Reaction (5) as suggested previously by Cardona [
24].
It this way the ratio 371/331 cannot be directly assigned to Reaction (5). Alternatively, the 371/217 ratio can be helpful in estimating the kinetics of Reaction (3).
The ratio of 371/217 has low values (ranges from 0.032 for Sample 3 to 0.067 for Sample 19). Chloride ions shift the steady state of Reaction (5) for samples immersed longer (ratio equal to 0.067 for Sample 19 and 0.032 for Sample 3), whilst for those with short immersion time changes are less significant (Sample 1—0.06 vs. Sample 17—0.05). On the other hand, for Sample 19 we observed very strong signals for SPS–H, SP-Na, and Cu[MPS]2, due to the long immersion time (60 s).
While analysing the layers deposited on the copper substrates (samples 21, 23, 29, and 31) we must consider that the amount of SPS in the solutions is significantly reduced due to the dissociative cleavage of disulphide bonds on the copper surfaces. Due to this reason, the intensity of SPSNa, SPS–H, and SPS is reduced, and it has an influence on the shift of Reactions (5) and (6). It can be clearly seen that Reaction (5) is shifted to the right side for Samples 21, 23, 29, and 31 containing Cl ions. For the same solutions but without chloride ions (Samples 5, 7, 13, and 15) the steady state is shifted to the left side. This unambiguously means that Cl ions are involved in the formation of Cu–SPS complexes in the layer deposited on the copper in the crystalline phase. On the other hand, we did not find fragments in the TOF-SIMS spectra that can be assigned to the Cu–SPS–Cl complexes.
The smaller mass fragments: CH2SO3−, CH2SO3H−, C2H3SO3−, and C3H5SO3− were identified in all samples but they are less informative than high mass fragments. The intensity of C2H3SO3− and C3H5SO3− demonstrates a very similar distribution, whereas the distribution of the protonated form CH2SO3H is slightly different. The CH2SO3H− fragment demonstrates a very good correlation with the CuS− fragment. It can strongly suggest that for proximity of sulfonate group and copper substrate hydrogen radical transfer to the sulfonate end is efficient and gives high yield of CH2SO3H−. Moreover, the CH2SO3− fragment can be yielded not only from Cu(0)SC3H6SO3H− but also from fragments: Cu(I)MPS, Cu(II)SPS. Similarly, C2H3SO3− and C3H5SO3− fragments demonstrate similar distribution, slightly different than CH2SO3−, which may suggest that they are yielded from different geometrical conformation than CH2SO3−.
Sulphate ions were identified in the form: SO4− and SO4H−. For the copper substrate for samples containing chloride ions (MPS/Cl and SPS/Cl) as well as SPS, the corresponding distribution of SO4− and SO4H− is similar showing minimum intensity for Samples 13–16, 21–24, and 29–32, meaning that after adsorption of MPS, SPS, and chloride ions there are no available adsorption sites for sulphate ions. On the other hand, for sample 31 sulphate ions can coexist with chloride and thiolate ions. This means that during dissociative adsorption of SPS competitive co-adsorption of sulphate and chloride ions occurs.
What is interesting is that for Samples 5 and 7 (MPS without chloride ions), adsorption of sulphate ions in the form SO
4H
− takes place. On the contrary for SPS, (Samples 13 and 15) sulphate ions are not adsorbed. It can strongly support the mobility of the SPS species on the copper surface that was identified before dissociation onto two MPS units’ [
10,
45,
46,
47] block adsorption sites for sulphate ions. On the other hand, MPS are immediately bonded to the copper leaving a number of adsorption sites for sulphate ions.
The last very important fragment is CuSC
3H
6SO
3− (
m/z = 217). This fragment can be assigned to: (1) Cu(0)SC
3H
6SO
3, (2) Cu(I)C
3H
6SO
3, and (3) -SC
3H
6SO
3Cu(II). Fragment (1) is yielded from MPS molecules chemically adsorbed on the copper. Fragment (2) is the product of Reaction (3). The third fragment can be yielded from the SPS–Cu complex (
m/z = 371) as well as from fragmentation of Cu[MPS]
2− (the product of Reaction (5)). In the latter case, it can be assigned to -Cu(I)C
3H
6SO
3. Moreover, the CuSC
3H
6SO
3− fragment can interfere with persulfate ion NaS
2O
8H
2− with the same
m/z = 217. Possible formation of persulphate ion may occur through the reaction of sulphate radical with hydrosulphate ion:
and then thiolate radical reacts with hydrosulphate ions:
Thiosulfate ions S2O82− in the TOF-SIMS spectra are identified in the form NaS2O8H2−.
To evaluate the possibilities of this reaction occurring, in Reactions (8) and (9) we deposited layers on the nitinol surface from solutions: (1) 0.1 M Na2SO4; (2) 0.1 M Na2SO4 + 0.1 M H2SO4; (3) Na2SO4 + 0.1 M MPS; (4) Na2SO4 + H2SO4 + MPS; (5) Na2SO4 + SPS; (6) Na2SO4+H2SO4+SPS; (7) 0.1 M Na2S2O8; (8) MPS + H2SO4; (9) SPS + H2SO4; (10) 0.1 M SPS; and (11) 0.1 M MPS, by using the dip-coating method. In the TOF-SIMS spectra we detected a peak at m/z = 217 for Samples 2, 4, 6, 7, 8, 9.
Identification of the peak at m/z = 217 for Sample 2 proves that during sputtering the Reaction (8) can occur. For the Samples 4, 6, 8, and 9 the existence of persulfate ion is determined by Reaction (9) as HSO4− ions from dissociation of sulphuric acid and •SC3H6SO3− from MPS or SPS are available. Overall, the presence of sulphuric acid in the solution is necessary for Reactions (8) and (9) as a source of hydrosulphate ions.
Hopefully, the TOF-SIMS method allows for detection of isotopes that provide an additional tool for identification. The ratio intensity for CuSC3H6SO3−/65CuSC3H6SO3− (m/z = 219) is equal to 60/32 whilst NaS2O8H2/Na34S2O8H2 (m/z = 219) is equal to 88/9. Based on isotopic analysis it is relatively easy to assign fragment m/z = 217 to CuSC3H6SO3 or NaS2O8H2.
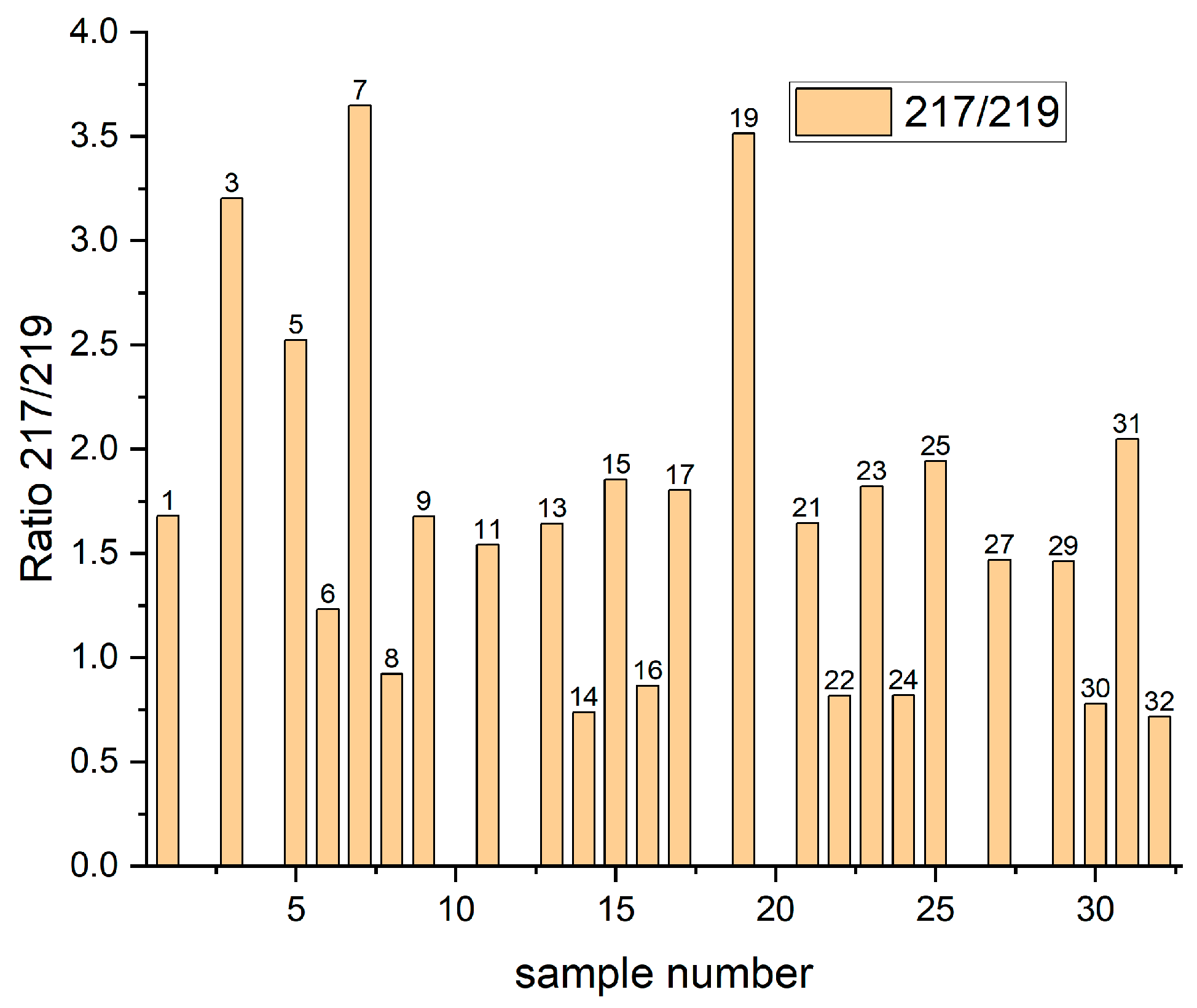
Figure 3 shows distribution of the intensity ratio of isotopes 217/219 for Samples 1–32. For Samples 2, 4, 10, 12 18, 20, 26, and 28 the
m/z = 217 fragment was not detected. All these layers were deposited on the nitinol substrate and were rinsed, meaning that Cu(I)C
3H
6SO
3 is highly soluble in water and is removed during rinsing.
The existence of
m/z = 217 for rinsed samples on the copper substrate (6, 8, 14, 16, 22, 24, 30, 32) can be assigned to Cu(0)C
3H
6SO
3 as MPS molecules that become chemically bonded to the copper and remain even during rinsing. We can observe (
Figure 3) that the intensity of the 217/219 ratio for these samples is slightly below one, only for Sample 6 is the intensity ratio higher than one. It is not clear why the amount of fragment 219 is higher than 217 for mentioned samples. It seems that some unidentified fragments determine the growth of the intensity of fragment
m/z = 219 for rinsed samples.
The unrinsed samples: 1, 9, 11, 13, 15, 17, 21, 23, 25, 27, 29, and 31 demonstrate similar, close to theoretical isotope distribution and we can assign the fragment with m/z = 217 to the ion Cu(I)SC3H6SO3− as a product of Reaction (3). For Samples 1, 17, 21, and 23 no other reactions are possible. For Samples 9, 11, 25, and 27 the fragment m/z = 217 can be assigned to –SC3H6SO3Cu (II) that refers to Cu–SPS complex. Moreover, Samples 3, 5, 7, and 19 demonstrate a significantly higher ratio of 217/219, meaning that the CuSC3H6SO3− fragment in this case is very likely overlapped by fragment Na2S2O8H2 corresponding to the thiosulfate ions S2O82 produced in Reactions (8) and (9).
2.2. Cyclic Voltammetry Measurements—Copper Electrodeposition from Base Solution without and with Additives: Cl, MPS, and SPS
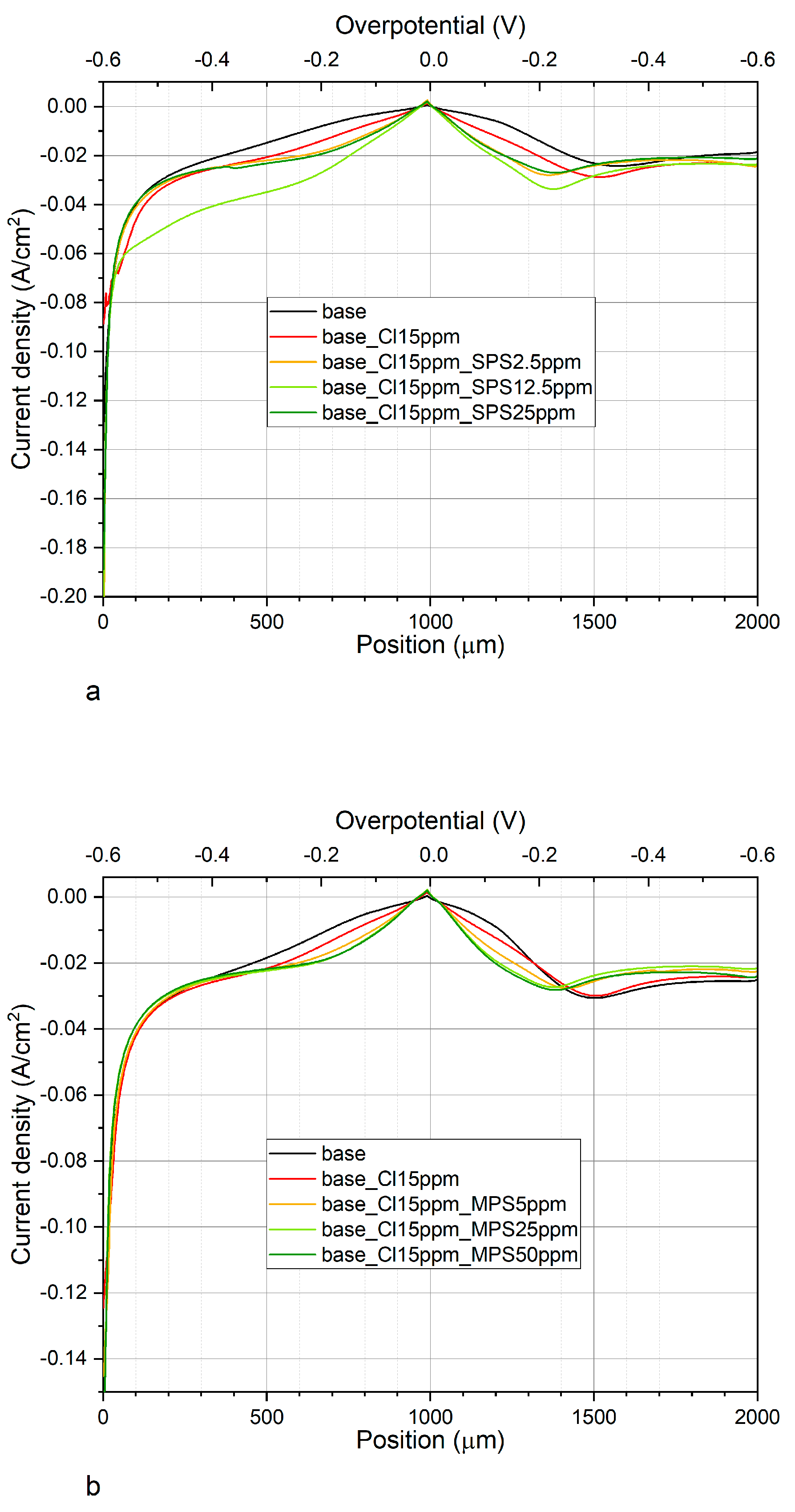
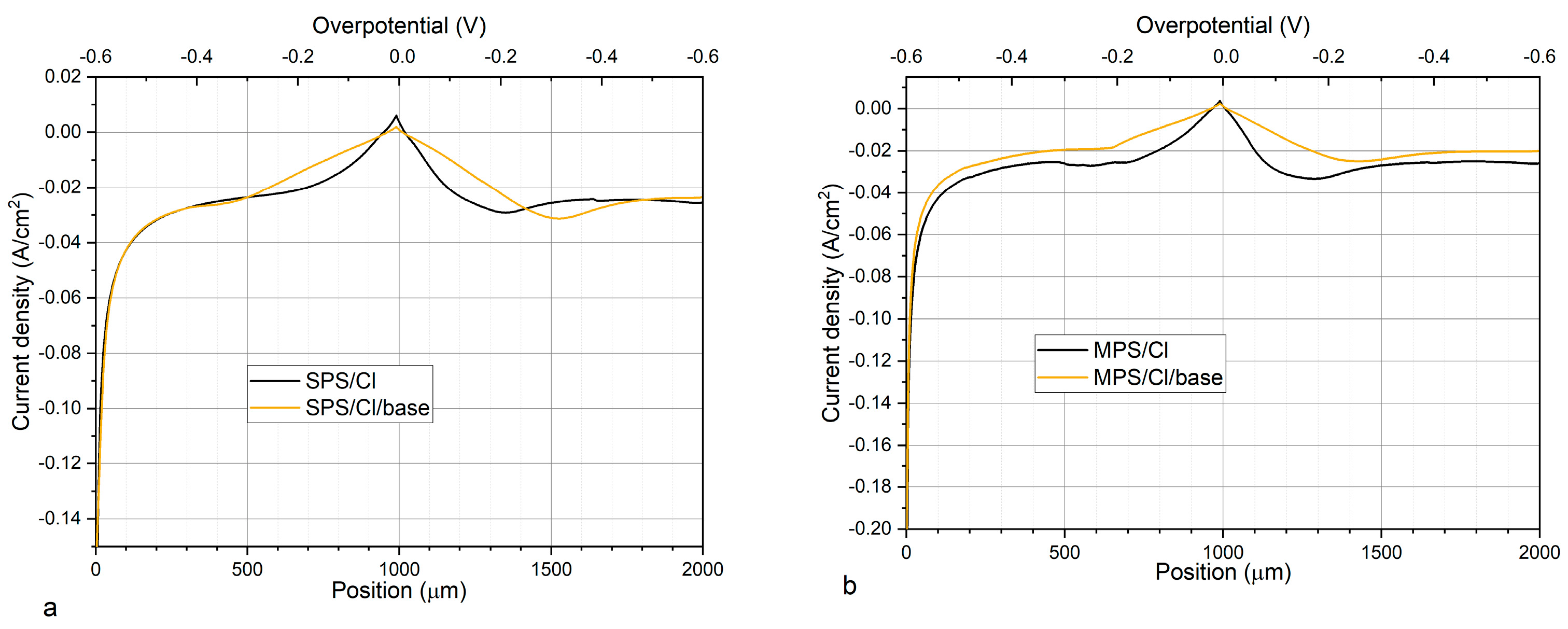
Figure 4a,b depicts CV curves while
Figure 5a,b shows exchange current densities, obtained for base electrolyte, and after addition of Cl, SPS, or MPS at different concentrations.
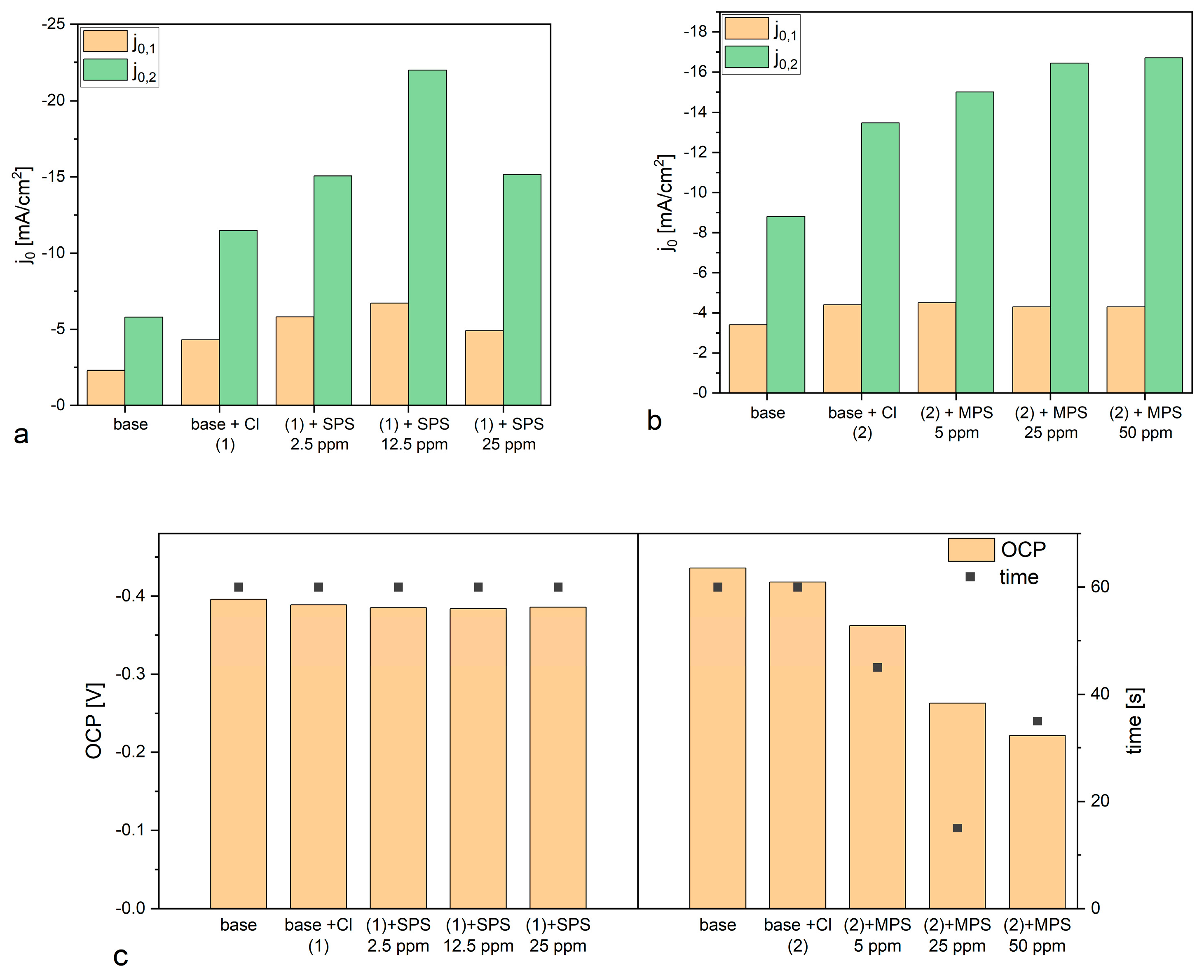
Figure 5a shows distribution of exchange current densities for the reduction of Cu
+ to the metallic Cu (j
0,1) and the reduction of Cu
2+ to Cu
+ (j
0,2) for base solution and after addition Cl and SPS at different concentrations. The exchange current density j
0,1 was calculated based on the CV curves by linear fitting of the CV curve for high overpotential region (η = −150 mV to −250 mV) while j
0,2 by fitting the CV curve for very low overpotentials (η < −20 mV) for forward scans. It is widely assumed that the reduction of Cu
+ to the metallic Cu is based on the inner-sphere mechanism [
8,
9].
On the other hand, the reduction of Cu
2+ to Cu
+ relies on the ion transport rather than electron transfer reactions and the ion migration reactions are determined by the reorganization of solvent and surface coverage [
48]. As it is depicted in
Figure 5a that addition of chloride ions as well as SPS in different concentrations to the base electrolyte increases the exchange current densities j
0,1 and j
0,2. However, the accelerating abilities are the greatest for Cl/SPS12.5ppm. Furthermore, the ratio of j
0,2/j
0,1 is varied in the following order: 2.51, 2.67, 2.6, 3.28, and 3.09 for base; and Cl, Cl/SPS2.5ppm, Cl/SPS12.5ppm, and Cl/SPS25ppm, respectively. This means that the accelerating abilities of Cl and Cl/SPS2.5ppm are determined by enhancement of inner-sphere reactions (j
0,1) and ion migration reactions (j
0,2) at the same level. Recently, it was suggested [
8] that exchange current density j
0,2 can strongly depend on working electrode rotation speed. Due to this reason, in the latter work [
8] where the electrode rotational speed was fixed to 800 rpm, the ratio of j
0,2/j
0,1 was substantially different than in our work since we electrodeposited copper without stirring. For the highest concentrations of SPS (12.5 and 25.0 ppm) the ratio of j
0,2/j
0,1 significantly increases, this strongly suggests the ion migration reactions determined by solvent reorganization (dehydration sphere) and surface coverage dominate the inner-sphere reaction. Moreover, the greatest values of j
0,1 and j
0,2 were calculated for Cl/SPS12.5ppm.
Figure 5b depicts the distribution of exchange current densities j
0,1 and j
0,2 for base, and with the additions of Cl and Cl/MPS solutions. The j
0,2/j
0,1 ratio for the base electrolyte is similar to what was obtained in the previous experiment, when accelerating abilities of SPS were examined, while exchange current densities j
0,1 and j
0,2 demonstrate higher values. The reason for greater accelerating abilities is unknown. We examined the copper surface chemistry after electrodeposition by TOF-SIMS (see the next section) and the results prove that the solution did not consist of any impurities and traces of Cl/SPS from the previous experiment. Larger accelerating abilities might be determined by different surface roughness of nitinol substrate. After addition of Cl to the base electrolyte, the j
0,2/j
0,1 ratio value increases from 2.6 (obtained for base) to 3.06, and to 3.3 and 3.8 after addition of MPS5ppm, MPS25ppm, respectively. At the highest concentration of MPS (50 ppm) the j
0,2/j
0,1 ratio is similar as for MPS25ppm. In comparison to SPS, it can clearly be seen that after addition of MPS the exchange current density j
0,1 is constant while j
0,2 increases. It strongly suggests that in our experimental condition during copper electrodeposition (the electrolyte was not stirred) the accelerating abilities of MPS rely only on the ion migration reactions determined by the solvent reorganization. Distribution of open circuit potential (OCP) defined after deposition of the copper layer at constant current densities j = −20 mA/cm
2 over 30 s is shown in
Figure 5c. For electrolytes containing different concentrations of SPS, the OCP value and the evaluation time needed to reach steady state, OCP was constant. Interestingly, the OCP decreased as a function of MPS concentration while the evaluation time of OCP was minimal for MPS25 ppm.
Our work shows that under open circuit conditions, SPS weakly interact with copper surfaces. On the other hand, strong interaction/adsorption is observed for MPS molecules. The lowest OCP potential was observed for the electrolyte with the greatest MPS concentration (50 ppm) while steady state was reached the fastest for MPS25ppm. It seems that it determines the mechanism of electrodeposition that relies mainly on the ion migration reactions, while for the solution consisting of SPS, inner-sphere mechanisms also play significant role. The above discussion, which is based on the cyclic voltammetry curves, is widely used for investigations on the mechanism of copper electrodeposition in the presence of organic additives such as MPS and SPS [
2,
3,
12,
49]. On the other hand, Gileadi et al. [
9] postulated that the Tafel slope extracted from the CV curve could not to be used to build a mechanistic model to distinguish different mechanisms of metal deposition.
Due to this reason, to gain more insight into the accelerating abilities of MPS and SPS we examined surface chemistry of samples by using the TOF-SIMS spectrometry and detailed discussion is provided in the next section.
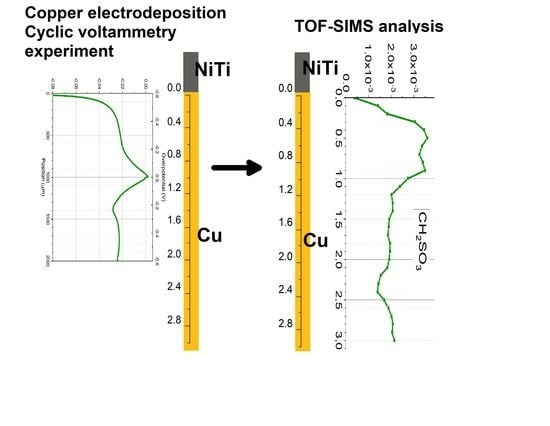
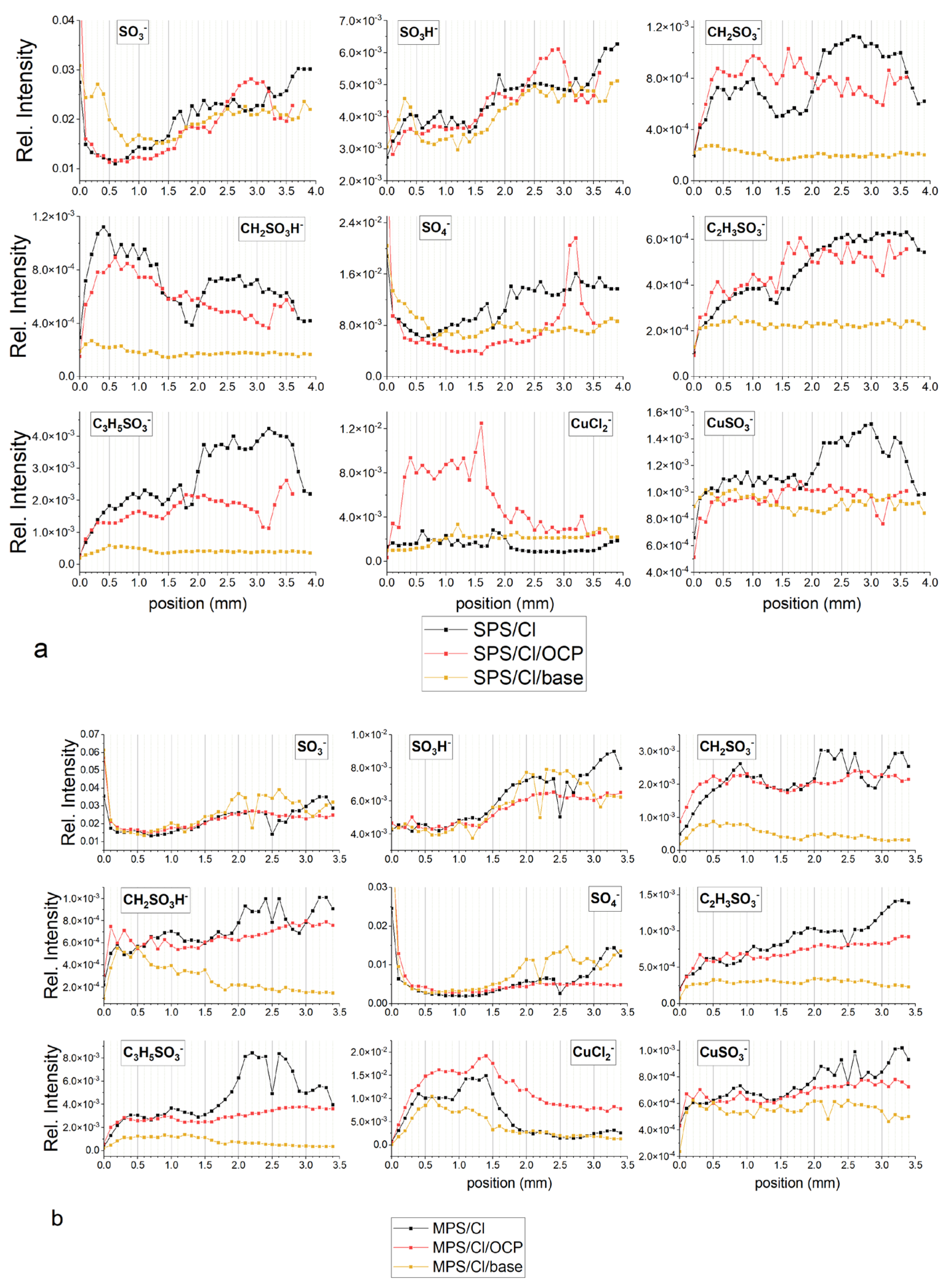
2.5. TOF-SIMS Measurements—Copper Electrodeposition from Base Solution with and without Additives: Cl, MPS, and SPS
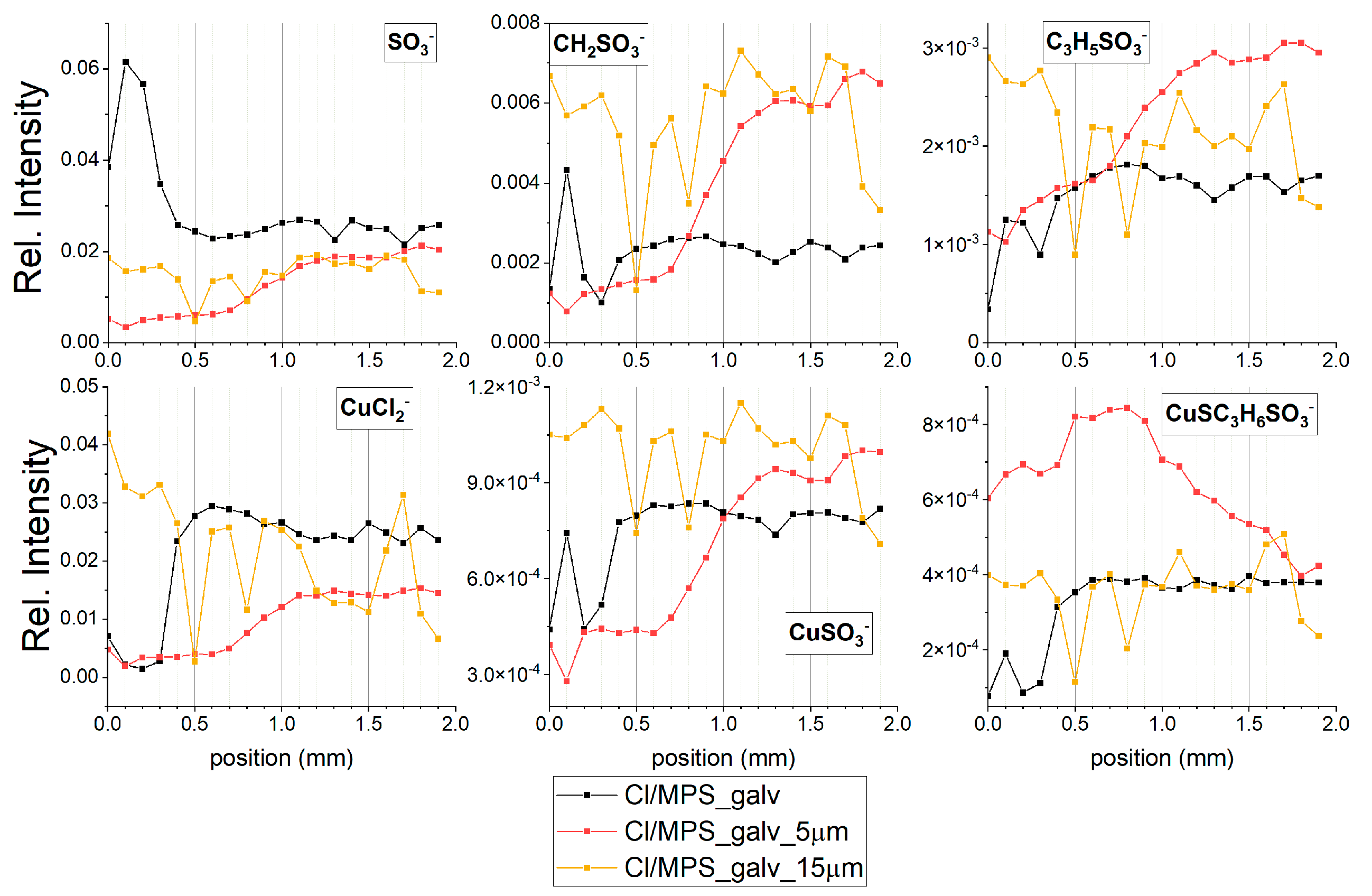
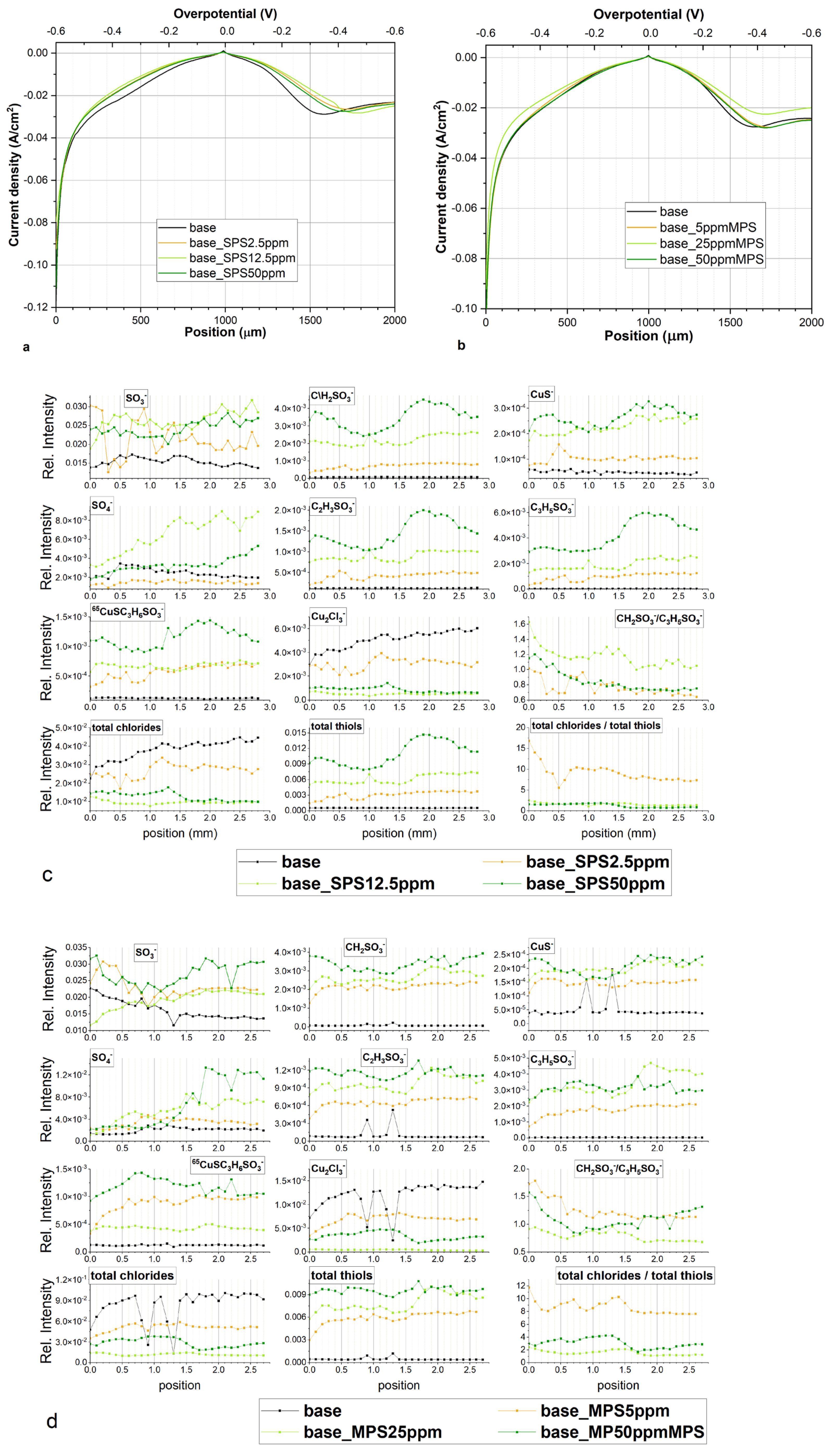
Figure 10a depicts the TOF-SIMS data of selected negative ions distributed along the wire length for samples obtained from the base electrolyte (black line), and solutions consisting of 15 ppm Cl (red) and 15 ppm Cl, with the addition of 2.5 ppm SPS (yellow), 12.5 ppm SPS (light green), and 25 ppm SPS (dark green). Relevant CV curves were shown and analysed in
Section 2.2. The forward scan in the anodic direction (more positive potential, wire position 0–1 mm) corresponds to the adsorption regime, as we can expect an increase of adsorption of additives during a shift potential to OCP. On the other hand, the reverse scan in the cathodic direction (wire position 1–2 mm) corresponds to the desorption part, as we can expect that desorption should be accelerated, when potential becomes more negative. The chloride ions were identified as CuCl
−, CuCl
2− and Cu
2Cl
3− fragments while thiolate ions were identified as CuS
−, CH
2SO
3−, C
2H
3SO
3−, C
3H
5SO
3−, and CuSC
3H
6SO
3−.
In the dip-coating experiment (
Section 2.1), it was demonstrated that the Cu(I)SC
3H
6SO
3− complex was identified as a fragment CuSC
3H
6SO
3− and was deposited as a thin layer on the copper surface; it is highly soluble after rinsing in an optimized way (see details in experimental section). About 20–30% of the primary amount of Cu(I)SC
3H
6SO
3 complex still remained on the copper surface and could be identified (
Figure 1a). The CuS
− fragment corresponds to the Cu–S bond and is yielded from adsorbed thiolate molecules on the copper surface. Potentially, the CuS
− fragment may also be yielded from the Cu(I)C
3H
6SO
3 complex. However, the previous dip-coating experiment demonstrates that the CuS
− fragment is mainly yielded from the thiolate molecules that are chemically bonded to the copper surface. This is determined by the fact that after rinsing of Samples 5 and 7 (Cu(I)C
3H
6SO
3 on the copper surface) in the dip-coating experiment, CuS
− intensity significantly increases while CuSC
3H
6SO
3− intensity decreases (Samples 6 and 8;
Figure 1a). A similar observation was made for MPS/Cl solutions by comparing Samples 21/22 (rinsed) and 23/24 (rinsed), respectively.
The existence of the CuS
− fragments proves that MPS molecules are bonded to the copper by the thiolate end, which is fully consistent with previous results [
35,
45,
46], as well as our dip-coating experiment (
Section 2.1).
The increase in potential towards the anodic direction for the forward scan was accompanied by an increase in the surface coverage of chloride ions, since the intensity of CuCl
2− and Cu
2Cl
3− increased (red line,
Figure 10a). After injection of 2.5 ppm SPS (yellow line,
Figure 10a), chloride surface coverage was reduced for nearly the whole potential and demonstrated stronger deviations in a function of applied potential. For the potential region corresponding to the 0–1.2 mm position, intensity of chloride significantly increased, while the rest of the potential region of the reverse scan exponentially reduced in similar way. In the dip-coating experiments (
Section 2.1) it was suggested that CuCl
2− ions can be a good marker of chloride ions, as well as of metallic copper. However, it is important question how to distinguish between a chloride adlayer and uncovered copper metallic surface for samples received in CV experiments.
Figures S2–S4 (Supplement Materials) show that increases in the intensity of the thiolate (CH
2SO
3−, C
3H
5SO
3−) and chloride (CuCl
2−) fragments are accompanied by a decrease in the intensity of Cu
−. This is consistent with the dip-coating experiment (see
Figure 1) and unambiguously proves that with the presence of a thiolate and chloride monolayer the area of the uncovered copper surface is reduced. In this way, the CuCl
2− fragment can be unambiguously assigned to the Cl
− adlayer.
Thiolate coverage is identified by the fragments: CuS
−, CH
2SO
3−, C
2H
3SO
3−, C
3H
5SO
3−, and CuSC
3H
6SO
3−. For high overpotentials (0–0.3 mm) the total thiolate surface coverage (
Figure 10a) was rather low and stable, while significant increases and synergy effect with chloride ions was observed for lower overpotentials (region 0.4–1.0 mm) that exhibit significant increases in thiolate coverage and chloride ions.
The turning point was observed after switching to the reverse scan (position 1.1 mm), when strong desorption of chloride and thiolate ions occurred while replacement of the desorbed chloride ions by sulphate ions was observed at position 1.2 mm (η = −120 mV). For the rest of the range of the reverse scan of chloride ions, desorption continued while desorption of thiolate ions was stopped at wire position 1.4 mm (η = −240 mV), accompanied by a slight increase (at position 1.5–2.0mm).
To compare the adsorption coverage of chloride to that of thiolate ions, the intensity of all forms of chloride (CuCl−, CuCl2−, and Cu2Cl3−) and thiolate fragments (CH2SO3−, CuS−, C2H3SO3−, C3H5SO3−, and CuSC3H6SO3−) were added up separately and assigned as total chlorides and total thiols, respectively.
A high and rather stable ratio of Cl/thiols for the wire position 0.2–0.8 mm (
Figure 10a) corresponds quite well to greater accelerating properties in comparison to the solution consisting of only chloride ions (see CV curve,
Figure 4a). Subsequently, the ratio Cl
−/thiols decreases and reaches a lower value than was observed for the corresponding overpotential for the forward scan in the 0.9–1.4 mm range. For lower overpotentials (0.8–1.2 mm), when strong co-adsorption of Cl and thiolate occurs, adsorption of thiolate is slightly dominated over chloride adsorption. For the reverse scan, when co-desorption started (1.1–1.2 mm) in a narrow potential window (−60 to −120 mV), the desorption rates of Cl
− and thiolate were similar, while for the rest potential region (1.3–2.0 mm), chloride ions were significantly more strongly desorbed than thiolate. It is interesting that the CH
2SO
3−/C
3H
5SO
3− ratio demonstrates roughly similar behaviour to the ratio of Cl/thiols. For the forward scan, the amount of CH
2SO
3− fragments dominated over C
3H
5SO
3− and exhibited a rather stable CH
2SO
3−/C
3H
5SO
3− ratio value around 2.7 while for the reverse scan the CH
2SO
3−/C
3H
5SO
3− ratio was significantly reduced. Recently, SHINER studies [
8] showed that the conformation gauche/trans ratio for the SPS/Cl system is strongly dependent on the applied potential. We also tried to evaluate the ratio of gauche/trans by Raman spectroscopy, and while the received Raman spectra were not reproducible and consisted of very regular background peaks that overlapped with the characteristic peaks, they did correspond to the thiolate gauche and trans conformations (results not shown). Due to this reason, full reproducibility of the whole potential range using Raman spectroscopy for our samples was not possible.
On the other hand, to the best of our knowledge, TOF-SIMS spectrometry has not been used for the determination of conformation changes in the adsorbed thiolate monolayer. At this point, we can safely assume that the CH
2SO
3−/C
3H
5SO
3− ratio can be used as an indicator of the ratio of gauche/trans conformation. However, the CH
2SO
3−/C
3H
5SO
3− ratio is not only determined by the total thiolate surface coverage depicted in
Figure 10a, since we should expect that the greater the total surface coverage the lower the gauche/trans ratio. However, under low thiolate coverage as took place for 2.5 ppm SPS, adsorbed thiolate molecules can rather be isolated by greater chloride ions and the gauche conformation can be easily attained in these molecular arrangements. Previously, it was shown that chloride coverage and distribution of pitting in the chloride adlayer can be determined by selective desorption of chloride ions in a function of applied potential, which plays an important role in reorientation of thiolate molecules [
12]. This fact can explain the significant reduction in CH
2SO
3−/C
3H
5SO
3− ratio for the reverse scan in the cathodic direction. During strong co-adsorption of chloride and thiolate ions (position 0.8–1.0 mm) due to the ordering of thiolate molecules, the contribution of trans conformation increases as the intensity of the C
3H
5SO
3− fragment rises more than that of CH
2SO
3−. Moreover, during co-desorption (1.1–1.4 mm) thiolates in gauche conformation are preferentially desorbed over trans conformation. What it is also important is that total chloride ions are also strongly desorbed during the reverse scan. This result determined the decreases in CH
2SO
3−/C
3H
5SO
3−ratio, since Cl ions are replaced by thiolate ions.
In the TOF-SIMS spectra, we did not find fragments that corresponded to the Cu–MPS–Cl complexes in any form of copper (metallic, Cu
+, or Cu
2+) on the electrode surface. Moreover, in the solution the only evidence was the presence of MPS–Cu
+–Cl complexes, which was supported by spectroscopic data [
3], while other investigations [
24] were in contradiction to this finding, demonstrating only the presence of Cu(I)MPS complexes. The latter finding was also supported by other investigations [
4]. On one hand, we cannot definitely rule out the occurrence of Cu
+(Cu
2+)/MPS/Cl
− complexes. Furthermore, electrostatic interaction of sulfonate ends with Cu
2+ cannot be directly examined by TOF-SIMS spectrometry due to lack of strong chemical Cu
2+/SO
3− bonds. What is important, is that we also did not find any evidence of existing Cu–MPS–Cl complexes in our dip-coating experiment (
Section 2.1). The latter observation provides more arguments against the existence of Cu–MPS–Cl complexes in the solution.
In order to gain more insight into the chemical state on the copper electrode, we performed CV experiments with greater SPS concentrations (12.5 ppm and 25 ppm) in the electrolyte solution.
After an injection of 10 ppm of SPS (total concentration of SPS 12.5 ppm) we observed a strong enhancement in accelerating abilities for the whole potential range (see discussion in the previous section) and significantly greater thiol coverage (light green curve) for the forward as well as reverse scan. For position 0.0–0.7 mm, total thiolate coverage is rather stable, then gradually decreases reaching minimum around 1.1 mm (overpotential η = −60 mV). Consecutively, thiolate coverage increases up to 1.5 mm and then is stable in the rest of potential range (position 1.5–2.0 mm). After switching off the current, under OCP conditions (position 2.0–2.8 mm) total thiol coverage is similar as observed at position 2.0 mm. Distribution of intensity of chloride ions in the range 0.7–2.0 mm is negatively correlated with thiolate, the greater chloride the lower thiolate coverage. It means that thiolate ions are partially replaced by chloride ions for low overpotential range during the forward scan while during the reverse scan in the region 1.2–2.0 mm, desorption of chloride ions is accompanied by thiolate adsorption. In comparison to solution consisting of 2.5 ppm SPS, chloride coverage is greater for the forward scan only while around OCP, while at position 0.9–1.0 mm is slightly lower. Moreover, the maximum chloride coverage at SPS 12.5 ppm is shifted to −60 mV for the reverse scan in comparison to solution of SPS 2.5 ppm. On the other hand, chloride coverage is very similar to the solution consisting of only chloride ions. Sulphate coverage is maintained on the low value for the forward as well as reverse scan, demonstrating an inverted distribution to chloride ions. The greater chloride coverage the lower sulphate ion coverage and vice versa.
After adding the next amount of SPS (total SPS concentration 25 ppm), thiolate and chloride coverage as well as the ratio chloride/thiolate is significantly lower than observed for 12.5 ppm SPS. This strongly suggests that the optimal molar ratio SPS/Cl is required for greater adsorption of Cl and SPS. For 25 ppm of SPS concentration, chloride and thiolate surface coverage is reduced while thiolate molecules demonstrate greater adsorption than chloride ions. The lower accelerating abilities may also relate to higher amounts of Cu(I)SC3H6SO3 accumulated over the thiolate layer. In this scenario, Cu(I)SC3H6SO3 plays the suppression role by blocking the access of Cu2+.
For 12.5 and 25 ppm SPS, the intensity of CuSC3H6SO3− fragments demonstrate a stable value around OCP (the region 0.7–1.2 mm). The intensity of fragments CH2SO3−, C2H3SO3−, C3H5SO3−, and CuS− in that potential region decreases. This is especially visible at 12.5 ppm SPS for CH2SO3 and CuS−. Different distribution of CuSC3H6SO3− and CH2SO3−, C2H3SO3−, C3H5SO3−, and CuS− suggests that the fragment CuSC3H6SO3− corresponds to the Cu(I) thiolate complex while the fragments CH2SO3−, C2H3SO3−, C3H5SO3−, and CuS− to the adsorbed thiolate molecules. It means that in this potential region, a significant amount of desorbed MPS molecules is converted into the Cu(I)SC3H6SO3 complex while the amount of the SPS is significantly reduced and this reaction is accelerated by the chloride ions.
Even more so, for SPS 12.5 ppm and SPS 25 ppm the ratio CH2SO3−/C3H5SO3− is significantly lower than was observed for SPS 2.5 ppm and exhibits a tendency to decrease during the reverse and forward scan. It is rational to assume that under greater thiolate and chloride surface coverage, the contribution of gauche conformation is significantly reduced, since adsorbed MPS molecules are tightly surrounded by chloride ions. On the other hand, the ratio of CH2SO3−/C3H5SO3− is unexpectedly greater for higher thiolate and chloride coverage (SPS 12.5 ppm) than observed in lower thiolate and chloride coverage (SPS 25 ppm). It strongly suggests that chloride ions play an important and positive role in the gauche orientation of thiolate molecules, even under high thiolate and chloride coverage.
This observation is supported by the chloride/thiol intensity ratio that demonstrates the same behaviour as the CH2SO3−/C3H5SO3− ratio, which exhibits a decreasing trend as SPS concentration increases.
Figure 10b depicts distribution of characteristic fragments for base solution, after addition of chloride ions and MPS at concentrations of 5, 25, and 50 ppm, respectively. Similarly, as observed in the experiment with the addition of SPS, the copper surface deposited from the base electrolyte does not contain chloride or thiolate ions. After addition of chloride ions, the fragment Cu
2Cl
3− demonstrates greater intensity as observed in the experiment with the addition of SPS. The maximum intensity of chloride ions is observed around OCP.
After addition of 5 ppm MPS, the intensity of chloride ions is significantly reduced for the whole range of overpotentials for forward and reverse scans. Similarly, the intensity of total thiols demonstrates greater value for the whole potential range of forward and reverse scans. This is different behaviour than was observed for 2.5 ppm SPS, when the distribution of intensity of chloride was more positively correlated with thiolate ions. Furthermore, the ratio of total chloride/total thiols is about two times lower than was observed for 2.5 ppm SPS.
The ratio CH
2SO
3−/C
3H
5SO
3− is nearly constant in the 0.0–1.2 mm range, while greater overpotentials slightly increases during the reverse scan. The exchange current density j
0,2 only slightly increases from 13.5 mA/cm
2 (Cl) to 15 mA/cm
2 for MPS 5 ppm. On the other hand, exchange current density j
0,1 remains unchanged while for SPS 2.5 ppm, j
0,2 rises from 11.5 mA/cm
2 (Cl only) to 15 mA/cm
2 for SPS 2.5 ppm. The most important difference between MPS and SPS at the lowest concentration is that chloride ions are strongly co-adsorbed with SPS ions in the range of low overpotentials (η < −120 mV). On the other hand, chloride ion coverage is stable in the broad potential range (0.3–1.2 mm) while thiolate coverage increases. Synergetic co-adsorption of chloride and SPS may be determined by preferential changes in the surface roughness (crystal orientation) of electrodeposited copper in the presence of Cl/SPS, which in turn increases the available adsorption centres for chloride ions. Moreover, the binding energy of SPS onto copper is lower than for MPS [
12] that may allow the formation of Cu
+–SPS
−–Cl
− complexes on the near-electrode surface under electrodeposition conditions. Determination of open circuit potential (see the previous section) demonstrates and proves the great adsorption abilities of MPS and weak direct interactions of SPS with copper without an external current source.
At greater MPS concentrations (25 and 50 ppm), chloride surface coverage demonstrates very similar distribution and is significantly reduced in comparison to the MPS 5 ppm. Thiolate surface coverage of CH2SO3−, C2H3SO3−, and C3H5SO3− CuS− increases and demonstrates significantly different distribution while the intensity of CuS− C3H5SO3− is more positively correlated with the distribution of chloride. For MPS 25 ppm, the intensity of the fragment CH2SO3− rapidly grows at the highest potentials (0.0–0.3 mm) then is stable and decreases from 0.8 mm. On the other hand, the intensity of CH2SO3− is similar in the 0.8–1.3 mm range, while for position 0.0–0.7 mm it is lower, and for 1.4 to 2.0 mm it is greater than was observed for MPS 25 ppm. The distribution of the fragment C3H5SO3− is similar for MPS 25 ppm and 50 ppm and grows up to position 1.5 mm and then gradually decreases. In consequence to the forward scan in the position from 0.0–0.7 mm, the ratio of CH2SO3−/C3H5SO3− is significantly higher for MPS 25 ppm than for MPS 50 ppm. For position 0.8–1.6 mm the ratio is very similar, while for the range from 1.7–2.0 mm it is slightly higher for MPS 25 ppm. The ratio of total chloride/total thiols for MPS 25 ppm and MPS 50 ppm is only very similar for the range 0.0–0.5 mm and is only slightly higher for MPS 25 ppm. Moreover, the intensity distribution of C3H5SO3− positively correlates with chloride ions demonstrating a significant increase from 0.9 to 1.3 mm while CH2SO3 in this region significantly decreases.
It is very interesting that distribution of CuS− is very similar to CH2SO3− while different from C3H5SO3− for MPS 25 ppm and MPS 50 ppm. In the previous considerations (the dip-coating paragraph) we considered that the CuS− fragment corresponds to the Cu–S bond. Since distribution of CuS− is very similar to CH2SO3−, it means that the CH2SO3− fragment is yielded from thiolate molecules that are in close proximity to the copper surface while the C3H5SO3− fragment that is yielded from the thiolate consists of a thiolate end located a greater distance from the copper.
Recently, it was shown that a lower ratio of Cl/MPS on the copper surface leads to a longer Cu–Cl distance [
20]. Distribution of intensity of CuS
− shows that Cu–S distance for MPS 50 ppm is strongly dependent on the potential (rapid reduction is observed at the position 0.8 mm) when significant number of thiolate molecules in gauche conformation is reoriented into trans conformation. It seems that the reorientation gauche/trans process does not directly depend on chloride surface coverage, while at OCP and for low overpotential for reverse scan it is significantly stabilized by the greater surface coverage since we observe the minimum intensity ratio CH
2SO
3−/C
3H
5SO
3− in the region from 1.0–1.5 mm for the reverse scan. A similar, less distinct effect can also be observed for MPS 25 ppm. A significantly higher gauche/trans ratio (CH
2SO
3−/C
3H
5SO
3−) in the region 0.0–0.7 mm can be observed for MPS 50 ppm but under lower chloride surface coverage it determines the same accelerating effect (see CV curve,
Figure 4b). This shows that the final accelerating effect (CV curve shape) is determined by the total chloride/total thiols and CH
2SO
3−/C
3H
5SO
3− ratios, total chloride, total thiols, CH
2SO
3− (gauche conformation), and C
3H
5SO
3− (trans conformation). For the rest of the overpotential region the total exchange current density is the result of the above factors.
In the range of 0.7–1.1 mm, CV curves for MPS 25 ppm and MPS 50 ppm are completely overlapped, since we can observe a similar ratio of chloride/thiolate, and intensity of total chloride, as well as CH2SO3− and C3H5SO3−.
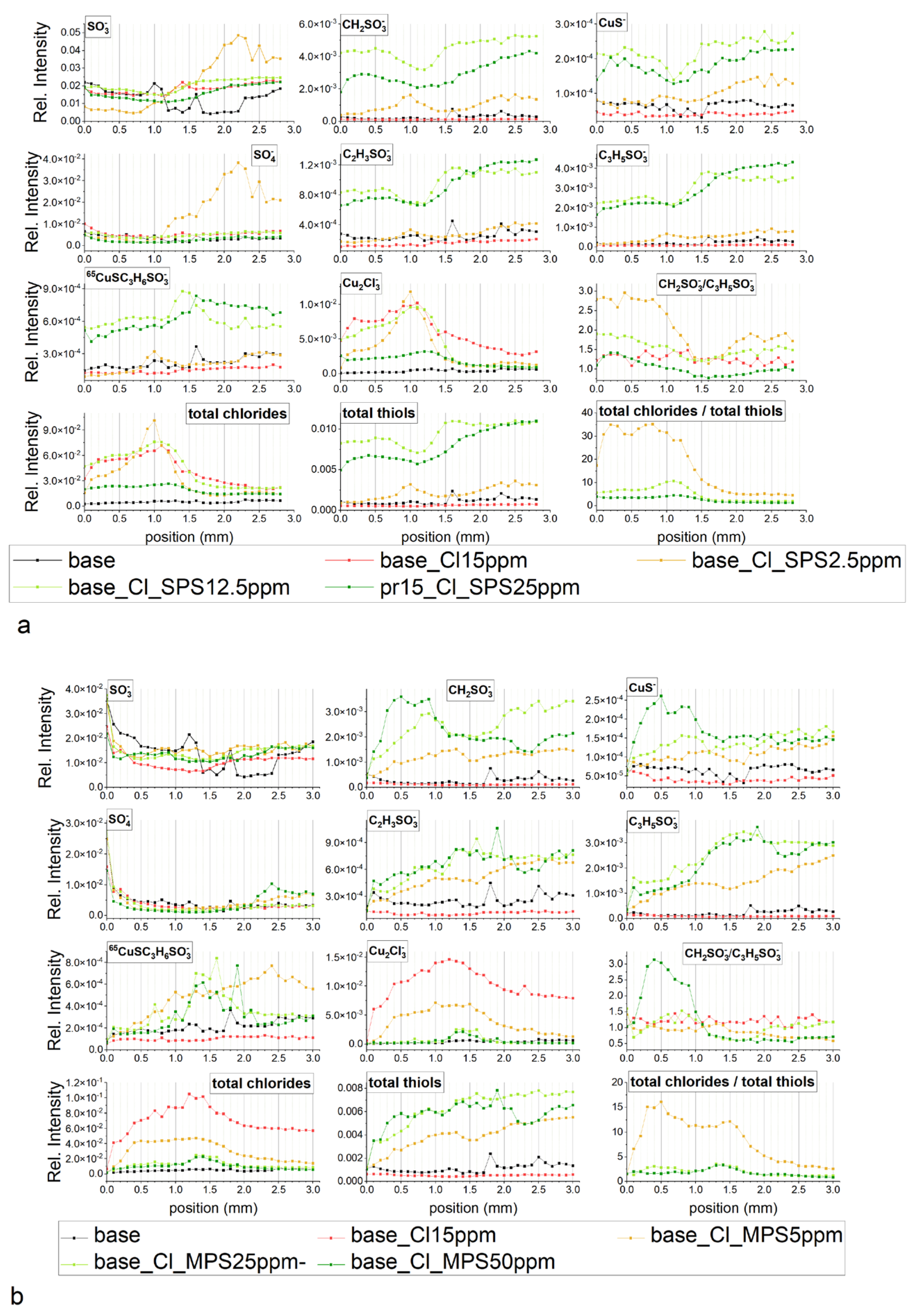
In the light of above results, we propose the mechanism of copper electrodeposition in the presence of Cl/SPS and Cl/MPS as follows.
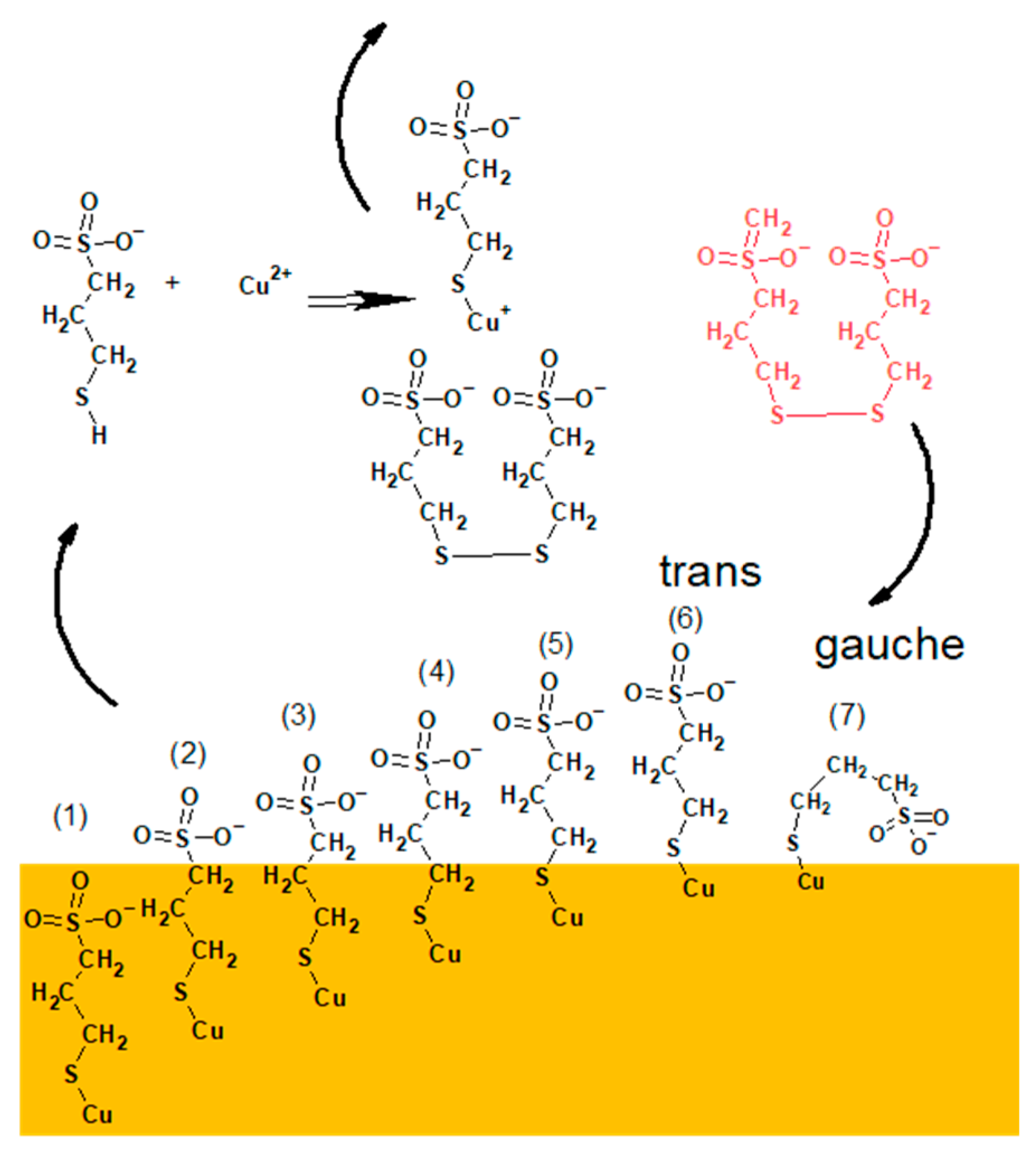
The proposed model of distribution of SPS/MPS into and on the copper surface is shown in
Figure 11.
In the first step, an SPS molecule is adsorbed on the copper surface and immediately dissociated onto two MPS units, since we did not find the existence of S–S bonds in the TOF-SIMS data (lack of fragments
m/z = 331 and
m/z = 371 identified in the dip-coating experiment, see paragraph). This observation is in full agreement with previous findings [
12].
The accumulation of MPS units on the copper surface is dependent on the concentration of SPS in the solution and applied potential. For the lowest concentration (SPS2.5ppm), the maximum MPS coverage is observed around OCP and is positively correlated with chloride ions, while for 12.5 ppm and 25 ppm lower overpotential decreases since some MPS units are replaced by chloride ions. Adsorbed MPS molecules exist in gauche (CH
2SO
3−) and trans (C
3H
5SO
3−) conformations. The relative amount of gauche/trans MPS units depends on the applied potential and demonstrates the highest value for SPS2.5ppm and decreases in SPS12.5ppm and SPS25ppm. The thiolate end in MPS in the gauche conformation is in closer proximity to the copper surface than in trans conformation. During the etching experiment (
Section 2.2), we demonstrated that both gauche and trans forms of MPS molecules were incorporated into the copper deposit while gauche conformation was more preferentially incorporated with the chloride ions. This is an essential difference in comparison to the model presented in
Figure 9 where MPS molecules were not incorporated into deposit.
Moreover, only small amounts of thiolate molecules are desorbed (
Figure 11) and the greater ones are incorporated into deposit. It depends on the SPS/MPS and Cl concentration in the solution. Incorporation of thiolate into deposit may be induced by the existence of a few monolayers of thiolate on the copper surface that significantly reduce MPS desorption. The presence of a few MPS/SPS monolayers makes it difficult for closer proximity of Cu(I)MPS and SPS (the second product of reaction shown in
Figure 11) to the copper surface. Instead of these SPS molecules, the SPS ones from the solution indicated by red colour in
Figure 11 are able to efficiently adsorb on the copper surface.
The greater density of MPS molecules in the first monolayer and the existence of subsequent monolayers (amounts) of SPS are absolutely necessary for inducing the closer proximity of SO
3−Cu
2+. This is determined by the incorporation of MPS molecules into the copper deposit as shown in
Figure 11. The number of direct interactions between SO
3− and Cu
2+ reduce the energy of solvation of Cu
2+ that accelerates the Cu
2+ reduction to Cu
+ and transfers the Cu
+ to the Cl adlayer [
8]. In consequence, the accelerating abilities are improved. The direct interactions between sodium cations and chloride ions on silver substrate were also proven by SERS studies [
41].















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}