
Synthesis and Spectroscopic Characterization of Selected Phenothiazines and Phenazines Rationalized Based on DFT Calculation

 ,
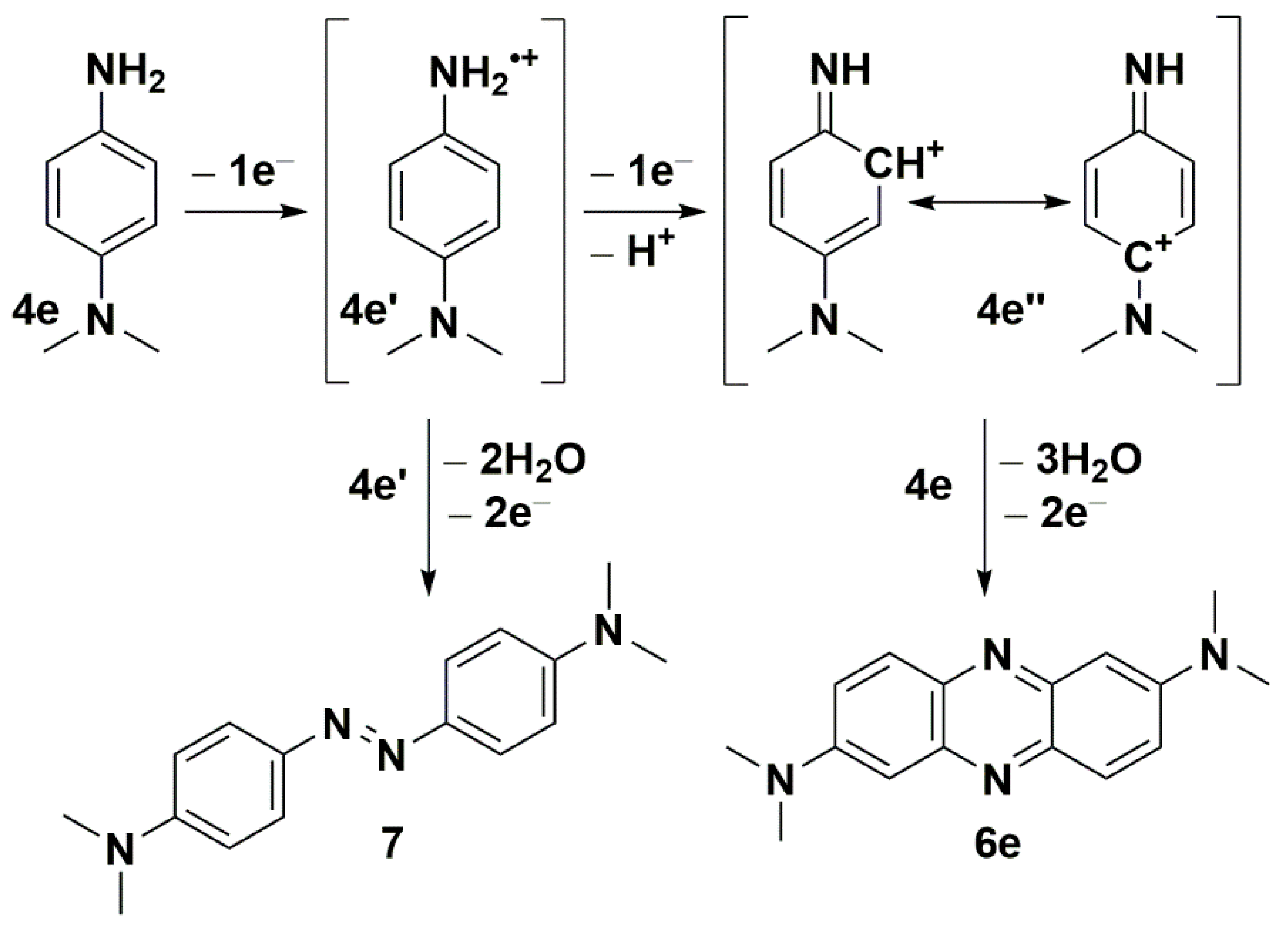
,  , , , ,
, , , , 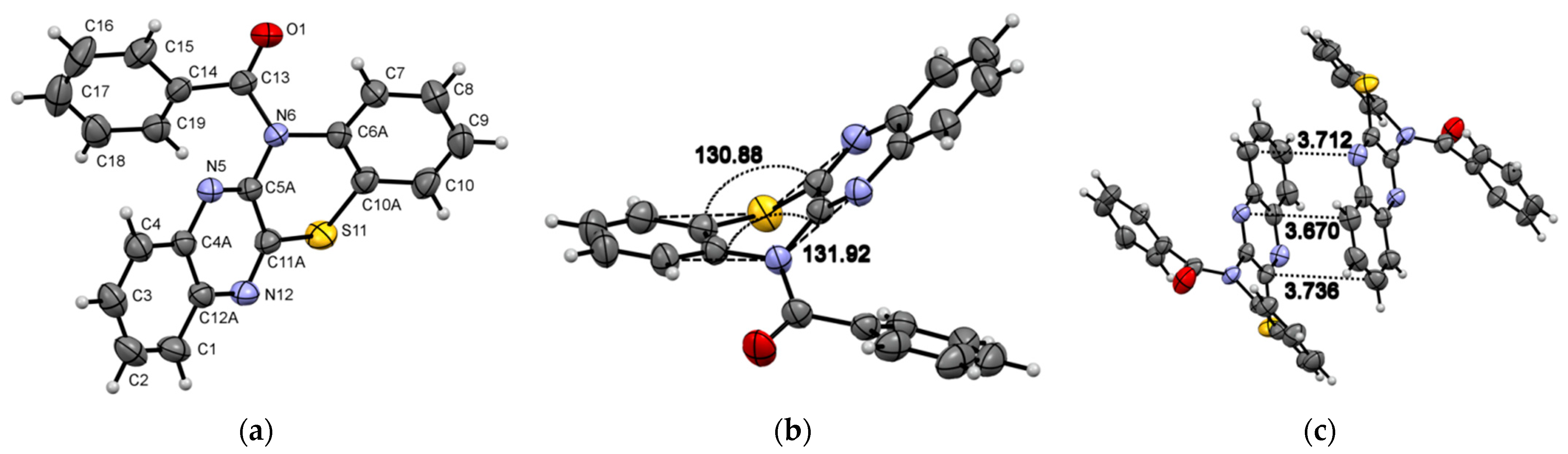{kind=link}
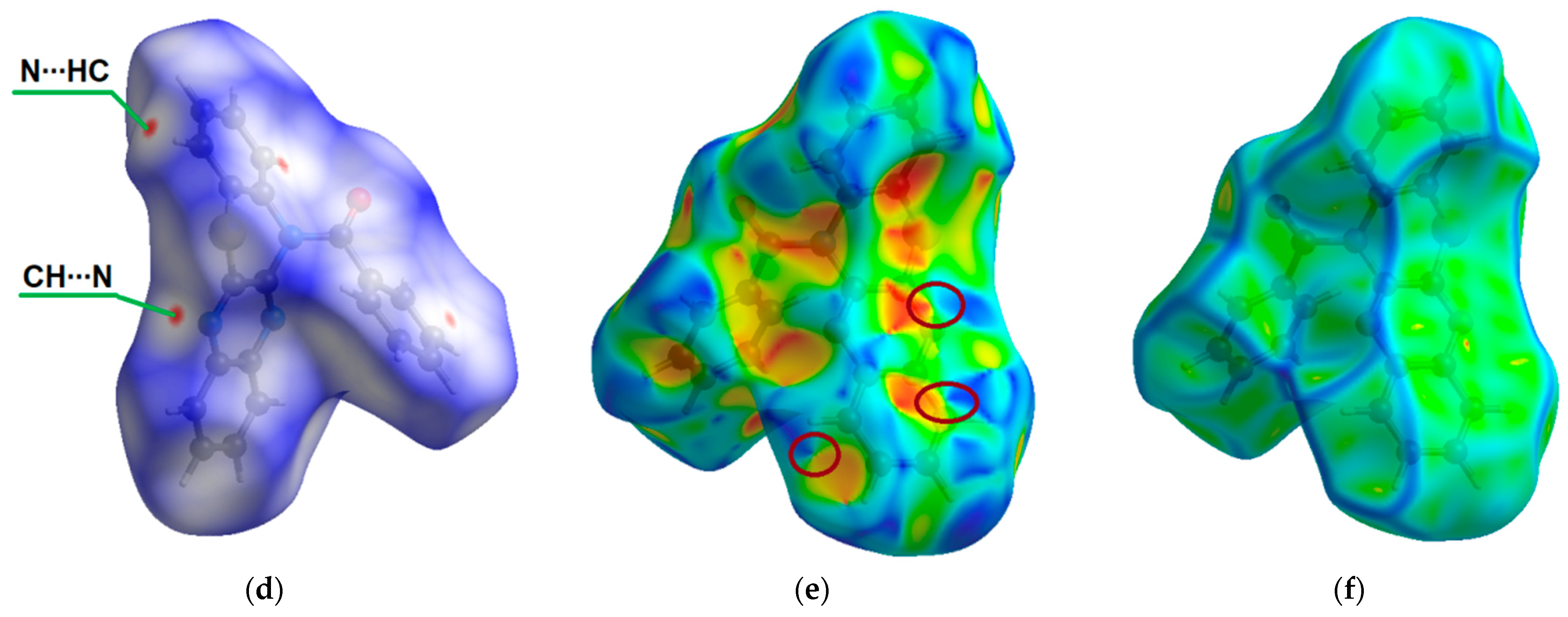{kind=link}
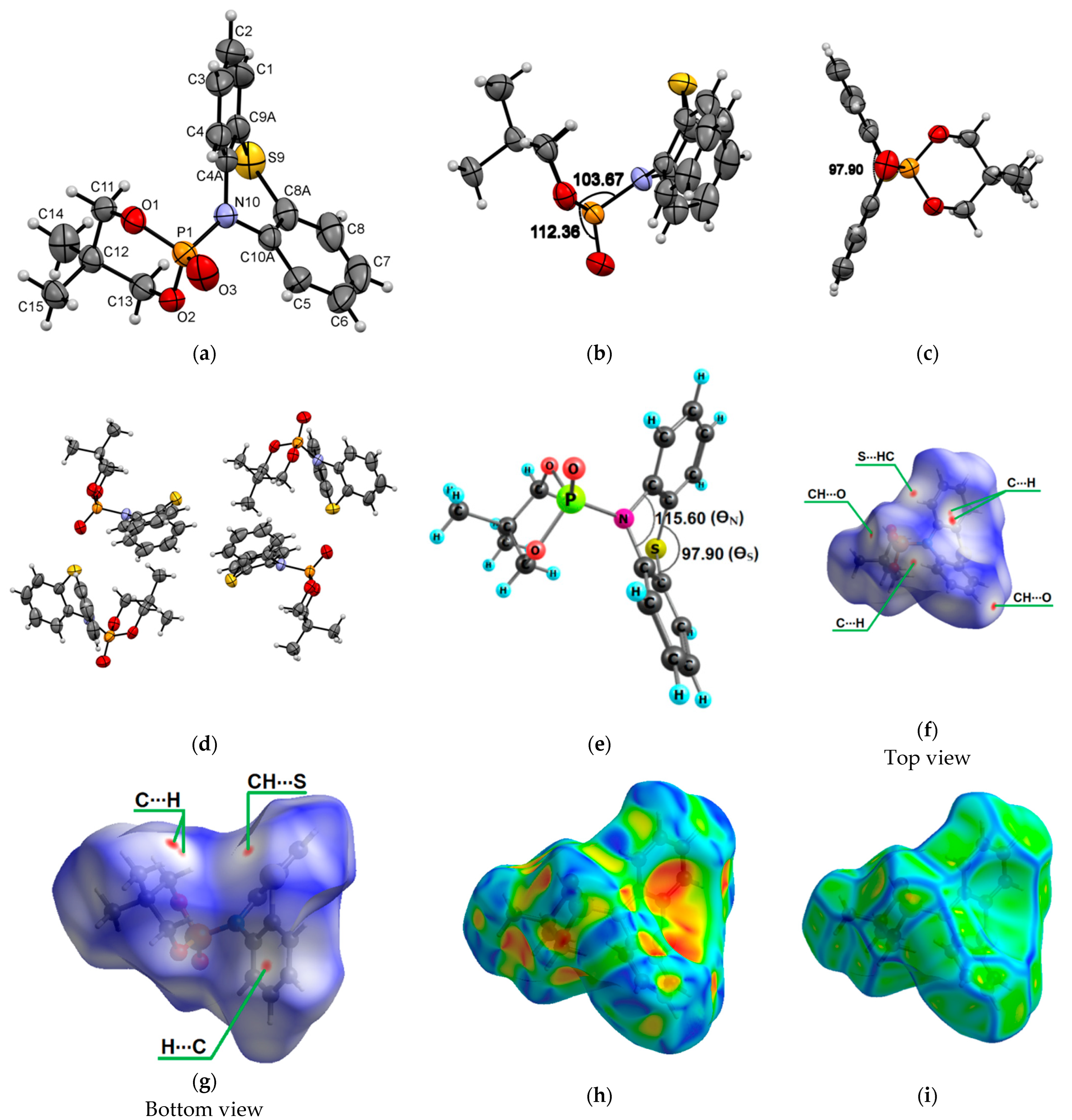{kind=link}
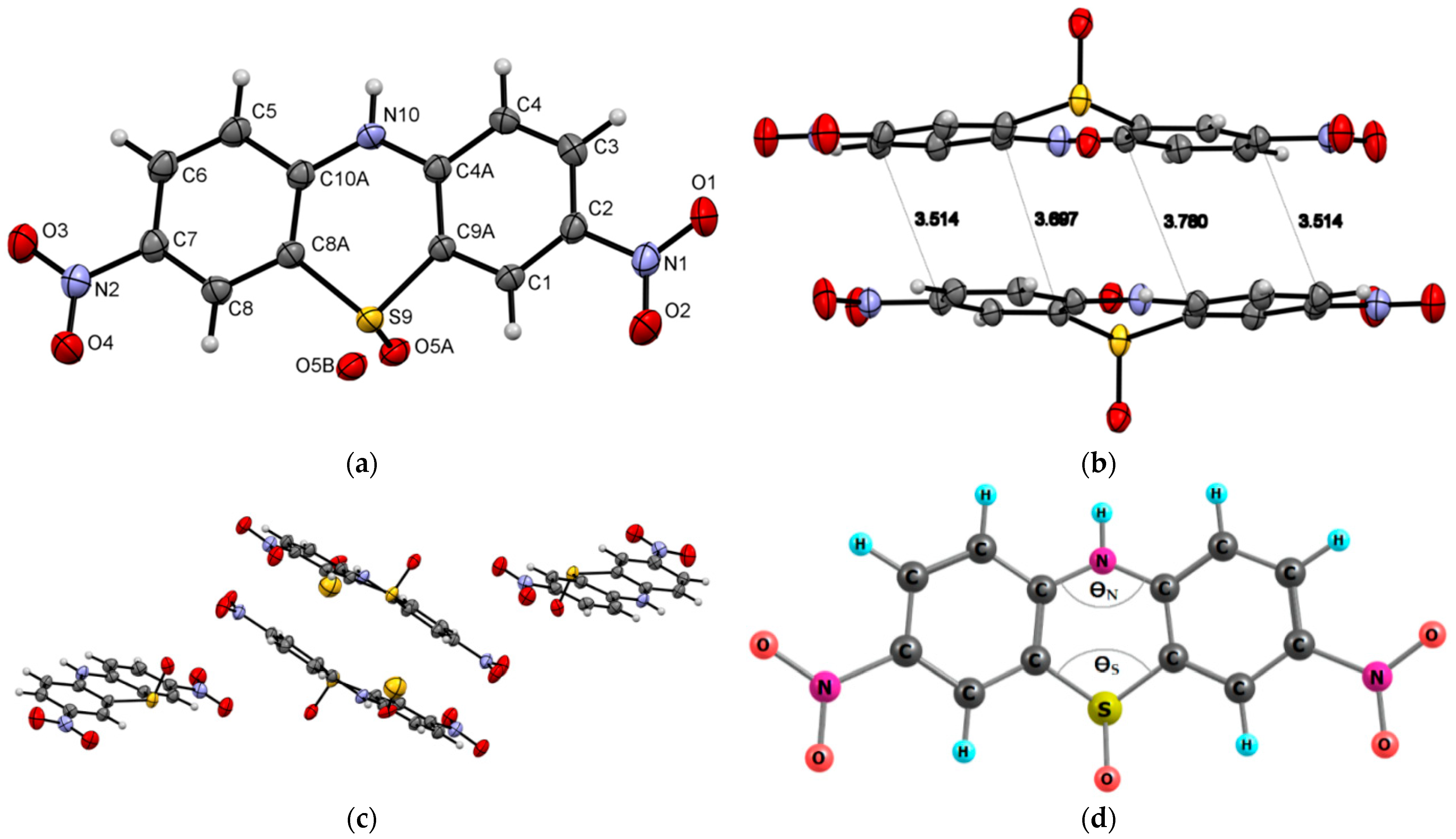{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
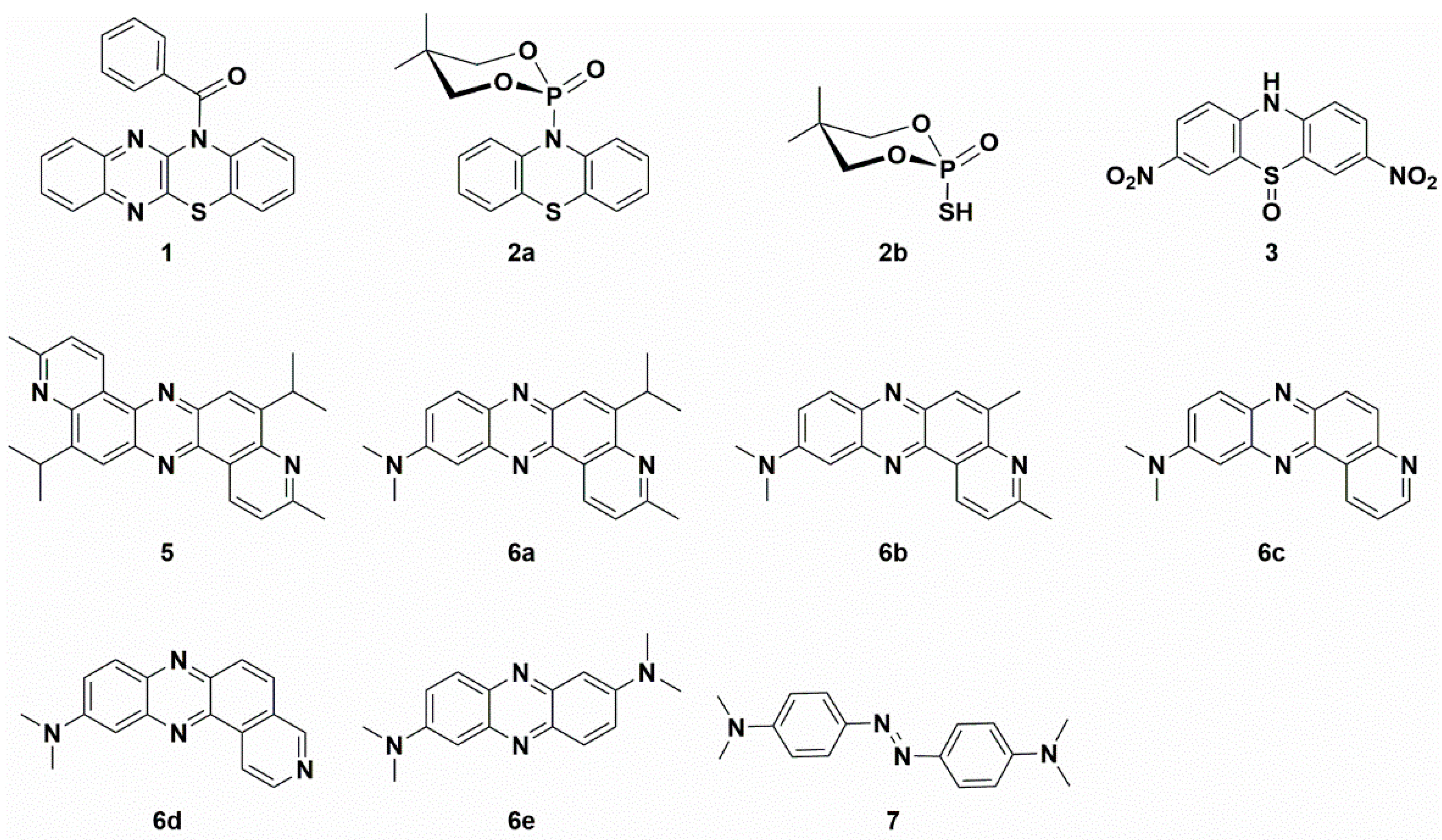
2.1. Synthesis and Structural Characterization of Studied Compounds
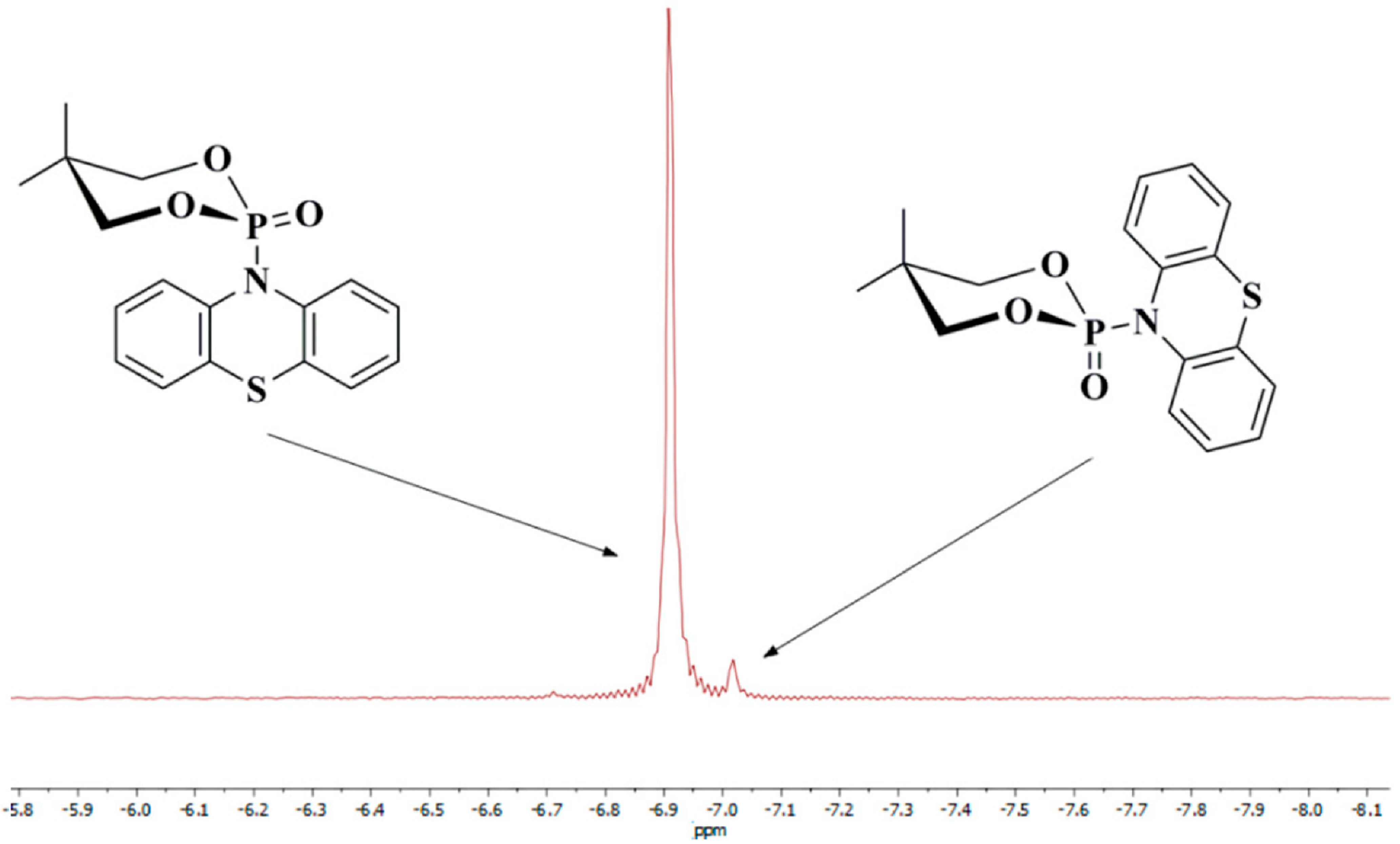
2.2. Anomeric Effect of Compound 2a
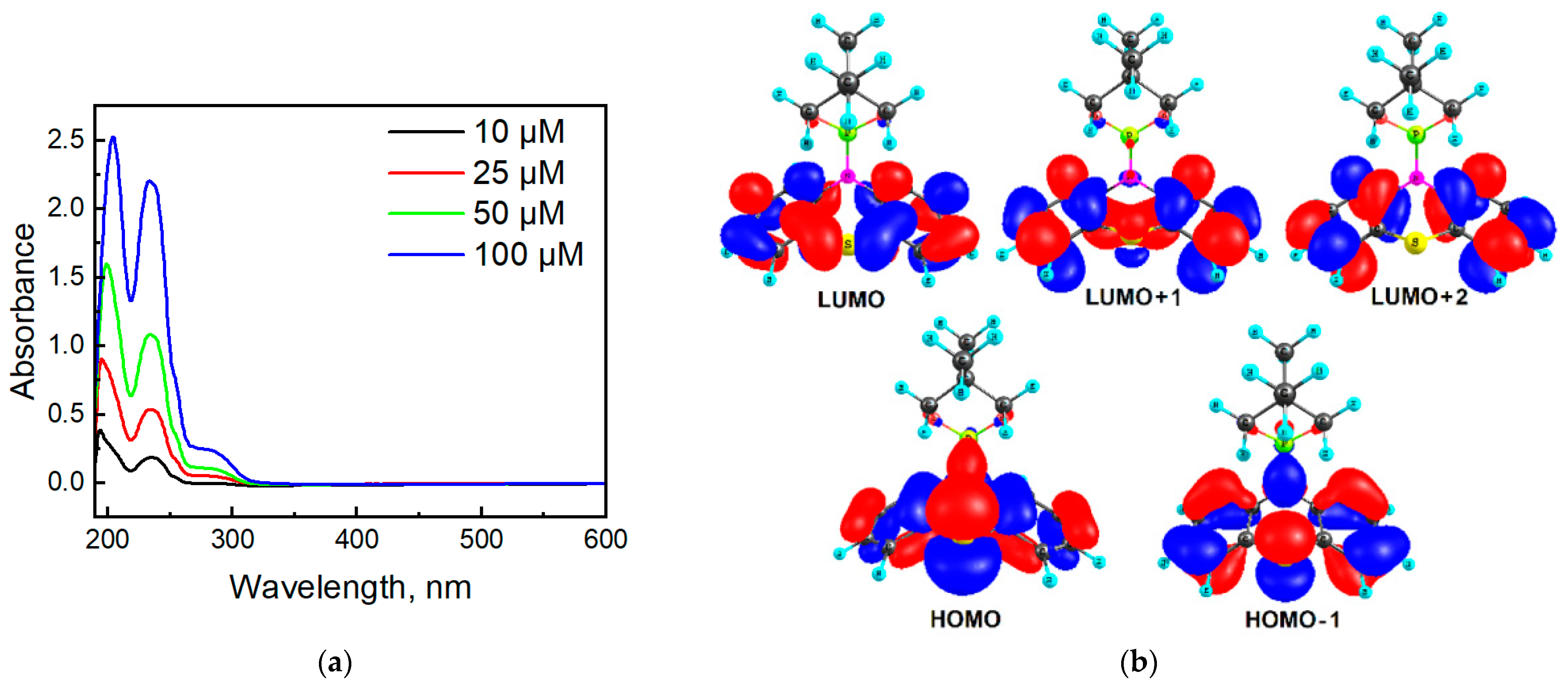
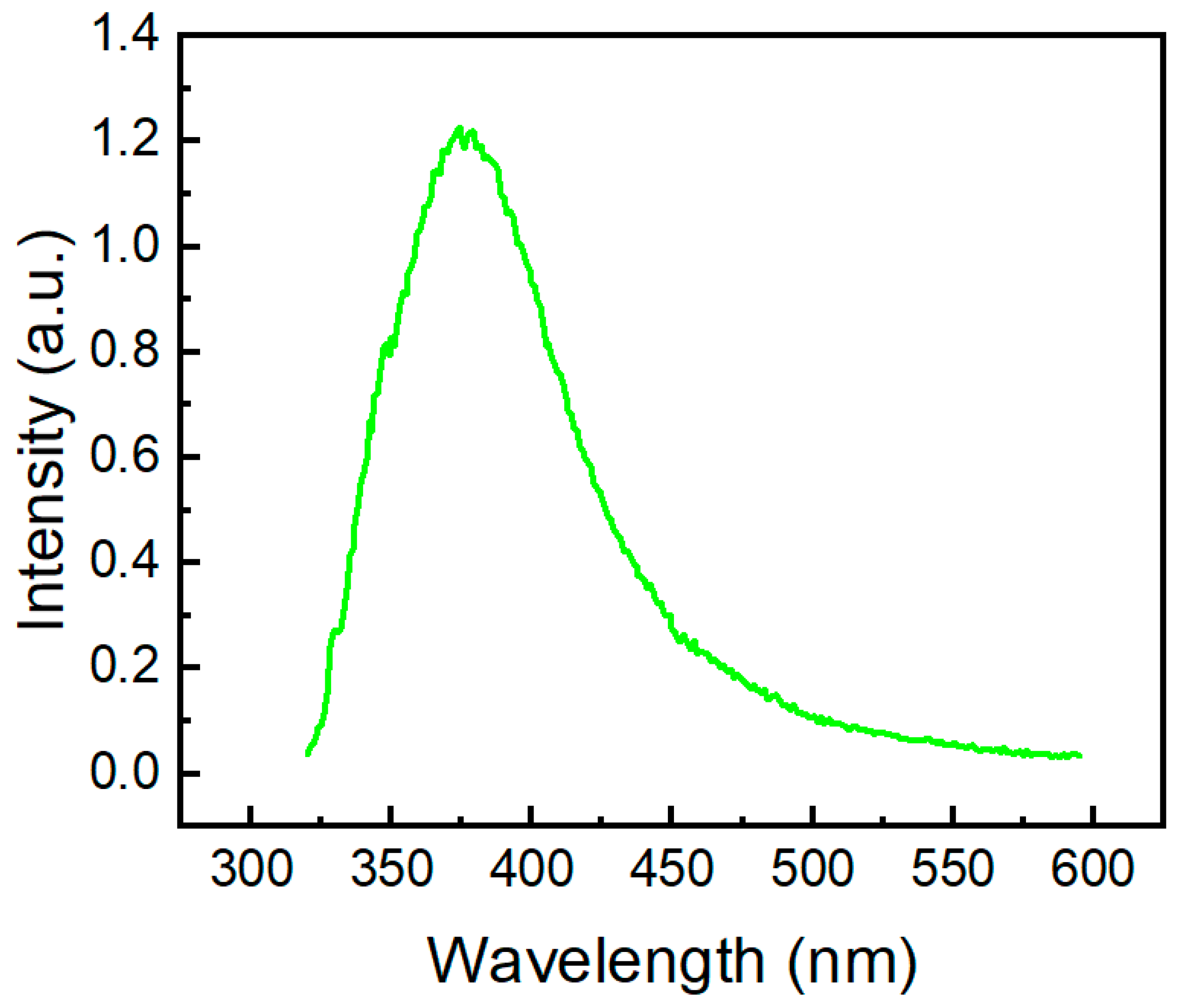
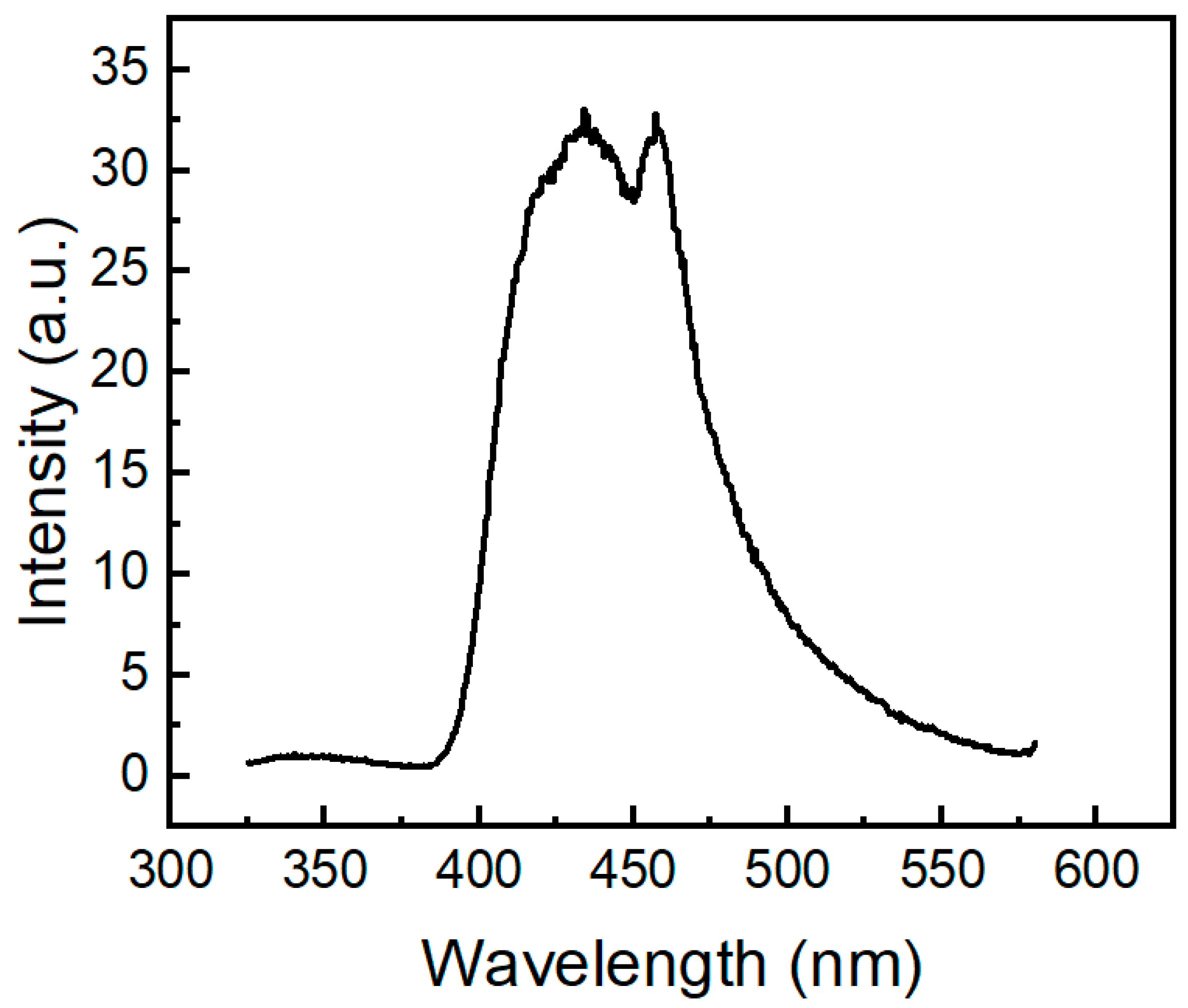
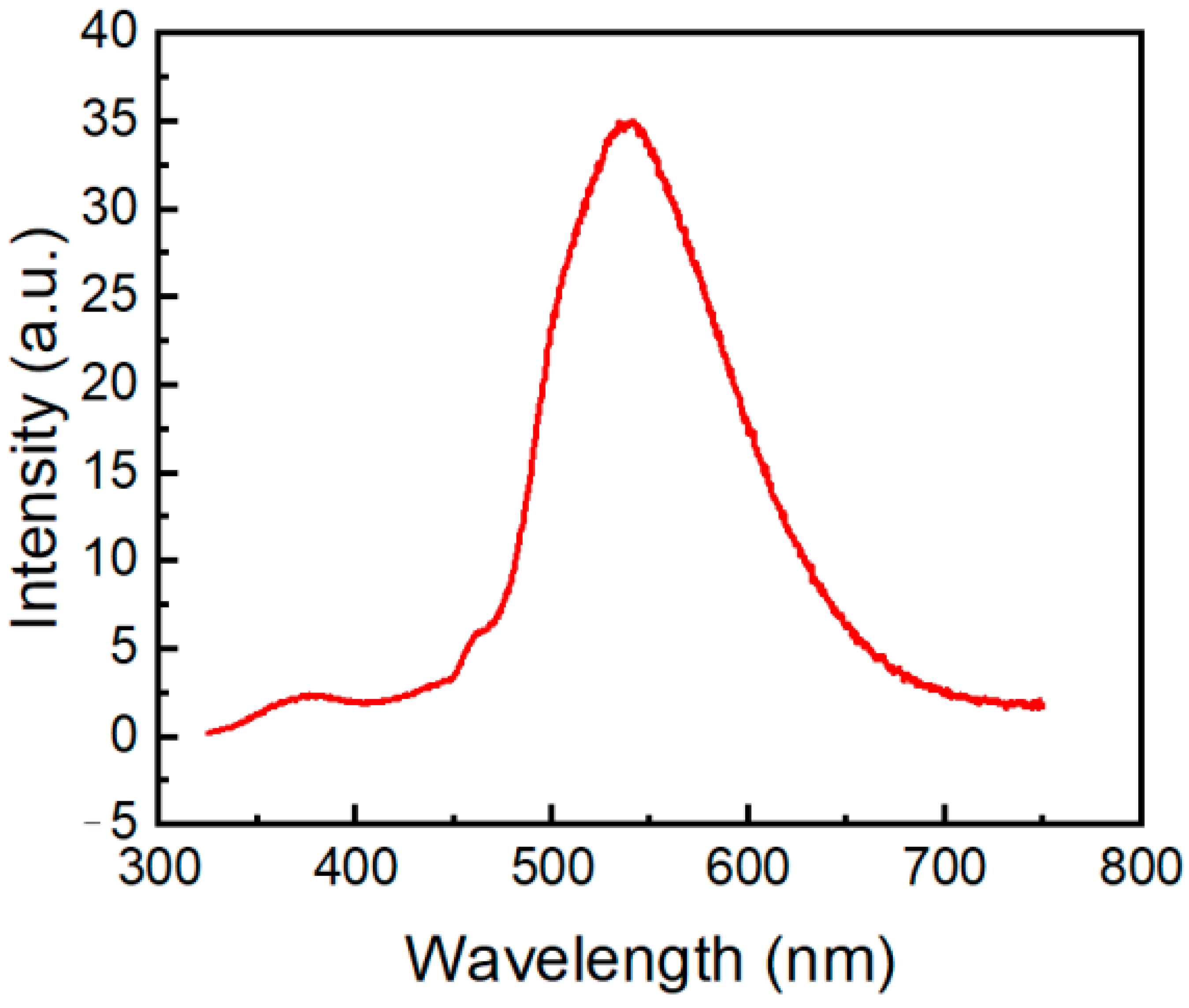
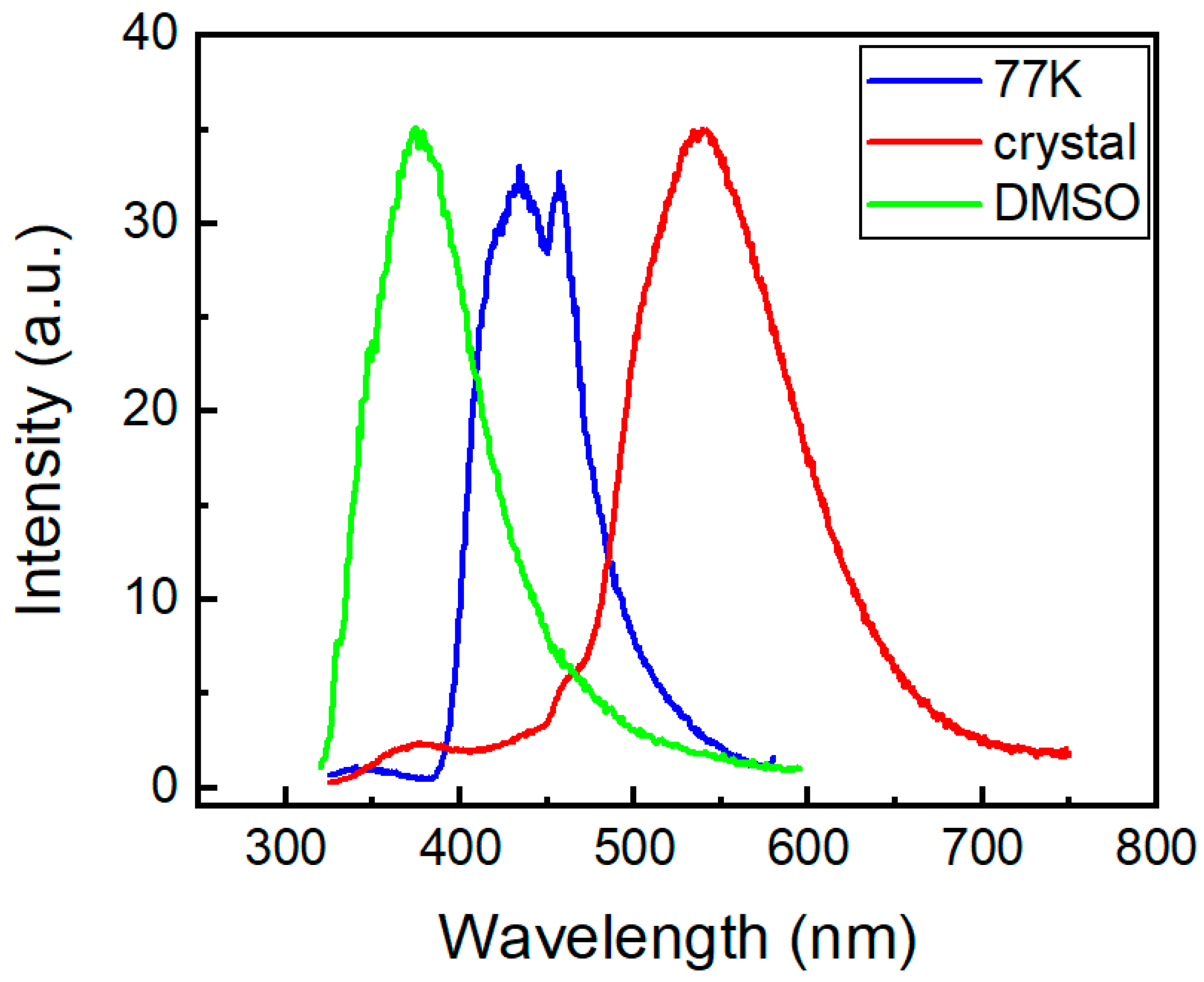
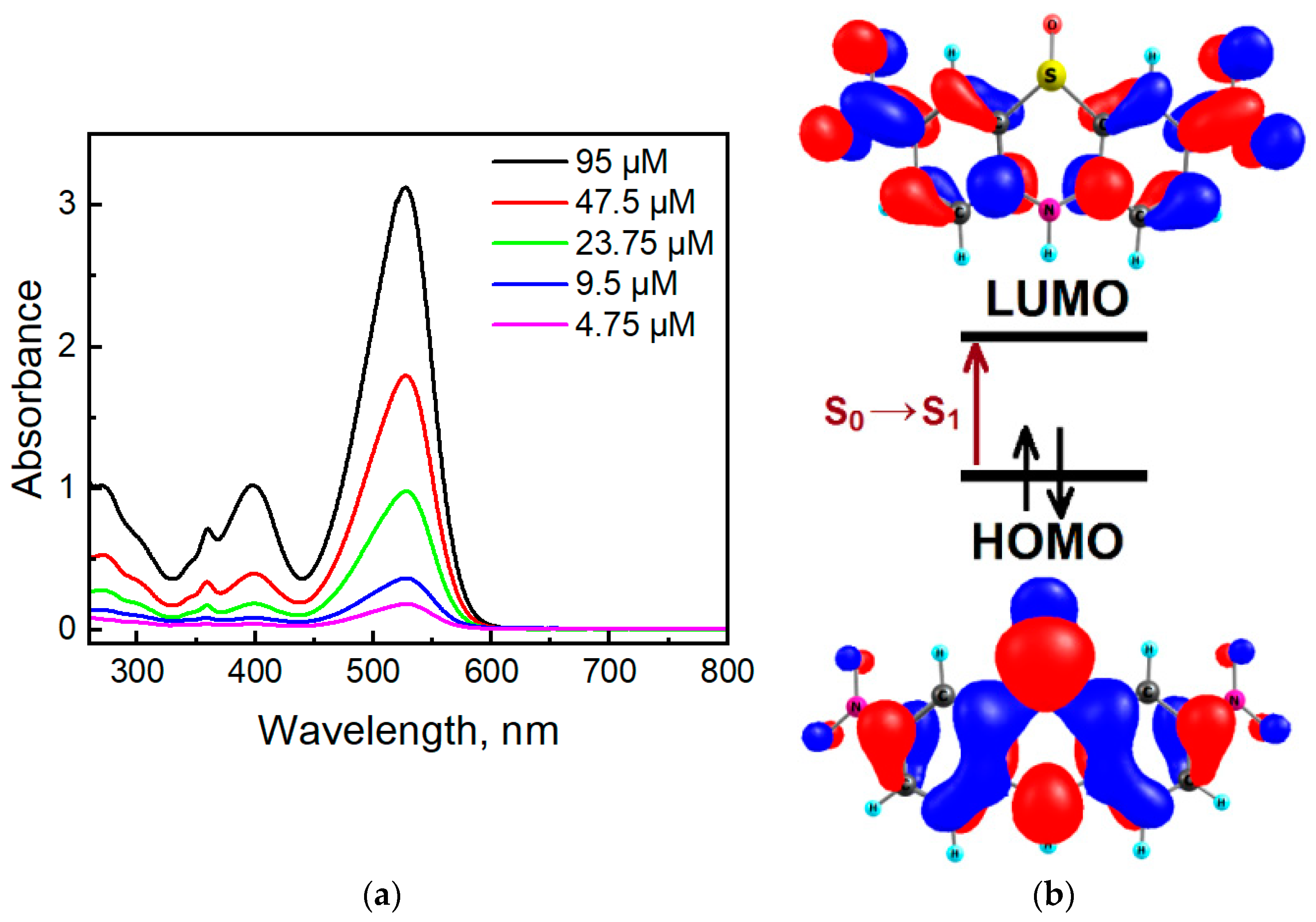
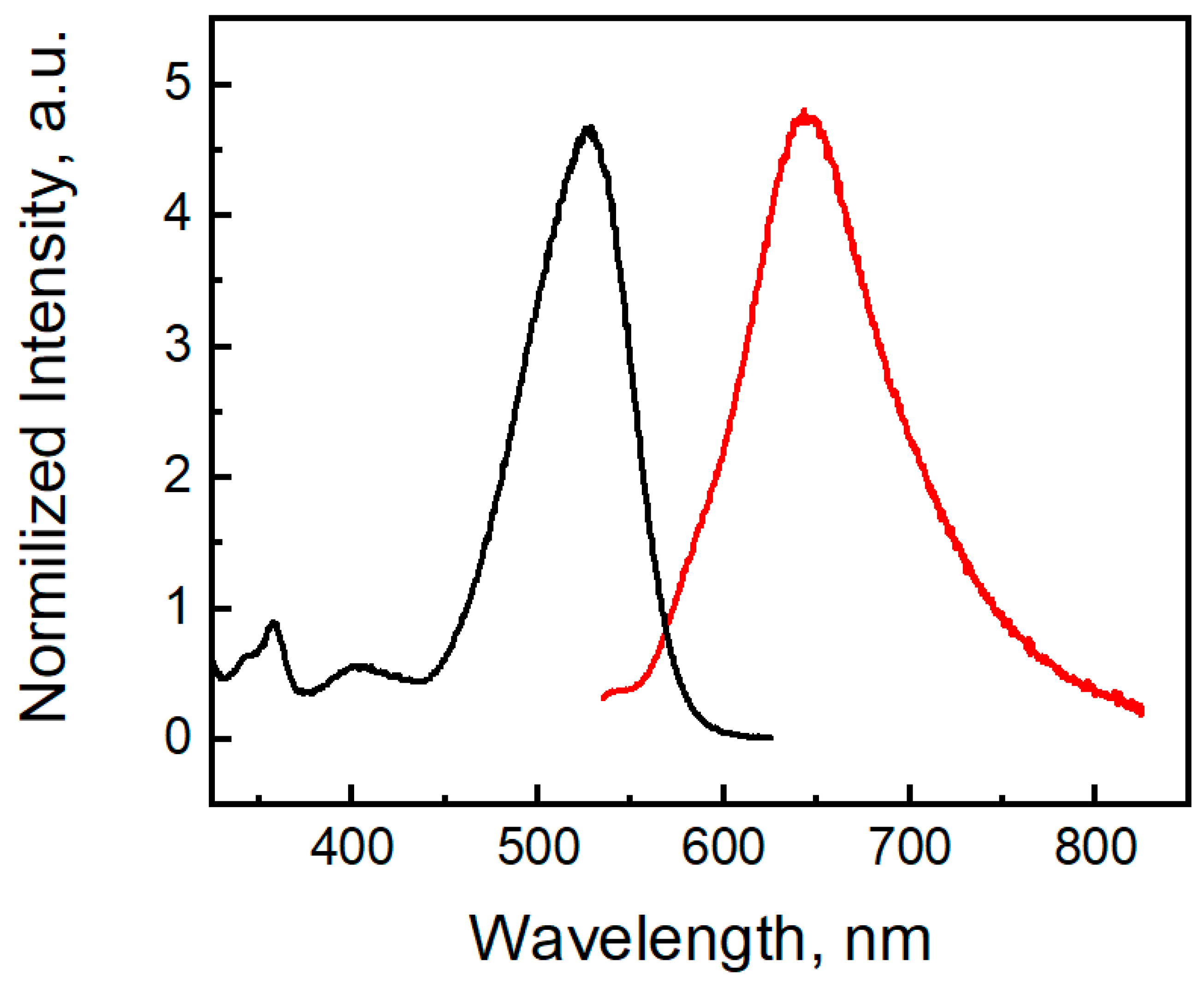
2.3. Photophysical Properties of the Phenothiazine Derivatives 2a and 3
3. Materials and Methods
3.1. Materials
3.2. Instrumentation
3.3. Synthesis of (4a,12a-Dihydro-12H-benzo[5,6][1,4]thiazino[2,3-b]quinoxalin-12-yl)(phenyl)methanone Followed Our Procedure Described in the Literature [23]
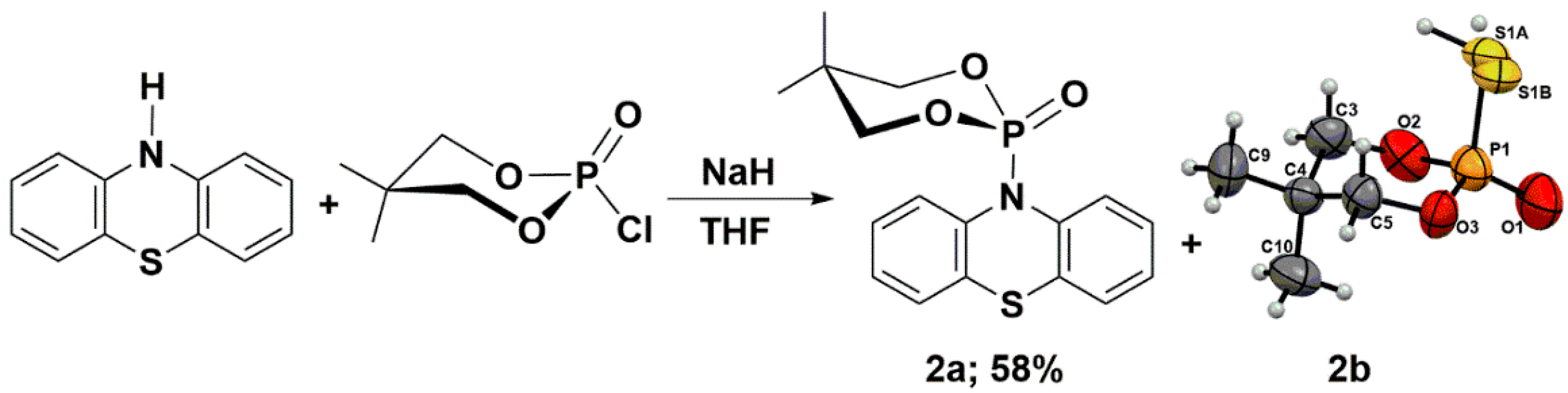
3.4. Phosphorylation of Phenothiazine
3.5. Synthesis of 3,7-Dinitro-10H-phenothiazine 5-oxide (3)
3.6. General Procedure for the Synthesis of 8-(Alkyl)-2-methylquinolin-5-amines
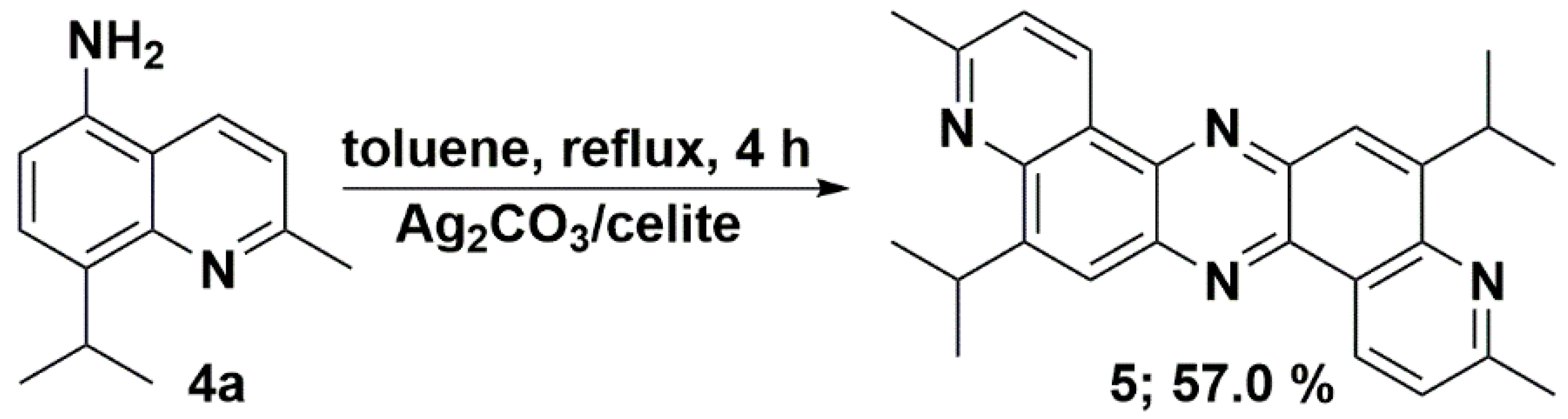
3.7. Synthesis of 5,12-Diisopropyl-3,10-dimethyldipyrido[3,2-a:3′,2′-h]phenazine (5)
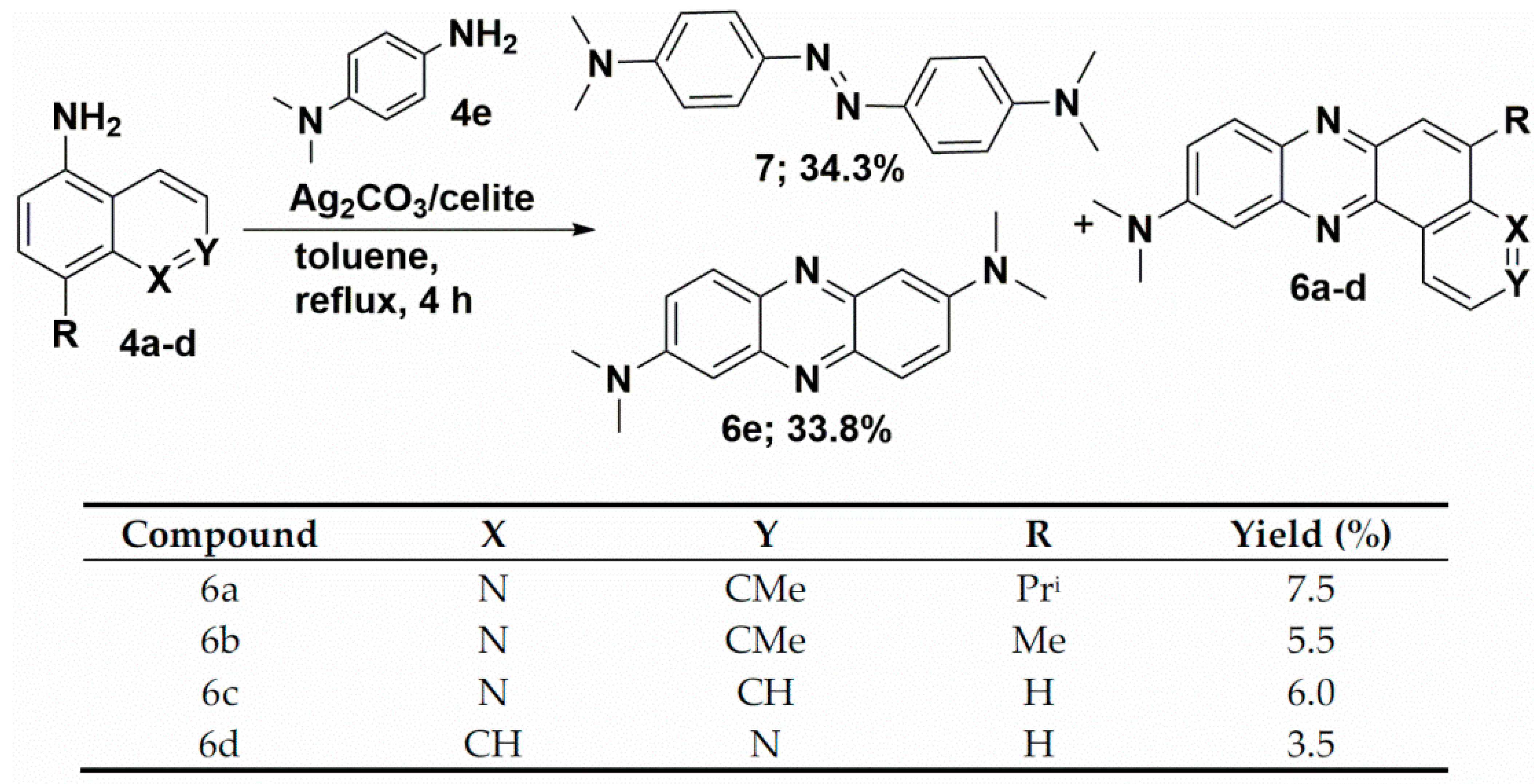
3.8. Synthesis of Unsymmetrical Phenazines (6a–d)
3.9. Synthesis of 6e and 7
3.10. Hirshfeld Surface Calculations
3.11. Density Functional Theory (DFT) Computations
3.12. X-ray Diffraction Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
| DFT | density-functional theory |
| LC-MS | liquid chromatography-mass spectrometry |
| DMF | dimethylformamide |
| THF | tetrahydrofuran |
| DMSO | dimethyl sulfoxide |
| MeCN | acetonitrile |
| TDDFT | time-dependent density-functional theory |
| HRMS | high-resolution mass spectrometry |
| ESI | electrospray ionisation |
| iPrOH | 2-propanol |
| Et2O | diethyl ether |
References
- Padnya, P.L.; Khadieva, A.I.; Stoikov, I.I. Current achievements and perspectives in synthesis and applications of 3,7-disubstituted phenothiazines as Methylene Blue analogues. Dye. Pigment. 2022, 208, 110806. [Google Scholar] [CrossRef]
- Che, Y.-X.; Qi, X.-N.; Lin, Q.; Yao, H.; Qu, W.-J.; Shi, B.; Zhang, Y.-M.; Wei, T.-B. Design strategies and applications of novel functionalized phenazine derivatives: A review. J. Mater. Chem. C 2022, 10, 11119–11174. [Google Scholar] [CrossRef]
- Posso, M.C.; Domingues, F.C.; Ferreira, S.; Silvestre, S. Development of Phenothiazine Hybrids with Potential Medicinal Interest: A Review. Molecules 2022, 27, 276. [Google Scholar] [CrossRef] [PubMed]
- Laursen, J.B.; Nielsen, J. Phenazine Natural Products: Biosynthesis, Synthetic Analogues, and Biological Activity. Chem. Rev. 2004, 104, 1663–1685. [Google Scholar] [CrossRef]
- Mavrodi, D.M.; Bonsall, R.F.; Delaney, S.M.; Soule, M.J.; Phillips, G.; Thomashow, L.S. Functional Analysis of Genes for Biosynthesis of Pyocyanin and Phenazine-1-Carboxamide from Pseudomonas aeruginosa PAO1. J. Bacteriol. 2001, 183, 6454–6465. [Google Scholar] [CrossRef] [Green Version]
- Dietz, K.; Keller, H.J.; Nóthe, D.; Wehe, D. Charge Transfer and Addition Products between Alkylated Phenazine Donors and Ethanetetracarbonitrile (TCNE): Crystal and Molecular Structure of the Dye 2,2’-(2,3,5,10-Tetrahydro-5,10-dimethylphenazine-2,3-diylidene)bis(propanedinitrile) (TMPP). J. Am. Chem. Soc. 1982, 104, 7581–7585. [Google Scholar] [CrossRef]
- Mosnaim, A.D.; Ranade, V.V.; Wolf, M.E.; Puente, J.; Antonieta, V.M. Phenothiazine molecule provides the basic chemical structure for various classes of pharmacotherapeutic agents. Am. J. Ther. 2006, 13, 261–273. [Google Scholar] [CrossRef]
- Franz, A.W.; Rominger, F.; Muller, T.J.J. Synthesis and electronic properties of sterically demanding N-arylphenothiazines and unexpected Buchwald-Hartwig aminations. J. Org. Chem. 2008, 73, 1795–1802. [Google Scholar] [CrossRef]
- Okafor, C.O. Studies in the heterocyclic series: The chemistry and biological activity of new aza and thia-phenothiazines, and related dibenzothiazepines. Phosphorus Sulfur. 1978, 4, 79–80. [Google Scholar] [CrossRef]
- Sailer, M.; Nonnenmacher, M.; Oeser, T.; Muller, T.J.J. Synthesis and electronic properties of 3-acceptor-substituted and 3,7-bisacceptor-substituted phenothiazines. Eur. J. Org. Chem. 2006, 2006, 423435. [Google Scholar] [CrossRef]
- Pluta, K.; Morak-Miodawska, B.; Jelen, M. Recent progress in biological activities of synthesized phenothiazines. Eur. J. Med. Chem. 2011, 46, 3179–3189. [Google Scholar] [CrossRef] [PubMed]
- Kramer, C.S.; Zeitler, K.; Muller, T.J.J. First synthesis and electronic properties of (hetero) aryl bridged and directly linked redox active phenothiazinyl dyads and triads. Tetrahedron Lett. 2001, 42, 8619–8624. [Google Scholar] [CrossRef]
- Chen, M.-C.; Lee, Y.-L.; Huang, Z.-X.; Chen, D.-G.; Chou, P.-T. Tuning Electron-Withdrawing Strength on Phenothiazine Derivatives: Achieving 100% Photoluminescence Quantum Yield by NO2 Substitution. Chem. Eur. J. 2020, 26, 7124–7130. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.; dos Santos, C.G.; Riposati, A.; Barbosa, L.R.; Di Mascio, P.; Itri, R.; Baptista, M.S.; Nascimento, O.R.; Nantes, I.L. Photochemically Generated Stable Cation Radical of Phenothiazine Aggregates in Mildly Acid Buffered Solutions. J. Phys. Chem. B 2006, 110, 12257–12265. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, K.; Asthana, A.; Tamrakar, R.K. Sensitive spectrophotometric method for determination of some phenothiazine drugs. Res. Chem. Intermed. 2015, 41, 7481–7495. [Google Scholar] [CrossRef]
- Murphy, C.M.; Ravner, H.; Smith, N.L. Mode of Action of Phenothiazine-Type Antioxidants. Ind. Eng. Chem. 1950, 42, 2479–2489. [Google Scholar] [CrossRef]
- Fukuzumi, K.; Ikeda, N.; Egawa, M. Phenothiazine derivatives as new antioxidants for the autoxidation of methyl linoleate and their reaction mechanisms. J. Am. Oil Chem. Soc. 1976, 53, 623–627. [Google Scholar] [CrossRef]
- Yamamura, T.; Suzuchi, K.; Yamaguchi, T.; Nishiyama, T. Synthetic Studies on Polypropionate-Derived 4-Pyrone-Containing Marine Natural Products. Bull. Chem. Soc. Jpn. 1997, 70, 413–419. [Google Scholar] [CrossRef]
- Cini, M.; Fariello, R.G.; Bianchetti, A.; Moretti, A. Studies on lipid peroxidation in the rat brain. Neurochem. Res. 1994, 19, 283–288. [Google Scholar] [CrossRef]
- Slater, T.F. The inhibitory effects in vitro of phenothiazines and other drugs on lipid-peroxidation systems in rat liver microsomes, and their relationship to the liver necrosis produced by carbon tetrachloride. Biochem. J. 1968, 106, 155–160. [Google Scholar] [CrossRef]
- Levy, L.B. Inhibition of acrylic acid polymerization by phenothiazine and p-methoxyphenol. J. Polym. Sci. 1985, 23, 1505–1515. [Google Scholar] [CrossRef]
- Nicolson, A. The Effect of Oxygen Concentration on Methylacrylic Acid Stability. Plant Oper. Prog. 1991, 10, 171–183. [Google Scholar] [CrossRef]
- Podsiadły, R.; Strzelczyk, R. N-substituted quinoxalinobenzothiazine/iodonium salt systems as visible photoinitiators for hybrid polymerization. Dye. Pigment. 2013, 97, 462–468. [Google Scholar] [CrossRef]
- Jin, R.; Bub, C.L.; Patureau, F.W. Phenothiazinimides: Atom-Efficient Electrophilic Amination Reagents. Org. Lett. 2018, 20, 2884–2887. [Google Scholar] [CrossRef]
- Milen, M.; Ábrányi-Balogh, P.; Balogh, G.; Drahos, L.; Keglevich, G. A study on the phosphorylation of indole, imidazole, carbazole, and phenothiazine derivatives. Phosphorus Sulfur Silicon Relat. Elem. 2012, 187, 1091–1100. [Google Scholar] [CrossRef]
- Prinz, H.; Chamasmani, B.; Vogel, K.; Böhm, K.J.; Aicher, B.; Gerlach, M.; Günther, E.G.; Amon, P.; Ivanov, I.; Müller, K. N-Benzoylated Phenoxazines and Phenothiazines: Synthesis, Antiproliferative Activity, and Inhibition of Tubulin Polymerization. J. Med. Chem. 2011, 54, 4247–4263. [Google Scholar] [CrossRef]
- Tian, S.; Ma, H.; Wang, X.; Lv, A.; Shi, H.; Geng, Y.; Li, J.; Liang, F.; Su, Z.-M.; An, Z.; et al. Utilizing d–pπ Bonds for Ultralong Organic Phosphorescence. Angew. Chem. Int. Ed. 2019, 58, 6645–6649. [Google Scholar] [CrossRef]
- Nycz, J.E.; Małecki, J.G.; Pajchel, Ł.; Ponikiewski, Ł.; Wiącek, M. One-pot synthesis of selected P-vinylbenzyls under solvent-free conditions. ChemistrySelect 2022, 7, e202200568. [Google Scholar] [CrossRef]
- Nycz, J.E. The synthesis of hypodiphosphoric acid and derivatives with P-P bond, including esters and diphosphine dioxides: A Review. Molecules 2021, 26, 7286. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilakaa, D. Hirshfeld surface analysis. Cryst. Eng. Comm. 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Nycz, J.E.; Szala, M.; Malecki, G.J.; Nowak, M.; Kusz, J. Synthesis, spectroscopy and computational studies of selected hydroxyquinolines and their analogues. Spectrochim. Acta A 2014, 117, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Malecki, G.; Nycz, J.E.; Ryrych, E.; Ponikiewski, L.; Nowak, M.; Kusz, J.; Pikies, J. Synthesis, spectroscopy and computational studies of some biologically important hydroxyhaloquinolines and their novel derivatives. J. Mol. Struct. 2010, 969, 130–138. [Google Scholar] [CrossRef]
- Nycz, J.E.; Malecki, G.; Ponikiewski, L.; Leboschka, M.; Nowak, M.; Kusz, J. Synthesis, spectroscopy and computational studies of some novel phosphorylated derivatives of quinoline-5,8-diones. J. Mol. Struct. 2011, 986, 39–48. [Google Scholar] [CrossRef]
- Wantulok, J.; Swoboda, D.; Nycz, J.E.; Książek, M.; Kusz, J.; Malecki, J.G.; Kubíček, V. Direct amination of nitroquinoline derivatives via nucleophilic displacement of aromatic hydrogen. Molecules 2021, 26, 1857. [Google Scholar] [CrossRef]
- Staab, A.H.; Elbl, K. Tetrakis(dialkylamino)aromatics and Their Charge Transfer Complexes. US Patent 4,999,441, 12 March 1991. [Google Scholar]
- Seth, K.; Roy, S.R.; Chakraborti, A.K. Synchronous double C–N bond formation via C–H activation for a novel synthetic route to phenazine. Chem. Commun. 2016, 52, 922–925. [Google Scholar] [CrossRef]
- Seth, K.; Roy, S.R.; Kumar, A.; Chakraborti, A.K. The palladium and copper contrast: A twist to products of different chemotypes and altered mechanistic pathways. Catal. Sci. Technol. 2016, 6, 2892–2896. [Google Scholar] [CrossRef]
- Thompson, D.C.; Eling, T.E. Reactive Intermediates Formed during the Peroxidative Oxidation of Anisidine Isomers. Chem. Res. Toxicol. 1991, 4, 474–481. [Google Scholar] [CrossRef]
- Stiborová, M.; Mikšanová, M.; Havlíček, V.; Schmeiser, H.H.; Frei, E. Mechanism of peroxidse-mediated oxidation of carcinogenic o-anisidine and its binding to DNA. Mutat. Res. 2002, 500, 49–66. [Google Scholar] [CrossRef]
- Mortzfeld, F.B.; Pietruszka, J.; Baxendale, I.R. A Simple and Efficient Flow Preparation of Pyocyanin a Virulence Factor of Pseudomonas aeruginosa. Eur. J. Org. Chem. 2019, 2019, 5424–5433. [Google Scholar] [CrossRef]
- Edward, J.T. Stability of glycosides to acid hydrolysis. Chem. Ind. 1955, 36, 1102–1104. [Google Scholar]
- Lemieux, R.U.; Chu, P. 133rd National Meeting of the American Chemical Society 31N; American Chemical Society: Washington, DC, USA, 1958. [Google Scholar]
- Gold, V.; Loening, K.L.; McNaught, A.D.; Sehmi, P. International Union of Pure and Applied Chemistry Compendium of Chemical Terminology IUPAC Recommendations; Blackwell Scientific Publications Limited: Malden, MA, USA, 1987. [Google Scholar]
- Bentrude, W.G.; Setzer, W.N.; Sopchik, A.E.; Bajwa, G.S.; Burright, D.D.; Hutchinson, J.P. Conformations of saturated six-membered-ring phosphorus heterocycles related to cyclophosphamide. NMR, x-ray, and infrared studies of 2-methoxy-2-oxo-1,3,2-oxazaphosphorinane and 2-thio-1,3,2-oxazaphosphorinane. J. Am. Chem. Soc. 1986, 108, 6669–6675. [Google Scholar] [CrossRef]
- Bentrude, W.G.; Hargis, J.H. Conformations of six-membered ring phosphorus heterocycles. I. Ring conformations and phosphorus configurations of isomeric six-membered ring phosphites. J. Am. Chem. Soc. 1970, 92, 7136–7144. [Google Scholar] [CrossRef]
- Cramer, C.J.; Truhlar, D.G.; French, A.D. Exo-anomeric effects on energies and geometries of different conformations of glucose and related systems in the gas phase and aqueous solution. Carbohyd. Res. 1997, 298, 105. [Google Scholar] [CrossRef]
- Manju, T.; Manoj, N.; Braunc, A.M.; Oliveros, E. Self sensitized photooxidation of N-methyl phenothiazine: Acidity control of the competition between electron and energy transfer mechanisms. Photochem. Photobiol. Sci. 2012, 11, 1744–1755. [Google Scholar] [CrossRef]
- García, C.; Oyola, R.; Piñero, L.E.; Arce, R.; Silva, J.; Sánchez, V. Substitution and Solvent Effects on the Photophysical Properties of Several Series of 10-Alkylated Phenothiazine Derivatives. J. Phys. Chem. A 2005, 109, 3360–3371. [Google Scholar] [CrossRef] [PubMed]
- Dilelio, M.C.; Kaufman, T.S.; Iglesias, B.A.; Silveira, C.C. Synthesis and evaluation of photophysical and electrochemical properties of vinyl chalcogenide derivatives of phenothiazines. Dye. Pigment. 2022, 198, 109982. [Google Scholar] [CrossRef]
- Schmidt, M.; Esser, B. Cavity-promotion by pillar[5]arenes expedites organic photoredox-catalysed reductive dehalogenations. Chem. Commun. 2021, 57, 9582–9585. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Huang, R.; Batsanov, A.S.; Pander, P.; Hsu, Y.-T.; Chi, Z.; Dias, F.B.; Martin, R.; Bryce, M.R. Intramolecular Charge Transfer Controls Switching Between Room Temperature Phosphorescence and Thermally Activated Delayed Fluorescence. Angew. Chem. Int. Ed. 2018, 57, 16407. [Google Scholar] [CrossRef] [Green Version]
- Yao, L.; Zhang, S.; Wang, R.; Li, W.; Shen, F.; Yang, B.; Ma, Y. Highly Efficient Near-Infrared Organic Light-Emitting Diode Based on a Butterfly-Shaped Donor–Acceptor Chromophore with Strong Solid-State Fluorescence and a Large Proportion of Radiative Excitons. Angew. Chem. Int. Ed. 2014, 53, 2119–2123. [Google Scholar] [CrossRef]
- Minaev, B.F.; Terpugova, A.F. Spin-orbit interaction in charge-transfer complexes. Sov. Phys. J. 1968, 12, 1260–1263. [Google Scholar] [CrossRef]
- Mitchell, S.C.; Kestell, P.; Steventon, G.B.; Waring, R.H. Fate of the anthelmintic, phenothiazine, in man. Xenobiotica 2002, 32, 771–782. [Google Scholar] [CrossRef]
- Hayashi, H.; Koizumi, T. Preparation and electrochemical behavior of N-substituted phenothiazine oxide. Heterocycles 2016, 92, 1441–1449. [Google Scholar] [CrossRef]
- Mellinger, T.J.; Keeler, C.E. Spectrofluorometric identification of phenothiazine drugs. Anal. Chem. 1963, 35, 554–558. [Google Scholar] [CrossRef]
- Nycz, J.E.; Wantulok, J.; Sokolova, R.; Pajchel, L.; Stankevič, M.; Szala, M.; Malecki, J.G.; Swoboda, D. Synthesis and electrochemical and spectroscopic characterization of 4,7-diamino-1,10-phenanthrolines and their precursors. Molecules 2019, 24, 4102. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Cao, X.; Shi, L.; Qi, F.; Guo, Z.; Lu, J.; Gu, H. A Highly Active Nano-Palladium Catalyst for the Preparation of Aromatic Azos under Mild Conditions. Org. Lett. 2011, 13, 5640–5643. [Google Scholar] [CrossRef]
- Mardirossian, N.; Head-Gordon, M. Thirty years of density functional theory in computational chemistry: An overview and extensive assessment of 200 density functional. Mol. Phys. 2017, 115, 2315–2372. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Perspective: Fifty years of density-functional theory in chemical physics. J. Chem. Phys. 2014, 140, 18A301. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Francl, M.M.; Petro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3666. [Google Scholar] [CrossRef] [Green Version]
- Runge, E.; Gross, E.K.U. Density-Functional Theory for Time-Dependent Systems. Phys. Rev. Lett. 1984, 52, 997–1000. [Google Scholar] [CrossRef]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic Interaction of a Solute with a Continuum. A Direct Utilizaion of Ab Initio Molecular Potentials for the Prevision of Solvent Effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- CrysAlisPro, Version 1.171.38.41q. Rigaku Oxford Diffraction. Applied Rigaku Technologies, Inc.: Austin, TX, USA, 2015.
- CrysAlisPro, Version 1.171.39.15e. Rigaku Oxford Diffraction. Applied Rigaku Technologies, Inc.: Austin, TX, USA, 2015.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wainwright, M. Photosensitisers in Biomedicine; John Wiley & Sons, Ltd.: Chichester, UK, 2009; pp. 54–58. [Google Scholar]
- Cheema, S.; Zhang, M.; Labine-Romain, M.; Lal, B.; Lavania, M.; Lee, M.; Li, X.; Lauro, F.M.; Beckmann, S.; Manefield, M. Neutral Red: The Synthetic Phenazine Full of Electrochemical Surprises. In Encyclopedia of Interfacial Chemistry; Elsevier: Amsterdam, The Netherlands, 2018; pp. 382–391. [Google Scholar]
- Lindgren, M.; Glimsdal, R.; Vestberg, R. Electronic states and phosphorescence of dendron functionalized platinum (II) acetylides. J. Lumin. 2007, 124, 302–310. [Google Scholar] [CrossRef]






















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swoboda, D.; Nycz, J.E.; Karaush-Karmazin, N.; Minaev, B.; Książek, M.; Kusz, J.; Podsiadły, R. Synthesis and Spectroscopic Characterization of Selected Phenothiazines and Phenazines Rationalized Based on DFT Calculation. Molecules 2022, 27, 7519. https://doi.org/10.3390/molecules27217519
Swoboda D, Nycz JE, Karaush-Karmazin N, Minaev B, Książek M, Kusz J, Podsiadły R. Synthesis and Spectroscopic Characterization of Selected Phenothiazines and Phenazines Rationalized Based on DFT Calculation. Molecules. 2022; 27(21):7519. https://doi.org/10.3390/molecules27217519
Chicago/Turabian StyleSwoboda, Daniel, Jacek E. Nycz, Nataliya Karaush-Karmazin, Boris Minaev, Maria Książek, Joachim Kusz, and Radosław Podsiadły. 2022. "Synthesis and Spectroscopic Characterization of Selected Phenothiazines and Phenazines Rationalized Based on DFT Calculation" Molecules 27, no. 21: 7519. https://doi.org/10.3390/molecules27217519