
Design, Synthesis, and Bioassay of 2′-Modified Kanamycin A

Abstract
:1. Introduction
2. Result and Discussion
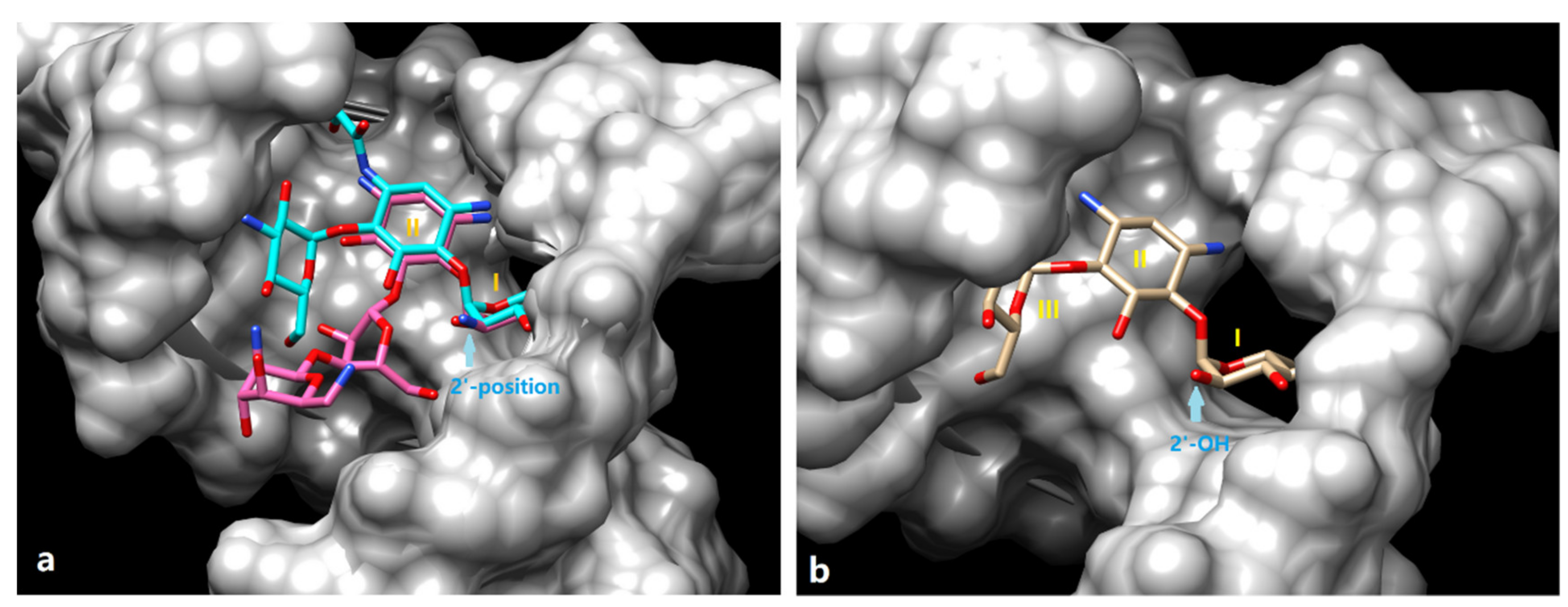
2.1. Design
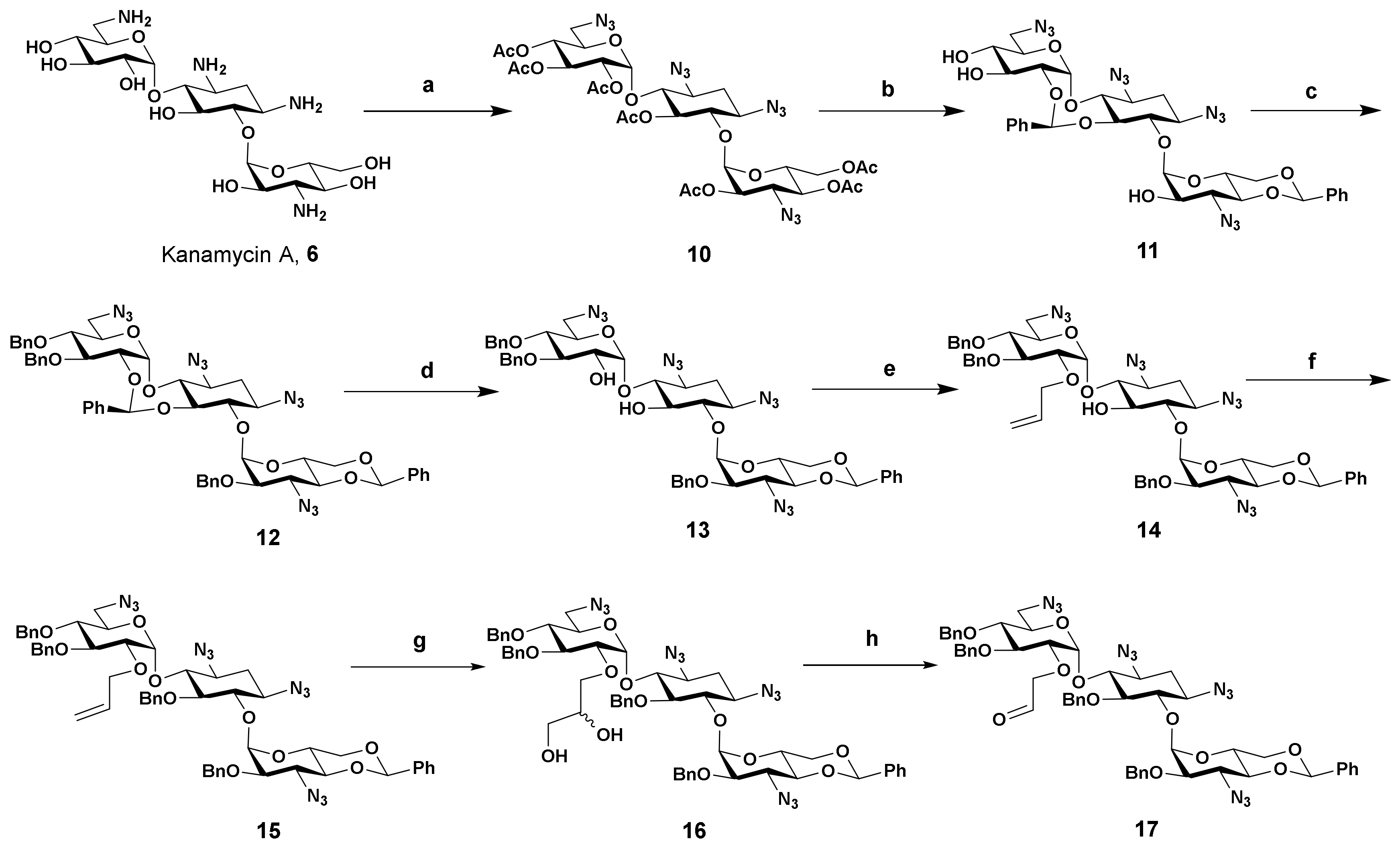
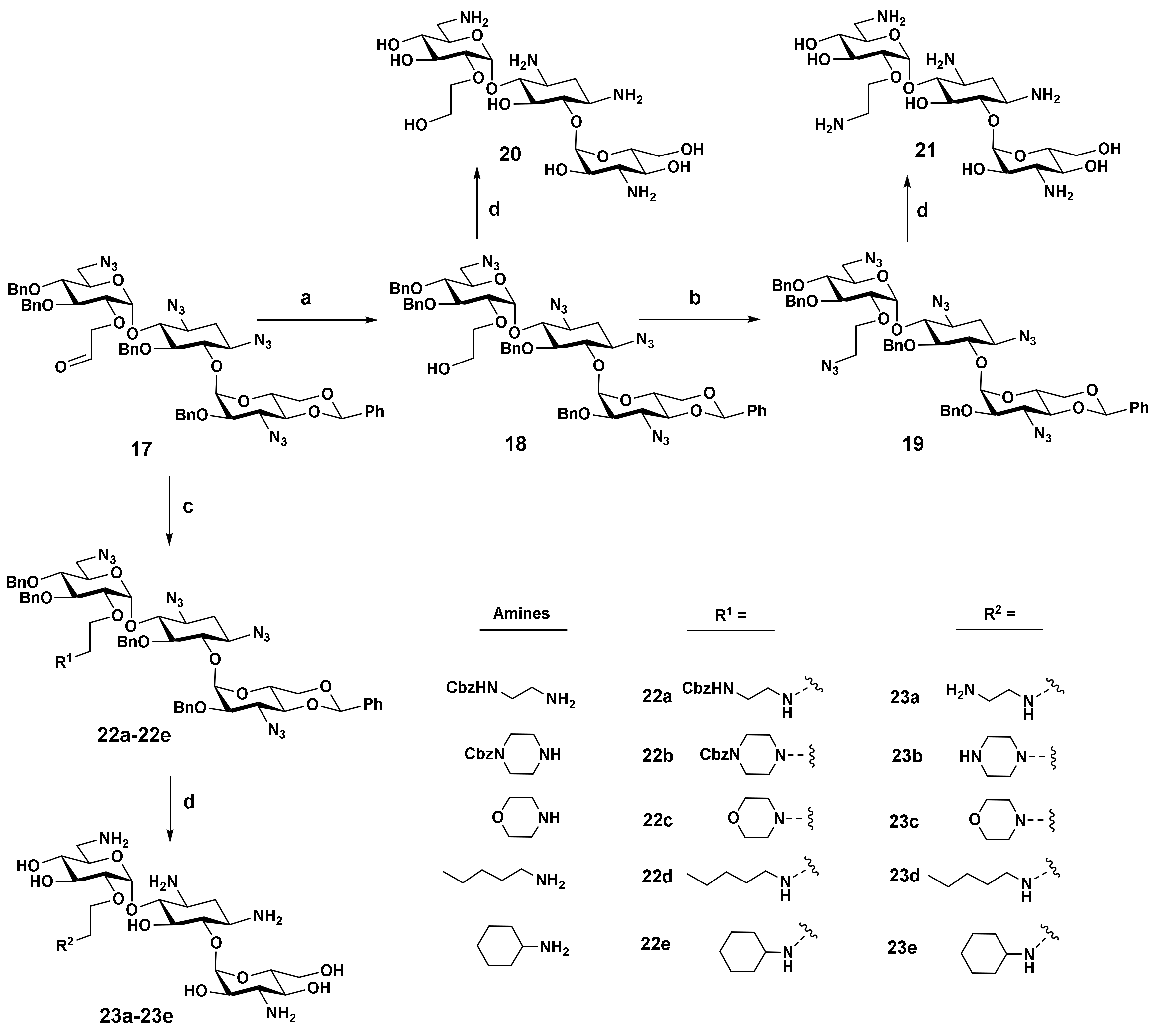
2.2. Synthesis
2.3. Bioactivity Assay
3. Materials and Methods
3.1. Chemistry
- 2′-O-Allyl-1,3,6′,3″-tetraazido-4″,6″-O-benzylidene-3′,4′,2″-tri-O-benzylkanamycin A (14): To a flask was added 13 (121 mg, 0.13 mmol), toluene (2 mL), freshly prepared silver (I) oxide (80 mg, 0.34 mmol), tetrabutylammonium iodide (10 mg, 0.03 mmol) and the allyl bromide (35mg, 0.29 mmol). The mixture was stirred overnight at room temperature and TLC showed the starting material disappeared. The reaction mixture was filtered and the filtrate was concentrated to residue. The gross product was purified by silica gel chromatography with the mixed solvent (petroleum/ethyl acetate from 15:1 to 6:1) as eluent to give the titled compound (112 mg, 0.12 mmol, 90% yield) as a colorless semisolid. 1H NMR (600 MHz, CDCl3) δ 7.50–7.49 (m, 2H), 7.43–7.42 (m, 2H), 7.39–7.26 (m, 16H), 5.91–5.85 (m, 1H), 5.52 (s, 1H), 5.25–5.22 (m, 2H), 5.18–5.16 (m, 1H), 5.09 (d, J = 3.6 Hz, 1H), 4.91–4.83 (m, 4H), 4.77 (d, J = 11.8 Hz, 1H), 4.70 (d, J = 1.7 Hz, 1H), 4.62 (d, J = 11.0 Hz, 1H), 4.48 (td, J1 = 5.0 Hz, J2 = 10.0 Hz, 1H), 4.34–4.31 (m, 1H), 4.22 (dd, J1 = 5.0 Hz, J2 = 10.0 Hz, 1H), 4.18–4.15 (m, 1H), 4.07–4.02 (m, 2H), 3.96 (t, J = 9.4 Hz, 1H), 3.65–3.54 (m, 5H), 3.50 (d, J1 = 3.6 Hz, J2 = 9.8 Hz, 1H), 3.45–3.63 (m, 4H), 3.30–3.21 (m, 2H), 2.43 (ddd, J1 = J2 = 4.5 Hz, J3 = 13.2 Hz, 1H), 1.60 (ddd, J1 = J2 = J3 = 12.6 Hz, 1H). 13C NMR (150 MHz, CDCl3) δ 138.17, 137.77, 137.18, 137.09, 133.14, 128.95, 128.54, 128.48, 128.22, 128.21, 128.13, 128.01, 127.90, 127.80, 127.78, 126.04, 119.43, 101.32, 101.20, 97.64, 86.51, 81.81, 80.20, 80.09, 79.79, 78.10, 77.54, 75.60, 75.31, 74.13, 74.08, 73.10, 71.09, 68.78, 62.78, 61.94, 60.38, 58.76, 51.19, 32.21. HRMS (ESI/APCI) calculated for (C49H54N12O11Na) [M + Na+]: 1009.3927, found: 1009.3962.
- 2′-O-Allyl-1,3,6′,3″-tetraazido-4″,6″-O-benzylidene-5,3′,4′,2″-tetra-O-benzylkanamycin A (15): To a stirred solution of 14 (226 mg, 0.23 mmol) in anhydrous DMF (3 mL) was added sodium hydride (26 mg, 60% in mineral oil, 0.65 mmol) and the resulting mixture was stirred for 30 min at room temperature, then benzyl bromide (59 mg, 0.35 mmol) was added in one portion. After stirring for 4 h, the reaction mixture was poured into water (50 mL) and the aqueous layer was extracted with ethyl acetate (3 × 15 mL). The combined organic phase was washed with brine, dried over anhydrous sodium sulfate, and concentrated under a vacuum. The residue was purified by silica gel column chromatography with the mixed solvent (petroleum ether/ethyl acetate 15:1 to 9:1) as eluent to give product 15 (217 mg, 0.20 mmol, 86% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.44–7.24 (m, 20H), 7.19–7.11 (m, 4H), 7.05 (t, J = 7.4 Hz, 1H), 5.55 (d, J = 3.7 Hz, 1H), 5.52 (d, J = 4.0 Hz, 1H), 5.51–5.43 (m, 1H), 5.28 (s, 1H), 5.10 (d, J = 12.4 Hz, 1H), 4.98–4.74 (m, 8H), 4.58 (d, J = 11.3 Hz, 1H), 4.31–4.27 (m, 1H), 4.02–3.92 (m, 3H), 3.86–3.80 (m, 2H), 3.75 (d, J = 9.4 Hz, 1H), 3.67–3.50 (m, 6H), 3.46–3.36 (m, 4H), 3.28 (dd, J1 = 3.8 Hz, J2 = 9.9 Hz, 1H), 3.22 (t, J = 9.8 Hz, 1H), 2.40 (ddd, J1 = J2 = 4.5 Hz, J3 = 13.2 Hz, 1H), 1.54 (ddd, J1 = J2 = J3 = 12.9 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 138.45, 138.04, 137.58, 137.27, 136.78, 134.25, 128.84, 128.59, 128.46, 128.44, 128.37, 128.19, 128.17, 127.92, 127.83, 127.74, 127.62, 127.10, 126.32, 125.19, 117.48, 101.39, 97.33, 96.32, 82.83, 81.81, 79.59, 78.70, 78.28, 78.11, 77.92, 77.15, 75.52, 74.96, 74.51, 73.44, 73.31, 70.76, 68.58, 62.89, 61.47, 60.17, 59.19, 51.37, 32.16. HRMS (ESI/APCI) calculated for (C56H60N12O11Na) [M + Na+]: 1099.4397, found: 1099.4412.
- 2′-O-(2,3-Dihydroxypropyl)-1,3,6′,3″-tetraazido-4″,6″-O-benzylidene-5,3′,4′,2″-tetra-O-benzylkanamycin A (16): To a flask was added 15 (213 mg, 0.20 mmol), potassium osmate (VI) dihydrate (4 mg, 0.01 mmol), N-methylmorpholine-N-oxide solution in water (94 mg, 0.40 mmol, 50% w/w), acetone (5 mL), and water (0.1 mL) in sequence. After stirring overnight at room temperature, TLC showed the reaction was completed. The reaction mixture was poured into aqueous solution of sodium thiosulfate pentahydrate (50 mL, 1% w/w) and stirred for 30 min, then the resulting mixture was extracted with ethyl acetate (3 × 10 mL). The organic layer was combined, dried over anhydrous sodium sulfate, and concentrated under a vacuum. The gross product was purified by silica gel chromatography with the mixed solvent (petroleum/ethyl acetate from 8:1 to 4:1) as the solvent to give the titled compound 16 (125 mg, 0.11 mmol, 57% yield) as a colorless semisolid. 1H NMR (400 MHz, CDCl3) δ 7.44–7.14 (m, 24H), 7.10 (t, J = 7.2 Hz, 1H), 5.54 (t, J = 4.0 Hz, 1H), 5.48 (t, J = 4.1 Hz, 1H), 5.34 (s, 1H), 5.03–4.94 (m, 2H), 4.86 (d, J = 11.2 Hz, 1H), 4.82–4.68 (m, 4H), 4.59 (d, J = 11.2 Hz, 1H), 4.21–4.08 (m, 2H), 3.94–3.87 (m, 2H), 3.79–3.42 (m, 9H), 3.41–3.15 (m, 8H), 3.08 (br, 1H), 2.43–2.35 (m, 1H), 1.68–1.58 (m, 1H). HRMS (ESI/APCI) calculated for (C56H62N12NaO13) [M + Na+]: 1133.4452, found: 1133.4487.
- 2′-O-(2-Oxoethyl)-1,3,6′,3″-tetraazido-4″,6″-O-benzylidene-5,3′,4′,2″-tetra-O-benzylkanamycin A (17): To a flask was added 16 (125 mg, 0.11 mmol), methanol (5 mL), and sodium periodate (47 mg, 0.22 mmol) and the resulting mixture was stirred at room temperature. After 4 h, TLC showed all starting material was consumed. The reaction mixture was poured into aqueous solution of sodium thiosulfate pentahydrate (50 mL, 0.4% w/w) and stirred for 30 min, then the resulting mixture was extracted with ethyl acetate (3 × 10 mL). The organic layer was combined, dried over anhydrous sodium sulfate, and concentrated under a vacuum. The resulting product 17 (122 mg, 0.11 mmol, 100%) was used in the following reactions directly without further purification. 1H NMR (400 MHz, CDCl3) δ 1H NMR (400 MHz, CDCl3) δ 9.09 (s, 1H), 7.44–7.16 (m, 22H), 7.10–7.04 (m, 3H), 5.56 (d, J = 3.8 Hz, 1H), 5.51 (d, J = 3.8 Hz, 1H), 5.28 (s, 1H), 5.07 (d, J = 12.5 Hz, 2H), 4.96 (d, J = 12.4 Hz, 1H), 4.86-4.76 (m, 4H), 4.68 (d, J = 11.2 Hz, 1H), 4.59 (d, J = 11.2 Hz, 1H), 4.29–4.25 (m, 1H), 4.03–3.96 (m, 3H), 3.86–3.79 (m, 2H), 3.74–3.61 (m, 3H), 3.59–3.49 (m, 3H), 3.46–3.38 (m, 5H), 3.25–3.19 (m, 2H), 2.41 (ddd, J1 = J2 = 4.7 Hz, J3 = 13.0 Hz, 1H), 1.68 (ddd, J1 = J2 = J3 = 12.6 Hz, 1H). HRMS (ESI/APCI) calculated for (C55H58N12NaO12) [M + Na+]: 1101.4189, found: 1101.4218.
- 2′-O-(2-Hydroxyethyl)-1,3,6′,3″-tetraazido-4″,6″-O-benzylidene-5,3′,4′,2″-tetra-O-benzylkanamycin A (18): The product 17 (110 mg, 0.10 mmol) was dissolved in the mixture of methanol (3 mL) and dichloromethane (2 mL), and to the solution was added sodium borohydride (15 mg, 0.40 mmol) in several portions while stirring at ice bath temperature. After 30 min, TLC showed the reaction was completed. The solvent was removed, and the gross product was purified with silica gel chromatography by using the mixed solvent (petroleum ether/ethyl acetate 20:1 to 10:1) as eluent to give product 18 (101 mg, 0.09 mmol, 92% yield) as a white semisolid. 1H NMR (400 MHz, CDCl3) δ 7.43–7.24 (m, 20H), 7.20–7.15 (m, 4H), 7.08 (t, J = 7.2 Hz, 1H), 5.53 (d, J = 4.2 Hz, 1H), 5.52 (d, J = 4.3 Hz, 1H), 5.31 (s, 1H), 5.05 (d, J = 12.3 Hz, 1H), 4.97 (d, J = 12.2 Hz, 1H), 4.88 (d, J = 11.2 Hz, 1H), 4.82–4.75 (m, 4H), 4.59 (d, J = 11.3 Hz, 1H), 4.25–4.21 (m, 1H), 4.06 (dd, J1 = 5.0 Hz, J2 = 10.2 Hz, 1H), 3.96-3.85 (m, 2H), 3.66–3.29 (m, 14 H), 3.24 (t, J = 9.8 Hz, 1H), 2.65 (t, J = 6.1 Hz, 1H), 2.39 (dt, J1 = 4.3 Hz, J2 = 13.2, Hz, 1H), 1.63 (ddd, J1 = J2 = J3 = 12.7 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 137.97, 137.83, 137.26, 137.23, 136.81, 128.87, 128.63, 128.58, 128.50, 128.47, 128.18, 128.16, 127.97, 127.93, 127.83, 127.76, 127.47, 126.27, 125.72, 101.41, 97.21, 96.43, 82.29, 81.35, 80.54, 79.60, 78.55, 78.43, 78.03, 77.76, 75.80, 75.03, 74.95, 73.45, 73.29, 71.01, 68.60, 62.91, 62.31, 61.40, 60.18, 59.92, 51.20, 32.35. HRMS (ESI/APCI) calculated for (C55H60N12NaO12) [M + Na+]: 1103.4346, found: 1103.4369.
- 2′-O-(2-Azidoethyl)-1,3,6′,3″-tetraazido-4″,6″-O-benzylidene-5,3′,4′,2″-tetra-O-benzylkanamycin A (19): To the solution of 18 (65 mg, 0.06 mmol) in pyridine (3 mL) was added p-toluene sulfonyl chloride (46 mg, 0.18 mmol) in one portion, and the resulting mixture was stirred overnight. Then, the solvent was removed, and the residue was purified by column chromatography on silica gel using petroleum ether/ethyl acetate (10:1 to 5:1) as eluent to afford the tosylate intermediate, which was mixed with sodium azide (12 mg, 0.18 mmol) and DMF (3 mL). After stirring at 80 °C for 4 h, the reaction mixture was poured into 50 mL of water and the aqueous layer was extracted with ethyl acetate (3 × 10 mL). The combined organic phase was dried over Na2SO4 and concentrated under a vacuum. The crude was purified by column chromatography on silica gel by using the mixture of petroleum ether and ethyl acetate (10:1 to 5:1) as eluent to afford 19 (56 mg, 0.05 mmol, 84% yield). 1H NMR (600 MHz, CDCl3) δ 7.43–7.38 (m, 4H), 7.35–7.24 (m, 16H), 7.18 (t, J = 7.7 Hz, 2H), 7.12–7.06 (m, 3H), 5.56–5.55 (m, 2H), 5.29 (s, 1H), 5.09 (d, J = 12.3 Hz, 1H), 4.99 (d, J = 12.3 Hz, 1H), 4.87–4.76 (m, 5H), 4.58 (d, J = 11.3 Hz, 1H), 4.29–4.26 (m, 1H), 4.01 (dd, J1 = 4.9 Hz, J2 = 10.1 Hz, 1H), 3.95 (t, J = 9.4 Hz, 1H), 3.87–3.82 (m, 2H), 3.75 (t, J = 9.4 Hz, 1H), 3.66 (t, J = 9.4 Hz, 1H), 3.61–3.51 (m, 5H), 3.46–3.36 (m, 4H), 3.25–3.22 (m, 2H), 3.15–3.11 (m, 1H), 3.04–2.96 (m, 2H), 2.40 (ddd, J1 = J2 = 4.6 Hz, J3 = 13.3, Hz, 1H), 1.64 (ddd, J1 = J2 = J3 = 12.7 Hz, 1H). 13C NMR (150 MHz, CDCl3) δ 138.39, 137.90, 137.54, 137.21, 136.74, 128.85, 128.58, 128.45, 128.41, 128.19, 127.92, 127.87, 127.82, 127.63, 127.60, 127.22, 126.29, 125.14, 101.39, 96.89, 96.32, 82.78, 81.58, 80.20, 79.55, 78.35, 78.01, 77.76, 75.41, 74.94, 74.46, 73.30, 70.76, 70.75, 68.57, 62.87, 61.46, 60.15, 59.25, 51.25, 50.81, 32.14. HRMS (ESI/APCI) calculated for (C55H59N15NaO11) [M + Na+]: 1128.4411, found: 1128.4442.
- 2′-O-[[2-[2-(benzyloxycarbonylamino)ethyl]amino]-ethyl]-1,3,6′,3″-tetraazido-4″,6″-O-benzylidene-5,3′,4′,2″-tetra-O-benzylkanamycin A (22a): To a flask was added 17 (53 mg, 0.05 mmol), 1,2-dichloroethane (3 mL), and 1-(benzyloxycarbonylamino)-2-aminoethane (48 mg, 0.25 mmol) in one portion. The resulting mixture was stirred for 30 min, then sodium triacetoxyborohyride (53 mg, 0.25 mmol) was added in portions. After stirring overnight at room temperature, TLC showed a minor amount of 17 remained. Then, another portion of sodium triacetoxyborohyride (32 mg, 0.15 mmol) was added. After stirring for another 8 h, the reaction was complete. The solvent was removed, and the residue was purified by silica gel chromatography (petroleum/ethyl acetate from 8:1 to 4:1) to give the titled compound 22a (38 mg, 0.03 mmol, 67% yield) as a colorless semisolid. 1H NMR (400 MHz, CDCl3) δ 7.45–7.09 (m, 29H), 7.06 (t, J = 7.2 Hz, 1H), 5.57 (d, J = 3.6 Hz, 1H), 5.53 (d, J = 3.6 Hz, 1H), 5.33 (s, 1H), 5.06 (s, 2H), 5.01–4.93 (m, 2H), 4.88–4.74 (m, 5H), 4.58 (d, J = 11.2 Hz, 1H), 4.26–4.20 (m, 1H), 4.02 (dd, J1 = 10.1, J2 = 4.8 Hz, 1H), 3.96–3.80 (m, 3H), 3.75 (t, J = 9.4 Hz, 1H), 3.68 (t, J = 9.4 Hz, 1H), 3.62–3.21 (m, 13H), 3.01 (br, 2H), 2.53–2.28 (m, 5H), 1.65 (ddd, J1 = J2 = J3 = 12.6 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 156.46, 138.30, 137.83, 137.28, 136.78, 136.64, 128.88, 128.58, 128.49, 128.18, 128.15, 128.10, 128.06, 127.98, 127.93, 127.89, 127.76, 127.53, 127.41, 126.23, 125.96, 101.35, 96.77, 96.24, 82.64, 81.23, 80.15, 79.66, 78.46, 78.15, 78.05, 77.70, 75.38, 74.96, 74.90, 73.30, 70.93, 68.61, 66.60, 62.94, 61.47, 60.09, 59.71, 51.29, 48.60, 48.34, 39.80, 32.19. HRMS (ESI/APCI) calculated for (C65H73N14O13) [M + H+]: 1257.5476, found: 1257.5442.
- 2′-O-[2-(4-benzyloxycarbonylpiperizyl)-ethyl]-1,3,6′,3″-tetraazido-4″,6″-O-benzylidene-5,3′,4′,2″-tetra-O-benzylkanamycin A (22b): This compound was synthesized through reductive amination by coupling 17 with 1-benzyloxycarbonylpiperize with the same procedure as described in the preparation of 22a. 55% yield, colorless semisolid. 1H NMR (400 MHz, CDCl3) δ 7.43–7.22 (m, 25H), 7.16–7.10 (m, 4H), 7.00 (t, J = 7.5 Hz, 1H), 5.56 (d, J = 3.6 Hz, 1H), 5.54 (d, J = 3.7 Hz, 1H), 5.28 (s, 1H), 5.10–5.06 (m, 3H), 4.93 (d, J = 12.1 Hz, 1H), 4.86–4.75 (m, 5H), 4.58 (d, J = 11.2 Hz, 1H), 4.31–4.27 (m, 1H), 3.97 (dd, J1 = 5.0 Hz, J2 = 10.3 Hz, 1H), 3.91 (t, J = 9.4 Hz, 1H), 3.84–3.72 (m, 3H), 3.67–3.36 (m, 10H), 3.30–3.20 (m, 7H), 2.40 (ddd, J1 = J2 = 4.4 Hz, J3 = 13.1Hz, 1H), 2.29–2.22 (m, 1H), 2.12–1.94 (m, 5H), 1.64 (ddd, J1 = J2 = J3 = 12.7 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 155.09, 138.45, 137.84, 137.46, 137.21, 136.73, 128.85, 128.59, 128.52, 128.47, 128.39, 128.18, 128.09, 127.92, 127.56, 127.29, 127.16, 126.25, 125.24, 101.25, 97.18, 96.28, 82.67, 81.52, 80.11, 79.55, 78.3, 77.94, 77.71, 75.16, 74.95, 73.3, 70.8, 68.52, 67.12, 62.83, 61.43, 60.12, 59.22, 57.30, 52.88, 51.25, 43.46, 32.16, 29.70. HRMS (ESI/APCI) calculated for (C67H75N14O13) [M + H+]: 1283.5633, found: 1283.5602.
- 2′-O-[2-(4-Morpholinyl)-ethyl]-1,3,6′,3″-tetraazido-4″,6″-O-benzylidene-5,3′,4′,2″-tetra-O-benzylkanamycin A (22c): This compound was synthesized through reductive amination by coupling 17 with morpholine with the same procedure as described in the preparation of 22a. 68% yield, colorless semisolid. 1H NMR (400 MHz, CDCl3) δ 7.44–7.21 (m, 20H), 7.17–7.10 (m, 4H), 7.01 (t, J = 7.4 Hz, 1H), 5.57 (d, J = 3.7 Hz, 1H), 5.54 (d, J = 3.7 Hz, 1H), 5.28 (s, 1H), 5.08 (d, J = 12.2 Hz, 1H), 4.93 (d, J = 12.2 Hz, 1H), 4.89–4.75 (m, 5H), 4.58 (d, J = 11.3 Hz, 1H), 4.32–4.27 (m, 1H), 3.99–3.90 (m, 2H), 3.85–3.71 (m, 3H), 3.67–3.36 (m, 14H), 3.31–3.20 (m, 3H), 2.40 (ddd, J1 = J2 = 4.5 Hz, J3 = 13.1, Hz, 1H), 2.30–2.23 (m, 1H), 2.12–2.02 (m, 3H), 1.64 (ddd, J1 = J2 = J3 = 13.0 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ138.51, 137.84, 137.46, 137.24, 136.76, 128.83, 128.61, 128.58, 128.45, 128.36, 128.18, 128.15, 127.91, 127.89, 127.50, 127.29, 127.13, 126.24, 125.37, 101.26, 97.08, 96.28, 82.61, 81.40, 80.12, 79.59, 78.34, 77.97, 77.70, 77.23, 77.02, 76.81, 75.09, 74.93, 74.49, 73.3, 70.85, 69.59, 68.54, 66.65, 62.85, 61.46, 60.12, 59.3, 57.7, 53.62, 51.28, 32.17, 29.69. HRMS (ESI/APCI) calculated for (C59H68N13O12) [M + H+]: 1150.5105, found: 1150.5063.
- 2′-O-(2-n-Pentylamino-ethyl)-1,3,6′,3″-tetraazido-4″,6″-O-benzylidene-5,3′,4′,2″-tetra-O-benzylkanamycin A (22d): This compound was synthesized through reductive amination by coupling 17 with n-pentylamine with the same procedure as described in the preparation of 22a. 64% yield, colorless semisolid. 1H NMR (400 MHz, CDCl3) δ 7.45–7.21 (m, 20H), 7.18–7.14 (m, 4H), 7.06 (t, J = 7.3 Hz, 1H), 5.56–5.54 (m, 2H), 5.30 (s, 1H), 5.05 (d, J = 12.1 Hz, 1H), 4.95 (d, J = 12.1 Hz, 1H), 4.86–4.75 (m, 5H), 4.57 (d, J = 11.2 Hz, 1H), 4.29–4.23 (m, 1H), 4.01 (dd, J1 = 4.8, J2 = 10.1 Hz, 1H), 3.92 (t, J = 9.3 Hz, 1H), 3.89–3.81 (m, 2H), 3.77 (t, J = 9.3 Hz, 1H), 3.70–3.20 (m, 13H), 2.54–2.51 (m, 1H), 2.46–2.22 (m, 4H), 1.65 (ddd, J1 = J2 = J3 = 12.6 Hz, 1H), 1.33–1.06 (m, 6H), 0.84 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 138.34, 137.83, 137.35, 137.17, 136.71, 128.84, 128.55, 128.43, 128.38, 128.14, 127.92, 127.85, 127.60, 127.43, 127.32, 126.24, 125.49, 101.33, 96.80, 96.28, 82.58, 81.18, 80.19, 79.54, 78.30, 78.00, 77.83, 77.53, 75.33, 74.90, 74.64, 73.27, 71.25, 70.83, 68.54, 62.87, 61.39, 60.11, 59.46, 51.28, 49.61, 49.14, 32.21, 29.37, 29.16, 22.49, 14.04. HRMS (ESI/APCI) calculated for (C60H72N13O11) [M + H+]: 1150.5469, found: 1150.5430.
- 2′-O-(2-Cyclohexylamino-ethyl)-1,3,6′,3″-tetraazido-4″,6″-O-benzylidene-5,3′,4′,2″-tetra-O-benzylkanamycin A (22e): This compound was synthesized through reductive amination by coupling 17 with cyclohexylamine with the same procedure as described in the preparation of 22a. 52% yield, colorless semisolid. 1H NMR (400 MHz, CDCl3) δ 7.45–7.22 (m, 20H), 7.19–7.15 (m, 4H), 7.08 (t, J = 7.3 Hz, 1H), 5.55–5.53 (m, 2H), 5.32 (s, 1H), 5.02 (d, J = 12.1 Hz, 1H), 4.94 (d, J = 12.0 Hz, 1H), 4.86–4.73 (m, 5H), 4.56 (d, J = 11.3 Hz, 1H), 4.26–4.20 (m, 1H), 4.01 (dd, J1 = 4.8 Hz, J2 = 10.1Hz, 1H), 3.94–3.83 (m, 3H), 3.78 (t, J = 9.3 Hz, 1H), 3.69–3.31 (m, 12H), 3.25 (t, J = 9.8 Hz, 1H), 2.64–2.50 (m, 2H), 2.38 (ddd, J1 = J2 = 4.4 Hz, J3 = 13.0 Hz, 1H), 2.19–2.12 (m, 1H), 1.71–1.51 (m, 4H), 1.12–0.80 (m, 6H). 13C NMR (100 MHz, CDCl3) δ 138.36, 137.83, 137.44, 137.26, 136.80, 128.95, 128.66, 128.52, 128.24, 128.02, 127.97, 127.74, 127.49, 126.32, 125.74, 101.44, 96.74, 96.36, 82.55, 81.03, 80.11, 79.63, 78.43, 78.12, 77.94, 77.83, 75.26, 74.89, 74.68, 73.37, 71.05, 68.63, 62.97, 61.47, 60.16, 59.60, 56.70, 51.37, 45.57, 32.26, 31.88, 25.64, 24.86. HRMS (ESI/APCI) calculated for (C61H72N13O11) [M + H+]: 1162.5469, found: 1162.5486.
- 2′-O-(2-Hydroxyethyl)-kanamycin A (20): 78% yield, white amorphous powder. 1H NMR (600 MHz, D2O) δ 5.70 (d, J = 3.8 Hz, 1H), 5.10 (d, J = 3.6 Hz, 1H), 3.99–3.95 (m, 1H), 3.93–3.80 (m, 8H), 3.77–3.70 (m, 4H), 3.67 (t, J = 10.1 Hz, 1H), 3.58–3.45 (m, 4H), 3.43–3.36 (m, 2H), 3.15 (dd, J1 = 8.1 Hz, J2 = 13.4 Hz, 1H), 2.50 (ddd, J1 = J2 = 4.2 Hz, J3 = 12.5 Hz, 1H), 1.94 (s, 12H), 1.90 (ddd, J1 = J2 = J3 = 12.5 Hz, 1H). 13C NMR (150 MHz, D2O) δ 180.83, 101.36, 95.18, 84.83, 79.81, 78.73, 73.94, 73.51, 73.34, 72.56, 71.64, 69.27, 68.88, 66.16, 61.30, 60.67, 55.65, 50.51, 48.36, 41.04, 28.46, 23.19. HRMS (ESI/APCI) calculated for (C20H41N4O12) [M + H]+ requires m/z 529.2715, found m/z 529.2734.
- 2′-O-(2-Aminoethyl)-kanamycin A (21): 58% yield, white amorphous powder. 1H NMR (600 MHz, D2O) δ 5.83 (d, J = 3.8 Hz, 1H), 5.11 (d, J = 3.7 Hz, 1H), 4.09–4.06 (m, 1H), 3.95–3.87 (m, 7H), 3.83 (dd, J1 = 2.2 Hz, J2 = 12.3 Hz, 1H), 3.77–3.73 (m, 2H), 3.67 (t, J = 10.1 Hz, 1H), 3.57–3.40 (m, 6H), 3.30–3.26 (m, 1H), 3.21–3.18 (m, 2H), 2.49 (ddd, J1 = J2 = 4.1 Hz, J3 = 12.6 Hz, 1H), 1.93 (s, 15H), 1.89 (ddd, J1 = J2 = J3 = 12.7 Hz, 1H). 13C NMR (150 MHz, D2O) δ 181.37, 101.31, 95.57, 84.95, 79.72, 78.29, 74.25, 73.65, 71.76, 71.69, 69.30, 68.84, 67.98, 66.27, 60.78, 55.67, 50.44, 48.92, 40.98, 39.94, 28.89, 23.53. HRMS (ESI/APCI) calculated for (C20H42N5O11) [M + H]+ requires m/z 528.2875, found m/z 528.2892.
- 2′-O-[2-(4-Mopholinyl)-ethyl]-kanamycin A (23c): 62% yield, white amorphous powder. 1H NMR (600 MHz, D2O) δ 5.89 (d, J = 3.7 Hz, 1H), 5.11 (d, J = 3.6 Hz, 1H), 4.17 (ddd, J1 = J2 = 4.4 Hz, J3 = 12.2 Hz, 1H), 4.02–3.73 (m, 14H), 3.67 (t, J = 10.1 Hz, 1H), 3.58–3.39 (m, 12H), 3.19 (dd, J1 = 7.7 Hz, J2 = 13.4 Hz, 1H), 2.50 (ddd, J1 = J2 = 4.2 Hz, J3 = 12.5 Hz, 1H), 1.97–1.91 (m, 16H). 13C NMR (150 MHz, D2O) δ 180.26, 100.56, 94.57, 84.09, 78.86, 76.79, 73.67, 72.88, 71.06, 70.71, 68.53, 68.06, 65.43, 63.94, 63.70, 59.93, 56.19, 54.88, 51.76, 49.59, 48.24, 40.22, 27.90, 22.56. HRMS (ESI/APCI) calculated for (C24H48N5O12) [M + H]+ requires m/z 598.3294, found m/z 598.3315.
- 2′-O-[2-[(2-Aminoethyl)amino]-ethyl]-kanamycin A (23a): 55% yield, white amorphous powder. 1H NMR (600 MHz, D2O) δ 5.84 (d, J = 3.7 Hz, 1H), 5.13 (d, J = 3.6 Hz, 1H), 4.09–4.05(m, 1H), 3.99–3.92 (m, 3H), 3.91–3.66 (m, 8H), 3.55–3.15 (m, 13H), 2.40 (ddd, J1 = J2 = 4.2 Hz, J3 = 12.7 Hz, 1H), 1.92 (s, 17H), 1.80 (ddd, J1 = J2 = J3 = 12.6 Hz, 1H). 13C NMR (150 MHz, D2O) δ 182.09, 101.16, 95.41, 85.46, 79.81, 79.43, 74.52, 73.57, 71.86, 71.78, 69.16, 68.97, 67.93, 66.37, 60.74, 55.70, 50.68, 49.06, 48.30, 45.48, 41.09, 37.26, 30.25, 23.95. HRMS (ESI/APCI) calculated for (C22H47N6O11) [M + H]+ requires m/z 571.3297, found m/z 571.3318
- 2′-O-[2-[(2-Piperizinylethyl)amino]-ethyl]-kanamycin A (23b): 60% yield, white amorphous powder. 1H NMR (600 MHz, D2O) δ 5.78 (d, J = 3.7 Hz, 1H), 5.10 (d, J = 3.6 Hz, 1H), 3.97-3.91 (m, 4H), 3.86-3.81 (m, 4H), 3.78–3.74 (m, 2H), 3.70–3.64 (m, 2H), 3.50–3.33 (m, 6H), 3.27 (t, J = 4.9 Hz, 4H), 3.17 (dd, J1 = 8.0 Hz, J2 = 13.4 Hz, 1H), 2.87–2.80 (m, 4H), 2.76–2.72 (m, 2H), 2.39 (ddd, J1 = J2 = 4.1 Hz, J3 = 12.7 Hz, 1H), 1.90 (s, 15H), 1.77 (ddd, J1 = J2 = J3 = 12.5 Hz, 1H). 13C NMR (150 MHz, D2O) δ 181.98, 101.17, 95.39, 85.46, 79.95, 79.54, 74.40, 73.54, 72.00, 71.76, 69.10, 68.93, 67.92, 66.28, 60.67, 57.12, 55.71, 50.68, 49.89, 48.94, 43.57, 41.06, 30.12, 23.87. HRMS (ESI/APCI) calculated for (C24H49N6O11) [M + H]+ requires m/z 597.3454, found m/z 597.3473.
- 2′-O-[2-(n-Pentylamino)-ethyl]-kanamycin A (23d): 68% yield, white amorphous powder. 1H NMR (600 MHz, D2O) δ 5.88 (d, J = 3.5 Hz, 1H), 5.14 (d, J = 3.4 Hz, 1H), 4.14–4.11 (m, 1H), 4.00–3.84 (m, 8H), 3.78–3.75 (m, 2H), 3.69 (t, J = 10.1 Hz, 1H), 3.57–3.41 (m, 6H), 3.36–3.33 (m, 1H), 3.29–3.25 (m, 1H), 3.22–3.18 (m, 1H), 3.09 (t, J = 7.7 Hz, 2H), 2.46 (ddd, J1 = J2 = 4.5 Hz, J3 = 12.5 Hz, 1H), 1.93 (s, 17H), 1.90 (ddd, J1 = J2 = J3 = 12.5 Hz, 1H), 1.73–1.68 (m, 2H), 1.39–1.32 (m, 4H), 0.90 (t, J = 6.9 Hz, 3H). 13C NMR (150 MHz, D2O) δ 181.71, 101.27, 95.52, 85.16, 79.68, 78.52, 74.64, 73.65, 71.87, 71.62, 69.24, 68.90, 66.66, 66.30, 60.77, 55.68, 50.57, 49.12, 48.34, 47.56, 41.09, 29.51, 28.57, 25.87, 23.71, 22.21, 13.77. HRMS (ESI/APCI) calculated for (C25H52N5O11) [M + H]+ requires m/z 598.3658, found m/z 598.3678.
- 2′-O-[2-(Cyclohexylamino)-ethyl]-kanamycin A (23e): 64% yield, white amorphous powder. 1H NMR (600 MHz, D2O) δ 5.84 (d, J = 3.7 Hz, 1H), 5.11 (d, J = 3.7 Hz, 1H), 4.12–4.09 (m, 1H), 3.96–3.80 (m, 8H), 3.77–3.65 (m, 3H), 3.55–3.34 (m, 7H), 3.29–3.25 (m, 1H), 3.21–3.12 (m, 2H), 2.42 (ddd, J1 = J2 = 4.5 Hz, J3 = 12.4 Hz, 1H), 2.08 (br, 2H), 1.92 (s, 12H), 1.85–1.79 (m, 3H), 1.69–1.64 (m, 1H), 1.40–1.27 (m, 4H), 1.21–1.14 (m, 1H). 13C NMR (150 MHz, D2O) δ 181.90, 101.25, 95.59, 85.29, 79.65, 79.08, 74.59, 73.63, 71.79, 71.63, 69.21, 68.89, 66.87, 66.28, 60.74, 58.02, 55.69, 50.59, 49.09, 44.59, 41.01, 29.81, 29.70, 29.50, 25.19, 24.68, 24.65, 23.88. HRMS (ESI/APCI) calculated for (C26H52N5O11) [M + H]+ requires m/z 610.3658, found m/z 610.3680.
3.2. Bioassay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Magnet, S.; Blanchard, J.S. Molecular Insights into Aminoglycoside Action and Resistance. Chem. Rev. 2005, 105, 477–497. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.S.; Tolmasky, M.E. Aminoglycoside Modifying Enzymes. Drug Resist. Updat. 2010, 13, 151–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garneau-Tsodikova, S.; Labby, K.J. Mechanisms of Resistance to Aminoglycoside Antibiotics: Overview and Perspectives. MedChemComm 2016, 7, 11–27. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.S.; Zhang, L.H. Aminoglycoside Mimetics as Small-Molecule Drugs Targeting RNA. Curr. Med. Chem. 2002, 9, 929–939. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, G.; Zhang, L.-H.; Ye, X.-S. Modifications of Aminoglycoside Antibiotics Targeting RNA. Med. Res. Rev. 2007, 27, 279–316. [Google Scholar] [CrossRef]
- Houghton, J.L.; Green, K.D.; Chen, W.; Garneau-Tsodikova, S. The Future of Aminoglycosides: The End or Renaissance? ChemBioChem 2010, 11, 880–902. [Google Scholar] [CrossRef] [Green Version]
- Obszynski, J.; Loidon, H.; Blanc, A.; Weibel, J.-M.; Pale, P. Targeted modifications of neomycin and paromomycin: Towards resistance-free antibiotics? Bioorg. Chem. 2022, 126, 105824. [Google Scholar] [CrossRef]
- Chandrika, N.T.; Garneau-Tsodikova, S. Comprehensive review of chemical strategies for the preparation of new aminoglycosides and their biological activities. Chem. Soc. Rev. 2018, 47, 1189–1249. [Google Scholar] [CrossRef]
- Moazed, D.; Noller, H.F. Interaction of Antibiotics with Functional Sites in 16S Ribosomal RNA. Nature 1987, 327, 389–394. [Google Scholar] [CrossRef]
- Woodcock, J.; Moazed, D.; Cannon, M.; Davies, J.; Noller, H.F. Interaction of Antibiotics with A- and P-site-specific Bases in 16S Ribosomal RNA. EMBO J. 1991, 10, 3099–3103. [Google Scholar] [CrossRef]
- Fourmy, D.; Recht, M.I.; Blanchard, S.C.; Puglisi, J.D. Structure of The A-site of Escherichia Coli 16S Ribosomal RNA Complexed with An Aminoglycoside Antibiotic. Science 1996, 274, 1367–1371. [Google Scholar] [CrossRef] [PubMed]
- Carter, A.P.; Clemons, W.M.; Brodersen, D.E.; Morgan-Warren, R.J.; Wimberly, B.T.; Ramakrishnan, V. Functional Insights from The Structure of The 30S Ribosomal Subunit and Its Interactions with Antibiotics. Nature 2000, 407, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Bastida, A.; Hidalgo, A.; Chiara, J.L.; Torrado, M.; Corzana, F.; Pérez-Cañadillas, J.M.; Groves, P.; Garcia-Junceda, E.; Gonzalez, C.; Jimenez-Barbero, J.; et al. Exploring the Use of Conformationally Locked Aminoglycosides as a New Strategy to Overcome Bacterial Resistance. J. Am. Chem. Soc. 2006, 128, 100–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanessian, S.; Szychowski, J.; Adhikari, S.S.; Vasquez, G.; Kandasamy, P.; Swayze, E.E.; Migawa, M.T.; Ranken, R.; François, B.; Wirmer-Bartoschek, J.; et al. Structure-Based Design, Synthesis, and A-Site rRNA Cocrystal Complexes of Functionally Novel Aminoglycoside Antibiotics: C2″ Ether Analogues of Paromomycin. J. Med. Chem. 2007, 50, 2352–2369. [Google Scholar] [CrossRef]
- Kondo, J.; Pachamuthu, K.; François, B.; Szychowski, J.; Hanessian, S.; Westhof, E. Crystal Structure of the Bacterial Ribosomal Decoding Site Complexed with a Synthetic Doubly Functionalized Paromomycin Derivative: A New Specific Binding Mode to an A-Minor Motif Enhances in vitro Antibacterial Activity. ChemMedChem 2007, 2, 1631–1638. [Google Scholar] [CrossRef]
- Yan, R.-B.; Yuan, M.; Wu, Y.; You, X.; Ye, X.-S. Rational design and synthesis of potent aminoglycoside antibiotics against resistant bacterial strains. Bioorg. Med. Chem. 2011, 19, 30–40. [Google Scholar] [CrossRef]
- Kanazawa, H.; Saavedra, O.M.; Maianti, J.P.; Young, S.A.; Izquierdo, L.; Smith, T.K.; Hanessian, S.; Kondo, J. Structure-Based Design of a Eukaryote-Selective Antiprotozoal Fluorinated Aminoglycoside. ChemMedChem 2018, 13, 1541–1548. [Google Scholar] [CrossRef] [Green Version]
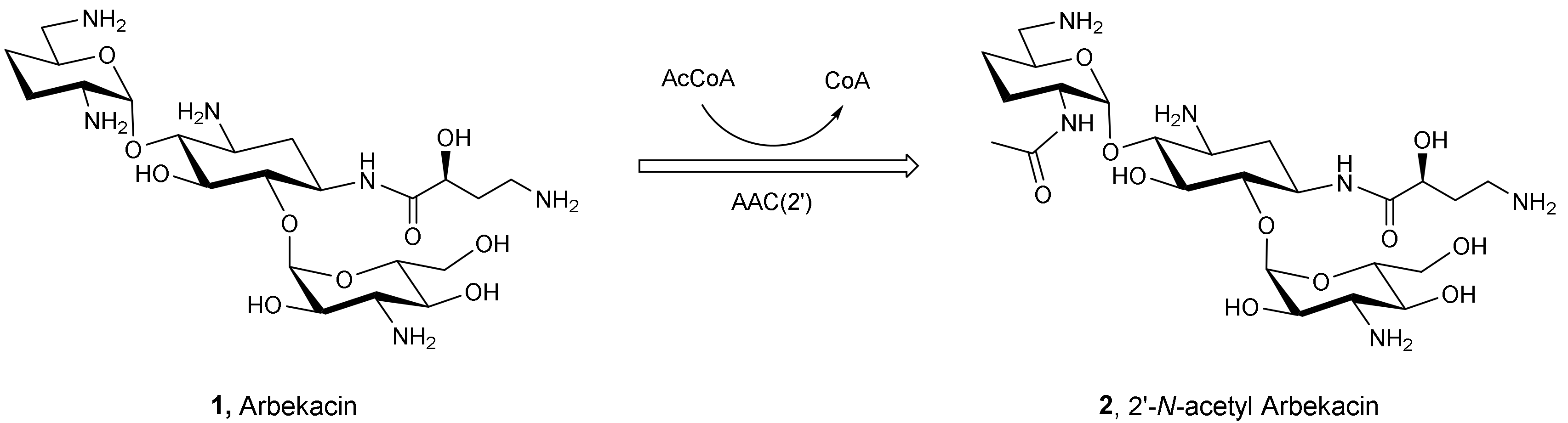
- Hotta, K.; Zhu, C.-B.; Ogata, T.; Sunada, A.; Ishikawa, J.; Mizuno, S.; Ikeda, Y.; Kondo, S. Enzymatic 2’-N-acetylation of arbekacin and antibiotic activity of its product. J. Antibiot. 1996, 49, 458–464. [Google Scholar] [CrossRef] [Green Version]
- Vicens, Q.; Westhof, E. Molecular recognition of aminoglycoside antibiotics by ribosomal RNA and resistance enzymes: An analysis of x-ray crystal structures. Biopolymers 2003, 70, 42–57. [Google Scholar] [CrossRef]
- François, B.; Russell, R.J.M.; Murray, J.B.; Aboul-Ela, F.; Masquida, B.; Vicens, Q.; Westhof, E. Crystal structures of complexes between aminoglycosides and decoding A site oligonucleotides: Role of the number of rings and positive charges in the specific binding leading to miscoding. Nucleic Acids Res. 2005, 33, 5677–5690. [Google Scholar] [CrossRef]
- Sati, G.C.; Sarpe, V.A.; Furukawa, T.; Mondal, S.; Mantovani, M.; Hobbie, S.N.; Vasella, A.; Böttger, E.C.; Crich, D. Modification at the 2′-Position of the 4,5-Series of 2-Deoxystreptamine Aminoglycoside Antibiotics to Resist Aminoglycoside Modifying Enzymes and Increase Ribosomal Target Selectivity. ACS Infect. Dis. 2019, 5, 1718–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.X.; Chen, Y.; Liang, Q.Z.; Li, H.; Jin, H.; Zhang, L.; Meng, X.; Li, Z. Design, Synthesis, and Antibacterial Activities of Conformationally Constrained Kanamycin A Derivatives. J. Org. Chem. 2013, 78, 400–409. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MIC (µg/mL) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compd | E. coli | S. aureus | K. pneumoniae | E. faecalis | P. aeruginosa | |||||
| ATCC 35218 | ATCC 25922 | ATCC 29213 | ATCC 25923 | ATCC 33591 | ATCC 700603 | ATCC 13883 | ATCC 29212 | ATCC 27853 | PAO1 | |
| Kan | 2 | 4 | 2 | 4 | 64 | 16 | 1 | 64 | >128 | 8 |
| 20 | 4 | 32 | 1 | 4 | 64 | 8 | 2 | 64 | >128 | 16 |
| 21 | 2 | 16 | 0.5 | 0.5 | 8 | 2 | 1 | 64 | 128 | 2 |
| 23a | 4 | >128 | 0.5 | 4 | 8 | 4 | 2 | 64 | 128 | 16 |
| 23b | 4 | 64 | 1 | 4 | 128 | 8 | 4 | 128 | >128 | >128 |
| 23c | 8 | 32 | 0.5 | 4 | 128 | 8 | 4 | 64 | >128 | 128 |
| 23d | 8 | 64 | 1 | 8 | 32 | 16 | 4 | 64 | >128 | 128 |
| 23e | 2 | 16 | 1 | 4 | 8 | 4 | 1 | 64 | >128 | 16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, R.; Li, X.; Liu, Y.; Ye, X. Design, Synthesis, and Bioassay of 2′-Modified Kanamycin A. Molecules 2022, 27, 7482. https://doi.org/10.3390/molecules27217482
Yan R, Li X, Liu Y, Ye X. Design, Synthesis, and Bioassay of 2′-Modified Kanamycin A. Molecules. 2022; 27(21):7482. https://doi.org/10.3390/molecules27217482
Chicago/Turabian StyleYan, Ribai, Xiaonan Li, Yuheng Liu, and Xinshan Ye. 2022. "Design, Synthesis, and Bioassay of 2′-Modified Kanamycin A" Molecules 27, no. 21: 7482. https://doi.org/10.3390/molecules27217482