
Conformational Essentials Responsible for Neurotoxicity of Aβ42 Aggregates Revealed by Antibodies against Oligomeric Aβ42

Abstract
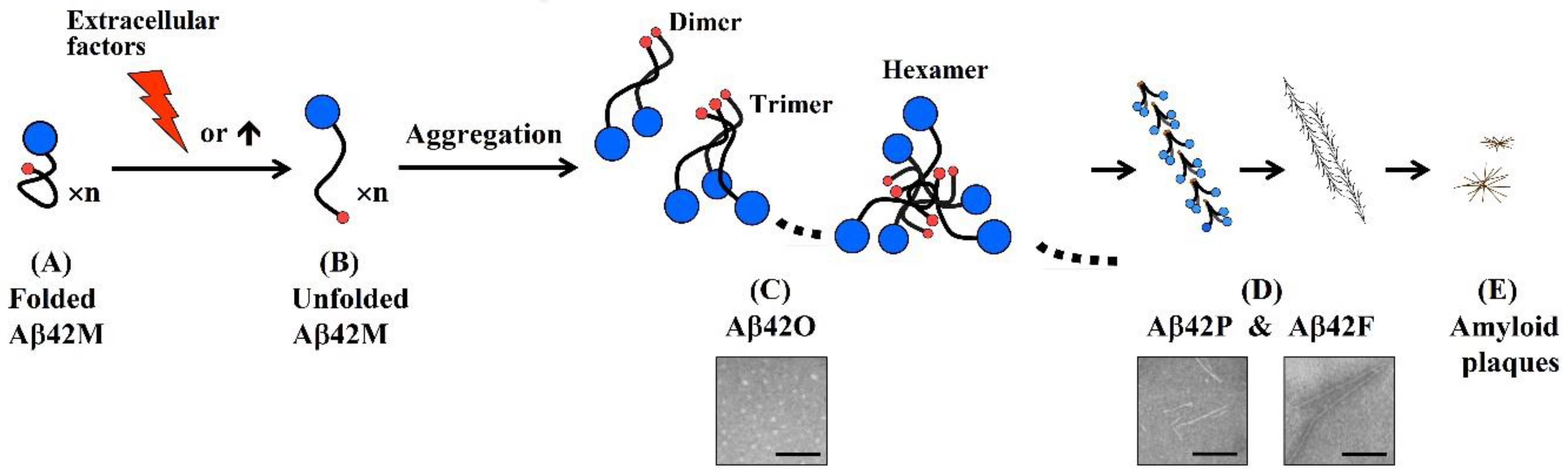
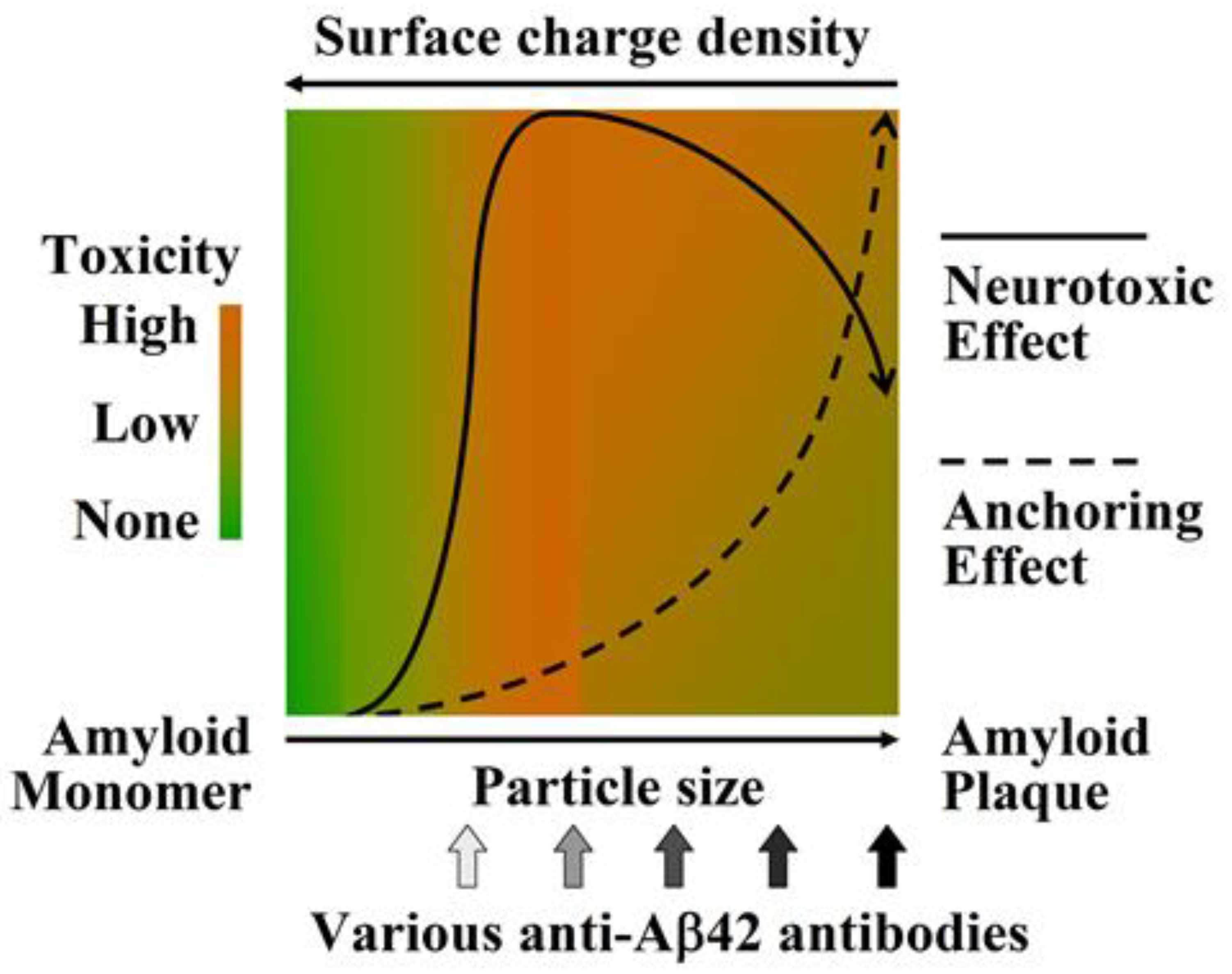
:1. Aβ42 Oligomers Are the Most Pathogenic Aβ Species
2. A Specific Integrated Conformation Underlies the Neurotoxicity of Aβ42Os
- (1)
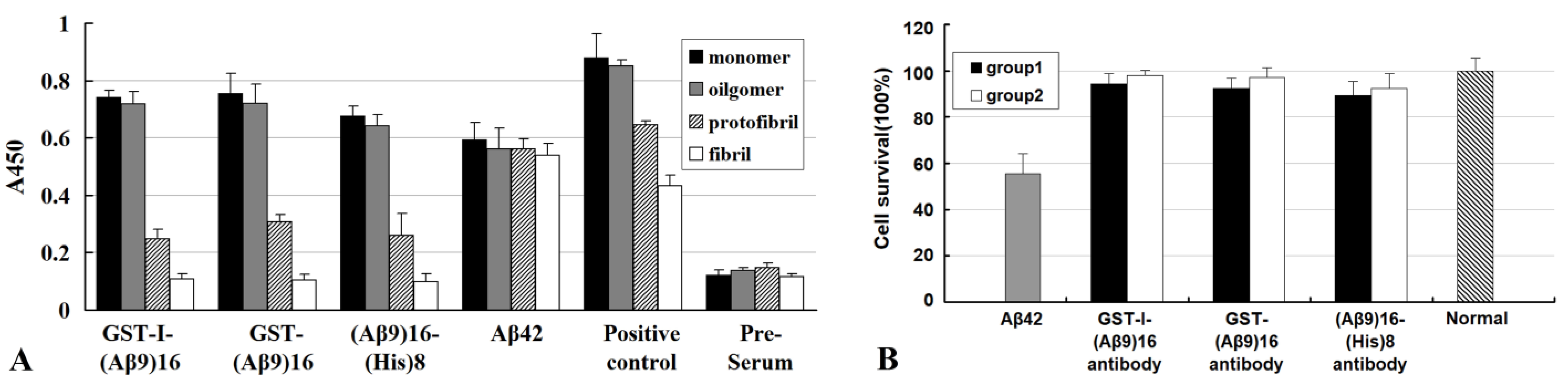
- An antibody molecule usually recognizes only the exposed portions of an antigenic unit. The high binding specificity of antibodies induced by various N-terminal fragments of Aβ42 for Aβ42O demonstrates that the proportion of surface-located N-terminal regions is much higher in Aβ42O than in Aβ42P or Aβ42F (Figure 2C–E). In protofibrils and fibrils, the N-terminal region of Aβ42 is most likely distributed on the surface and inside in a closely juxtaposed manner, as shown in Figure 2D. Thus, the solubility of Aβ42P and Aβ42F is much lower than that of Aβ42O because of the hydrophilicity of the N-terminal region and hydrophobicity of the C-terminal region of the Aβ42 chain.
- (2)
- The integrated conformation of Aβ42 aggregate species is closely related to its toxic activity; therefore, the binding specificity of an antibody against different Aβ42 aggregate species largely determines its efficacy in blocking or neutralizing the neurotoxicity of Aβ42 aggregates.
- (3)
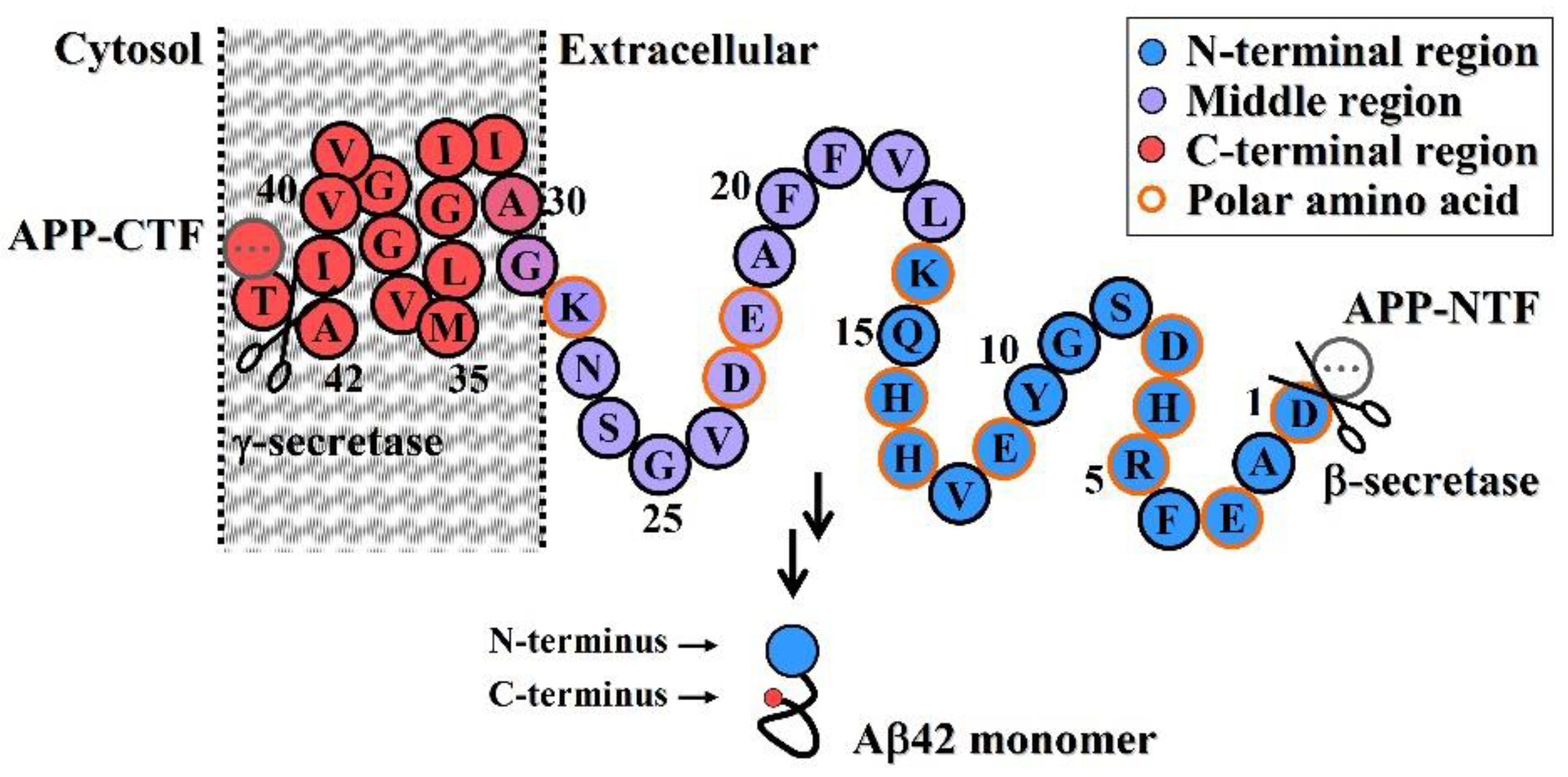
- Neuroprotective efficacy of antibodies induced by various N-terminal fragments of Aβ42 reveals that the exposed N-terminal region, approximately the first 16 amino acids of Aβ42 (DAEFRHDSGYEVHHQK) (Figure 1), appears to be the major structural element constituting the effector site responsible for Aβ42O neurotoxicity [33,34,35,36]. It is speculated that the N-terminally integrated structures of Aβ42O appear to be directly involved in binding to the membrane receptors and/or membrane structures of neural cells, thereby acting as alternative ligands to competitively or non-competitively disrupt some normal signaling pathways.
- (4)
- The C-terminal and central regions of an Aβ42 chain and their interactions indirectly affect the N-terminal integration structure, so they are also structural factors affecting Aβ42O toxicity. Any factor that disrupts the central and C-terminal regions of the Aβ42 chain may indirectly affect the integrated conformation of the N-terminus of Aβ42O, thereby affecting the toxicity of Aβ42O.
3. Structure−Toxicity Relationships of Aβ42 Aggregates Revealed by Passive Immunization
4. Discussion and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| Aβ42 | amyloid β-protein (1-42) |
| APP | amyloid precursor protein |
| Aβ42M/Aβ42O/Aβ42P/Aβ42F | Aβ42 monomer/oligomer/protofibril/fibril |
| scFv | single-chain variable fragment |
| VH or VL | heavy or light chain variable domain |
| ECM | extracellular matrix |
References
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [Green Version]
- Copani, A. The underexplored question of β-amyloid monomers. Eur. J. Pharmacol. 2017, 817, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Li, H.; Zhang, T.; Wang, Z.; Li, H.; Zhang, Y. Nonlinear and mixed inhibitory effect of matrine on the cytotoxicity of oligomeric amyloid-β protein. Neurochem. Int. 2020, 137, 104746. [Google Scholar] [CrossRef] [PubMed]
- Qiu, T.; Liu, Q.; Chen, Y.X.; Zhao, Y.F.; Li, Y.M. Aβ42 and Aβ40: Similarities and differences. J. Pept. Sci. 2015, 21, 522–529. [Google Scholar] [CrossRef]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Amyloid β oligomers (AβOs) in Alzheimer’s disease. J. Neural Transm. 2018, 125, 177–191. [Google Scholar] [CrossRef]
- Vandendriessche, C.; Balusu, S.; Van Cauwenberghe, C.; Brkic, M.; Pauwels, M.; Plehiers, N.; Bruggeman, A.; Dujardin, P.; Van Imschoot, G.; Van Wonterghem, E.; et al. Importance of extracellular vesicle secretion at the blood-cerebrospinal fluid interface in the pathogenesis of Alzheimer’s disease. Acta Neuropathol. Commun. 2021, 9, 143. [Google Scholar] [CrossRef]
- Mairet-Coello, G.; Polleux, F. Involvement of ‘stress-response’ kinase pathways in Alzheimer’s disease progression. Curr. Opin. Neurobiol. 2014, 27, 110–117. [Google Scholar] [CrossRef] [Green Version]
- Siqueira, L.D.; Celes, A.P.M.; Santos, H.D.; Ferreira, S.T. A Specialized Nutritional Formulation Prevents Hippocampal Glial Activation and Memory Impairment Induced by Amyloid-β Oligomers in Mice. J. Alzheimers Dis. 2021, 83, 1113–1124. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Barage, S.H.; Sonawane, K.D. Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides 2015, 52, 1–18. [Google Scholar] [CrossRef]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimers Dis. 2018, 64, S567–S610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, C.; Bascuñán, D.; Opazo, C.; Aguayo, L.G. Differential Membrane Toxicity of Amyloid-β Fragments by Pore Forming Mechanisms. J. Alzheimers Dis. 2016, 51, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Ciudad, S.; Puig, E.; Botzanowski, T.; Meigooni, M.; Arango, A.S.; Do, J.; Mayzel, M.; Bayoumi, M.; Chaignepain, S.; Maglia, G.; et al. Aβ(1-42) tetramer and octamer structures reveal edge conductivity pores as a mechanism for membrane damage. Nat. Commun. 2020, 11, 3014. [Google Scholar] [CrossRef]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Cellular Receptors of Amyloid β Oligomers (AβOs) in Alzheimer’s Disease. Int. J. Mol. Sci. 2018, 19, 1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.J.; Nam, E.; Lee, H.J.; Savelieff, M.G.; Lim, M.H. Towards an understanding of amyloid-β oligomers: Characterization, toxicity mechanisms, and inhibitors. Chem. Soc. Rev. 2017, 46, 310–323. [Google Scholar] [CrossRef]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [Green Version]
- Finch, M.S.; Bagit, A.; Marko, D.M. Amyloid beta 42 oligomers induce neuronal and synaptic receptor dysfunctions. J. Physiol. 2020, 598, 3545–3546. [Google Scholar] [CrossRef]
- Fani, G.; Mannini, B.; Vecchi, G.; Cascella, R.; Cecchi, C.; Dobson, C.M.; Vendruscolo, M.; Chiti, F. Aβ Oligomers Dysregulate Calcium Homeostasis by Mechanosensitive Activation of AMPA and NMDA Receptors. ACS Chem. Neurosci. 2021, 12, 766–781. [Google Scholar] [CrossRef]
- Ito, S.; Ménard, M.; Atkinson, T.; Brown, L.; Whitfield, J.; Chakravarthy, B. Relative expression of the p75 neurotrophin receptor, tyrosine receptor kinase A, and insulin receptor in SH-SY5Y neuroblastoma cells and hippocampi from Alzheimer’s disease patients. Neurochem. Int. 2016, 101, 22–29. [Google Scholar] [CrossRef]
- Coulson, E.J. Does the p75 neurotrophin receptor mediate Abeta-induced toxicity in Alzheimer’s disease? J. Neurochem. 2006, 98, 654–660. [Google Scholar] [CrossRef]
- Takahashi, R.H.; Yokotsuka, M.; Tobiume, M.; Sato, Y.; Hasegawa, H.; Nagao, T.; Gouras, G.K. Accumulation of cellular prion protein within β-amyloid oligomer plaques in aged human brains. Brain Pathol. 2021, 31, e12941. [Google Scholar] [CrossRef] [PubMed]
- Scott-McKean, J.J.; Surewicz, K.; Choi, J.K.; Ruffin, V.A.; Salameh, A.I.; Nieznanski, K.; Costa, A.C.S.; Surewicz, W.K. Soluble prion protein and its N-terminal fragment prevent impairment of synaptic plasticity by Aβ oligomers: Implications for novel therapeutic strategy in Alzheimer’s disease. Neurobiol. Dis. 2016, 91, 124–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinders, N.R.; Pao, Y.; Renner, M.C.; da Silva-Matos, C.M.; Lodder, T.R.; Malinow, R.; Kessels, H.W. Amyloid-β effects on synapses and memory require AMPA receptor subunit GluA. Proc. Natl. Acad. Sci. USA 2016, 113, E6526–E6534. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Song, C.; Zheng, C.; Chen, X.; Zhang, Y. Extracellular Amyloid β-protein (1–42) Oligomers Anchor Brain Cells and Make them inert as an Unconventional Integrin-Coupled Ligand. Cell Mol. Neurobiol. 2022. [Google Scholar] [CrossRef]
- Bayer, A.J.; Bullock, R.; Jones, R.W.; Wilkinson, D.; Paterson, K.R.; Jenkins, L.; Millais, S.B.; Donoghue, S. Evaluation of the safety and immunogenicity of synthetic Abeta42 (AN1792) in patients with AD. Neurology 2005, 64, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Pride, M.; Seubert, P.; Grundman, M.; Hagen, M.; Eldridge, J.; Black, R.S. Progress in the active immunotherapeutic approach to Alzheimer’s disease: Clinical investigations into AN1792-associated meningoencephalitis. Neurodegener. Dis. 2008, 5, 194–196. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, D.H.; Ghochikyan, A.; Vasilevko, V.; Tran, M.; Petrushina, I.; Sadzikava, N.; Babikyan, D.; Kesslak, P.; Kieber-Emmons, T.; Cotman, C.W.; et al. Adjuvant-dependent modulation of Th1 and Th2 responses to immunization with beta-amyloid. Int. Immunol. 2003, 15, 505–514. [Google Scholar] [CrossRef]
- Ghochikyan, A.; Mkrtichyan, M.; Petrushina, I.; Movsesyan, N.; Karapetyan, A.; Cribbs, D.H.; Agadjanyan, M.G. Prototype Alzheimer’s disease epitope vaccine induced strong Th2-type anti-Abeta antibody response with Alum to Quil A adjuvant switch. Vaccine 2006, 24, 2275–2282. [Google Scholar] [CrossRef] [Green Version]
- Vandenberghe, R.; Riviere, M.E.; Caputo, A.; Sovago, J.; Maguire, R.P.; Farlow, M.; Marotta, G.; Sanchez-Valle, R.; Scheltens, P.; Ryan, J.M.; et al. Active Aβ immunotherapy CAD106 in Alzheimer’s disease: A phase 2b study. Alzheimers Dement. 2017, 3, 10–22. [Google Scholar] [CrossRef]
- Mantile, F.; Prisco, A. Vaccination against β-Amyloid as a Strategy for the Prevention of Alzheimer’s Disease. Biology 2020, 9, 425. [Google Scholar] [CrossRef]
- Wang, C.Y.; Wang, P.N.; Chiu, M.J.; Finstad, C.L.; Lin, F.; Lynn, S.; Tai, Y.H.; De Fang, X.; Zhao, K.; Hung, C.H.; et al. UB-311, a novel UBITh® amyloid β peptide vaccine for mild Alzheimer’s disease. Alzheimers Dement. 2017, 3, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Hull, M.; Sadowsky, C.; Arai, H.; Le Prince Leterme, G.; Holstein, A.; Booth, K.; Peng, Y.; Yoshiyama, T.; Suzuki, H.; Ketter, N.; et al. Long-Term Extensions of Randomized Vaccination Trials of ACC-001 and QS-21 in Mild to Moderate Alzheimer’s Disease. Curr. Alzheimer Res. 2017, 14, 696–708. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Huang, X.; Wang, J.; Zhang, Y. Specific and efficient anti-Aβ42 antibodies induced by sixteen tandem repeats of Aβ9. J. Neuroimmunol. 2010, 227, 18–25. [Google Scholar] [CrossRef]
- Huang, X.; Wang, J.; Cui, L.; Zou, X.; Zhang, Y. Recombinant GST-I-A beta 28-induced efficient serum antibody against A beta. J. Neurosci. Methods 2010, 186, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Wiessner, C.; Wiederhold, K.H.; Tissot, A.C.; Frey, P.; Danner, S.; Jacobson, L.H.; Jennings, G.T.; Lüönd, R.; Ortmann, R.; Reichwald, J.; et al. The second-generation active Aβ immunotherapy CAD106 reduces amyloid accumulation in APP transgenic mice while minimizing potential side effects. J. Neurosci. 2011, 31, 9323–9331. [Google Scholar] [CrossRef]
- Davtyan, H.; Ghochikyan, A.; Petrushina, I.; Hovakimyan, A.; Davtyan, A.; Poghosyan, A.; Marleau, A.M.; Movsesyan, N.; Kiyatkin, A.; Rasool, S.; et al. Immunogenicity, efficacy, safety, and mechanism of action of epitope vaccine (Lu AF20513) for Alzheimer’s disease: Prelude to a clinical trial. J. Neurosci. 2013, 33, 4923–4934. [Google Scholar] [CrossRef] [Green Version]
- Gardberg, A.S.; Dice, L.T.; Ou, S.; Rich, R.L.; Helmbrecht, E.; Ko, J.; Wetzel, R.; Myszka, D.G.; Patterson, P.H.; Dealwis, C. Molecular basis for passive immunotherapy of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 15659–15664. [Google Scholar] [CrossRef] [Green Version]
- Miles, L.A.; Wun, K.S.; Crespi, G.A.; Fodero-Tavoletti, M.T.; Galatis, D.; Bagley, C.J.; Beyreuther, K.; Masters, C.L.; Cappai, R.; McKinstry, W.J.; et al. Amyloid-beta-anti-amyloid-beta complex structure reveals an extended conformation in the immunodominant B-cell epitope. J. Mol. Biol. 2008, 377, 181–192. [Google Scholar] [CrossRef]
- Basi, G.S.; Feinberg, H.; Oshidari, F.; Anderson, J.; Barbour, R.; Baker, J.; Comery, T.A.; Diep, L.; Gill, D.; Johnson-Wood, K.; et al. Structural correlates of antibodies associated with acute reversal of amyloid beta-related behavioral deficits in a mouse model of Alzheimer disease. J. Biol. Chem. 2010, 285, 3417–3427. [Google Scholar] [CrossRef] [Green Version]
- Lozupone, M.; Solfrizzi, V.; D’Urso, F.; Di Gioia, I.; Sardone, R.; Dibello, V.; Stallone, R.; Liguori, A.; Ciritella, C.; Daniele, A.; et al. Anti-amyloid-β protein agents for the treatment of Alzheimer’s disease: An update on emerging drugs. Expert Opin. Emerg. Drugs 2020, 25, 319–335. [Google Scholar] [CrossRef]
- Loureiro, J.C.; Pais, M.V.; Stella, F.; Radanovic, M.; Teixeira, A.L.; Forlenza, O.V.; de Souza, L.C. Passive antiamyloid immunotherapy for Alzheimer’s disease. Curr. Opin. Psychiatry 2020, 33, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Frisardi, V.; Imbimbo, B.P.; Seripa, D.; Paris, F.; Santamato, A.; D’Onofrio, G.; Logroscino, G.; Pilotto, A.; Solfrizzi, V. Anti-β-amyloid immunotherapy for Alzheimer’s disease: Focus on bapineuzumab. Curr. Alzheimer Res. 2011, 8, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Tayeb, H.O.; Murray, E.D.; Price, B.H.; Tarazi, F.I. Bapineuzumab and solanezumab for Alzheimer’s disease: Is the ‘amyloid cascade hypothesis’ still alive? Expert Opin. Biol. Ther. 2013, 13, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- La Porte, S.L.; Bollini, S.S.; Lanz, T.A.; Abdiche, Y.N.; Rusnak, A.S.; Ho, W.H.; Kobayashi, D.; Harrabi, O.; Pappas, D.; Mina, E.W.; et al. Structural basis of C-terminal β-amyloid peptide binding by the antibody ponezumab for the treatment of Alzheimer’s disease. J. Mol. Biol. 2012, 421, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Landen, J.W.; Zhao, Q.; Cohen, S.; Borrie, M.; Woodward, M.; Billing, C.B., Jr.; Bales, K.; Alvey, C.; McCush, F.; Yang, J.; et al. Safety and pharmacology of a single intravenous dose of ponezumab in subjects with mild-to-moderate Alzheimer disease: A phase I, randomized, placebo-controlled, double-blind, dose-escalation study. Clin. Neuropharmacol. 2013, 36, 14–23. [Google Scholar] [CrossRef]
- Siemers, E.R.; Sundell, K.L.; Carlson, C.; Case, M.; Sethuraman, G.; Liu-Seifert, H.; Dowsett, S.A.; Pontecorvo, M.J.; Dean, R.A.; Demattos, R. Phase 3 solanezumab trials: Secondary outcomes in mild Alzheimer’s disease patients. Alzheimers Dement. 2016, 12, 110–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imbimbo, B.P.; Ottonello, S.; Frisardi, V.; Solfrizzi, V.; Greco, A.; Seripa, D.; Pilotto, A.; Panza, F. Solanezumab for the treatment of mild-to-moderate Alzheimer’s disease. Expert Rev. Clin. Immunol. 2012, 8, 135–149. [Google Scholar] [CrossRef]
- Ultsch, M.; Li, B.; Maurer, T.; Mathieu, M.; Adolfsson, O.; Muhs, A.; Pfeifer, A.; Pihlgren, M.; Bainbridge, T.W.; Reichelt, M.; et al. Structure of Crenezumab Complex with Aβ Shows Loss of β-Hairpin. Sci. Rep. 2016, 6, 39374. [Google Scholar] [CrossRef] [Green Version]
- Bohrmann, B.; Baumann, K.; Benz, J.; Gerber, F.; Huber, W.; Knoflach, F.; Messer, J.; Oroszlan, K.; Rauchenberger, R.; Richter, W.F.; et al. Gantenerumab: A novel human anti-Aβ antibody demonstrates sustained cerebral amyloid-β binding and elicits cell-mediated removal of human amyloid-β. J. Alzheimers Dis. 2012, 28, 49–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrowitzki, S.; Deptula, D.; Thurfjell, L.; Barkhof, F.; Bohrmann, B.; Brooks, D.J.; Klunk, W.E.; Ashford, E.; Yoo, K.; Xu, Z.X.; et al. Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch. Neurol. 2012, 69, 198–207. [Google Scholar] [CrossRef]
- Yang, T.; Dang, Y.; Ostaszewski, B.; Mengel, D.; Steffen, V.; Rabe, C.; Bittner, T.; Walsh, D.M.; Selkoe, D.J. Target engagement in an alzheimer trial: Crenezumab lowers amyloid β oligomers in cerebrospinal fluid. Ann. Neurol. 2019, 86, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, H.; Honig, L.S.; Lin, H.; Sink, K.M.; Blondeau, K.; Quartino, A.; Dolton, M.; Carrasco-Triguero, M.; Lian, Q.; Bittner, T.; et al. Safety, Tolerability, and Pharmacokinetics of Crenezumab in Patients with Mild-to-Moderate Alzheimer’s Disease Treated with Escalating Doses for up to 133 Weeks. J. Alzheimers Dis. 2020, 76, 967–979. [Google Scholar] [CrossRef]
- Adolfsson, O.; Pihlgren, M.; Toni, N.; Varisco, Y.; Buccarello, A.L.; Antoniello, K.; Lohmann, S.; Piorkowska, K.; Gafner, V.; Atwal, J.K.; et al. An effector-reduced anti-β-amyloid (Aβ) antibody with unique aβ binding properties promotes neuroprotection and glial engulfment of Aβ. J. Neurosci. 2012, 32, 9677–9689. [Google Scholar] [CrossRef] [PubMed]
- Kastanenka, K.V.; Bussiere, T.; Shakerdge, N.; Qian, F.; Weinreb, P.H.; Rhodes, K.; Bacskai, B.J. Immunotherapy with Aducanumab Restores Calcium Homeostasis in Tg2576 Mice. J. Neurosci. 2016, 36, 12549–12558. [Google Scholar] [CrossRef] [Green Version]
- Padda, I.S.; Parmar, M. Aducanumab. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022. [Google Scholar]
- Logovinsky, V.; Satlin, A.; Lai, R.; Swanson, C.; Kaplow, J.; Osswald, G.; Basun, H.; Lannfelt, L. Safety and tolerability of BAN2401—A clinical study in Alzheimer’s disease with a protofibril selective Aβ antibody. Alzheimers Res. Ther. 2016, 8, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolar, M.; Abushakra, S.; Hey, J.A.; Porsteinsson, A.; Sabbagh, M. Aducanumab, gantenerumab, BAN2401, and ALZ-801-the first wave of amyloid-targeting drugs for Alzheimer’s disease with potential for near term approval. Alzheimers Res. Ther. 2020, 12, 95. [Google Scholar] [CrossRef]
- Mintun, M.A.; Lo, A.C.; Duggan Evans, C.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.; Sims, J.R.; Brys, M.; et al. Donanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef] [PubMed]
- Alawode, D.O.T.; Heslegrave, A.J.; Fox, N.C.; Zetterberg, H. Donanemab removes Alzheimer’s plaques: What is special about its target? Lancet Healthy Longev. 2021, 2, e395–e396. [Google Scholar] [CrossRef]
- Bouter, Y.; Liekefeld, H.; Pichlo, S.; Westhoff, A.C.; Fenn, L.; Bakrania, P.; Bayer, T.A. Donanemab detects a minor fraction of amyloid-β plaques in post-mortem brain tissue of patients with Alzheimer’s disease and Down syndrome. Acta Neuropathol. 2022, 143, 601–603. [Google Scholar] [CrossRef]
- Bemani, P.; Mohammadi, M.; Hakakian, A. ScFv Improvement Approaches. Protein Pept. Lett. 2018, 25, 222–229. [Google Scholar] [CrossRef]
- Bitencourt, A.L.B.; Campos, R.M.; Cline, E.N.; Klein, W.L.; Sebollela, A. Antibody Fragments as Tools for Elucidating Structure-Toxicity Relationships and for Diagnostic/Therapeutic Targeting of Neurotoxic Amyloid Oligomers. Int. J. Mol. Sci. 2020, 21, 8920. [Google Scholar] [CrossRef] [PubMed]
- van Dyck, C.H. Anti-Amyloid-β Monoclonal Antibodies for Alzheimer’s Disease: Pitfalls and Promise. Biol. Psychiatry 2018, 83, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Güell-Bosch, J.; Lope-Piedrafita, S.; Esquerda-Canals, G.; Montoliu-Gaya, L.; Villegas, S. Progression of Alzheimer’s disease and effect of scFv-h3D6 immunotherapy in the 3xTg-AD mouse model: An in vivo longitudinal study using Magnetic Resonance Imaging and Spectroscopy. NMR Biomed. 2020, 33, e4263. [Google Scholar] [CrossRef] [PubMed]
- Esquerda-Canals, G.; Roda, A.R.; Martí-Clúa, J.; Montoliu-Gaya, L.; Rivera-Hernández, G.; Villegas, S. Treatment with scFv-h3D6 Prevented Neuronal Loss and Improved Spatial Memory in Young 3xTg-AD Mice by Reducing the Intracellular Amyloid-β Burden. J. Alzheimers Dis. 2019, 70, 1069–1091. [Google Scholar] [CrossRef]
- Sebollela, A.; Cline, E.N.; Popova, I.; Luo, K.; Sun, X.; Ahn, J.; Barcelos, M.A.; Bezerra, V.N.; Lyra, E.S.N.M.; Patel, J.; et al. A human scFv antibody that targets and neutralizes high molecular weight pathogenic amyloid-β oligomers. J. Neurochem. 2017, 142, 934–947. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, N.; Ma, J.; Gu, Z.; Yu, L.; Fu, X.; Liu, X.; Wang, J. Effects of an amyloid-beta 1-42 oligomers antibody screened from a phage display library in APP/PS1 transgenic mice. Brain Res. 2016, 1635, 169–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kou, J.; Kim, H.; Pattanayak, A.; Song, M.; Lim, J.E.; Taguchi, H.; Paul, S.; Cirrito, J.R.; Ponnazhagan, S.; Fukuchi, K. Anti-Amyloid-β Single-Chain Antibody Brain Delivery Via AAV Reduces Amyloid Load But May Increase Cerebral Hemorrhages in an Alzheimer’s Disease Mouse Model. J. Alzheimers Dis. 2011, 27, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Pattanayak, A.; Song, M.; Kou, J.; Taguchi, H.; Paul, S.; Ponnazhagan, S.; Lalonde, R.; Fukuchi, K. Muscle-directed anti-Aβ single-chain antibody delivery via AAV1 reduces cerebral Aβ load in an Alzheimer’s disease mouse model. J. Mol. Neurosci. 2013, 49, 277–288. [Google Scholar] [CrossRef] [Green Version]
- Zameer, A.; Kasturirangan, S.; Emadi, S.; Nimmagadda, S.V.; Sierks, M.R. Anti-oligomeric Abeta single-chain variable domain antibody blocks Abeta-induced toxicity against human neuroblastoma cells. J. Mol. Biol. 2008, 384, 917–928. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Y.; Huai, Y.; Zhang, Y.J. Functional Characteristics and Molecular Mechanism of a New scFv Antibody Against Aβ42 Oligomers and Immature Protofibrils. Mol. Neurobiol. 2015, 52, 1269–1281. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, X.; Liu, J.; Zhang, Y. The protective effects and underlying mechanism of an anti-oligomeric Aβ42 single-chain variable fragment antibody. Neuropharmacology 2015, 99, 387–395. [Google Scholar] [CrossRef]
- Zhang, X.; Huai, Y.; Cai, J.; Song, C.; Zhang, Y. Novel antibody against oligomeric amyloid-β: Insight into factors for effectively reducing the aggregation and cytotoxicity of amyloid-β aggregates. Int. Immunopharmacol. 2019, 67, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huai, Y.; Zhang, X.; Song, C.; Cai, J.; Zhang, Y. The Mode of Action of an Anti-Oligomeric Amyloid β-Protein Antibody Affects its Protective Efficacy. Neurotox. Res. 2019, 35, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Lacor, P.N.; Buniel, M.C.; Chang, L.; Fernandez, S.J.; Gong, Y.; Viola, K.L.; Lambert, M.P.; Velasco, P.T.; Bigio, E.H.; Finch, C.E.; et al. Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J. Neurosci. 2004, 24, 10191–10200. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Binding Sites | Target | Reference |
|---|---|---|---|
| ScFv-h3D6 | not reported | Aβ42 monomers, oligomers, and fibrils | [64,65] |
| NUsc1 | not reported | Aβ42 oligomers | [66] |
| 11A5 | not reported | Aβ42 oligomers (34 kDa) | [67] |
| ScFv59 | not reported | Aβ42 oligomers and amyloid plaques | [68,69] |
| A4 | not reported | Aβ42 oligomers | [70] |
| AS | Aβ1–15, Aβ20–33 (by molecular docking) | Aβ42 oligomers and protofibrils (25–55 kDa) | [71] |
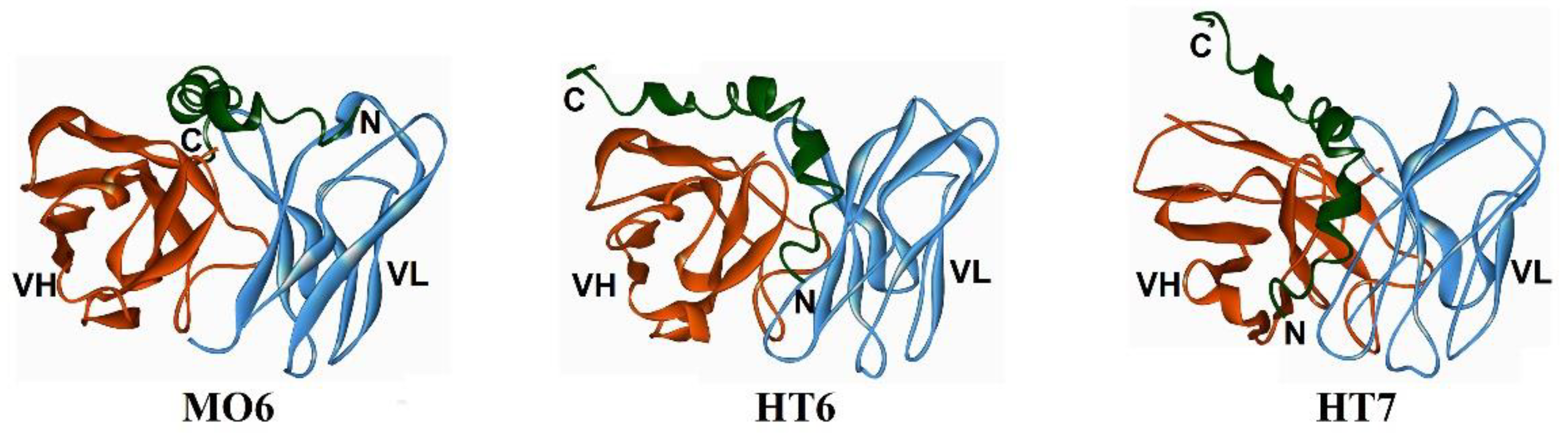
| MO6 | Aβ3–4, Aβ15–42 by molecular docking) | Aβ42 oligomers and immature fibrils (18–37 kDa) | [72] |
| HT6 | Aβ1–14, Aβ21–30 (by molecular docking) | Aβ42 oligomers and immature fibrils (18–45 kDa) | [73] |
| HT7 | Aβ1–21/26 (by molecular docking) | Aβ42 oligomers and immature fibrils (23–55 kDa) | [74] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, C.; Zhang, T.; Zhang, Y. Conformational Essentials Responsible for Neurotoxicity of Aβ42 Aggregates Revealed by Antibodies against Oligomeric Aβ42. Molecules 2022, 27, 6751. https://doi.org/10.3390/molecules27196751
Song C, Zhang T, Zhang Y. Conformational Essentials Responsible for Neurotoxicity of Aβ42 Aggregates Revealed by Antibodies against Oligomeric Aβ42. Molecules. 2022; 27(19):6751. https://doi.org/10.3390/molecules27196751
Chicago/Turabian StyleSong, Chuli, Tianyu Zhang, and Yingjiu Zhang. 2022. "Conformational Essentials Responsible for Neurotoxicity of Aβ42 Aggregates Revealed by Antibodies against Oligomeric Aβ42" Molecules 27, no. 19: 6751. https://doi.org/10.3390/molecules27196751