

Green and Efficient Determination of Fluoroquinolone Residues in Edible Green Fruits and Leafy Vegetables by Ultrasound-Assisted Extraction Followed by HPLC-MS/MS
 , , , and
, , , and 
Abstract
:
1. Introduction
2. Results and Discussion
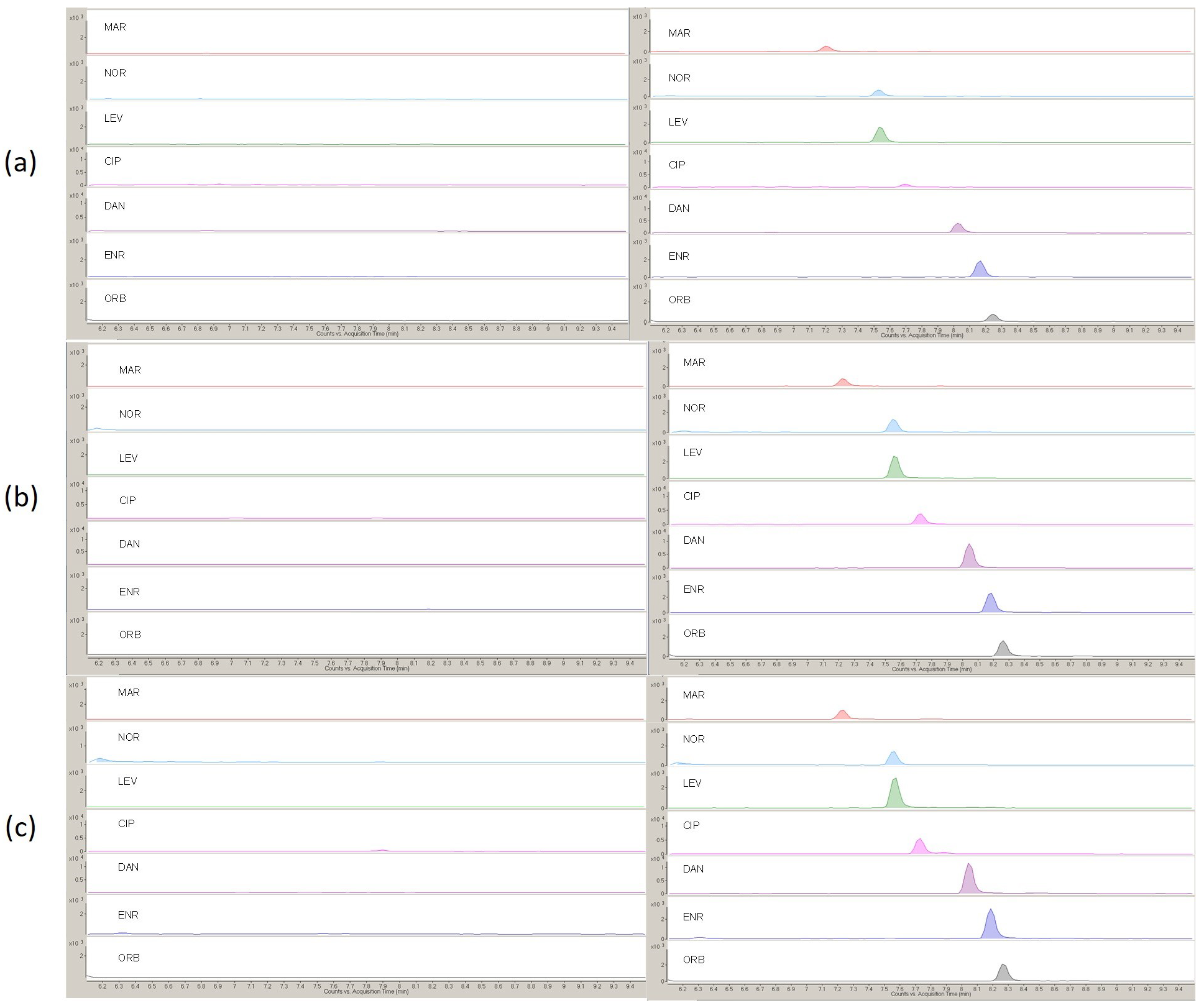
2.1. Development of UAE Procedure and Cleanup
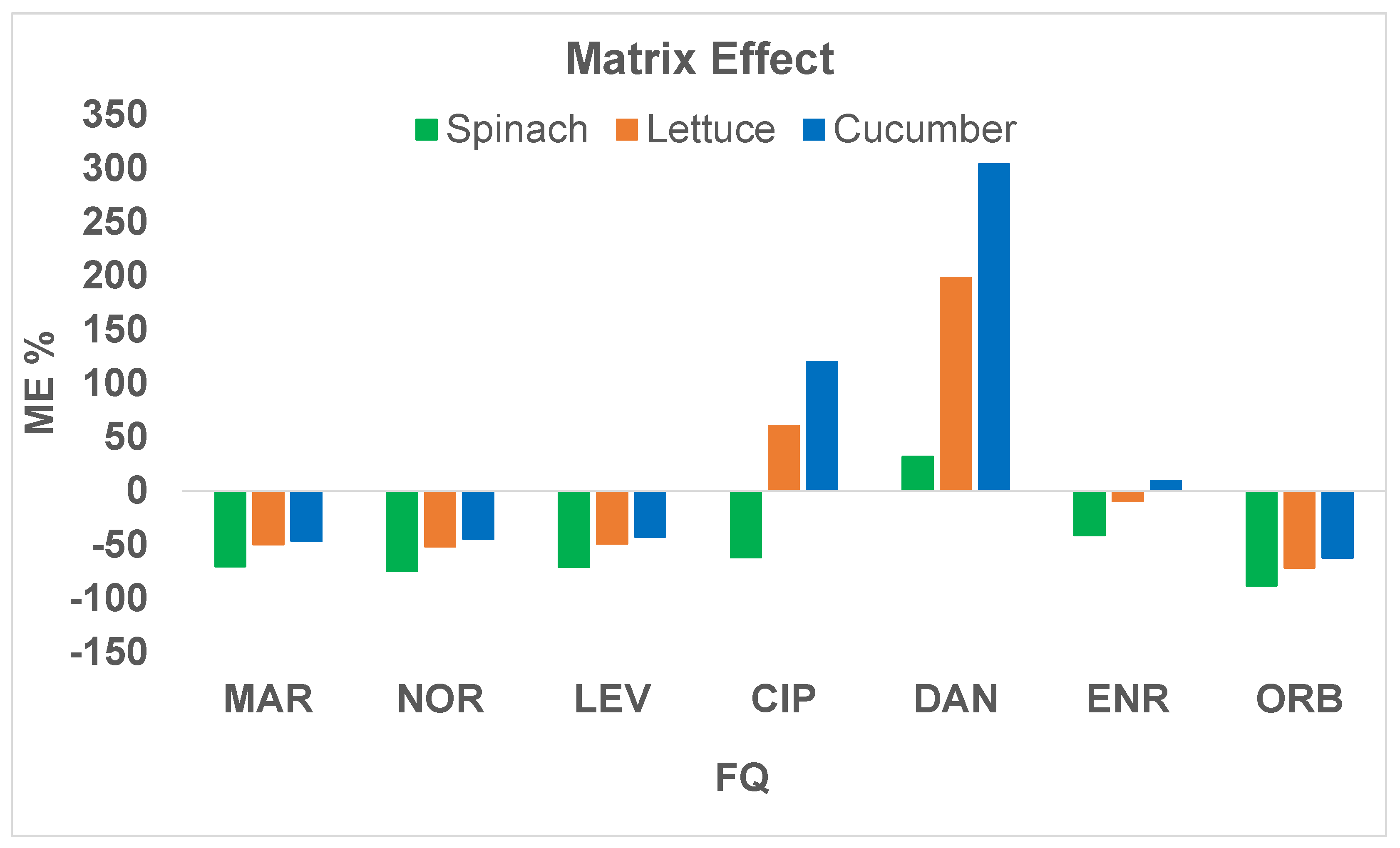
2.2. Analytical Evaluation of the Method
2.3. Comparison of the Developed Procedure with Recent UAE-Based Methods in Edible Green Fruits and Leafy Vegetables
2.4. Determination of FQs in Commercial Green Fruits and Leafy Vegetables
3. Materials and Methods
3.1. Chemicals and Materials
3.2. Edible Green Fruits and Leafy Vegetable Samples
3.3. UAE Procedure and Cleanup
3.4. HPLC-ESI-MS/MS
3.5. Analytical Evaluation of the Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Li, X.; Xie, Y.-F.; Li, C.-L.; Zhao, H.-N.; Wang, N.; Wang, J.-F. Investigation of residual fluoroquinolones in a soil–vegetable system in an intensive vegetable cultivation area in Northern China. Sci. Total Environ. 2014, 468–469, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Bian, K.; Liu, Y.; Su, Y.; Zhou, T.; Song, X.; He, L. Development of a modified QUick, Easy, CHeap, Effective, Rugged and Safe method for the determination of multi-class antimicrobials in vegetables by liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2014, 1368, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Kokoszka, K.; Kobus, A.; Bajkacz, S. Optimization of a Method for Extraction and Determination of Residues of Selected Antimicrobials in Soil and Plant Samples Using HPLC-UV-MS/MS. Int. J. Environ. Res. Public Health 2021, 18, 1159. [Google Scholar] [CrossRef]
- Hu, Y.; Habibul, N.; Hu, Y.-Y.; Meng, F.-L.; Sheng, G.-P. Chemical speciation of ciprofloxacin in aqueous solution regulates its phytotoxicity and uptake by rice (Oryza sativa L.). Sci. Total Environ. 2021, 771, 144787. [Google Scholar] [CrossRef]
- Speltini, A.; Sturini, M.; Maraschi, F.; Viti, S.; Sbarbada, D.; Profumo, A. Fluoroquinolone residues in compost by green enhanced microwave-assisted extraction followed by ultra performance liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2015, 1410, 44–50. [Google Scholar] [CrossRef]
- Feng, Y.; Zhang, W.-J.; Liu, Y.-W.; Xue, J.-M.; Zhang, S.-Q.; Li, Z.-J. A Simple, Sensitive, and Reliable Method for the Simultaneous Determination of Multiple Antibiotics in Vegetables through SPE-HPLC-MS/MS. Molecules 2018, 23, 1953. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Deng, Y.; Zheng, J.; Zhang, Y.; Yang, L. Trends in Analytical Chemistry The Application of the QuEChERS Methodology in the Determination of Antibiotics in Food: A Review. Trends Anal. Chem. 2019, 118, 517–537. [Google Scholar] [CrossRef]
- Albero, B.; Tadeo, J.L.; Miguel, E.; Pérez, R.A. Rapid determination of antibiotic residues in cereals by liquid chromatography triple mass spectrometry. Anal. Bioanal. Chem. 2019, 411, 6129–6139. [Google Scholar] [CrossRef]
- Tadić, Đ.; Matamoros, V.; Bayona, J.M. Simultaneous determination of multiclass antibiotics and their metabolites in four types of field-grown vegetables. Anal. Bioanal. Chem. 2019, 411, 5209–5222. [Google Scholar] [CrossRef] [PubMed]
- Albero, B.; Tadeo, J.L.; Delgado, M.D.M.; Miguel, E.; Pérez, R.A. Analysis of Multiclass Antibiotics in Lettuce by Liquid Chromatography–Tandem Mass Spectrometry to Monitor Their Plant Uptake. Molecules 2019, 24, 4066. [Google Scholar] [CrossRef]
- Chen, J.; Ying, G.-G.; Deng, W.-J. Antibiotic Residues in Food: Extraction, Analysis, and Human Health Concerns. J. Agric. Food Chem. 2019, 67, 7569–7586. [Google Scholar] [CrossRef] [PubMed]
- Margenat, A.; You, R.; Cañameras, N.; Carazo, N.; Díez, S.; Bayona, J.M.; Matamoros, V. Occurrence and human health risk assessment of antibiotics and trace elements in Lactuca sativa amended with different organic fertilizers. Environ. Res. 2020, 190, 109946. [Google Scholar] [CrossRef] [PubMed]
- Sturini, M.; Speltini, A.; Maraschi, F.; Rivagli, E.; Profumo, A. Solvent-free microwave-assisted extraction of fluoroquinolones from soil and liquid chromatography-fluorescence determination. J. Chromatogr. A 2010, 1217, 7316–7322. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Naeem, M.; Chaudhry, M.N.; Iqbal, M.A. Accumulation of Residual Antibiotics in the Vegetables Irrigated by Pharmaceutical Wastewater. Expo. Health 2016, 8, 107–115. [Google Scholar] [CrossRef]
- Yu, X.; Liu, H.; Pu, C.; Chen, J.; Sun, Y.; Hu, L. Determination of multiple antibiotics in leafy vegetables using QuEChERS-UHPLC-MS/MS. J. Sep. Sci. 2018, 41, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Sturini, M.; Speltini, A.; Pretali, L.; Fasani, E.; Profumo, A. Solid-phase extraction and HPLC determination of fluoroquinolones in surface waters. J. Sep. Sci. 2009, 32, 3020–3028. [Google Scholar] [CrossRef]
- Davydov, N.; Zairov, R.; Mustafina, A.; Syakayev, V.; Tatarinov, D.; Mironov, V.; Eremin, S.; Konovalov, A.; Mustafin, M. Determination of fluoroquinolone antibiotics through the fluorescent response of Eu(III) based nanoparticles fabricated by layer-by-layer technique. Anal. Chim. Acta 2013, 784, 65–71. [Google Scholar] [CrossRef]
- Pan, M.; Wong, C.K.C.; Chu, L.M. Distribution of Antibiotics in Wastewater-Irrigated Soils and Their Accumulation in Vegetable Crops in the Pearl River Delta, Southern China. J. Agric. Food Chem. 2014, 62, 11062–11069. [Google Scholar] [CrossRef]
- Kipper, K.; Lillenberg, M.; Herodes, K.; Nei, L.; Haiba, E. Simultaneous Determination of Fluoroquinolones and Sulfonamides Originating from Sewage Sludge Compost. Sci. World J. 2017, 2017, 9254072. [Google Scholar] [CrossRef] [Green Version]
- Migliore, L.; Cozzolino, S.; Fiori, M. Phytotoxicity to and uptake of enrofloxacin in crop plants. Chemosphere 2003, 52, 1233–1244. [Google Scholar] [CrossRef]
- He, Z.; Wang, Y.; Xu, Y.; Liu, X. Determination of Antibiotics in Vegetables Using QuEChERS-Based Method and Liquid Chromatography-Quadrupole Linear Ion Trap Mass Spectrometry. Food Anal. Methods 2018, 11, 2857–2864. [Google Scholar] [CrossRef]
- López-Lorente, I.; Pena-Pereira, F.; Pedersen-Bjergaard, S.; Zuin, V.G.; Ozkan, S.A.; Psillakis, E. The ten principles of green sample preparation. TrAC Trends Anal. Chem. 2022, 148, 116530. [Google Scholar] [CrossRef]
- FDA. Guidelines for the Validation of Chemical Methods for the FDA Foods Program, 3rd ed.; FDA: Silver Spring, MD, USA, 2019. [Google Scholar]
- Bhatta, S.; Janezic, T.S.; Ratti, C. Freeze-Drying of Plant-Based Foods. Foods 2020, 9, 87. [Google Scholar] [CrossRef] [Green Version]
- Zhong, M.; Wang, T.; Huang, J.; Yu, G. Application Agilent HPLC-MS/MS 1200 Series; Agilent Technologies: Santa Clara, CA, USA, 2020. [Google Scholar]
- Kruve, A.; Rebane, R.; Kipper, K.; Oldekop, M.-L.; Evard, H.; Herodes, K.; Ravio, P.; Leito, I. Tutorial review on validation of liquid chromatography–mass spectrometry methods: Part I. Anal. Chim. Acta 2015, 870, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Kruve, A.; Rebane, R.; Kipper, K.; Oldekop, M.-L.; Evard, H.; Herodes, K.; Ravio, P.; Leito, I. Tutorial review on validation of liquid chromatography–mass spectrometry methods: Part II. Anal. Chim. Acta 2015, 870, 8–28. [Google Scholar] [CrossRef] [PubMed]
- Kurbanoglu, S.; Uslu, B.; Ozkan, S.A. Validation of Analytical Methods for the Assessment of Hazards in Food; Elsevier Inc.: Amsterdam, The Netherlands, 2018; ISBN 9780128149560. [Google Scholar]
- Feng, Y.Z. NordVal Protocol n◦2 Guide in Validation of Alternative Proprietary Chemical Methods. J. Food Saf. Qual. 2014, 5, 2595–2601. [Google Scholar]
- Merlo, F.; Suppini, S.; Maraschi, F.; Profumo, A.; Speltini, A. A simple and fast multiclass method for determination of steroid hormones in berry fruits, root and leafy vegetables. Talanta Open 2022, 5, 100081. [Google Scholar] [CrossRef]
- Speltini, A.; Merlo, F.; Maraschi, F.; Marrubini, G.; Faravelli, A.; Profumo, A. Magnetic Micro-Solid-Phase Extraction Using a Novel Carbon-Based Composite Coupled with HPLC–MS/MS for Steroid Multiclass Determination in Human Plasma. Molecules 2021, 26, 2061. [Google Scholar] [CrossRef]
- Speltini, A.; Merlo, F.; Maraschi, F.; Villani, L.; Profumo, A. HA-C@silica sorbent for simultaneous extraction and clean-up of steroids in human plasma followed by HPLC-MS/MS multiclass determination. Talanta 2021, 221, 121496. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Spinach | Lettuce | Cucumber | ||||||
|---|---|---|---|---|---|---|---|---|---|
| HQC | MQC | LQC | HQC | MQC | LQC | HQC | MQC | LQC | |
| MAR | 92(13) | 91(6) | 92(12) | 91(12) | 96(2) | 92(4) | 105(13) | 86(13) | 106(14) |
| NOR | 80(16) | 74(3) | 80(12) | 81(15) | 116(9) | 103(5) | 100(8) | 90(20) | 114(4) |
| LEV | 84(9) | 84(16) | 96(10) | 90(3) | 106(5) | 93(16) | 97(8) | 88(11) | 107(4) |
| CIP | 75(7) | 81(7) | 91(5) | 80(7) | 88(18) | 90(16) | 89(8) | 83(19) | 111(6) |
| DAN | 75(12) | 70(11) | 86(9) | 95(7) | 96(12) | 89(12) | 88(4) | 85(16) | 103(3) |
| ENR | 81(10) | 73(8) | 67(6) | 90(2) | 105(3) | 96(9) | 80(15) | 86(15) | 99(4) |
| ORB | 80(5) | 95(8) | 81(13) | 89(10) | 93(18) | 94(18) | 84(16) | 75(5) | 100(11) |
| Analyte | Spinach | Lettuce | Cucumber | |||
|---|---|---|---|---|---|---|
| MDL | MQL | MDL | MQL | MDL | MQL | |
| MAR | 1 | 4 | 1 | 2 | 2 | 5 |
| NOR | 3 | 10 | 2 | 5 | 1 | 3 |
| LEV | 2 | 5 | 1 | 2 | 1 | 3 |
| CIP | 2 | 5 | 1 | 3 | 2 | 5 |
| DAN | 1 | 2 | 1 | 3 | 1 | 2 |
| ENR | 1 | 3 | 1 | 2 | 1 | 3 |
| ORB | 1 | 3 | 1 | 3 | 1 | 2 |
| Matrix | Analyte | UAE | Cleanup | Recovery (%) | MQL (ng g−1) | Ref. |
|---|---|---|---|---|---|---|
| Cabbage Spinach Radish Corn Rice 1 g (d.w.) | NOR | 3 × 15 min × 30 mL acidified ACN-acetone (1:1 v/v) | HLB™ SPE on 1:200 diluted extract | 81–87 | 1.17 | [18] |
| Potato Carrot Lettuce Wheat 250 mg (d.w.) | NOR CIP Ofloxacin | 1 × 5 min × 10 mL ACN−1% CH3COOH (1:1 v/v) | HLB™ SPE on 1:15 diluted extract | 66–93 | 5–40 | [19] |
| Lettuce Tomato Cauliflower Bean 1 g (f.w.) | ENR Ofloxacin | 2 × 15 min × 10 mL MeOH | Strata™-X SPE on 1:10 diluted extract | 30–125 | 0.4–9.2 | [9] |
| Lettuce 0.2 g (d.w.) | CIP ENR | 2 × 15 min × 5 mL ACN-MeOH−0.5% HCOOH (65:15:20, v/v/v) | C18 d-SPE | 70–90 | 10–24 | [10] |
| Spinach Lettuce Cucumber 0.25–0.5 (d.w.) | MAR NOR LEV CIP DAN ENR ORB | 3 × 2 min × 6 mL 20% w/v Mg(NO3)2 2% v/v NH3 | HLB™ SPE on ~1:3 diluted extract | 67–116 | 2–10 | This work |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merlo, F.; Centenaro, D.; Maraschi, F.; Profumo, A.; Speltini, A. Green and Efficient Determination of Fluoroquinolone Residues in Edible Green Fruits and Leafy Vegetables by Ultrasound-Assisted Extraction Followed by HPLC-MS/MS. Molecules 2022, 27, 6595. https://doi.org/10.3390/molecules27196595
Merlo F, Centenaro D, Maraschi F, Profumo A, Speltini A. Green and Efficient Determination of Fluoroquinolone Residues in Edible Green Fruits and Leafy Vegetables by Ultrasound-Assisted Extraction Followed by HPLC-MS/MS. Molecules. 2022; 27(19):6595. https://doi.org/10.3390/molecules27196595
Chicago/Turabian StyleMerlo, Francesca, Dario Centenaro, Federica Maraschi, Antonella Profumo, and Andrea Speltini. 2022. "Green and Efficient Determination of Fluoroquinolone Residues in Edible Green Fruits and Leafy Vegetables by Ultrasound-Assisted Extraction Followed by HPLC-MS/MS" Molecules 27, no. 19: 6595. https://doi.org/10.3390/molecules27196595