
Sirtuin 1-Activating Compounds: Discovery of a Class of Thiazole-Based Derivatives

 ,
,  , , , ,
, , , ,
Abstract
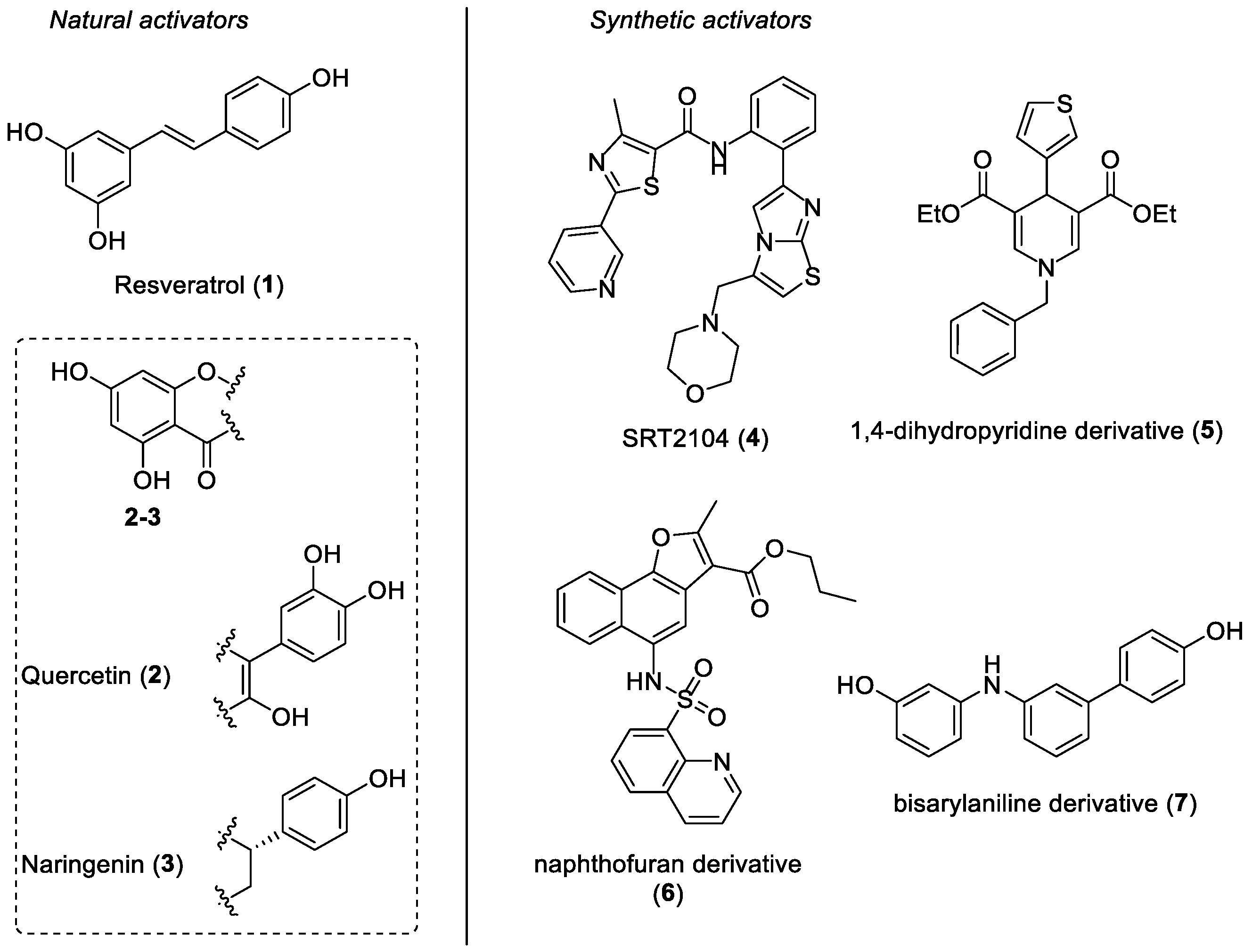
:1. Introduction
2. Results and Discussion
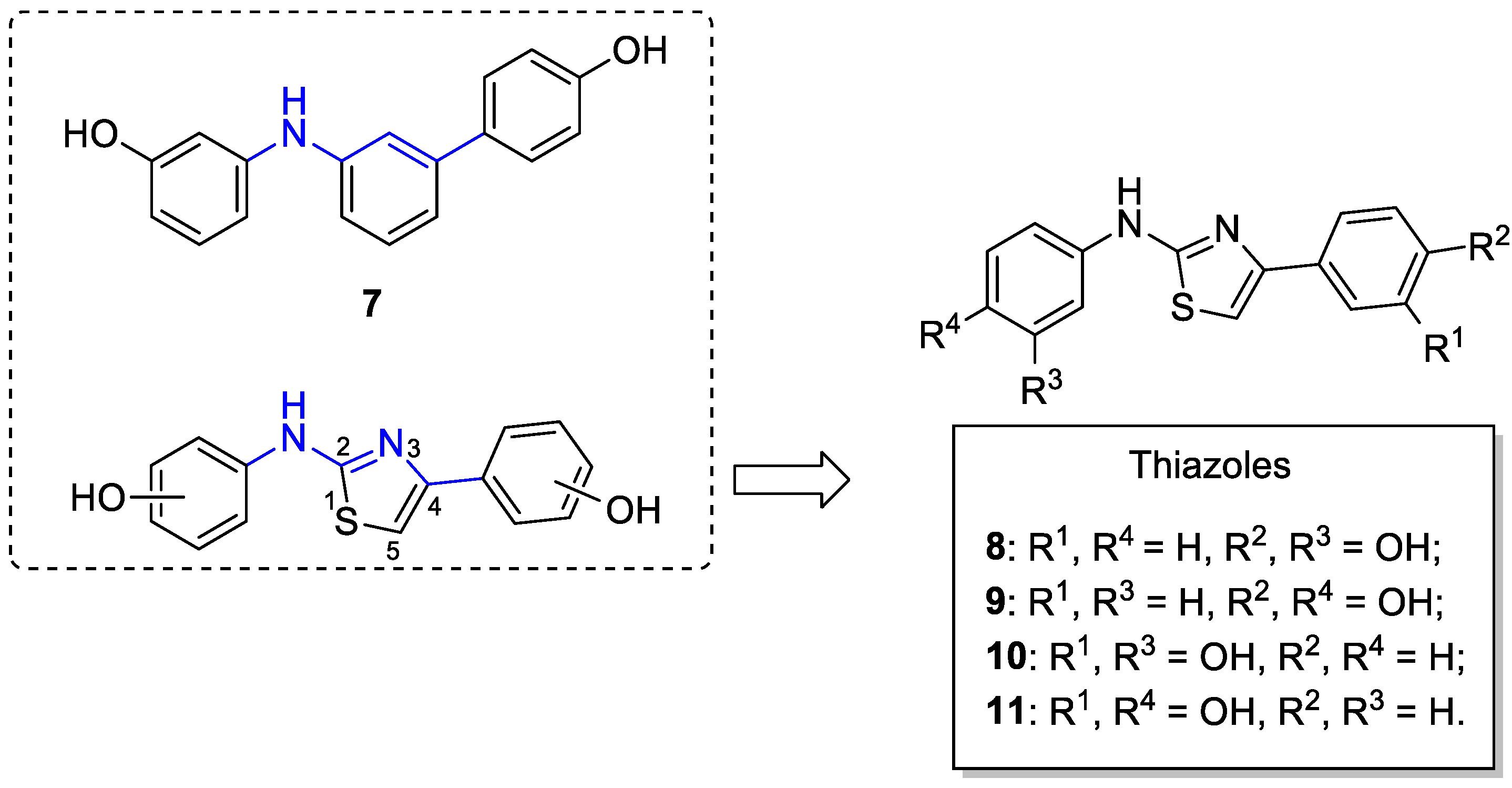
2.1. Chemistry
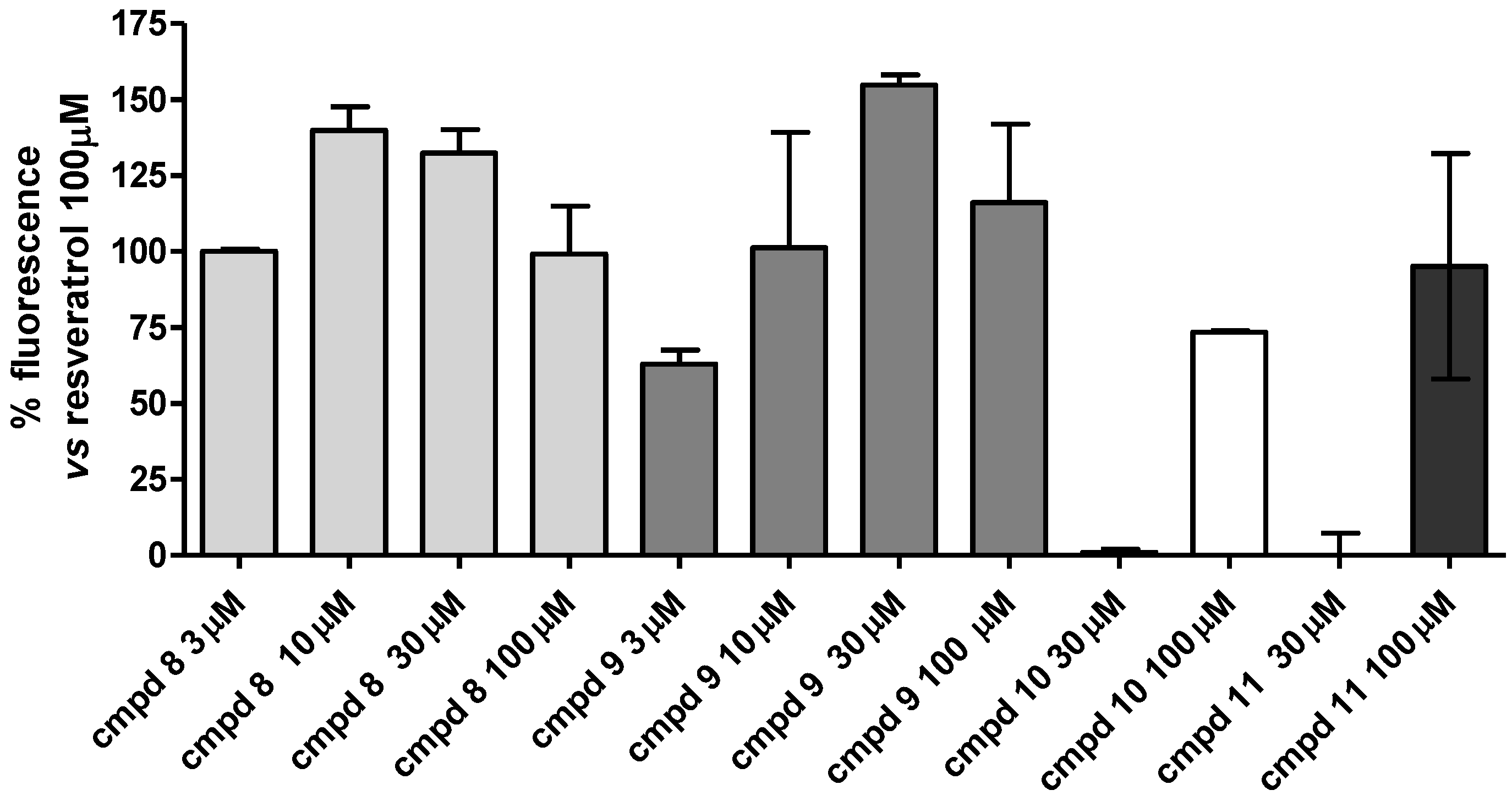
2.2. Enzymatic Assays
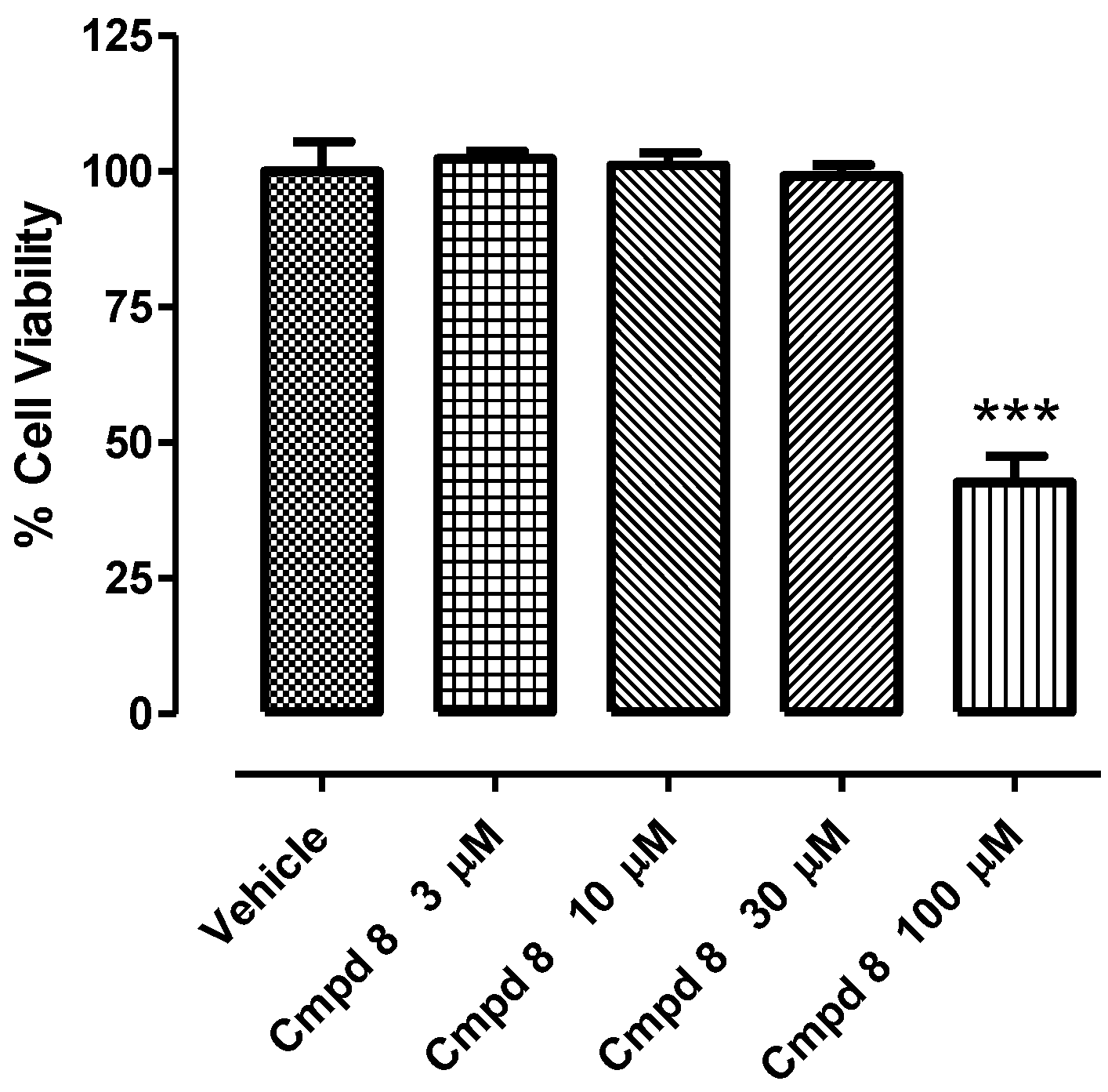
2.3. In Vitro H9c2 Cytotoxicity of Compound 8
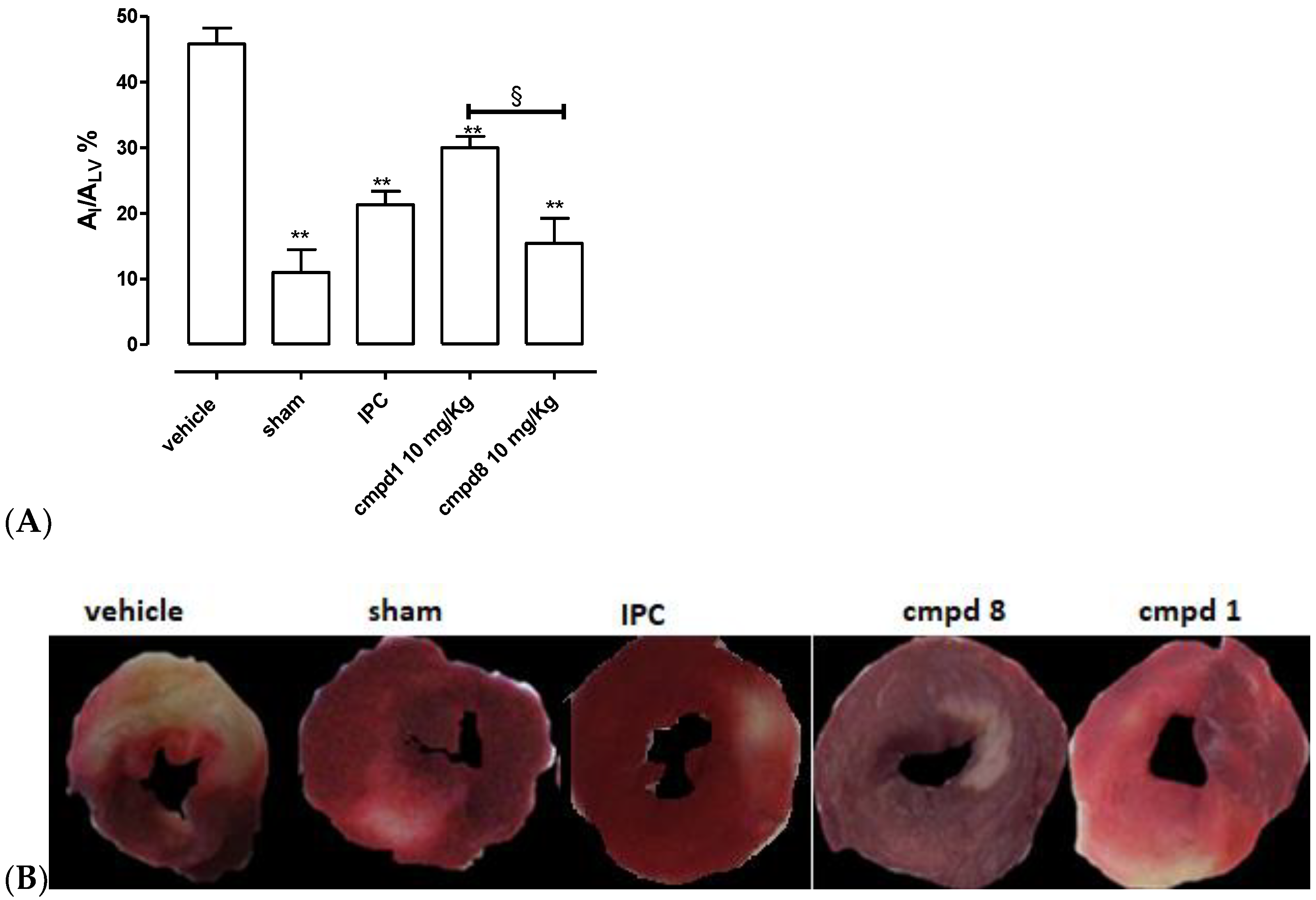
2.4. Cardioprotective Activity of Compound 8 in an In Vivo Acute Myocardial Infarct Model
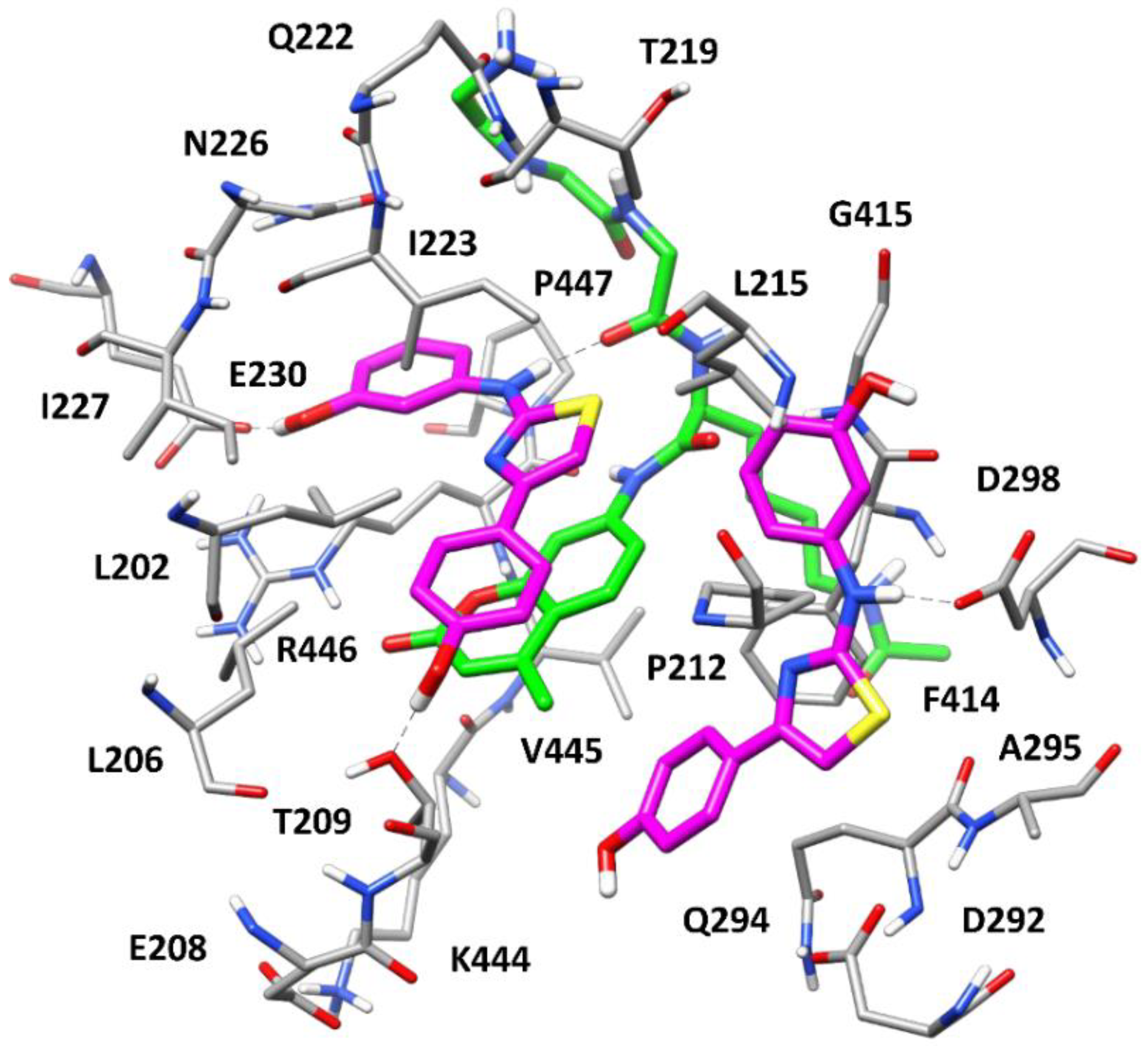
2.5. Molecular Modeling Studies
3. Materials and Methods
3.1. Synthesis. General Procedures and Materials
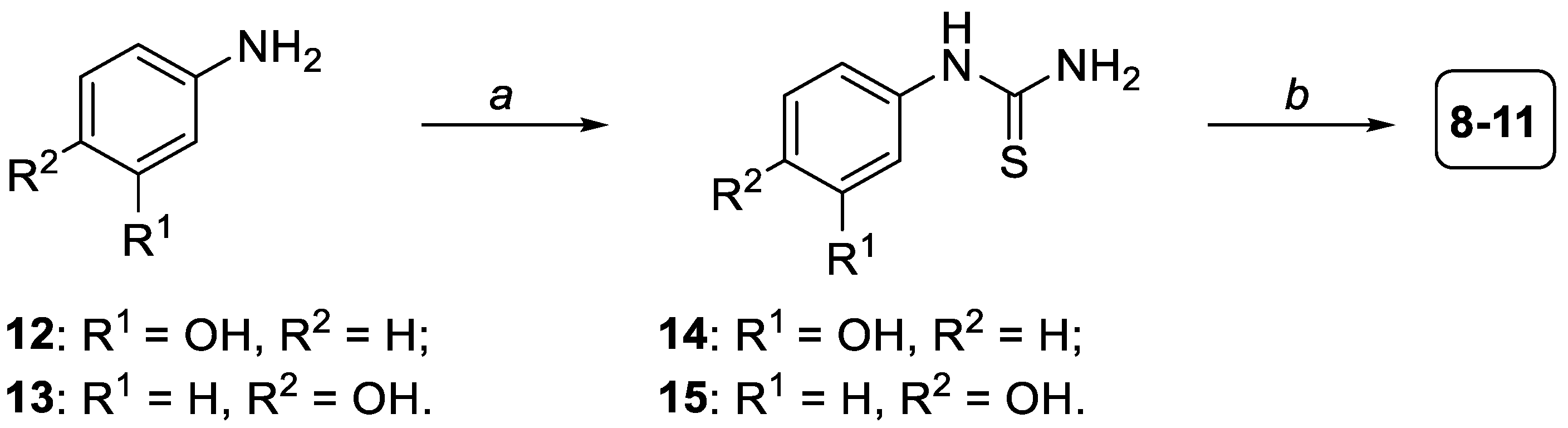
3.1.1. General Procedure for the Formation of Compounds 14,15
3.1.2. General Procedure for the Formation of Compounds 8–11
3.2. In Vitro Screening on Isolated SIRT1 Enzyme
3.3. In Vitro H9c2 Toxicity of Compound 8
3.4. In Vivo Acute Myocardial Infarction
3.5. Docking Calculations
3.6. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Frye, R.A. Phylogenetic Classification of Prokaryotic and Eukaryotic Sir2-like Proteins. Biochem. Biophys. Res. Commun. 2000, 273, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Guarente, L. Sirtuins in Aging and Disease. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Long Island, NY, USA, 2007; Volume 72, pp. 483–488. [Google Scholar]
- Haigis, M.C.; Sinclair, D.A. Mammalian Sirtuins: Biological Insights and Disease Relevance. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 253–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanno, M.; Sakamoto, J.; Miura, T.; Shimamoto, K.; Horio, Y. Nucleocytoplasmic Shuttling of the NAD+-Dependent Histone Deacetylase SIRT1. J. Biol. Chem. 2007, 282, 6823–6832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauve, A.A.; Schramm, V.L. Sir2 Regulation by Nicotinamide Results from Switching between Base Exchange and Deacetylation Chemistry. Biochemistry 2003, 42, 9249–9256. [Google Scholar] [CrossRef]
- Sauve, A.A.; Celic, I.; Avalos, J.; Deng, H.; Boeke, J.D.; Schramm, V.L. Chemistry of Gene Silencing: The Mechanism of NAD+-Dependent Deacetylation Reactions. Biochemistry 2001, 40, 15456–15463. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K. SIRT1: Regulation of Longevity via Autophagy. Cell. Signal. 2009, 21, 1356–1360. [Google Scholar] [CrossRef]
- Chen, C.; Zhou, M.; Ge, Y.; Wang, X. SIRT1 and Aging Related Signaling Pathways. Mech. Ageing Dev. 2020, 187, 111215. [Google Scholar] [CrossRef]
- Alves-Fernandes, D.K.; Jasiulionis, M.G. The Role of SIRT1 on DNA Damage Response and Epigenetic Alterations in Cancer. Int. J. Mol. Sci. 2019, 20, 3153. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.; Ubaid, S. Role of Silent Information Regulator 1 (SIRT1) in Regulating Oxidative Stress and Inflammation. Inflammation 2020, 43, 1589–1598. [Google Scholar] [CrossRef]
- Li, X. SIRT1 and Energy Metabolism. Acta Biochim. Biophys. Sin. 2013, 45, 51–60. [Google Scholar] [CrossRef]
- Wu, Y.-J.; Fang, W.-J.; Pan, S.; Zhang, S.-S.; Li, D.-F.; Wang, Z.-F.; Chen, W.-G.; Yin, Q.; Zuo, J. Regulation of Sirt1 on Energy Metabolism and Immune Response in Rheumatoid Arthritis. Int. Immunopharmacol. 2021, 101, 108175. [Google Scholar] [CrossRef] [PubMed]
- Karbasforooshan, H.; Karimi, G. The Role of SIRT1 in Diabetic Cardiomyopathy. Biomed. Pharmacother. 2017, 90, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.R.P.; Milner, J. SIRT1, Metabolism and Cancer. Curr. Opin. Oncol. 2012, 24, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Quan, N.; Sun, W.; Chen, X.; Cates, C.; Rousselle, T.; Zhou, X.; Zhao, X.; Li, J. Cardiomyocyte-Specific Deletion of Sirt1 Gene Sensitizes Myocardium to Ischaemia and Reperfusion Injury. Cardiovasc. Res. 2018, 114, 805–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granchi, C.; Minutolo, F. Activators of Sirtuin-1 and Their Involvement in Cardioprotection. Curr. Med. Chem. 2018, 25, 4432–4456. [Google Scholar] [CrossRef]
- Iside, C.; Scafuro, M.; Nebbioso, A.; Altucci, L. SIRT1 Activation by Natural Phytochemicals: An Overview. Front. Pharmacol. 2020, 11, 1225. [Google Scholar] [CrossRef]
- Han, D.; Wang, J.; Ma, S.; Chen, Y.; Cao, F. SIRT1 as a Promising Novel Therapeutic Target for Myocardial Ischemia Reperfusion Injury and Cardiometabolic Disease. Curr. Drug Targets 2017, 18, 1746–1753. [Google Scholar] [CrossRef]
- Wang, D.; Cao, H.; Wang, X.; Wang, J.; Wang, M.; Zhang, J.; Wang, L. SIRT1 Is Required for Exercise-Induced Beneficial Effects on Myocardial Ischemia/Reperfusion Injury. J. Inflamm. Res. 2021, 14, 1283–1296. [Google Scholar] [CrossRef]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.-L.; et al. Small Molecule Activators of Sirtuins Extend Saccharomyces Cerevisiae Lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef]
- Wood, J.G.; Regina, B.; Lavu, S.; Hewitz, K.; Helfand, S.L.; Tatar, M.; Sinclair, D. Sirtuin Activators Mimic Caloric Restriction and Delay Ageing in Metazoans. Nature 2004, 430, 686–689. [Google Scholar] [CrossRef]
- Yu, C.; Shin, Y.G.; Chow, A.; Li, Y.; Kosmeder, J.W.; Lee, Y.S.; Hirschelman, W.H.; Pezzuto, J.M.; Mehta, R.G.; van Breemen, R.B. Human, Rat, and Mouse Metabolism of Resveratrol. Pharm. Res. 2002, 19, 1907–1914. [Google Scholar] [CrossRef] [PubMed]
- Henderson, R.F. Species Differences in the Metabolism of Olefins: Implications for Risk Assessment. Chem. Biol. Interact. 2001, 135–136, 53–64. [Google Scholar] [CrossRef]
- Bononi, G.; Flori, L.; Citi, V.; Acciai, C.; Nocilla, V.; Martelli, A.; Poli, G.; Tuccinardi, T.; Granchi, C.; Testai, L.; et al. New Synthetic Analogues of Natural Polyphenols as Sirtuin 1-Activating Compounds. Pharmaceuticals 2022, 15, 339. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Lu, L.; Liu, Y.; Ma, J.; Yang, L.; Li, L.; Guo, H.; Yu, S.; Ren, J.; Bai, H.; et al. Quercetin Improve Ischemia/Reperfusion-Induced Cardiomyocyte Apoptosis in Vitro and in Vivo Study via SIRT1/PGC-1α Signaling. J. Cell. Biochem. 2019, 120, 9747–9757. [Google Scholar] [CrossRef] [PubMed]
- Testai, L.; Piragine, E.; Piano, I.; Flori, L.; Da Pozzo, E.; Miragliotta, V.; Pirone, A.; Citi, V.; Di Cesare Mannelli, L.; Brogi, S.; et al. The Citrus Flavonoid Naringenin Protects the Myocardium from Ageing-Dependent Dysfunction: Potential Role of SIRT1. Oxid. Med. Cell. Longev. 2020, 2020, 4650207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, E.; Wald, J.; Lavu, S.; Roberts, J.; Beaumont, C.; Haddad, J.; Elliott, P.; Westphal, C.; Jacobson, E. Pharmacokinetics and Tolerability of SRT2104, a First-in-Class Small Molecule Activator of SIRT1, after Single and Repeated Oral Administration in Man. Br. J. Clin. Pharmacol. 2013, 75, 186–196. [Google Scholar] [CrossRef] [Green Version]
- Libri, V.; Brown, A.P.; Gambarota, G.; Haddad, J.; Shields, G.S.; Dawes, H.; Pinato, D.J.; Hoffman, E.; Elliot, P.J.; Vlasuk, G.P.; et al. A Pilot Randomized, Placebo Controlled, Double Blind Phase I Trial of the Novel SIRT1 Activator SRT2104 in Elderly Volunteers. PLoS ONE 2012, 7, e51395. [Google Scholar] [CrossRef]
- Mercken, E.M.; Mitchell, S.J.; Martin-Montalvo, A.; Minor, R.K.; Almeida, M.; Gomes, A.P.; Scheibye-Knudsen, M.; Palacios, H.H.; Licata, J.J.; Zhang, Y.; et al. SRT2104 Extends Survival of Male Mice on a Standard Diet and Preserves Bone and Muscle Mass. Aging Cell 2014, 13, 787–796. [Google Scholar] [CrossRef]
- Jiang, M.; Zheng, J.; Peng, Q.; Hou, Z.; Zhang, J.; Mori, S.; Ellis, J.L.; Vlasuk, G.P.; Fries, H.; Suri, V.; et al. Sirtuin 1 Activator SRT2104 Protects Huntington’s Disease Mice. Ann. Clin. Transl. Neurol. 2014, 1, 1047–1052. [Google Scholar] [CrossRef] [Green Version]
- Venkatasubramanian, S.; Noh, R.M.; Daga, S.; Langrish, J.P.; Joshi, N.V.; Mills, N.L.; Hoffmann, E.; Jacobson, E.W.; Vlasuk, G.P.; Waterhouse, B.R.; et al. Cardiovascular Effects of a Novel SIRT1 Activator, SRT2104, in Otherwise Healthy Cigarette Smokers. J. Am. Heart Assoc. 2013, 2, e000042. [Google Scholar] [CrossRef]
- Venkatasubramanian, S.; Noh, R.M.; Daga, S.; Langrish, J.P.; Mills, N.L.; Waterhouse, B.R.; Hoffmann, E.; Jacobson, E.W.; Lang, N.N.; Frier, B.M.; et al. Effects of the Small Molecule SIRT1 Activator, SRT2104 on Arterial Stiffness in Otherwise Healthy Cigarette Smokers and Subjects with Type 2 Diabetes Mellitus. Open Heart 2016, 3, e000402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valente, S.; Mellini, P.; Spallotta, F.; Carafa, V.; Nebbioso, A.; Polletta, L.; Carnevale, I.; Saladini, S.; Trisciuoglio, D.; Gabellini, C.; et al. 1,4-Dihydropyridines Active on the SIRT1/AMPK Pathway Ameliorate Skin Repair and Mitochondrial Function and Exhibit Inhibition of Proliferation in Cancer Cells. J. Med. Chem. 2016, 59, 1471–1491. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Chen, Q.-Q.; Huang, Y.; Li, K.-H.; Geng, X.-J.; Wang, T.; Lin, Q.-S.; Yao, R.-S. Design, Synthesis and Pharmacological Evaluation of Naphthofuran Derivatives as Potent SIRT1 Activators. Front. Pharmacol. 2021, 12, 653233. [Google Scholar] [CrossRef]
- Heng, S.; Gryncel, K.R.; Kantrowitz, E.R. A Library of Novel Allosteric Inhibitors against Fructose 1,6-Bisphosphatase. Bioorg. Med. Chem. 2009, 17, 3916–3922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bursavich, M.G.; Parker, D.P.; Willardsen, J.A.; Gao, Z.-H.; Davis, T.; Ostanin, K.; Robinson, R.; Peterson, A.; Cimbora, D.M.; Zhu, J.-F.; et al. 2-Anilino-4-Aryl-1,3-Thiazole Inhibitors of Valosin-Containing Protein (VCP or P97). Bioorg. Med. Chem. Lett. 2010, 20, 1677–1679. [Google Scholar] [CrossRef]
- Xu, R.-Y.; Xu, X.-W.; Deng, Y.-Z.; Ma, Z.-X.; Li, X.-R.; Zhao, L.; Qiu, L.-J.; Liu, H.-Y.; Chen, H.-P. Resveratrol Attenuates Myocardial Hypoxia/Reoxygenation-Induced Cell Apoptosis through DJ-1-Mediated SIRT1-P53 Pathway. Biochem. Biophys. Res. Commun. 2019, 514, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Fourny, N.; Lan, C.; Sérée, E.; Bernard, M.; Desrois, M. Protective Effect of Resveratrol against Ischemia-Reperfusion Injury via Enhanced High Energy Compounds and ENOS-SIRT1 Expression in Type 2 Diabetic Female Rat Heart. Nutrients 2019, 11, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantazi, E.; Zaouali, M.A.; Bejaoui, M.; Folch-Puy, E.; Ben Abdennebi, H.; Roselló-Catafau, J. Role of Sirtuins in Ischemia-Reperfusion Injury. World J. Gastroenterol. 2013, 19, 7594–7602. [Google Scholar] [CrossRef] [Green Version]
- Cao, D.; Wang, M.; Qiu, X.; Liu, D.; Jiang, H.; Yang, N.; Xu, R.-M. Structural Basis for Allosteric, Substrate-Dependent Stimulation of SIRT1 Activity by Resveratrol. Genes Dev. 2015, 29, 1316–1325. [Google Scholar] [CrossRef] [Green Version]
- Li, G.-Y.; Vaulina, D.D.; Li, J.-J.; Fedorova, O.S.; Wang, H.-E.; Liu, R.-S.; Krasikova, R.N.; Chen, C.-L. Synthesis and Biological Evaluation of 2-(3,4-Dimethoxyphenyl)-6-(2-[18F]Fluoroethoxy)Benzothiazole ([18F]FEDBT) for PET Imaging of Breast Cancer. Bioorg. Med. Chem. Lett. 2017, 27, 3460–3463. [Google Scholar] [CrossRef]
- Calderone, V.; Testai, L.; Martelli, A.; Rapposelli, S.; Digiacomo, M.; Balsamo, A.; Breschi, M.C. Anti-Ischemic Properties of a New Spiro-Cyclic Benzopyran Activator of the Cardiac Mito-KATP Channel. Biochem. Pharmacol. 2010, 79, 39–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flori, L.; Petrarolo, G.; Brogi, S.; La Motta, C.; Testai, L.; Calderone, V. Identification of Novel SIRT1 Activators Endowed with Cardioprotective Profile. Eur. J. Pharm. Sci. 2021, 165, 105930. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C.; Bononi, G.; Ferrisi, R.; Gori, E.; Mantini, G.; Glasmacher, S.; Poli, G.; Palazzolo, S.; Caligiuri, I.; Rizzolio, F.; et al. Design, Synthesis and Biological Evaluation of Second-Generation Benzoylpiperidine Derivatives as Reversible Monoacylglycerol Lipase (MAGL) Inhibitors. Eur. J. Med. Chem. 2021, 209, 112857. [Google Scholar] [CrossRef] [PubMed]
- Testai, L.; Martelli, A.; Marino, A.; D’Antongiovanni, V.; Ciregia, F.; Giusti, L.; Lucacchini, A.; Chericoni, S.; Breschi, M.C.; Calderone, V. The Activation of Mitochondrial BK Potassium Channels Contributes to the Protective Effects of Naringenin against Myocardial Ischemia/Reperfusion Injury. Biochem. Pharmacol. 2013, 85, 1634–1643. [Google Scholar] [CrossRef]
- Suzuki, K.; Hayashi, R.; Ichikawa, T.; Imanishi, S.; Yamada, T.; Inomata, M.; Miwa, T.; Matsui, S.; Usui, I.; Urakaze, M.; et al. SRT1720, a SIRT1 Activator, Promotes Tumor Cell Migration, and Lung Metastasis of Breast Cancer in Mice. Oncol. Rep. 2012, 27, 1726–1732. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | % SIRT1 Activation |
|---|---|
| Resveratrol (1) | 100 |
| 7 | 62.7 ± 2.7 |
| 8 | 99.1 ± 15.8 |
| 9 | 116.0 ± 25.9 |
| 10 | 73.4 ± 0.5 |
| 11 | 95.1 ± 37.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bononi, G.; Citi, V.; Lapillo, M.; Martelli, A.; Poli, G.; Tuccinardi, T.; Granchi, C.; Testai, L.; Calderone, V.; Minutolo, F. Sirtuin 1-Activating Compounds: Discovery of a Class of Thiazole-Based Derivatives. Molecules 2022, 27, 6535. https://doi.org/10.3390/molecules27196535
Bononi G, Citi V, Lapillo M, Martelli A, Poli G, Tuccinardi T, Granchi C, Testai L, Calderone V, Minutolo F. Sirtuin 1-Activating Compounds: Discovery of a Class of Thiazole-Based Derivatives. Molecules. 2022; 27(19):6535. https://doi.org/10.3390/molecules27196535
Chicago/Turabian StyleBononi, Giulia, Valentina Citi, Margherita Lapillo, Alma Martelli, Giulio Poli, Tiziano Tuccinardi, Carlotta Granchi, Lara Testai, Vincenzo Calderone, and Filippo Minutolo. 2022. "Sirtuin 1-Activating Compounds: Discovery of a Class of Thiazole-Based Derivatives" Molecules 27, no. 19: 6535. https://doi.org/10.3390/molecules27196535