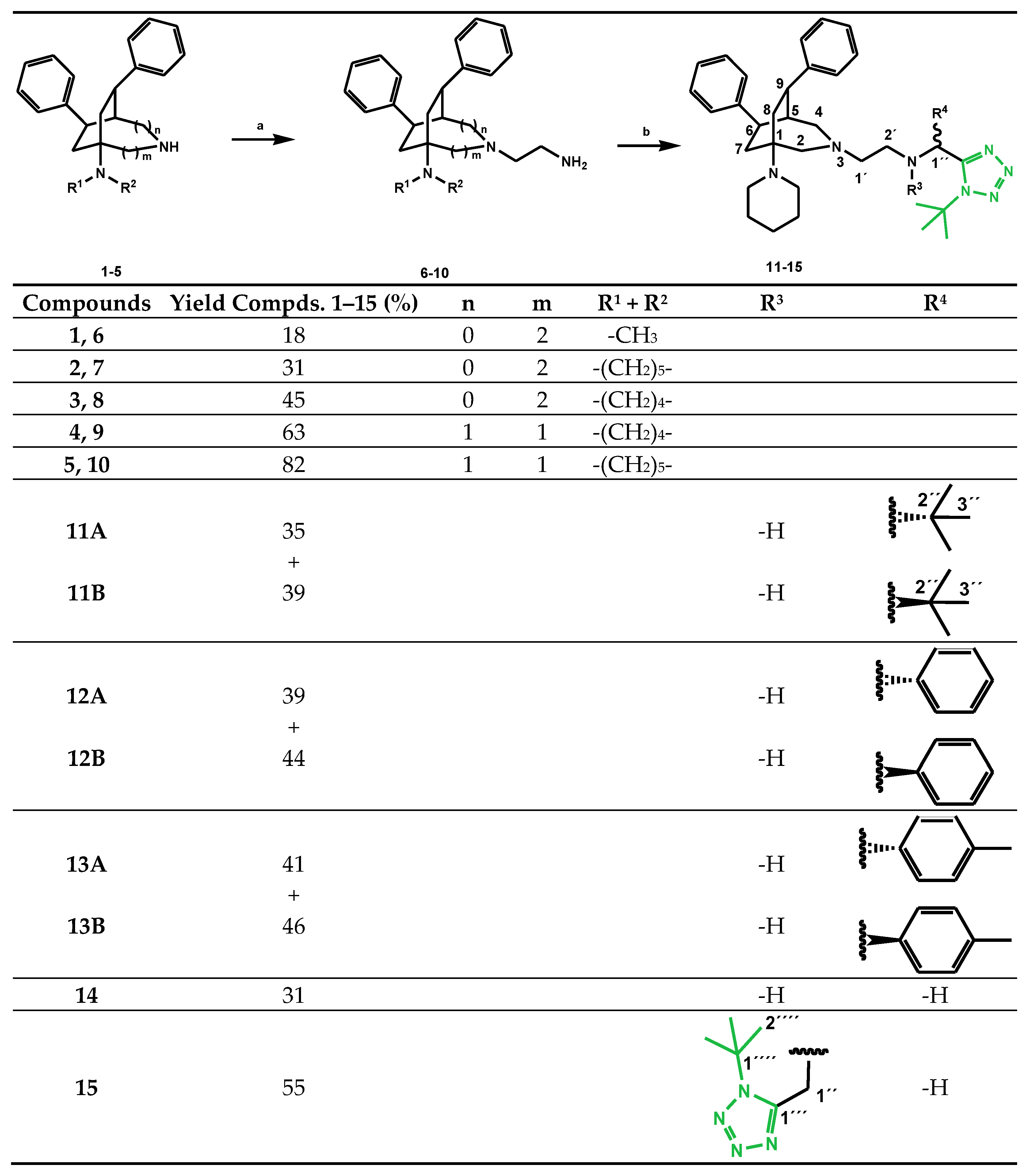
4.2.1. General Procedure for the Synthesis of rac-(7R,8R)-2-[5-(dialkylamino)-7,8-diphenyl-2-azabicyclo[3.2.2]nonan-2-yl]ethan-1-amines 6–8 and rac-(6R,9R)-2-[1-(dialkylamino)-6, 9-diphenyl-3-azabicyclo[3.2.2]nonan-3-yl]ethan-1-amines 9 and 10
The bicyclononane 1–5 was dissolved in 20 mL dry ethanol and cooled on an ice bath with stirring under an atmosphere of argon. A solution of chloroacetamide in 12 mL dry ethanol was added. The mixture was refluxed for 48 h at 100 °C, allowed to cool to room temperature, diluted with water and alkalized with 2N NaOH. It was then extracted 4 times with diethyl ether. The combined organic phases were washed with water, dried over anhydrous sodium sulfate, filtered and finally the solvent was removed in vacuo. The obtained crude products were purified, if necessary, by column chromatography giving the corresponding 2-substituted acetamide derivate as colorless resin. It was suspended in 20 mL dry diethyl ether under stirring and cooling in an ice bath. LiAlH4 was added in portions and the mixture was refluxed overnight. The reaction was cautiously quenched with ice water; 2N NaOH was added and the mixture was extracted 5 times with CH2Cl2; the combined organic phases were washed twice with water, dried over anhydrous sodium sulfate, filtered and finally the solvent was removed in vacuo giving colorless oils.
rac-(7R,8R)-2-[5-(Dimethylamino)-7,8-diphenyl-2-azabicyclo[3.2.2]nonan-2-yl]ethan-1-amine (6)
The reaction of 0.932 g 2-azabicyclo-nonane 1 (2.91 mmol) and 0.272 g chloroacetamide (2.91 mmol) gave after 48 h a crude product. It was suspended in dry diethyl ether and reacted with 0.270 g LiAlH4 (7.115 mmol) to a residue which was purified by multiple column chromatography (first time on neutral aluminum oxide, CH2Cl2/MeOH = 29 + 1; second time on neutral aluminum oxide, CH2Cl2/MeOH = 9 + 1; and finally CH2Cl2/MeOH = 1 + 1) yielding 0.056 g 6 (18%) as colorless oil. IR = 3024, 2930, 2823, 2780, 1600, 1494, 1450, 1154, 1093, 1034, 757, 700; UV (CH2Cl2, (log ε)): 233.5 (3.848); 1H NMR (CDCl3, 400 MHz): δ = 1.90–1.95 (m, 2H, 4-H), 2.02 (dd, J = 13.5, 7.7 Hz, 1H, 6-H), 2.12–2.26 (m, 2H, 2’-H, 9-H), 2.29–2.39 (m, 3H, 2’-H, 6-H, 9-H), 2.37 (s, 6H, N(CH3)2), 2.44–2.50 (m, 2H, 1’-H), 2.72 (d, J = 3.5 Hz, 1H, 1-H), 2.75–2.82 (m, 1H, 3-H), 2.96 (dt, J = 13.0, 5.6 Hz, 1H, 3-H), 3.26 (ddd, J = 11.0, 7.4, 3.5 Hz, 1H, 8-H), 3.45 (dd, J = 10.3, 7.7 Hz, 1H, 7-H), 7.14–7.36 (m, 10H, aromatic H); 13C NMR (CDCl3, 100 MHz): δ = 31.30 (C-4), 34.67 (C-6), 36.33 (C-9), 37.78 (C-8), 37.95 (N(CH3)2), 39.59 (C-2’), 39.75 (C-7), 48.58 (C-3), 58.06 (C-5), 59.18 (C-1’), 69.00 (C-1), 126.19, 126.23, 127.55, 128.10, 128.70, 128.72 (aromatic C), 144.56, 145.70 (aromatic Cq). HRMS (EI+) calcd for C24H33N3: 363.2675; found: 363.2685.
rac-(7R,8R)-2-[7,8-Diphenyl-5-(piperidin-1-yl)-2-azabicyclo[3.2.2]nonan-2-yl]ethan-1-amine (7)
The reaction of 0.200 g 2-azabicyclo-nonane 2 (0.556 mmol) and 0.052 g chloroacetamide (0.556 mmol) gave after 48 h a crude product. It was suspended in dry diethyl ether and reacted with 0.073 g LiAlH4 (1.912 mmol) to a residue, which was purified by multiple column chromatography (first time on neutral aluminum oxide, CH2Cl2/MeOH = 49 + 1; and finally on neutral aluminum oxide CH2Cl2/MeOH = 1 + 1) yielding 0.038 g 7 (31%) as colorless oil. IR = 3024, 2927, 2852, 1600, 1494, 1451, 1153, 1098, 1031, 757, 700; UV (CH2Cl2, (log ε)): 232.5 (3.881); 1H NMR (CDCl3, 400 MHz): δ = 1.44–1.52 (m, 2H, CH2), 1.62–1.71 (m, 4H, 2CH2), 1.93–2.07 (m, 3H, 4-H, 6-H), 2.16–2.48 (m, 7H, 1’-H, 2’-H, 6-H, 9-H), 2.63–2.73 (m, 4H, N(CH2)2), 2.77 (d, J = 2.9 Hz, 1H, 1-H), 2.77–2.82 (m, 1H, 3-H), 2.97 (dt, J = 13.2, 5.7 Hz, 1H, 3-H), 3.25 (ddd, J = 10.9, 7.7, 3.2 Hz, 1H, 8-H), 3.43 (br t, J = 9.1 Hz, 1H, 7-H), 7.15–7.38 (m, 10H, aromatic H); 13C NMR (CDCl3, 100 MHz): δ = 24.78 (CH2), 26.42 (2CH2), 32.62 (C-4), 34.48 (C-6), 35.99 (C-9), 38.17 (C-8), 39.63 (C-2’), 40.37 (C-7), 46.28 (N(CH2)2), 48.50 (C-3), 58.95 (C-5), 59.34 (C-1’), 68.86 (C-1), 126.08, 126.14, 127.49, 128.03, 128.54, 128.64 (aromatic C), 144.51, 145.72 (aromatic Cq). HRMS (EI+) calcd for C27H37N3: 403.2987; found: 403.2996.
rac-(7R,8R)-2-[7,8-Diphenyl-5-(pyrrolidin-1-yl)-2-azabicyclo[3.2.2]nonan-2-yl]ethan-1-amine (8)
The reaction of 0.270 g 2-azabicyclo-nonane 3 (0.750 mmol) and 0.070 g chloroacetamide (0.750 mmol) gave after 48 h a crude product. It was suspended in dry diethyl ether and reacted with 0.119 g LiAlH4 (3.135 mmol), yielding 0.118 g 8 (45%) as colorless oil. IR = 3024, 2926, 2854, 1600, 1494, 1450, 1116, 1031, 755, 700; UV (CH2Cl2, (log ε)): 232.5 (3.838); 1H NMR (CDCl3, 400 MHz): δ = 1.74–1.81 (m, 4H, (CH2)2), 1.95–2.01 (m, 2H, 4-H), 2.08 (dd, J = 13.8, 8.0 Hz, 1H, 6-H), 2.16–2.38 (m, 5H, 2’-H, 6-H, 9-H), 2.42 (t, J = 5.4 Hz, 2H, 1’-H), 2.74–2.83 (m, 6H, 1-H, 3-H, N(CH2)2), 2.95 (dt, J = 12.8, 5.5 Hz, 1H, 3-H), 3.25 (ddd, J = 10.9, 7.6, 3.3 Hz, 1H, 8-H), 3.45 (dd, J = 9.7, 8.3 Hz, 1H, 7-H), 7.14–7.39 (m, 10H, aromatic H); 13C NMR (CDCl3, 100 MHz): δ = 23.58 ((CH2)2), 33.20 (C-4), 35.52 (C-6), 36.64 (C-9), 37.83 (C-8), 39.59 (C-2’), 39.79 (C-7), 45.10 (N(CH2)2), 48.63 (C-3), 56.71 (C-5), 60.06 (C-1’), 69.01 (C-1), 125.83, 125.97, 127.50, 127.79, 128.52, 128.68 (aromatic C), 144.64, 146.00 (aromatic Cq). HRMS (EI+) calcd for C26H35N3: 389.2831; found: 389.2837.
rac-(6R,9R)-2-[6,9-Diphenyl-1-(pyrrolidin-1-yl)-3-azabicyclo[3.2.2]nonan-3-yl]ethan-1-amine (9)
The reaction of 0.874 g 3-azabicyclo-nonane 4 (2.5 mmol) and 0.236 g chloroacetamide (2.5 mmol) gave after 48 h a crude product. It was suspended in dry diethyl ether and reacted with 0.300 g LiAlH4 (7.9 mmol), yielding 0.625 g 9 (63%) as colorless oil. IR = 3420, 2953, 2624, 2484, 1600, 1496, 1452, 1031, 753, 702; 1H NMR (CDCl3, 400 MHz): δ = 1.76 (br s, 4H, (CH2)2), 1.87–2.05 (m, 3H, 5-H, 7-H, 8-H), 2.29–2.35 (m, 2H, 4-H, 7-H), 2.49–2.61 (m, 2H, 1’-H), 2.63–2.90 (m, 8H, 2-H, 8-H, N(CH2)2, 2’-H), 2.97–3.03 (m, 2H, 2-H, 4-H), 3.30–3.36 (m, 2H, 6-H, 9-H), 7.16–7.42 (m, 10H, aromatic H); 13C NMR (CDCl3, 100 MHz): δ = 23.52 ((CH2)2), 34.84 (C-8), 38.04 (C-7), 38.81 (C-9), 39.39 (C-2’), 44.56 (C-5), 44.91 (C-6), 45.32 (N(CH2)2), 58.03 (C-1), 58.78 (C-2), 60.72 (C-4), 61.12 (C-1’), 126.01, 126.11, 126.87, 128.17, 128.27, 128.54 (aromatic C), 144.92, 146.64 (aromatic Cq); HRMS (EI+): calcd for C26H35N3: 389.2831; found: 389.2834.
rac-(6R,9R)-2-[6,9-Diphenyl-1-(piperidin-1-yl)-3-azabicyclo[3.2.2]nonan-3-yl]ethan-1-amine (10)
The reaction of 1.14 g 3-azabicyclo-nonane 5 (3.16 mmol) and 0.295 g chloroacetamide (3.16 mmol) gave after 48 h a crude product. It was suspended in dry diethyl ether and reacted with 0.413 g LiAlH4 (10.88 mmol), yielding 0.80 g 10 (82%) as colorless oil. 1H NMR (CDCl3) δ = 1.39–1.48 (m, 2H, CH2), 1.54–1.64 (m, 4H, 2CH2), 1.84–1.94 (m, 2H, 7-H, 8-H), 1.96–2.01 (m, 1H, 5-H), 2.24–2.29 (m, 1H, 7-H), 2.31 (d, J = 11.5 Hz, 1H, 4-H), 2.49–2.65 (m, 8H, 1’-H, 2-H, 8-H, N(CH2)2), 2.77–2.91 (m, 2H, 2’-H), 2.95 (d, J = 11.8 Hz, 1H, 2-H), 3.01 (dd, J = 11.4, 5.2 Hz, 1H, 4-H), 3.26–3.31 (m, 2H, 6-H, 9-H), 7.15–7.42 (m, 10H, aromatic H); 13C NMR (CDCl3) δ = 25.03 (CH2), 26.74 (2CH2), 34.04 (C-8), 37.39 (C-7), 39.24 (C-9), 39.48 (C-2’), 44.18 (C-5), 45.01 (C-6), 46.52 (N(CH2)2), 58.60 (C-2), 59.81 (C-1), 60.80 (C-4), 61.22 (C-1’), 126.05, 126.13, 126.80, 128.20, 128.30, 128.56 (aromatic C), 145.04, 146.70 (aromatic Cq); HRMS (EI+) calcd for C27H37N3: 403.2987; found: 403.2979.
4.2.2. General Procedure for the Synthesis of N-[(1-tert-butyl-1H-tetrazol-5-yl)methyl]-2-[6,9-diphenyl-1-(piperidin-1-yl)-3-azabicyclo[3.2.2]nonan-3-yl]ethan-1-amines 11–15
2-(3-Azabicyclononan-3-yl)ethan-1-amine 10 was dissolved in dry MeOH in an atmosphere of Ar. The corresponding aldehyde was added and the reaction batch was stirred for 1 h at ambient temperature. Subsequently, tert-butylisocyanide and trimethylsilylazide were added and the reaction was stirred for an additional 20 h at ambient temperature in an atmosphere of Ar. Subsequently, the solvent was evaporated in vacuo yielding crude products 11–15, which were separated by column chromatography into their isomers (silica, diethyl ether/dioxane/MeOH = 25 + 1 + 1).
rac-(1R)-1-(1-tert-Butyl-1H-tetrazol-5-yl)-2,2-dimethyl-N-{2-[(6R,9R)-6,9-diphenyl-1-(piperidin-1-yl)-3-azabicyclo[3.2.2]nonan-3-yl]ethyl}propan-1-amine (11A) and rac-(1R)-1-(1-tert-Butyl-1H-tetrazol-5-yl)-2,2-dimethyl-N-{2-[(6S,9S)-6,9-diphenyl-1-(piperidin-1-yl)-3-azabicyclo[3.2.2]nonan-3-yl]ethyl}propan-1-amine (11B)
The reaction batch of 0.330 g 2-(3-azabicyclononan-3-yl)ethan-1-amine 10 (0.81 mmol), 0.079 g trimethylacetaldehyde (0.930 mmol), 0.072 g tert-butylisocyanide (0.830 mmol) and 0.104 g trimethylsilylazide (0.870 mmol) in 4.5 mL dry MeOH was stirred for 20 h. Subsequently, the solvent was evaporated in vacuo. Solvent chromatography gave 0.171 g (35%) of 11A and 0.191 g (39%) of 11B.
11A: IR = 2933, 1601, 1495, 1451, 1392, 1297, 1241, 1103, 1031, 909, 814, 744, 699; 1H NMR (CDCl3, 400 MHz): δ = 1.09 (s, 9H, 3’’-H), 1.39–1.47 (m, 2H, CH2), 1.51–1.62 (m, 4H, 2CH2), 1.80 (s, 9H, 2’’’’-H), 1.80–1.94 (m, 3H, 5-H, 7-H, 8-H), 2.16–2.24 (m, 1H, 7-H), 2.30 (d, J = 11.4 Hz, 1H, 4-H), 2.40–2.65 (m, 10H, 1’-H, 2-H, 2’-H, N(CH2)2), 2.84 (d, J = 12.8 Hz, 1H, 2-H), 2.89 (dd, J = 11.4, 4.8 Hz, 1H, 4-H), 3.21–3.30 (m, 2H, 6-H, 9-H), 4.02 (s, 1H, 1’’-H), 7.17 (t, J = 7.3 Hz, 1H, aromatic H), 7.19–7.44 (m, 9H, aromatic H); 13C NMR (CDCl3, 100 MHz): δ = 25.04 (CH2), 26.70 (2CH2), 27.21 (C-3’’), 31.41 (C-2’’’’), 33.71 (C-8), 36.29 (C-2’’), 37.66 (C-7), 39.34 (C-9), 44.42 (C-5), 44.83 (C-6), 46.39 (C-2’), 46.50 (N(CH2)2), 57.83 (C-2), 57.97 (C-1’), 59.50 (C-1), 61.47 (C-4), 62.01 (C-1’’’’), 62.45 (C-1’’), 125.90, 126.05, 126.84, 128.17, 128.51, 128.54 (aromatic C), 145.13, 146.79 (aromatic Cq), 157.25 (C-1’’’); HRMS (EI+) calcd for C37H55N7: 597.4519; found: 597.4533.
11B: IR = 2932, 2793, 1601, 1495, 1452, 1393, 1374, 1304, 1239, 1103, 1032, 911, 866, 811, 743, 699; 1H NMR (CDCl3, 400 MHz): δ = 1.07 (s, 9H, 3’’-H), 1.40–1.48 (m, 2H, CH2), 1.52–1.61 (m, 4H, 2CH2), 1.79 (s, 9H, 2’’’’-H), 1.80–1.89 (m, 2H, 7-H, 8-H), 1.91 (br s, 1H, 5-H), 2.20–2.31 (m, 2H, 4-H, 7-H), 2.36–2.42 (m, 1H, 2’-H), 2.47–2.63 (m, 9-H, 1’-H, 2-H, 2’-H, 8-H, N(CH2)2), 2.90 (d, J = 12.1 Hz, 1H, 2-H), 2.88–2.94 (m, 1H, 4-H), 3.19–3.30 (m, 2H, 6-H, 9-H), 4.00 (s, 1H, 1’’-H), 7.17 (d, J = 7.2 Hz, 1H, aromatic H), 7.19–7.40 (m, 9H, aromatic H); 13C NMR (CDCl3, 100 MHz): δ = 25.06 (CH2), 26.75 (2CH2), 27.24 (C-3’’), 31.38 (C-2’’’’), 34.18 (C-8), 36.18 (C-2’’), 37.58 (C-7), 39.48 (C-9), 44.14 (C-5), 44.79 (C-6), 46.58 (N(CH2)2), 46.67 (C-2’), 58.14 (C-1’), 58.33 (C-2), 59.67 (C-1), 60.74 (C-4), 62.15 (C-1’’’’), 62.60 (C-1’’), 126.01, 126.07, 126.79, 128.17, 128.52 (aromatic C), 145.18, 146.74 (aromatic Cq), 157.32 (C-1’’’); HRMS (EI+) C37H55N7: 597.4519; found: 597.4554.
rac-N-[(1R)-(1-tert-Butyl-1H-tetrazol-5-yl])(phenyl)methyl]-2-[(6R,9R)-6,9-diphenyl-1-(piperidin-1-yl)-3-azabicyclo[3.2.2]nonan-3-yl]ethan-1-amine (12A) and rac-N-[(1R)-1-tert-Butyl-1H-tetrazol-5-yl])(phenyl)methyl]-2-[(6S,9S)-6,9-diphenyl-1-(piperidin-1-yl)-3-azabicyclo[3.2.2]nonan-3-yl]ethan-1-amine (12B)
The reaction batch of 0.371 g 2-(3-azabicyclononan-3-yl)ethan-1-amine 10 (0.920 mmol), 0.079 g benzaldehyde (0.920 mmol), 0.099 g tert-butylisocyanide (1.21 mmol) and 0.143 g trimethylsilylazide (1.21 mmol) in 4.5 mL dry MeOH was stirred for 20 h. Subsequently, the solvent was evaporated in vacuo. Solvent chromatography gave 0.221 g (39%) of 12A and 0.250 g of 12B (44%).
12A: IR = 2931, 1600, 1494, 1452, 1374, 1236, 1105, 1030, 851, 744, 700; 1H NMR (CDCl3, 400 MHz): δ = 1.38–1.44 (m, 2H, CH2), 1.50–1.57 (m, 4H, 2CH2), 1.63 (s, 9H, 2’’’’-H), 1.79–1.89 (m, 2H, 7-H, 8-H), 1.93 (br s, 1H, 5-H), 2.21–2.29 (m, 1H, 7-H), 2.31 (d, J = 11.4 Hz, 1H, 4-H), 2.44–2.78 (m, 10H, 1’-H, 2-H, 2’-H, 8-H, N(CH2)2), 2.87 (d, J = 12.6 Hz, 1H, 2-H), 2.93 (dd, J = 11.4, 5.1 Hz, 1H, 4-H), 3.21–3.29 (m, 2H, 6-H, 9-H), 5.31 (s, 1H, 1’’-H), 7.15 (t, J = 7.3 Hz, 1H, aromatic H), 7.18–7.41 (m, 14H, aromatic H); 13C NMR (CDCl3, 100 MHz): δ = 24.98 (CH2), 26.69 (2CH2), 29.99 (C-2’’’’), 34.05 (C-8), 37.43 (C-7), 39.24 (C-9), 44.33 (C-5), 44.82 (C-6), 45.28 (C-2’), 46.45 (N(CH2)2), 57.74 (C-1’), 58.16 (C-2), 59.31 (C-1’’), 59.53 (C-1), 61.02 (C-4), 61.26 (C-1’’’’), 126.03, 126.07, 126.79, 128.08, 128.26, 128.33, 128.41, 128.52, 128.96 (aromatic C), 138.58, 144.74, 146.68 (aromatic Cq), 155.56 (C-1’’’); HRMS (EI+) calcd for C39H51N7: 617.4206; found: 617.4250.
12B: IR = 2930, 1672, 1601, 1494, 1452, 1374, 1237, 1105, 1031, 746, 700; 1H NMR (CDCl3, 400 MHz): δ = 1.41–1.49 (m, 2H, CH2), 154–1.62 (m, 4H, 2CH2), 1.65 (s, 9H, 2’’’’-H), 1.82–1.92 (m, 2H, 7-H, 8-H), 1.94 (br s, 1H, 5-H), 2.20–2.28 (m, 1H, 7-H), 2.31 (d, J = 11.4 Hz, 1H, 4-H), 2.54–2.67 (m, 9H, 1’-H, 2-H, 2’-H, 8-H, N(CH2)2), 2.70–2.79 (m, 1H, 2’-H), 2.87 (dd, J = 11.4, 5.0 Hz, 1H, 4-H), 2.93 (d, J = 12.5 Hz, 1H, 2-H), 3.22–3.28 (m, 1H, 9-H), 3.30 (d, J = 9.7 Hz, 1H, 6-H), 5.33 (s, 1H, 1’’-H), 7.11–7.40 (m, 15H, aromatic H); 13C NMR (CDCl3, 100 MHz): δ = 25.01 (CH2), 26.72 (2CH2), 30.00 (C-2’’’’), 33.87 (C-8), 37.39 (C-7), 39.09 (C-9), 44.28 (C-5), 44.75 (C-6), 45.57 (C-2’), 46.57 (N(CH2)2), 57.97 (C-1’), 58.98 (C-2), 59.26 (C-1’’), 59.60 (C-1), 60.56 (C-4), 61.17 (C-1’’’’), 125.88, 126.05, 126.83, 128.04, 128.14, 128.26, 128.35, 128.51, 128.95 (aromatic C), 138.81, 144.93, 146.76 (aromatic Cq), 155.61 (C-1’’’); HRMS (EI+) calcd for C39H51N7: 617.4206; found: 617.4246.
Crystal Structure Determination of 12B
Crystals of
12B (m.p.: 161.5–162.5 °C) for crystal structure analysis were obtained from a solution in ethyl acetate via slow evaporation of solvent. All the measurements were performed using monochromatized Mo K
a radiation at 100 K: C
39H
51N
7,
Mr 617.86, monoclinic, space group C 2, a = 28.755(2) Å, b = 6.5924(5) Å, c = 22.8112(16) Å, b = 128.182(8)°, V = 3399.0(5) Å
3, Z = 4, d
calc = 1.207 g cm
−3, m = 0.073 mm
−1. A total of 26,894 reflections were collected (Q
max = 27.0°), from which 7256 were unique (R
int = 0.0772), with 5244 having I > 2s(I). The structure was solved by direct methods (SHELXS-97) [
17] and refined by full-matrix least-squares techniques against
F2 (SHELXL-2014/6) [
18]. The nonhydrogen atoms were refined with anisotropic displacement parameters without any constraints. Owing to the absence of heavier elements, the absolute structure of the chiral molecule could not be determined reliably from the diffraction data and was chosen arbitrarily. The H atom bonded to N30 was taken from a difference Fourier map and refined without any positional constraints with an individual isotropic displacement parameter. The H atoms of the tertiary C–H groups were refined with individual isotropic displacement parameter and all X–C–H angles equal at a C–H distance of 1.00 Å. The H atoms of the CH
2 groups were refined with common isotropic displacement parameters for the H atoms of the same group and idealized geometry with approximately tetrahedral angles and C–H distances of 0.99 Å. The H atoms of the phenyl rings were put at the external bisectors of the C–C–C angles at C–H distances of 0.95 Å, and common isotropic displacement parameters were refined for the H atoms of the same ring. The H atoms of the methyl groups were refined with common isotropic displacement parameters for the H atoms of the same group and idealized geometries with tetrahedral angles, enabling rotations around the C–C bonds, and C–H distances of 0.98 Å. For 443 parameters, final
R indices of R
1 = 0.0513 and wR
2 = 0.1010 (GOF = 1.007) were obtained. The largest peak in a difference Fourier map was 0.215 e/Å
−3 rac-N-[(1R)-(1-tert-Butyl-1H-tetrazol-5-yl)(p-tolyl)methyl]-2-[(6R,9R)-6,9-diphenyl-1-(piperidin-1-yl)-3-azabicyclo[3.2.2]nonan-3-yl]ethan-1-amine (13A) and rac-N-[(1R)-(1-tert-butyl-1H-tetrazol-5-yl)(p-tolyl)methyl]-2-[(6S,9S)-6,9-diphenyl-1-(piperidin-1-yl)-3-azabicyclo[3.2.2]nonan-3-yl]ethan-1-amine (13B)
The reaction batch of 0.398g 2-(3-azabicyclononan-3-yl)ethan-1-amine 10 (0.986 mmol), 0.118 g p-tolylaldehyde (0.982 mmol), 0.110 g tert-butylisocyanide (1.31 mmol) and 0.151 g trimethylsilylazide (1.31 mmol) in 4.5 mL dry MeOH was stirred for 20 h. Subsequently, the solvent was evaporated in vacuo. Solvent chromatography gave 0.255 g (41%) of 13A and 0.286 g (46%) of 13B.
13A: IR = 2928, 1653, 1601, 1494, 1452, 1375, 1237, 1105, 1031, 862, 793, 745, 700; 1H NMR (CDCl3, 400 MHz): δ = 1.37–1.44 (m, 2H, CH2), 1.49–1.57 (m, 4H, 2CH2), 1.63 (s, 9H, 2’’’’-H), 1.80 (br t, J = 12.6 Hz, 1H, 8-H), 1.86 (br t, J = 12.8 Hz, 1H, 7-H), 1.92 (br s, 1H, 5-H), 2.20–2.31 (m, 2H, 4-H, 7-H), 2.31 (s, 3H, CH3), 2.44–2.54 (m, 5H, 2-H, N(CH2)2), 2.55–2.78 (m, 5H, 1’-H, 2’-H, 8-H), 2.86 (d, J = 12.6 Hz, 1H, 2-H), 2.93 (dd, J = 11.4, 4.8 Hz, 1H, 4-H), 3.21–3.29 (m, 2H, 6-H, 9-H), 5.27 (s, 1H, 1’’-H), 7.12–7.40 (m, 14H, aromatic H); 13C NMR (CDCl3, 100 MHz): δ = 21.06 (CH3), 25.01 (CH2), 26.72 (2CH2), 29.99 (C-2’’’’), 34.09 (C-8), 37.46 (C-7), 39.27 (C-9), 44.35 (C-5), 44.84 (C-6), 45.19 (C-2’), 46.44 (N(CH2)2), 57.74 (C-1’), 58.11 (C-2), 59.01 (C-1’’), 59.49 (C-1), 61.04 (C-4), 61.22 (C-1’’’’), 126.01, 126.06, 126.78, 127.96, 128.24, 128.42, 128.50, 129.61 (aromatic C), 135.57, 138.07, 144.75, 146.70 (aromatic Cq), 155.69 (C-1’’’); HRMS (EI+) calcd for C40H53N7: 631.4362; found: 631.4405.
13B: IR = 2930, 1636, 1601, 1495, 1451, 1374, 1237, 1105, 1031, 803, 746, 699; 1H NMR (CDCl3, 400 MHz): δ = 1.40-1.49 (m, 2H, CH2), 1.52–1.63 (m, 4H, 2CH2), 1.64 (s, 9H, 2’’’’-H), 1.82–1.92 (m, 2H, 7-H, 8-H), 1.94 (br s, 1H, 5-H), 2.20–2.32 (m, 2H, 4-H, 7-H), 2.33 (s, 3H, CH3), 2.52–2.76 (m, 10H, 1’-H, 2-H, 2’-H, 8-H, N(CH2)2), 2.87 (dd, J = 11.4, 4.9 Hz, 1H, 4-H), 2.92 (d, J = 12.7 Hz, 1H, 2-H), 3.24 (br td, J = 9.8, 2.7 Hz, 1H, 9-H), 3.29 (t, J = 9.4 Hz, 1H, 6-H), 5.29 (s, 1H, 1’’-H), 7.12–7.40 (m, 14H, aromatic H); 13C NMR (CDCl3, 100 MHz): δ = 21.08 (CH3), 25.04 (CH2), 26.76 (2CH2), 30.03 (C-2’’’’), 33.96 (C-8), 37.38 (C-7), 39.14 (C-9), 44.29 (C-5), 44.74 (C-6), 45.53 (C-2’), 46.58 (N(CH2)2), 58.00 (C-1’), 58.98 (C-1’’), 59.04 (C-2), 59.63 (C-1), 60.50 (C-4), 61.15 (C-1’’’’), 125.89, 126.06, 126.85, 127.39, 127.94, 128.14, 128.39, 128.51, 129.60 (aromatic C), 135.82, 138.06, 144.97, 146.79 (aromatic Cq), 155.76 (C-1’’’); HRMS (EI+) calcd for C40H53N7: 631.4362; found: 631.4387.
rac-(6R,9R)-N-[(1-tert-Butyl-1H-tetrazol-5-yl)methyl]-2-[6,9-diphenyl-1-(piperidin-1-yl)-3-azabicyclo[3.2.2]nonan-3-yl]ethan-1-amine (14) and rac-(6R,9R)-N,N-bis[(1-tert-Butyl-1H-tetrazol-5-yl)methyl]-2-[6,9-diphenyl-1-(piperidin-1-yl)-3-azabicyclo[3.2.2]nonan-3-yl]ethan-1-amine (15)
The reaction batch of 0.255g 2-(3-azabicyclononan-3-yl)ethan-1-amine 10 (0.632 mmol), 0.021 g paraformaldehyde (0.699 mmol), 0.071 g tert-butylisocyanide (0.844 mmol) and 0.097 g trimethylsilylazide (0.842 mmol) in 4.5 mL dry MeOH was stirred for 20 h. Subsequently, the solvent was evaporated in vacuo. Solvent chromatography gave 0.106 g of 14 (31%) and 0.130 mg of 15 (55%).
14: IR = 2931, 1636, 1495, 1456, 1373, 1237, 1110, 1031, 911, 731, 700; 1H NMR (CDCl3, 400 MHz): δ = 1.42–1.50 (m, 2H, CH2), 1.55–1.65 (m, 4H, 2CH2), 1.77 (s, 9H, 2’’’’-H), 1.85–1.96 (m, 2H, 7-H, 8-H), 2.05 (br s, 1H, 5-H), 2.26–2.32 (m, 1H, 7-H), 2.32 (d, J = 11.3 Hz, 1H, 4-H), 2.55–2.75 (m, 9H, 1’-H, 2-H, 2’-H, 8-H, N(CH2)2), 2.78–2.86 (m, 1H, 2’-H), 2.97 (d, J = 12.6 Hz, 1H, 2-H), 3.01 (dd, J = 11.3, 5.1 Hz, 1H, 4-H), 3.26–3.33 (m, 2H, 6-H, 9-H), 4.13 (s, 2H, 1’’-H), 7.15 (t, J = 7.2 Hz, 1H, aromatic H), 7.20–7.40 (m, 9H, aromatic H); 13C NMR (CDCl3, 100 MHz): δ = 24.95 (CH2), 26.67 (2CH2), 29.55 (C-2’’’’), 33.95 (C-8), 37.14 (C-7), 38.74 (C-9), 43.86 (C-5), 44.44 (C-1’’), 44.95 (C-6), 46.53 (C-2’), 46.59 (N(CH2)2), 57.38 (C-1’), 58.76 (C-2), 59.73(C-1), 60.50 (C-4), 61.30 (C-1’’’’), 126.03, 126.19. 126.81, 128.08, 128.23, 128.60 (aromatic C), 144.79, 146.58 (aromatic Cq), 152.92 (C-1’’’); HRMS (EI+) calcd for C33H47N7: 541.3893; found: 541.3937.
15: IR = 2933, 1635, 1453, 1374, 1234, 1106, 731, 700; 1H NMR (CDCl3, 400 MHz): δ = 1.41–1.49 (m, 2H, CH2), 1.52–1.64 (m, 4H, 2CH2), 1.66 (s, 18H, 2’’’’-H), 1.82–1.93 (m, 2H, 7-H, 8-H), 1.97 (br s, 1H, 5-H), 2.21–2.29 (m, 1H, 7-H), 2.33 (d, J = 11.4 Hz, 1H, 4-H), 2.47–2.64 (m, 5H, 8-H, N(CH2)2), 2.65–2.80 (m, 3H, 1’-H, 2-H), 2.93 (d, J = 12.8 Hz, 1H, 2-H), 2.97 (dd, J = 11.4, 4.9 Hz, 1H, 4-H), 3.14–3.21 (m, 2H, 2’-H), 3.21–3.30 (m, 2H, 6-H, 9-H), 4.39 (s, 4H, 1’’-H), 7.12–7.37 (m, 10H, aromatic H); 13C NMR (CDCl3, 100 MHz): δ = 24.87 (CH2), 26.63 (2CH2), 29.59 (C-2’’’’), 34.08 (C-8), 37.04 (C-7), 38.93 (C-9), 43.91 (C-5), 44.75 (C-6), 46.53 (N(CH2)2), 47.58 (C-1’’), 52.13 (C-2’), 55.71 (C-1’), 59.29 (C-2), 59.68 (C-1), 60.36 (C-4), 61.25 (C-1’’’’), 126.06, 126.16, 126.74, 128.12, 128.23, 128.55 (aromatic C), 144.77, 146.44 (aromatic Cq), 151.38 (C-1’’’); HRMS (EI+) calcd for C39H57N11: 679.4799; found: 679.4833.
4.2.3. General Procedure for the Synthesis of Benzenesulfonamides 18–20
Azabicyclo-nonanes 3, 7, 8 were dissolved in dry CH2Cl2. Then, 4-DMAP or DIPEA and the respective aromatic sulfonyl chloride were added under stirring. The mixture was refluxed for 20 h at 50 °C. Subsequently, the reaction batch was shaken with 2 N aq NaOH, washed with water until the aqueous phase reacted neutral, dried over anhydrous sodium sulfate, filtered and finally the solvent was removed in vacuo giving crude products, which were purified by column chromatography yielding compounds 18–20.
rac-(7R,8R)-2-(4-Chlorobenzenesulfonyl)-7,8-diphenyl-5-(pyrrolidin-1-yl)-2-azabicyclo[3.2.2]nonane (18)
The reaction of 0.333 g 2-azabicyclo-nonane 3 (0.96 mmol), 0.361 g 4-chlorobenzenesulfonyl chloride (1.71 mmol) and 1.92 g 4-DMAP (1.57 mmol) in 14 mL CH2Cl2 abs. gave a crude product, which was purified by column chromatography (aluminum oxide neutral, CH/EtAc = 3 + 1 and in addition 1% diethylamine) yielding 0.240 g 18 (48%) as colorless oil. IR = 2961, 2360, 1585, 1496, 1475, 1449, 1394, 1339, 1278, 1160, 1091, 1038, 1012, 974, 925, 864, 827, 760, 699; 1H NMR (CDCl3) δ = 1.74–1.82 (m, 4H, (CH2)2), 2.03–2.21 (m, 4H, 4-H, 6-H, 9-H), 2.24–2.36 (m, 2H, 6-H, 9-H), 2.66–2.79 (m, 4H, N(CH2)2), 3.26–3.44 (m, 3H, 3-H, 7-H, 8-H), 3.93 (dt, J = 13.7, 4.2 Hz, 1H, 3-H), 4.20 (d, J = 3.3 Hz, 1H, 1-H), 7.02 (d, J = 7.1 Hz, 2H, aromatic H), 7.07-7.41 (m, 10H, aromatic H), 7.47 (d, J = 7.5 Hz, 2H, aromatic H); 13C NMR (CDCl3) δ = 23.56 ((CH2)2), 31.97 (C-4), 33.85 (C-9), 36.67 (C-6), 38.30 (C-8), 42.41 (C-3), 45.23 (N(CH2)2), 46.73 (C-7), 56.43 (C-5), 63.62 (C-1), 126.54, 126.88, 127.53, 127.66, 127.97, 128.48, 128.81, 128.96 (aromatic C), 138.15, 139.03, 141.43, 143.46 (aromatic Cq); HRMS (EI+) calcd for C30H33ClN2O2S: 520.1951; found: 520.1979.
rac-(7R,8R)-4-Methyl-N-[2-(7,8-diphenyl-5-(piperidin-1-yl)-2-azabicyclo[3.2.2]nonan -2-yl)ethyl]benzenesulfonamide (19)
The reaction of 0.143 g 2-azabicyclo-nonane 7 (0.35 mmol), 0.137 g toluene-4-sulfonyl chloride (0.72 mmol) and 0.118 g DIPEA (0.91 mmol) in 5 mL CH2Cl2 abs. gave a crude product, which was purified by column chromatography (aluminum oxide neutral, CH/EtAc = 5 + 1 → 1 + 1 finished by CH/EtAc/MeOH 1 + 1 + 0.1) yielding 0.120 g 19 (63%) as colorless oil. IR = 2929, 1635, 1600, 1495, 1450, 1330, 1160, 1093, 815, 756, 701, 665; 1H NMR (CDCl3) δ = 1.39–1.49 (m, 2H, CH2), 1.55–1.64 (m, 4H, 2CH2), 1.78–1.92 (m, 3H, 4-H, 6-H), 2.08 (br t, J = 12.1 Hz, 1H, 9-H), 2.19–2.32 (m, 3H, 1’-H, 6-H, 9-H), 2.36 (s, 3H, CH3), 2.37–2.44 (m, 1H, 1’-H), 2.48–2.63 (m, 6H, 2’-H, 3-H, N(CH2)2), 2.55 (d, J = 3.6 Hz, 1H, 1-H), 2.71-2.84 (m, 2H, 2’-H, 3-H), 3.23 (ddd, J = 11.4, 8.1, 3.4 Hz, 1H, 8-H), 3.27 (t, J = 9.1 Hz, 1H, 7-H), 4.40 (br, 1H, NH), 7.14 (d, J = 8.0 Hz, 2H, aromatic H), 7.19–7.36 (m, 10H, aromatic H), 7.41 (d, J = 8.0 Hz, 2H, aromatic H); 13C NMR (CDCl3) δ = 21.44 (CH3), 25.01 (CH2), 26.81 (2CH2), 32.01 (C-4), 35.23 (C-6), 35.35 (C-9), 38.65 (C-8), 40.56 (C-2’), 41.08 (C-7), 46.22 (N(CH2)2), 47.79 (C-3), 55.54 (C-1’), 57.94 (C-5), 68.81 (C-1), 126.25, 126.55, 126.88, 127.30, 128.25, 128.40, 128.69, 128.35 (aromatic C), 137.13, 142.86, 144.21, 145.61 (aromatic Cq); HRMS (EI+) calcd for C34H43N3O2S: 557.3076; found: 557.3071.
rac-(7R,8R)-4-Methyl-N-{2-[7,8-diphenyl-5-(pyrrolidin-1-yl)-2-azabicyclo[3.2.2]no nan-2-yl]ethyl}benzenesulfonamide (20)
The reaction of 0.390 g 2-azabicyclo-nonane 8 (1.00 mmol), 0.366 g toluene-4-sulfonyl chloride (1.92 mmol) and 0.243 g 4-DMAP (1.99 mmol) in 15 mL CH2Cl2 abs. gave a crude product, which was purified by multiple column chromatography (first time on neutral aluminum oxide, CH/EtAc = 1 + 1 and in addition 1% diethylamine; second time on neutral aluminum oxide, CH/EtAc = 3+1+1% diethylamine) yielding 0.210 g 20 (39%) as colorless oil. IR = 2931, 2871, 1626, 1599, 1494, 1450, 1340, 1331, 1161, 1093, 1031, 939, 814, 755, 701; 1H NMR (CDCl3) δ = 1.78 (br s, 4H, (CH2)2), 1.91 (br t, J = 6.0 Hz, 2H, 4-H), 2.02 (dd, J = 13.6, 8.0 Hz, 1H, 6-H), 2.16–2.29 (m, 4H, 1’-H, 6-H, 9-H), 2.37 (s, 3H, CH3), 2.39–2.44 (m, 1H, 1’-H), 2.45–2.54 (m, 1H, 2’-H), 2.52 (d, J = 3.3 Hz, 1H, 1-H), 2.54–2.61 (m, 1H, 3-H), 2.70-2.81 (m, 6H, 2’-H, 3-H, N(CH2)2), 3.22–3.29 (m, 1H, 8-H), 3.32 (br t, J = 9.0 Hz, 1H, 7-H), 4.26 (br, 1H, NH), 7.15–7.44 (m, 14H, aromatic H); 13C NMR (CDCl3) δ = 21.46 (CH3), 23.63 ((CH2)2), 32.65 (C-4), 35.84 (C-6), 36.33 (C-9), 38.06 (C-8), 40.07 (C-7), 40.59 (C-2’), 45.15 (N(CH2)2), 47.83 (C-3), 55.86 (C-1’), 56.52 (C-5), 69.22 (C-1), 126.25, 126.57, 126.91, 127.47, 128.38, 128.48, 128.71, 129.35 (aromatic C), 137.26, 142.87, 144.33, 145.70 (aromatic Cq); HRMS (EI+) calcd for C33H41N3O2S: 543.2919; found: 543.2964.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}