
New 11-Methoxymethylgermacranolides from the Whole Plant of Carpesium divaricatum

Abstract
:1. Introduction
2. Results and Discussion
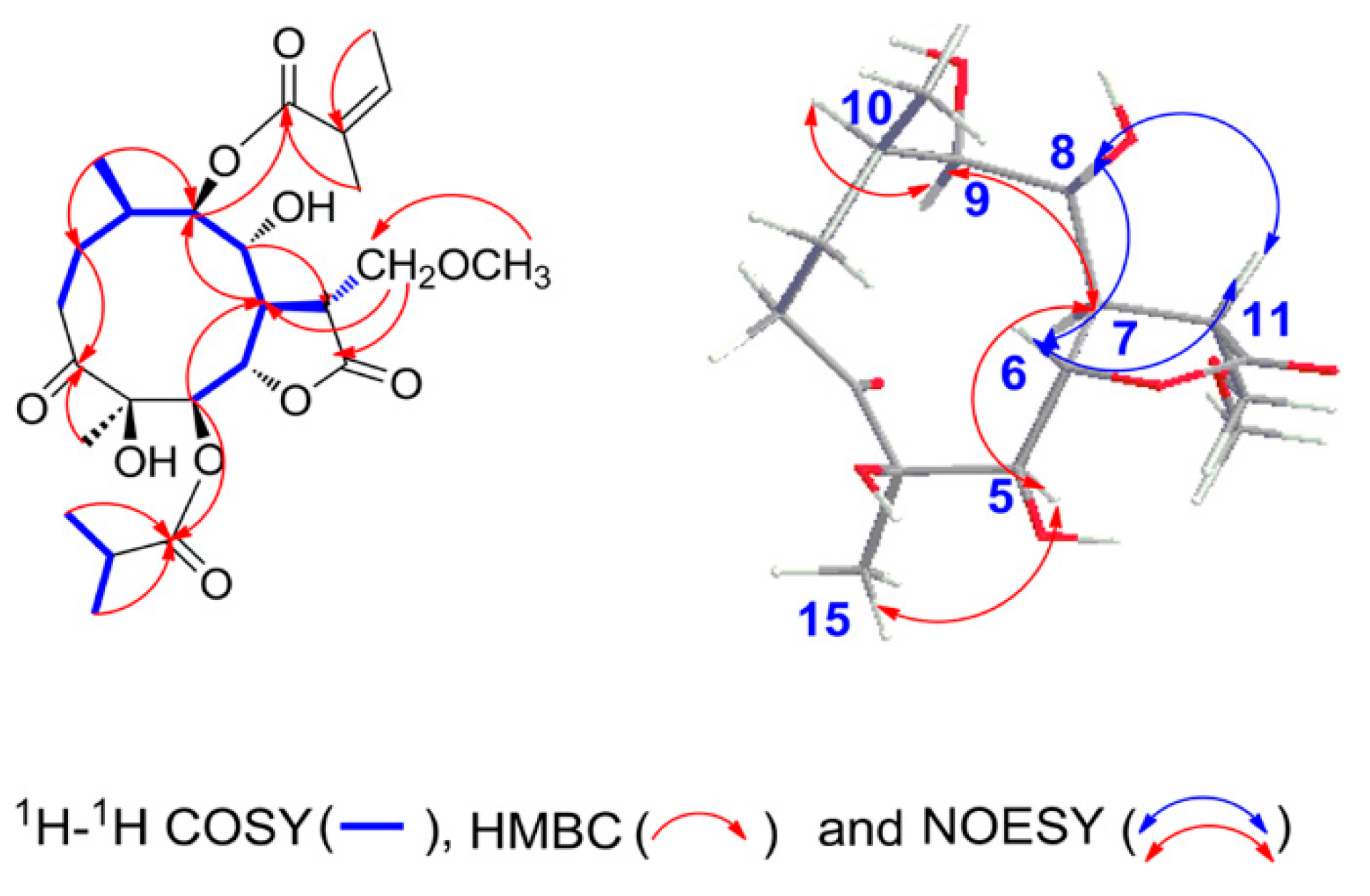
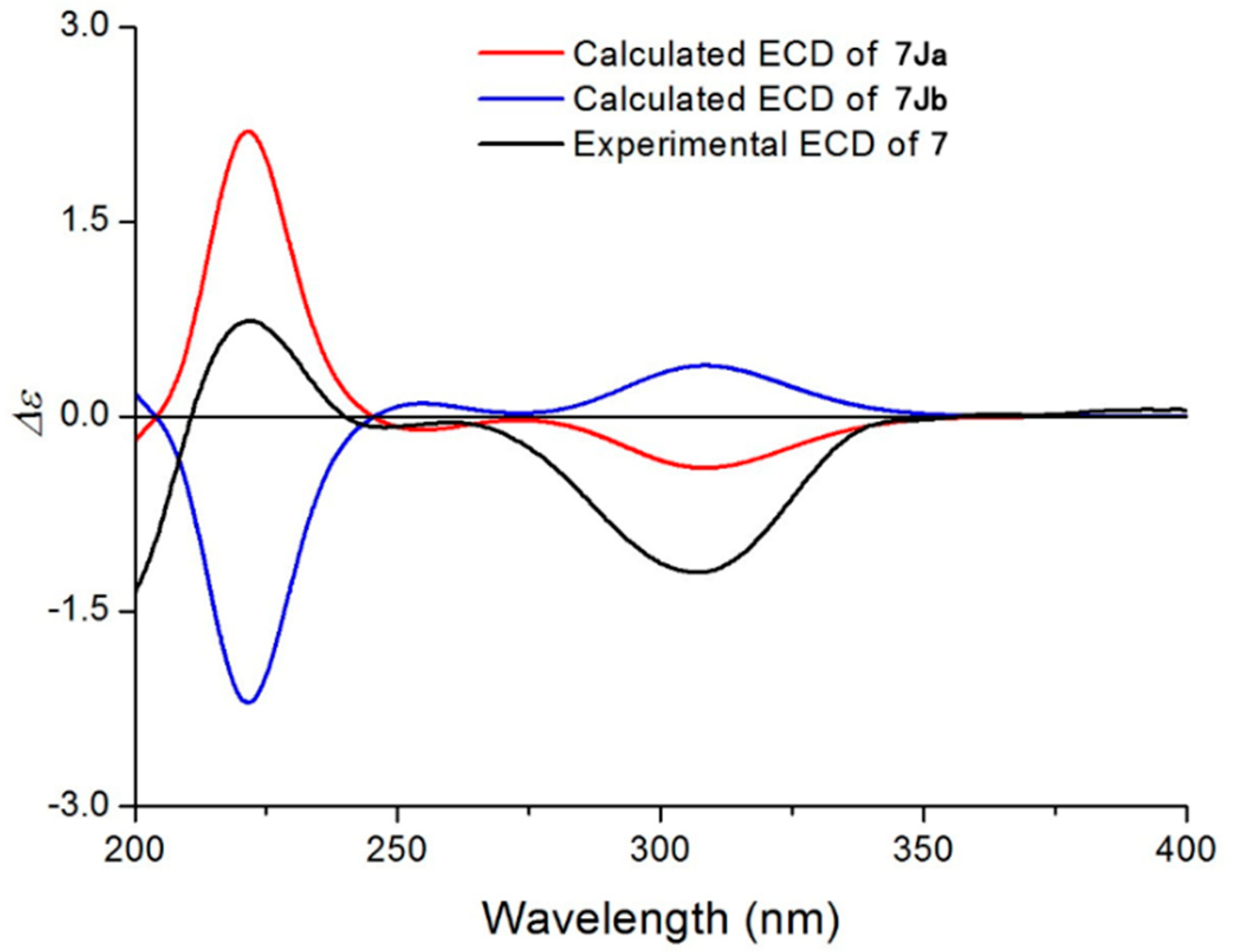
2.1. Structural Elucidation of the Isolated Compounds
2.2. Cytotoxic Activity
2.3. Analysis of the Macrophages Culture Supernatants PGE2 Levels
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Spectral Data
- 11-methoxymethylcardivarolide H (1)
- 11-methoxymethylcardivarolide I (2)
- 11-methoxymethylincaspitolide D (3)
- 11-methoxymethylcardivarolide J (4)
- 11-methoxymethylcardivarolide K (5)
- 11-methoxymethylcardivarolide L (6)
- 11-methoxyldivarolide G (7)
- 11-methoxyldivarolide F (8)
3.5. X-ray Crystal Structure Analysis
3.6. Biological Activity Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Tu, Y.Y. The discovery of artemisinin (qinghaosu) and gifts from Chinese medicine. Nat. Med. 2011, 17, 1217–1220. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lu, Y.X.; Ding, Y.H.; Zhai, J.D.; Ji, Q.; Ma, W.W.; Yang, M.; Fan, H.X.; Long, J.; Tong, Z.S.; et al. Guaianolide sesquiterpene lactones, a source to discover agents that selectively inhibit acute myelogenous leukemia stem and progenitor cells. J. Med. Chem. 2012, 55, 8757–8769. [Google Scholar] [CrossRef] [PubMed]
- Ghantous, A.; Sinjab, A.; Herceg, Z.; Darwiche, N. Parthenolide: From plant shoots to cancer roots. Drug Discov. Today 2013, 18, 894–905. [Google Scholar] [CrossRef]
- Zhu, N.L.; Tang, C.P.; Xu, C.H.; Ke, C.Q.; Lin, G.; Jenis, J.; Yao, S.; Liu, H.C.; Ye, Y. Cytotoxic germacrane-type sesquiterpene lactones from the whole plant of Carpesium lipskyi. J. Nat. Prod. 2019, 82, 919–927. [Google Scholar] [CrossRef]
- Zhang, J.P.; Wang, G.W.; Tian, X.H.; Yang, Y.X.; Liu, Q.X.; Chen, L.P.; Li, H.L.; Zhang, W.D. The genus Carpesium: A review of its ethnopharmacology, phytochemistry and pharmacology. J. Ethnopharmacol. 2015, 163, 173–191. [Google Scholar] [CrossRef]
- Kim, E.J.; Jin, H.K.; Kim, Y.K.; Lee, H.Y.; Lee, S.Y.; Lee, K.R.; Zee, O.P.; Han, J.W.; Lee, H.W. Suppression by a sesquiterpene lactone from Carpesium divaricatum of inducible nitric oxide synthase by inhibiting nuclear factor-κB activation. Biochem. Pharmacol. 2001, 61, 903–910. [Google Scholar] [CrossRef]
- Kim, D.K.; Beak, N.I.; Choi, S.U.; Lee, C.O.; Lee, K.R.; Zee, O.P. Four new cytotoxic germacranolides from Carpesium divaricatum. J. Nat. Prod. 1997, 60, 1199–1202. [Google Scholar] [CrossRef]
- Chung, I.M.; Seo, S.H.; Kang, E.Y.; Park, W.H.; Park, S.D.; Moon, H.I. Antiplasmodial activity of isolated compounds from Carpesium divaricatum. Phytother. Res. 2010, 24, 451–453. [Google Scholar] [CrossRef]
- Maruyama, M. Sesquiterpene lactones from Carpesium divaricatum. Phytochemistry 1990, 29, 547–550. [Google Scholar] [CrossRef]
- Kim, D.K.; Lee, K.R.; Zee, O.P. Sesquiterpene lactones from Carpesium divaricatum. Phytochemistry 1997, 46, 1245–1247. [Google Scholar] [CrossRef]
- Zhang, T.; Si, J.G.; Zhang, Q.B.; Ding, G.; Zou, Z.M. New highly oxygenated germacranolides from Carpesium divaricatum and their cytotoxic activity. Sci. Rep. 2016, 6, 27237–27245. [Google Scholar] [CrossRef]
- Zhang, T.; Si, J.G.; Zhang, Q.B.; Chen, J.H.; Ding, G.; Zhang, H.W.; Jia, H.M.; Zou, Z.M. Three new highly oxygenated germacranolides from Carpesium divaricatum and their cytotoxic activity. Molecules 2018, 23, 1078. [Google Scholar] [CrossRef]
- Zhang, T.; Chen, J.H.; Si, J.G.; Ding, G.; Zhang, Q.B.; Zhang, H.W.; Jia, H.M.; Zou, Z.M. Isolation, structure elucidation, and absolute configuration of germacrane isomers from Carpesium divaricatum. Sci. Rep. 2018, 8, 12418–12429. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, Q.B.; Fu, L.; Li, L.Y.; Ma, L.Y.; Si, J.G.; Zhang, H.W.; Wei, J.H.; Yu, S.S.; Zou, Z.M. New antiproliferative germacranolides from Carpesium divaricatum. RSC Adv. 2019, 9, 11493–11502. [Google Scholar] [CrossRef]
- Liu, Q.X.; Yang, Y.X.; Zhang, J.P.; Chen, L.P.; Shen, Y.H.; Li, H.L.; Zhang, W.D. Isolation, structure elucidation, and absolute configuration of highly oxygenated germacranolides from Carpesium cernuum. J. Nat. Prod. 2016, 10, 2479–2486. [Google Scholar] [CrossRef]
- Li, F.S.; Zhan, Z.L.; Liu, F.; Yang, Y.N.; Li, L.; Feng, Z.M.; Jiang, J.S.; Zhang, P.C. Polyflavanostilbene A, a new flavanol-fused stilbene glycoside from Polygonum cuspidatum. Org. Lett. 2013, 15, 674–677. [Google Scholar] [CrossRef]
- Gao, Y.; Hou, R.; Liu, F.; Cai, R.L.; Feng, L.; Peng, C.; Qi, Y. Highly specific and sensitive immunoassay for the measurement of prostaglandin E2 in biological fluids. Bioanalysis 2015, 7, 2597–2607. [Google Scholar] [CrossRef]
- Ma, J.; Chen, M.; Xia, S.K.; Shu, W.; Guo, Y.; Wang, Y.H.; Xu, Y.; Bai, X.M.; Zhang, L.; Zhang, H.; et al. Prostaglandin E2 promotes liver cancer cell growth by the upregulation of FUSE-binding protein 1 expression. Int. J. Oncol. 2013, 42, 1093–1104. [Google Scholar] [CrossRef]
- Peng, Y.F.; Shi, J.D.; Du, X.L.; Wang, L.; Klocker, H.; Mo, L.J.; Mo, Z.N.; Zhang, J. Prostaglandin E2 induces stromal cell-derived factor-1 expression in prostate stromal cells by activating protein kinase A and transcription factor Sp1. Int. J. Biochem. Cell Biol. 2021, 45, 521–530. [Google Scholar] [CrossRef]
- Kurahashi, Y.; Sugahara, M.; Ago, H.; Aoyama, S.; Takahashi, N.; Takio, K.; Katsukawa, M.; Yamamoto, S.; Miyano, M. Crystallization and preliminary diffraction studies of prostaglandin E2-specific monoclonal antibody Fab fragment in the ligand complex. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2008, 64, 1027–1030. [Google Scholar] [CrossRef] [Green Version]
- Mnich, S.J.; Veenhuizen, A.W.; Monahan, J.B.; Sheehan, K.C.; Lynch, K.R.; Isakson, P.C.; Portanova, J.P. Characterization of a monoclonal antibody that neutralizes the activity of prostaglandin E2. J. Immunol. 1995, 155, 4437–4444. [Google Scholar]
- Wang, B.J.; Won, S.J.; Su, Z.R.; Yu, C.L. Free radical scavenging and apoptotic effects of Cordyceps sinensis fractionated by supercritical carbon dioxide. Food Chem. Toxicol. 2006, 43, 543–552. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 a | 2 a | 3 a | 4 a | 5 a | 6 a | 7 b | 8 a |
|---|---|---|---|---|---|---|---|---|
| 1 | 1.86 m, 1.55 m | 1.81 m, 1.54 m | 1.82 m, 1.57 m | 1.85 m, 1.57 m | 1.82 m, 1.54 m | 1.82 m, 1.51 o | 1.86 m, 1.57 m | 1.87 m, 1.55 m |
| 2 | 3.89 dd (12.6, 3.6), 2.00 o | 3.87 dd (12.6, 3.6), 2.09 o | 3.89 br d (12.0), 2.10 o | 3.91 br d (12.6), 2.00 m | 3.87 dd (12.6, 3.6), 2.13 m | 3.87 dd (12.6, 3.6), 2.09 o | 3.92 dd (12.0, 2.0), 2.15 m | 3.92 dd (12.6, 3.6), 1.97 o |
| 5 | 5.44 d (9.6) | 5.43 dd (9.0, 1.8) | 5.43 br d (9.6) | 5.43 dd (9.0, 1.2) | 5.44 d (9.6) | 5.44 dd (9.6, 1.8) | 5.56 dd (9.5, 1.5) | 5.54 dd (9.0, 1.8) |
| 6 | 4.52 dd (9.6, 9.0) | 4.52 dd (9.0, 9.0) | 4.50 dd (9.6, 9.0) | 4.52 dd (9.0, 9.0) | 4.50 dd (9.6, 9.0) | 4.54 dd (9.6, 9.0) | 4.60 dd (9.0, 8.5) | 4.58 dd (9.0, 9.0) |
| 7 | 2.60 dd (9.0, 9.0) | 2.56 dd (9.0, 9.0) | 2.57 dd (9.0, 9.0) | 2.59 dd (9.0, 9.0) | 2.57 dd (9.0, 8.4) | 2.57 dd (9.0, 8.4) | 2.62 dd (9.0, 8.5) | 2.62 dd (9.0, 9.0) |
| 8 | 4.53 o | 4.47 br d (10.2) | 4.55 dd (8.4, 8.4) | 4.54 br d (10.2) | 4.47 br d (10.8) | 4.48 br d (10.2) | 4.52 d (10.5) | 4.55 br d (10.2) |
| 9 | 4.94 d (10.2) | 4.85 o | 4.82 o | 4.95 d (10.2) | 4.85 o | 4.85 o | 4.87 o | 4.95 d (10.2) |
| 10 | 2.15 m | 2.12 m | 2.12 m | 2.15 m | 2.10 o | 2.12 m | 2.16 m | 2.16 m |
| 11 | 3.31 ddd (9.0, 6.0, 3.6) | 3.26 ddd (10.8, 7.8, 4.2) | 3.25 ddd (9.0, 5.4, 4.2) | 3.29 ddd (10.2, 7.8, 4.2) | 3.26ddd (9.0, 7.8, 4.2) | 3.25 ddd (9.0, 7.8, 3.6) | 3.32 o | 3.33 ddd (9.0, 7.8, 3.0) |
| 13a | 3.67 dd (9.6, 4.2) | 3.65 dd (9.6, 4.2) | 3.66 dd (9.6, 4.2) | 3.66 dd (9.6, 4.2) | 3.65 dd (10.2, 4.2) | 3.66 dd (10.2, 4.2) | 3.70 dd (9.5, 4.5) | 3.70 dd (10.2, 4.2) |
| 13b | 3.43 dd (9.6, 4.2) | 3.40 dd (9.6, 4.2) | 3.40 dd (9.6, 4.2) | 3.42 dd (9.6, 4.2) | 3.41 dd (10.2, 4.2) | 3.40 dd (10.2, 4.2) | 3.43 dd (9.5, 4.0) | 3.45 dd (10.2, 4.2) |
| 14 | 0.88 d (6.6) | 0.87 d (7.2) | 0.86 d (7.2) | 0.87 d (7.2) | 0.87 d (6.6) | 0.87 d (7.2) | 0.90 d (7.0) | 0.89 d (6.6) |
| 15 | 1.18 s | 1.17 s | 1.18 s | 1.18 s | 1.18 s | 1.18 s | 1.22 s | 1.19 s |
| 16 | 3.35 s | 3.34 s | 3.34 s | 3.35 s | 3.34 s | 3.34 s | 3.37 s | 3.36 s |
| 2′ | 2.65 m | 2.64 m | 2.64 o | 2.65 m | 2.31 d (6.6), 2.23 d (6.6) | 2.47 m | ||
| 3′ | 1.20 d (7.2) | 1.20 d (7.2) | 1.19 d (6.6) | 1.20 d (7.2) | 2.10 o | 1.73 m, 1.51 o | 6.17 qq (7.0, 1.5) | 6.13 o |
| 4′ | 1.19 d (7.2) | 1.19 d (7.2) | 1.18 d (6.6) | 1.19 d (7.2) | 0.97 d (6.6) | 1.17 d (7.2) | 1.95 qq (1.5, 1.5) | 1.92 s |
| 5′ | 0.97 d (6.6) | 0.94 t (7.2) | 0.97 dq (7.0, 1.5) | 1.97 d (9.0) | ||||
| 2′′ | 2.28 d (7.2), 2.27 d (6.6) | 2.64 o | 2.27 o, 2.27 o | 2.28 d (7.2), 2.27 d (7.2) | 2.31 d (7.0), 2.30 d (7.0) | |||
| 3′′ | 6.12 q (7.2) | 2.09 o | 1.20 d (7.2) | 5.63 dq (3.6, 1.8), 6.12 dq (3.6, 1.8) | 2.10 o | 2.09 o | 2.13 m | 6.13 o |
| 4′′ | 1.93 s | 0.98 d (6.6) | 1.16 d (7.2) | 1.96 br s | 0.97 d (6.6) | 0.97 d (7.2) | 1.01 d (6.5) | 1.92 s |
| 5′′ | 1.98 br d (7.2) | 0.97 d (6.6) | 0.97 d (6.6) | 0.97 d (6.6) | 1.00 d (6.5) | 1.97 d (9.0) |
| No. | 1 a | 2 a | 3 a | 4 a | 5 a | 6 a | 7 b | 8 a |
|---|---|---|---|---|---|---|---|---|
| 1 | 25.3 | 25.3 | 25.3 | 25.3 | 25.4 | 25.3 | 25.4 | 25.6 |
| 2 | 31.6 | 31.6 | 31.7 | 31.7 | 31.9 | 31.5 | 31.6 | 31.6 |
| 3 | 217.4 | 217.5 | 217.5 | 217.5 | 217.4 | 217.4 | 217.7 | 217.7 |
| 4 | 80.4 | 80.4 | 80.4 | 80.4 | 80.3 | 80.3 | 80.4 | 80.4 |
| 5 | 79.0 | 79.0 | 79.0 | 79.0 | 79.0 | 78.9 | 78.9 | 78.9 |
| 6 | 80.3 | 80.3 | 80.3 | 80.3 | 80.3 | 80.3 | 80.4 | 80.4 |
| 7 | 39.3 | 39.2 | 39.2 | 39.2 | 39.2 | 39.2 | 39.2 | 39.3 |
| 8 | 67.3 | 67.1 | 67.1 | 67.1 | 67.1 | 67.1 | 67.1 | 67.3 |
| 9 | 79.3 | 79.5 | 79.3 | 79.3 | 79.5 | 79.5 | 79.3 | 79.3 |
| 10 | 29.4 | 29.2 | 29.4 | 29.4 | 29.3 | 29.3 | 29.4 | 29.4 |
| 11 | 40.2 | 40.2 | 40.1 | 40.1 | 40.2 | 40.2 | 40.2 | 40.2 |
| 12 | 177.0 | 177.0 | 177.0 | 177.0 | 176.9 | 176.8 | 177.0 | 177.1 |
| 13 | 69.7 | 69.7 | 69.7 | 69.7 | 69.8 | 69.8 | 69.7 | 69.7 |
| 14 | 20.0 | 20.0 | 19.9 | 19.9 | 20.0 | 20.0 | 20.0 | 20.0 |
| 15 | 23.5 | 23.6 | 23.6 | 23.6 | 23.7 | 23.6 | 23.6 | 23.6 |
| 16 | 58.0 | 57.9 | 57.9 | 57.9 | 58.0 | 58.0 | 58.0 | 58.0 |
| 1′ | 176.3 | 176.3 | 176.3 | 176.3 | 172.3 | 176.0 | 167.1 | 167.1 |
| 2′ | 33.9 | 33.9 | 33.9 | 33.9 | 42.8 | 41.1 | 127.5 | 127.5 |
| 3′ | 18.0 | 18.0 | 18.0 | 18.0 | 25.3 | 26.5 | 138.1 | 138.0 |
| 4′ | 17.9 | 17.9 | 17.9 | 17.9 | 21.4 | 15.7 | 19.3 | 19.5 |
| 5′ | 21.5 | 10.8 | 14.6 | 14.6 | ||||
| 1′′ | 168.0 | 173.4 | 177.4 | 177.4 | 173.4 | 173.4 | 173.4 | 168.0 |
| 2′′ | 128.0 | 43.1 | 34.1 | 136.6 | 43.1 | 43.1 | 43.1 | 128.0 |
| 3′′ | 137.4 | 25.4 | 18.5 | 129.4 | 25.3 | 25.4 | 25.4 | 137.4 |
| 4′′ | 19.5 | 21.4 | 17.8 | 17.1 | 21.4 | 21.4 | 21.4 | 19.2 |
| 5′′ | 14.6 | 21.4 | 21.4 | 21.5 | 21.4 | 14.6 |
| Compound | IC50 (μM) | ||
|---|---|---|---|
| Hep G2 | MCF-7 | A549 | |
| 1 | >40 | >40 | >40 |
| 2 | >40 | >40 | >40 |
| 3 | >40 | >40 | >40 |
| 4 | >40 | 37.32 ± 0.24 | >50 |
| 5 | >40 | >40 | >40 |
| 6 | >40 | >40 | >40 |
| 7 | >40 | >40 | >40 |
| 8 | >40 | >40 | >40 |
| cis-platin | 16.20 ± 0.24 | 22.80 ± 0.83 | 27.07 ± 0.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Zhang, H.; Lin, C.; Fu, L.; Zou, Z. New 11-Methoxymethylgermacranolides from the Whole Plant of Carpesium divaricatum. Molecules 2022, 27, 5991. https://doi.org/10.3390/molecules27185991
Zhang T, Zhang H, Lin C, Fu L, Zou Z. New 11-Methoxymethylgermacranolides from the Whole Plant of Carpesium divaricatum. Molecules. 2022; 27(18):5991. https://doi.org/10.3390/molecules27185991
Chicago/Turabian StyleZhang, Tao, Haixin Zhang, Chunyu Lin, Lu Fu, and Zhongmei Zou. 2022. "New 11-Methoxymethylgermacranolides from the Whole Plant of Carpesium divaricatum" Molecules 27, no. 18: 5991. https://doi.org/10.3390/molecules27185991