
Multilayer Graphtriyne Membranes for Separation and Storage of CO2: Molecular Dynamics Simulations of Post-Combustion Model Mixtures
 , , , ,
, , , ,  ,
,
Abstract
:1. Introduction
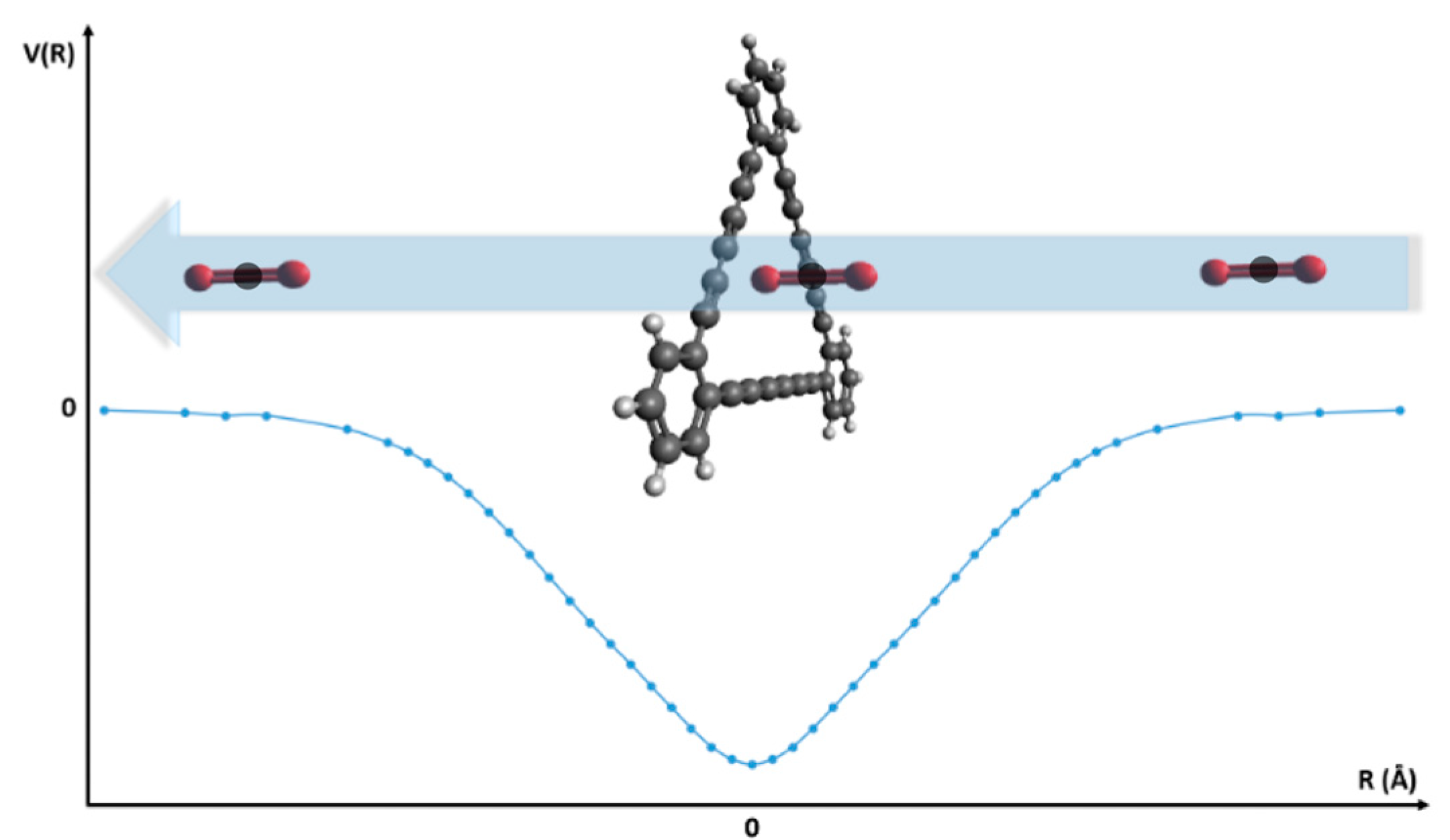
2. Methods
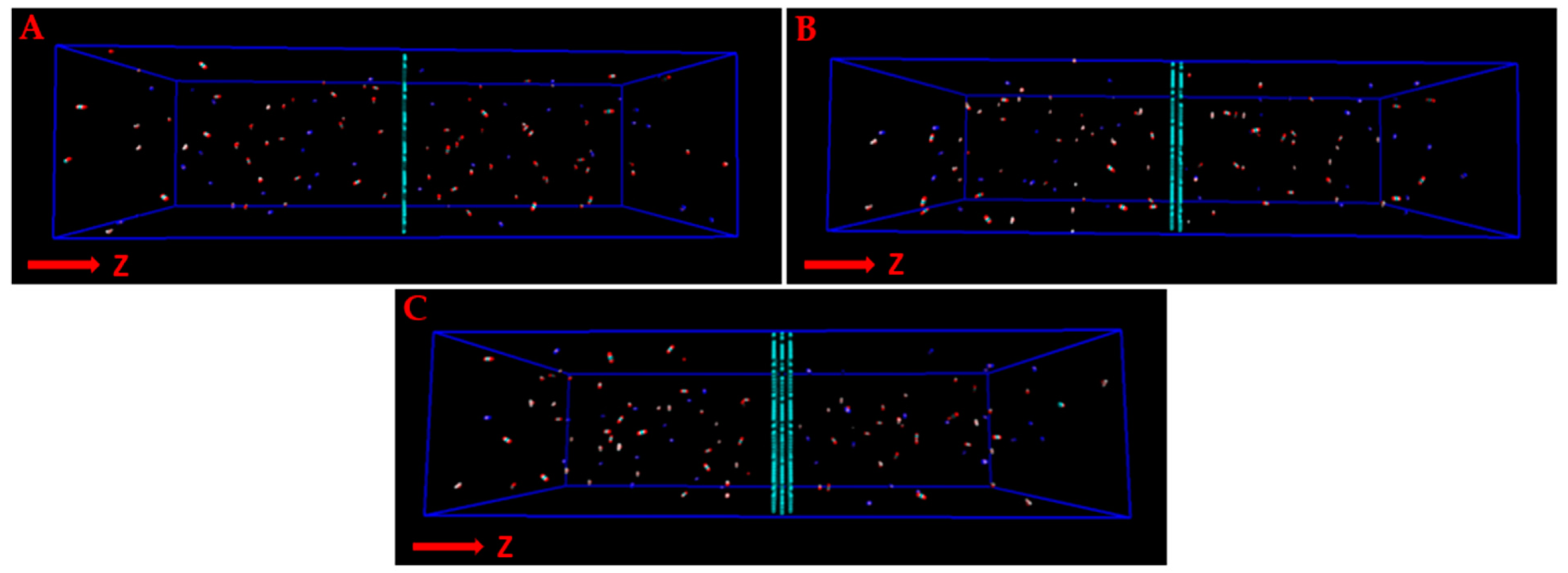
3. Results and Discussion
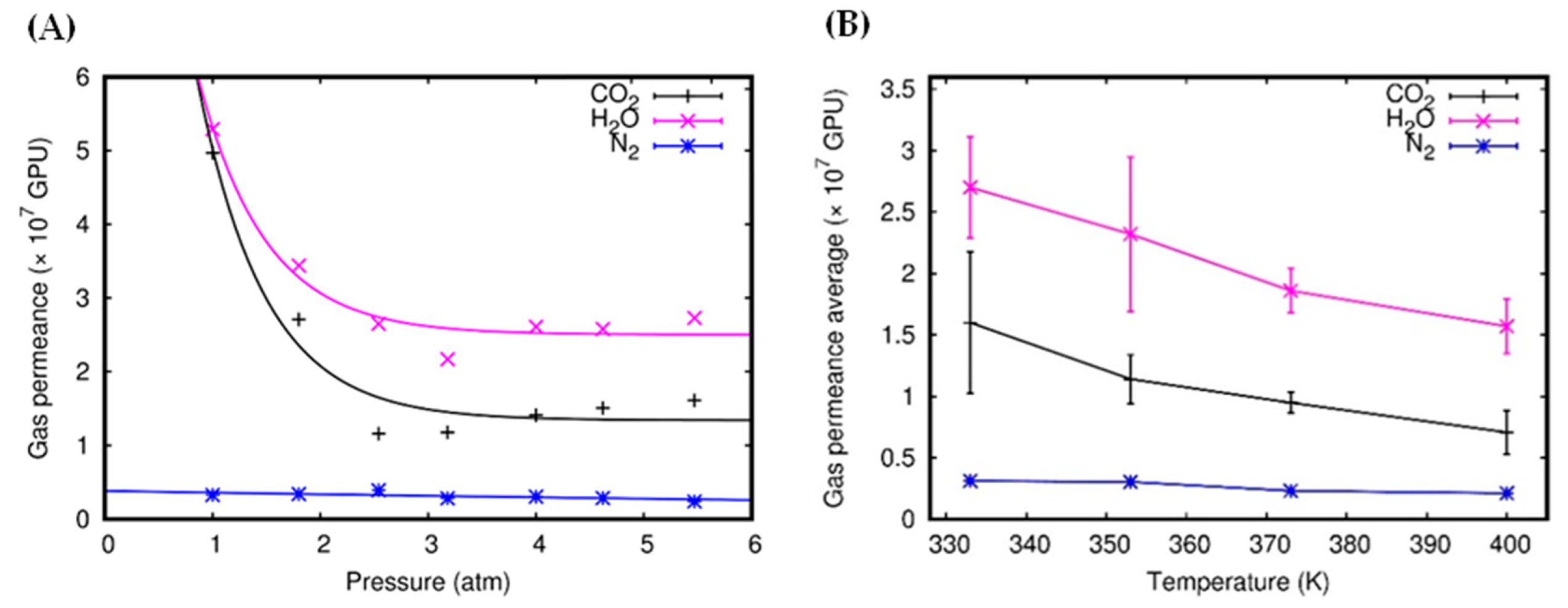
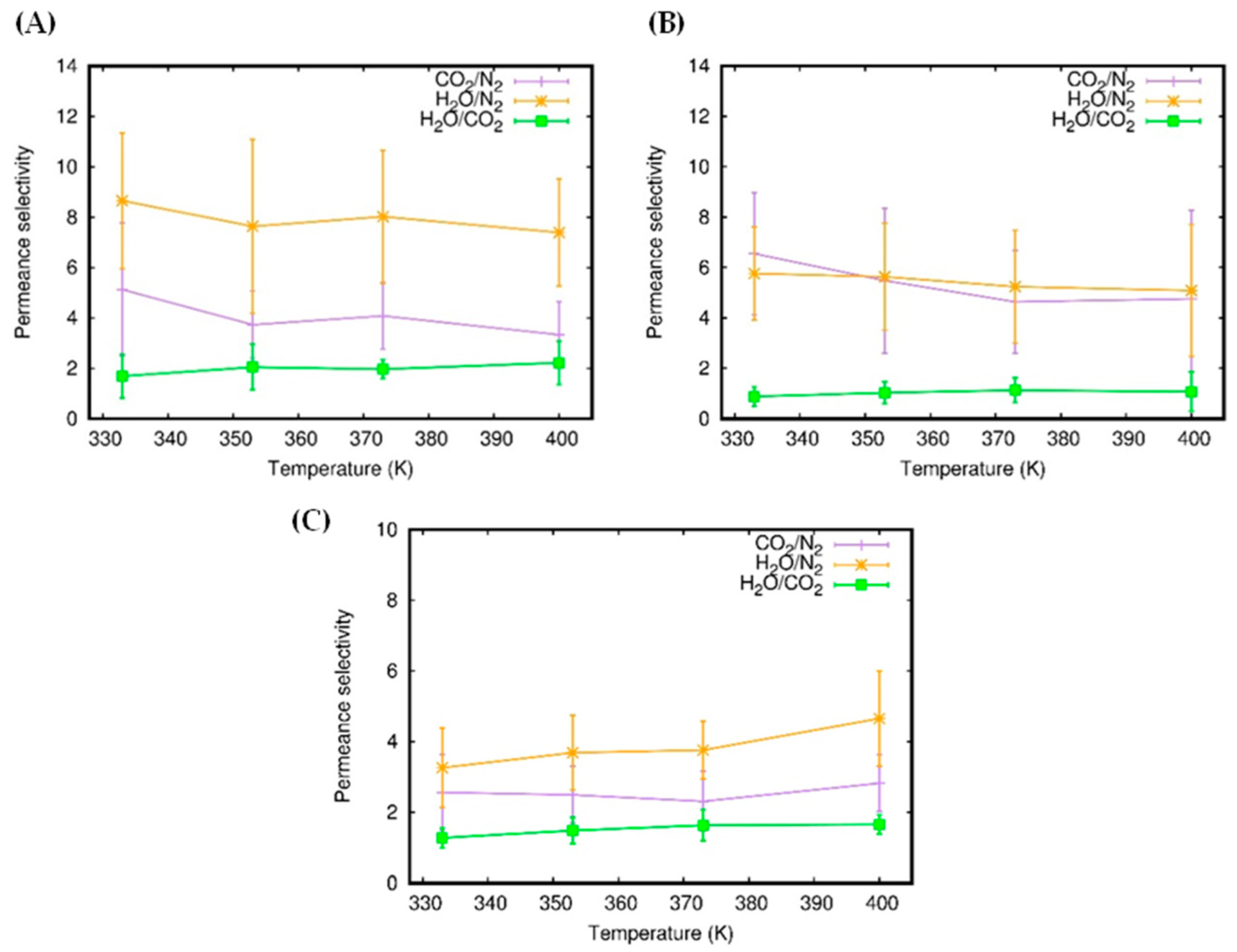
3.1. Gas Permeability
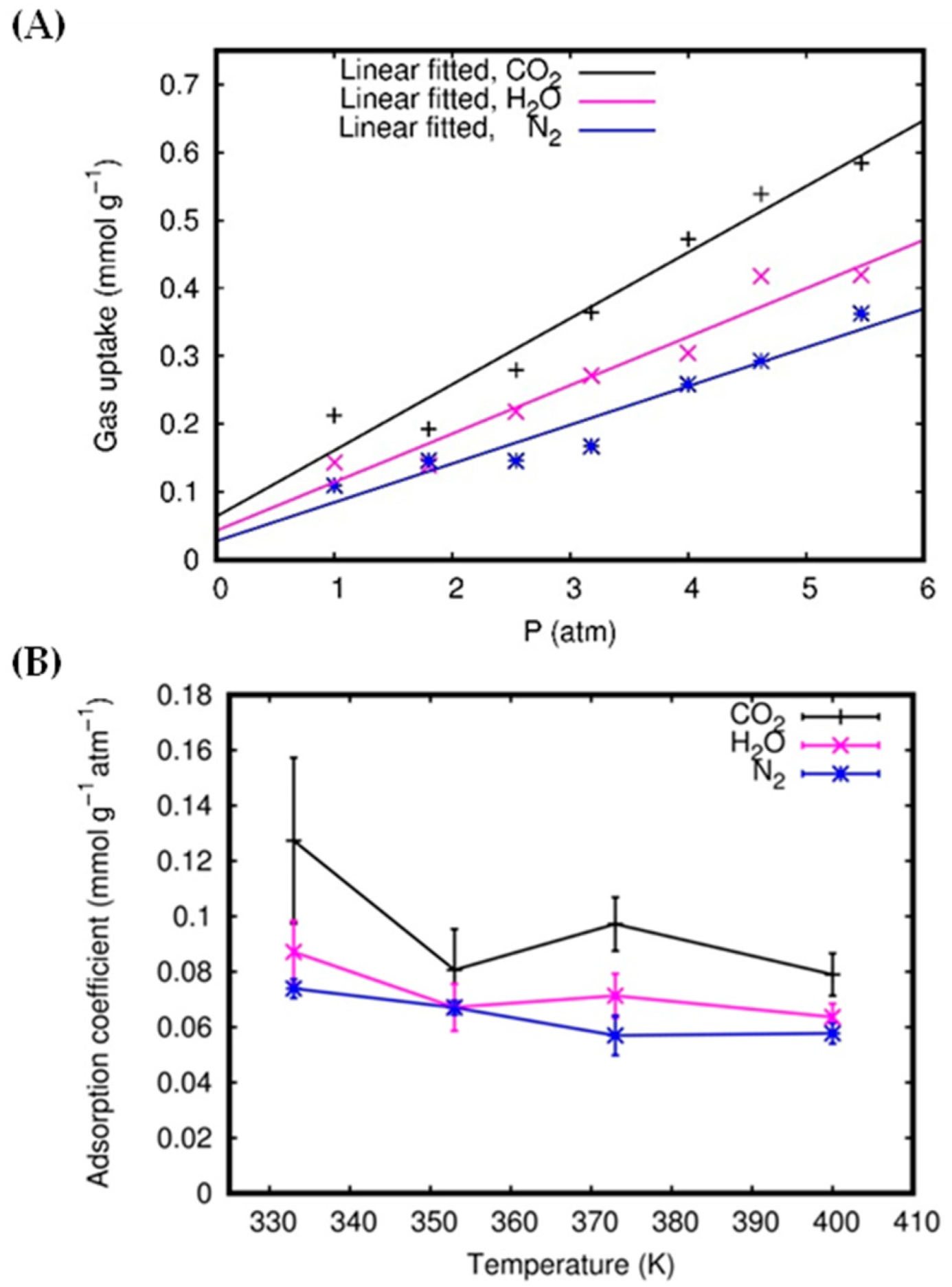
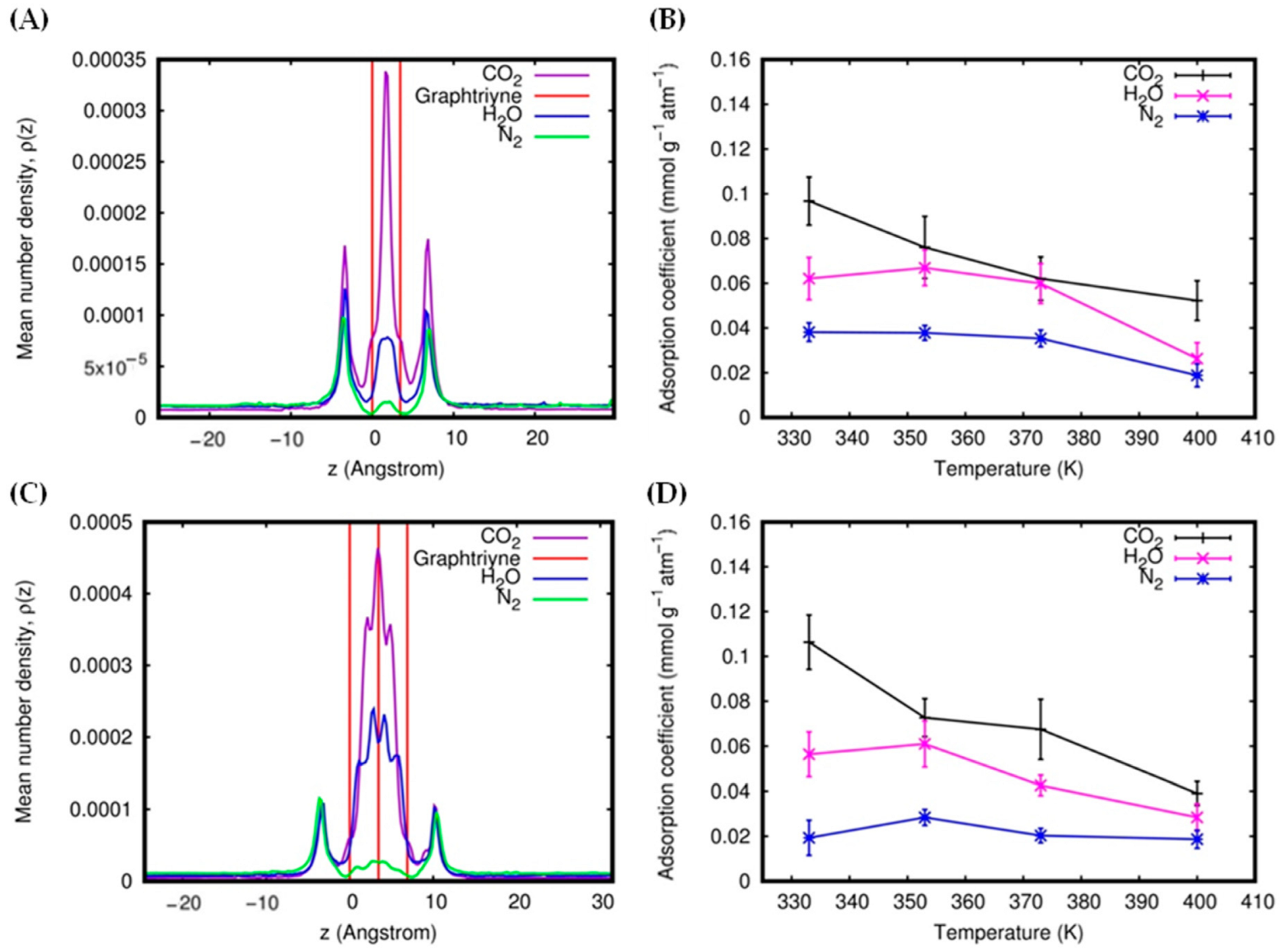
3.2. Gas Adsorption
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- United States Environmental Protection Agency (EPA). Climate Change Indicators in the United States: Global Greenhouse Gas Emissions. 2021. Available online: http://www.epa.gov/climate-indicators (accessed on 2 May 2022).
- World Resources Institute (WRI). Climate Analysis Indicators Tool (CAIT) 2.0: WRI’s Climate Data Explorer. 2019. Available online: http://cait.wri.org (accessed on 2 May 2022).
- Huck, J.M.; Lin, L.-C.; Berger, A.H.; Shahrak, M.N.; Martin, R.L.; Bhown, A.S.; Haranczyk, M.; Reuter, K.; Smit, B. Evaluating Different Classes of Porous Materials for Carbon Capture. Energy Environ. Sci. 2014, 7, 4132–4146. [Google Scholar] [CrossRef]
- Bui, M.; Adjiman, C.S.; Bardow, A.; Anthony, E.J.; Boston, A.; Brown, S.; Fennell, P.S.; Fuss, S.; Galindo, A.; Hackett, L.A.; et al. Carbon Capture and Storage (CCS): The Way Forward. Energy Environ. Sci. 2018, 11, 1062–1176. [Google Scholar] [CrossRef]
- Li, J.R.; Yu, J.; Lu, W.; Sun, L.B.; Sculley, J.; Balbuena, P.B.; Zhou, H.C. Porous Materials with Pre-Designed Single-Molecule Traps for CO2 Selective Adsorption. Nat. Commun. 2013, 4, 1538. [Google Scholar] [CrossRef]
- Smit, B. Carbon Capture and Storage: Introductory Lecture. Faraday Discuss. 2016, 192, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, G.; Krungleviciute, V.; Guo, Z.X.; Yildrim, T. Exceptional CO2 Capture in a Hieararchically Porous Carbon with Simultaneous High Surface Area and Pore Volume. Energy Environ. Sci. 2014, 7, 335–342. [Google Scholar] [CrossRef]
- Ganesan, A.; Shaijumon, M.M. Activated Graphene-Derived Porous Carbon with Exceptional Gas Adsorption Properties. Microporous Mesoporous Mater. 2016, 220, 21–27. [Google Scholar] [CrossRef]
- Ghosh, S.; Sevilla, M.; Fuertes, A.B.; Andreoli, E.; Ho, J.; Barron, A.R. Defining a Performance Map of Porous Carbon Sorbents for High-Pressure Carbon Dioxide Uptake and Carbon Dioxide-Methane Selectivity. J. Mater. Chem. A 2016, 4, 14739–14751. [Google Scholar] [CrossRef]
- Faginas-Lago, N.; Apriliyanto, Y.B.; Lombardi, A. Confinement of CO2 Inside Carbon Nanotubes. Eur. Phys. J. D 2021, 75, 1–10. [Google Scholar] [CrossRef]
- Kim, J.; Lin, L.C.; Swisher, J.A.; Haranczyk, M.; Smit, B. Predicting Large CO2 Adsorption in Aluminosilicate Zeolites for Postcombustion Carbon Dioxide Capture. J. Am. Chem. Soc. 2012, 134, 18940–18943. [Google Scholar] [CrossRef]
- Liu, B.; Smit, B. Molecular Simulation Studies of Separation of CO2/N2, CO2/CH4, and CH4/N2 by ZIFs. J. Phys. Chem. C 2010, 114, 8515–8522. [Google Scholar] [CrossRef]
- Lin, L.-C.; Kim, J.; Kong, X.; Scott, E.; McDonald, T.M.; Long, J.R.; Reimer, J.A.; Smit, B. Understanding CO2 Dynamics in Metal–Organic Frameworks with Open Metal Sites. Angew. Chem. 2013, 125, 4506–4509. [Google Scholar] [CrossRef]
- Queen, W.L.; Hudson, M.R.; Bloch, E.D.; Mason, J.A.; Gonzalez, M.I.; Lee, J.S.; Gygi, D.; Howe, J.D.; Lee, K.; Darwish, T.A.; et al. Comprehensive Study of Carbon Dioxide Adsorption in The Metal–Organic Frameworks M2(dobdc) (M = Mg, Mn, Fe, Co, Ni, Cu, Zn). Chem. Sci. 2014, 5, 4569–4581. [Google Scholar] [CrossRef]
- Liu, B.; Smit, B. Comparative Molecular Simulation Study of CO2/N2 and CH4/N2 Separation in Zeolites and Metal-Organic Frameworks. Langmuir 2009, 25, 5918–5926. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Huang, W.; Miao, J.; Xia, Q.; Zhang, Z.; Wang, H.; Li, Z. Enhanced Separation Performance of a Novel Composite Material Gro@MIL-101 for CO2/CH4 Binary Mixture. Chem. Eng. J. 2015, 266, 339–344. [Google Scholar] [CrossRef]
- Schrier, J. Carbon Dioxide Separation with a Two-Dimensional Polymer Membrane. ACS Appl. Mater. Interfaces 2012, 4, 3745–3752. [Google Scholar] [CrossRef]
- Xiang, Z.; Mercado, R.; Huck, J.M.; Wang, H.; Guo, Z.; Wang, W.; Cao, D.; Haranczyk, M.; Smit, B. Systematic Tuning and Multifunctionalization of Covalent Organic Polymers for Enhanced Carbon Capture. J. Am. Chem. Soc. 2015, 137, 13301–13307. [Google Scholar] [CrossRef]
- Sun, L.B.; Kang, Y.H.; Shi, Y.Q.; Jiang, Y.; Liu, X.Q. Highly Selective Capture of the Greenhouse Gas CO2 in Polymers. ACS Sustain. Chem. Eng. 2015, 3, 3077–3085. [Google Scholar] [CrossRef]
- Apriliyanto, Y.B.; Darmawan, N.; Faginas-Lago, N.; Lombardi, A. Two-dimensional Diamine-linked Covalent Organic Frameworks for CO2/N2 Capture and Separation: Theoretical Modeling and Simulations. Phys. Chem. Chem. Phys. 2020, 22, 25918–25929. [Google Scholar] [CrossRef]
- Liu, H.; Liu, B.; Lin, L.-C.; Chen, G.; Wu, Y.; Wang, J.; Gao, X.; Lv, Y.; Pan, Y.; Zhang, X.; et al. A Hybrid Absorption–Adsorption Method to Efficiently Capture Carbon. Nat. Commun. 2014, 5, 5147. [Google Scholar] [CrossRef]
- Koenig, S.P.; Wang, L.; Pellegrino, J.; Bunch, S. Selective Molecular Sieving Trough Porous Graphene. Nat. Nanotechnol. 2012, 7, 728–732. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Dai, S.; Jiang, D. Insights into CO2/N2 Separation Trough Nanoporous Graphene from Molecular Dynamics. Nanoscale 2013, 5, 9984–9987. [Google Scholar] [CrossRef]
- Wu, T.; Xue, Q.; Ling, C.; Shan, M.; Liu, Z.; Tao, Y.; Li, X. Fluorine-Modified Porous Graphene as Membrane for CO2/N2 Separation: Molecular Dynamic and First-Principles Simulations. J. Phys. Chem. C 2014, 118, 7369–7376. [Google Scholar] [CrossRef]
- Tao, Y.; Xue, Q.; Liu, Z.; Shan, M.; Ling, C.; Wu, T.; Li, X. Tunable Hydrogen Separation in Porous Graphene Membrane: First-Principle and Molecular Dynamic Simulation. ACS Appl. Mater. Interfaces 2014, 6, 8048–8058. [Google Scholar] [CrossRef]
- Meng, Z.; Zhang, X.; Zhang, Y.; Gao, H.; Wang, Y.; Shi, Q.; Rao, D.; Liu, Y.; Deng, K.; Lu, R. Graphdiyne as a High-Efficiency Membrane for Separating Oxygen from Harmful Gases: A First-Principles Study. ACS Appl. Mater. Interfaces 2016, 8, 28166–28170. [Google Scholar] [CrossRef] [PubMed]
- Bartolomei, M.; Carmona-Novillo, E.; Giorgi, G. First Principles Investigation of Hydrogen Physical Adsorption on Graphynes’ Layers. Carbon 2015, 95, 1076–1081. [Google Scholar] [CrossRef]
- Apriliyanto, Y.B.; Battaglia, S.; Evangelisti, S.; Faginas-Lago, N.; Leininger, T.; Lombardi, A. Toward a Generalized Hückel Rule: The Electronic Structure of Carbon Nanocones. J. Phys. Chem. A 2021, 125, 9819–9825. [Google Scholar] [CrossRef] [PubMed]
- Joos, L.; Huck, J.M.; Speybroeck, V.V.; Smit, B. Cutting the Cost of Carbon Capture: A Case for Carbon Capture and Utilization. Faraday Discuss. 2016, 192, 391–414. [Google Scholar] [CrossRef] [PubMed]
- Braun, E.; Zurhelle, A.F.; Thijssen, W.; Schnell, S.K.; Lin, L.C.; Kim, J.; Thompson, J.A.; Smit, B. High-Throughput Computational Screening of Nanoporous Adsorbents for CO2 Capture from Natural Gas. Mol. Syst. Des. Eng. 2016, 1, 175–188. [Google Scholar] [CrossRef]
- Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham, T.E., III; DeBolt, S.; Ferguson, D.; Seibel, G.; Kollman, P. AMBER, a Package of Computer Programs for Applying Molecular Mechanics, Normal Mode Analysis, Molecular Dynamics and Free Energy Calculations to Simulate the Structural and Energetic Properties of Molecules. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Rappé, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A., III; Skiff, W.M. UFF, a Full Periodic Table Force Fields for Molecular Mechanics and Molecular Dynamics Simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Vekeman, J.; Cuesta, I.G.; Faginas-Lago, N.; Wilson, J.; Sánchez-Marín, J.; de Merás, A.S. Potential Models for the Simulation of Methane Adsorption on Graphene: Development and CCSD (T) Benchmarks. Phys. Chem. Chem. Phys. 2018, 20, 25518–25530. [Google Scholar] [CrossRef] [PubMed]
- Bartolomei, M.; Carmona-Novillo, E.; Hernández, M.I.; Campos-Martínez, J.; Pirani, F.; Giorgi, G.; Yamashita, K. Penetration Barrier of Water through Graphynes’ Pores: First-Principles Predictions and Force Field Optimization. J. Phys. Chem. Lett. 2014, 5, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Boyd, P.G.; Moosavi, S.M.; Witman, M.; Smit, B. Force-Field Prediction of Materials Properties in Metal-Organic Frameworks. J. Phys. Chem. Lett. 2017, 8, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Mercado, R.; Vlaisavljevich, B.; Lin, L.-C.; Lee, K.; Lee, Y.; Mason, J.A.; Xiao, D.J.; Gonzalez, M.I.; Kapelewski, M.T.; Neaton, J.B.; et al. Force Field Development from Periodic Density Functional Theory Calculations for Gas Separation Applications Using Metal–Organic Frameworks. J. Phys. Chem. C 2016, 120, 12590–12604. [Google Scholar] [CrossRef]
- Lin, L.C.; Lee, K.; Gagliardi, L.; Neaton, J.B.; Smit, B. Force-Field Development from Electronic Structure Calculations with Periodic Boundary Conditions: Applications to Gaseous Adsorption and Transport in Metal–Organic Frameworks. J. Chem. Theory Comput. 2014, 10, 1477–1488. [Google Scholar] [CrossRef]
- Lim, J.R.; Yang, C.T.; Kim, J.; Lin, L.C. Transferability of CO2 Force Fields for Prediction of Adsorption Properties in All-Silica Zeolites. J. Phys. Chem. C 2018, 122, 10892–10903. [Google Scholar] [CrossRef]
- DuBay, K.H.; Hall, M.L.; Hughes, T.F.; Wu, C.; Reichman, D.R.; Friesner, R.A. Accurate Force Field Development for Modeling Conjugated Polymers. J. Chem. Theory Comput. 2012, 8, 4556–4569. [Google Scholar] [CrossRef]
- Ambrosetti, A.; Silvestrelli, P.L. Gas Separation in Nanoporous Graphene from First Principle Calculations. J. Phys. Chem. C 2014, 118, 19172–19179. [Google Scholar] [CrossRef]
- Du, H.; Li, J.; Zhang, J.; Su, G.; Li, X.; Zhao, Y. Separation of Hydrogen and Nitrogen Gases with Porous Graphene Membrane. J. Phys. Chem. C 2011, 115, 23261–23266. [Google Scholar] [CrossRef]
- Lin, S.; Buehler, M.J. Mechanics and Molecular Filtration Performance of Graphyne Nanoweb Membranes for Selective Water Purification. Nanoscale 2013, 5, 11801–11807. [Google Scholar] [CrossRef]
- James, A.; John, C.; Owais, C.; Myakala, S.N.; Shekar, S.C.; Choudhuri, J.R.; Swathi, R.S. Graphynes: Indispensable Nanoporous Architectures in Carbon Flatland. RSC Adv. 2018, 8, 22998–23018. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Gao, X.; Liu, R.; Xie, Z.; Yang, J.; Zhang, S.; Zhang, G.; Liu, H.; Li, Y.; Zhang, J.; et al. Synthesis of Graphdiyne Nanowalls Using Acetylenic Coupling Reaction. J. Am. Chem. Soc. 2015, 137, 7596–7599. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhu, Y.; Yi, D.; Zhou, J.; Zhang, S.; Yin, C.; Ding, F.; Zhang, S.; Yi, X.; Wang, J.; et al. Ultrathin Graphdiyne Film on Graphene through Solution-Phase Van Der Waals Epitaxy. Sci. Adv. 2018, 4, eaat6378. [Google Scholar] [CrossRef]
- Sakamoto, R.; Shiotsuki, R.; Wada, K.; Fukui, N.; Maeda, H.; Komeda, J.; Sekine, R.; Harano, K.; Nishihara, H. A Pyrazine-Incorporated Graphdiyne Nanofilm as a Metal-Free Electrocatalyst for The Hydrogen Evolution Reaction. J. Mater. Chem. A 2018, 6, 22189–22194. [Google Scholar] [CrossRef]
- Bao, H.; Wang, L.; Li, C.; Luo, J. Structural Characterization and Identification of Graphdiyne and Graphdiyne-Based Materials. ACS Appl. Mater. Interfaces 2019, 11, 2717–2729. [Google Scholar] [CrossRef]
- Gao, J.; Li, J.; Chen, Y.; Zuo, Z.; Li, Y.; Liu, H.; Li, Y. Architecture and Properties of a Novel two-Dimensional Carbon Material-graphtetrayne. Nano Energy 2018, 43, 192–199. [Google Scholar] [CrossRef]
- Bartolomei, M.; Giorgi, G. A Novel Nanoporous Graphite Based on Graphynes: First-Principles Structure and Carbon Dioxide Preferential Physisorption. ACS Appl. Mater. Interfaces 2016, 8, 27996–28003. [Google Scholar] [CrossRef]
- Apriliyanto, Y.B.; Faginas Lago, M.N.; Lombardi, A.; Evangelisti, S.; Bartolomei, M.; Leininger, T.; Pirani, F. Nanostructure Selectivity for Molecular Adsorption and Separation: The Case of Graphyne Layers. J. Phys. Chem. C 2018, 122, 16195–16208. [Google Scholar] [CrossRef]
- Albertí, M.; Aguilar, A.; Cappelletti, D.; Laganà, A.; Pirani, F. On the Development of an Effective Model Potential to Describe Water Interaction in Neutral and Ionic Clusters. Int. J. Mass Spectrom. 2009, 280, 50–56. [Google Scholar] [CrossRef]
- Albertí, M.; Pirani, F.; Laganà, A. Carbon Dioxide Clathrate Hydrates: Selective Role of Intermolecular Interactions and Action of the SDS Catalyst. J. Phys. Chem. A 2013, 117, 6991–7000. [Google Scholar] [CrossRef]
- Lombardi, A.; Pirani, F.; Laganà, A.; Bartolomei, M. Energy Transfer Dynamics and Kinetics of Elementary Processes (Promoted) by Gas-Phase CO2-N2 Collisions: Selectivity Control by the Anisotropy of the Interaction. J. Comput. Chem. 2016, 37, 1463–1475. [Google Scholar] [CrossRef] [PubMed]
- Bartolomei, M.; Pirani, F.; Laganà, A.; Lombardi, A. A Full Dimensional Grid Empowered Simulation of the CO2 + CO2 Processes. J. Comput. Chem. 2012, 33, 1806–1819. [Google Scholar] [CrossRef] [PubMed]
- Pirani, F.; Brizi, S.; Roncaratti, L.F.; Casavecchia, P.; Cappelletti, D.; Vecchiocattivi, F. Beyond the Lennard-Jones Model: A Simple and Accurate Potential Function Probed by High Resolution Scattering Data Useful for Molecular Dynamics Simulations. Phys. Chem. Chem. Phys. 2008, 10, 5489–5503. [Google Scholar] [CrossRef] [PubMed]
- Lago, N.F.; Larrañaga, F.H.; Albertí, M. On the Suitability of the ILJ Function to Match Different Formulations of the Electrostatic Potential for Water-Water Interactions. Eur. Phys. J. D 2009, 55, 75–85. [Google Scholar] [CrossRef]
- Faginas-Lago, N.; Yeni, D.; Huarte, F.; Wang, Y.; Alcamí, M.; Martin, F. Adsorption of Hydrogen Molecules on Carbon Nanotubes Using Quantum Chemistry and Molecular Dynamics. J. Phys. Chem. A 2016, 120, 6451–6458. [Google Scholar] [CrossRef]
- Wilson, J.; Faginas-Lago, N.; Vekeman, J.; Cuesta, I.G.; Sánchez-Marín, J.; de Merás, A.S. Modeling the Interaction of Carbon Monoxide with Flexible Graphene: From Coupled Cluster Calculations to Molecular-Dynamics Simulations. Chem. Phys. Chem. 2018, 19, 774–783. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577. [Google Scholar] [CrossRef]
- Smith, W.; Yong, C.; Rodger, P. DL_POLY: Application to Molecular Simulation. Mol. Simul. 2002, 28, 385–471. [Google Scholar] [CrossRef]
- Elliott, J.R.; Lira, C.T. Introductory Chemical Engineering Thermodynamics, 2nd ed.; Prentice Hall: Upper Saddle River, NJ, USA, 2012. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Molec. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Liu, Y.; Wilcox, J. Molecular Simulation Studies of CO2 Adsorption by Carbon Model Compounds for Carbon Capture and Sequestration Applications. Environ. Sci. Technol. 2013, 47, 95–101. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature (K) | Gas | System | Total Adsorption Selectivity | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 1.00 atm | 1.80 atm | 2.54 atm | 3.18 atm | 4.00 atm | 4.62 atm | 5.47 atm | |||
| 333 | CO2/N2 | Single layer | 4.97 | 2.81 | 1.65 | 1.63 | 1.88 | 2.24 | 2.30 |
| Bilayer | 3.43 | 4.58 | 4.67 | 3.67 | 4.14 | 3.26 | 2.90 | ||
| Trilayer | 7.61 | 8.92 | 7.64 | 5.62 | 4.99 | 4.82 | 5.95 | ||
| H2O/N2 | Single layer | 2.44 | 1.80 | 1.30 | 1.10 | 1.38 | 1.48 | 1.39 | |
| Bilayer | 3.00 | 2.84 | 2.40 | 1.65 | 2.10 | 1.86 | 2.12 | ||
| Trilayer | 3.73 | 3.07 | 3.74 | 2.38 | 2.65 | 2.23 | 2.91 | ||
| CO2/H2O | Single layer | 2.04 | 1.56 | 1.28 | 1.48 | 1.36 | 1.52 | 1.66 | |
| Bilayer | 1.14 | 1.62 | 1.95 | 2.22 | 1.97 | 1.75 | 1.37 | ||
| Trilayer | 2.04 | 2.91 | 2.04 | 2.36 | 1.89 | 2.16 | 2.05 | ||
| 353 | CO2/N2 | Single layer | 4.43 | 1.53 | 2.43 | 1.78 | 1.63 | 1.73 | 1.70 |
| Bilayer | 3.19 | 4.24 | 3.64 | 4.19 | 2.79 | 2.78 | 2.60 | ||
| Trilayer | 19.45 | 8.85 | 6.29 | 4.73 | 4.60 | 4.10 | 4.45 | ||
| H2O/N2 | Single layer | 2.10 | 1.40 | 1.21 | 1.13 | 1.11 | 1.19 | 1.25 | |
| Bilayer | 2.45 | 1.69 | 2.08 | 2.06 | 1.88 | 1.93 | 1.99 | ||
| Trilayer | 1.76 | 4.31 | 3.13 | 2.20 | 2.64 | 2.57 | 2.53 | ||
| CO2/H2O | Single layer | 2.11 | 1.09 | 2.01 | 1.57 | 1.46 | 1.45 | 1.35 | |
| Bilayer | 1.30 | 2.51 | 1.75 | 2.03 | 1.48 | 1.44 | 1.31 | ||
| Trilayer | 11.08 | 2.05 | 2.01 | 2.16 | 1.74 | 1.59 | 1.76 | ||
| 373 | CO2/N2 | Single layer | 2.30 | 1.34 | 2.07 | 2.37 | 1.97 | 1.98 | 1.70 |
| Bilayer | 5.16 | 3.04 | 3.40 | 2.67 | 2.62 | 2.62 | 2.33 | ||
| Trilayer | 2.19 | 2.92 | 4.12 | 5.34 | 4.16 | 3.78 | 2.90 | ||
| H2O/N2 | Single layer | 1.38 | 0.95 | 1.56 | 1.70 | 1.20 | 1.48 | 1.17 | |
| Bilayer | 0.81 | 3.12 | 2.05 | 1.42 | 2.04 | 1.59 | 1.84 | ||
| Trilayer | 2.68 | 2.99 | 2.27 | 2.80 | 2.34 | 2.35 | 2.30 | ||
| CO2/H2O | Single layer | 1.67 | 1.44 | 1.33 | 1.40 | 1.65 | 1.34 | 1.45 | |
| Bilayer | 6.35 | 0.97 | 1.65 | 1.88 | 1.28 | 1.65 | 1.26 | ||
| Trilayer | 0.82 | 0.97 | 1.81 | 1.92 | 1.75 | 1.61 | 1.26 | ||
| 400 | CO2/N2 | Single layer | 2.66 | 2.23 | 1.71 | 1.96 | 1.83 | 1.53 | 1.69 |
| Bilayer | 1.08 | 3.96 | 3.12 | 2.26 | 2.68 | 2.54 | 2.36 | ||
| Trilayer | 5.26 | 7.16 | 7.80 | 3.66 | 3.54 | 2.95 | 3.53 | ||
| H2O/N2 | Single layer | 1.05 | 1.72 | 0.98 | 1.39 | 1.25 | 1.17 | 1.13 | |
| Bilayer | 1.87 | 2.00 | 1.65 | 1.57 | 1.75 | 1.54 | 1.57 | ||
| Trilayer | 1.49 | 3.74 | 3.60 | 2.22 | 2.15 | 1.63 | 2.08 | ||
| CO2/H2O | Single layer | 2.53 | 1.30 | 1.74 | 1.41 | 1.46 | 1.31 | 1.50 | |
| Bilayer | 0.58 | 1.98 | 1.89 | 1.44 | 1.53 | 1.66 | 1.50 | ||
| Trilayer | 3.53 | 1.91 | 2.16 | 1.65 | 1.65 | 1.81 | 1.70 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Apriliyanto, Y.B.; Faginas-Lago, N.; Evangelisti, S.; Bartolomei, M.; Leininger, T.; Pirani, F.; Pacifici, L.; Lombardi, A. Multilayer Graphtriyne Membranes for Separation and Storage of CO2: Molecular Dynamics Simulations of Post-Combustion Model Mixtures. Molecules 2022, 27, 5958. https://doi.org/10.3390/molecules27185958
Apriliyanto YB, Faginas-Lago N, Evangelisti S, Bartolomei M, Leininger T, Pirani F, Pacifici L, Lombardi A. Multilayer Graphtriyne Membranes for Separation and Storage of CO2: Molecular Dynamics Simulations of Post-Combustion Model Mixtures. Molecules. 2022; 27(18):5958. https://doi.org/10.3390/molecules27185958
Chicago/Turabian StyleApriliyanto, Yusuf Bramastya, Noelia Faginas-Lago, Stefano Evangelisti, Massimiliano Bartolomei, Thierry Leininger, Fernando Pirani, Leonardo Pacifici, and Andrea Lombardi. 2022. "Multilayer Graphtriyne Membranes for Separation and Storage of CO2: Molecular Dynamics Simulations of Post-Combustion Model Mixtures" Molecules 27, no. 18: 5958. https://doi.org/10.3390/molecules27185958