
Modified Protein-Water Interactions in CHARMM36m for Thermodynamics and Kinetics of Proteins in Dilute and Crowded Solutions
 ,
,
Abstract
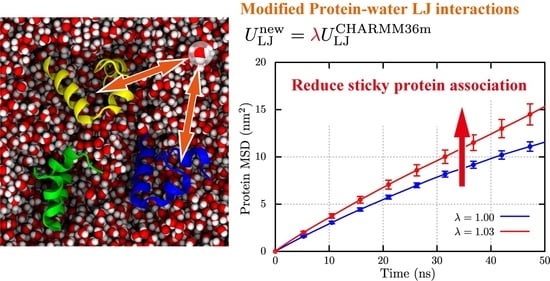
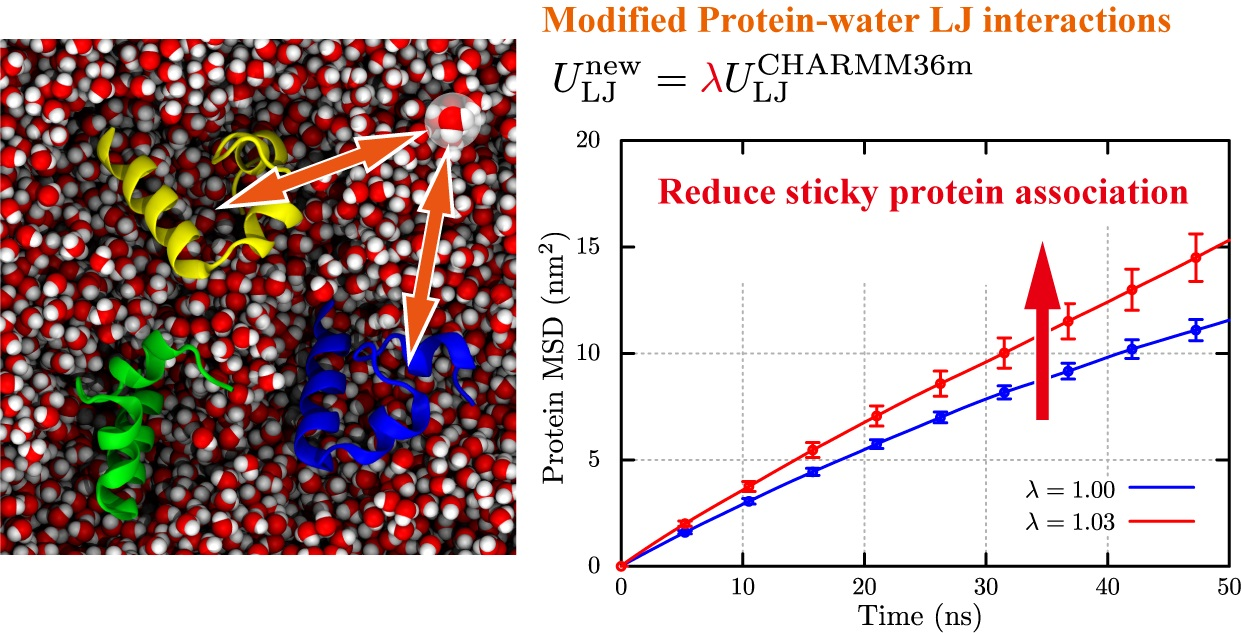
:
1. Introduction
2. Results
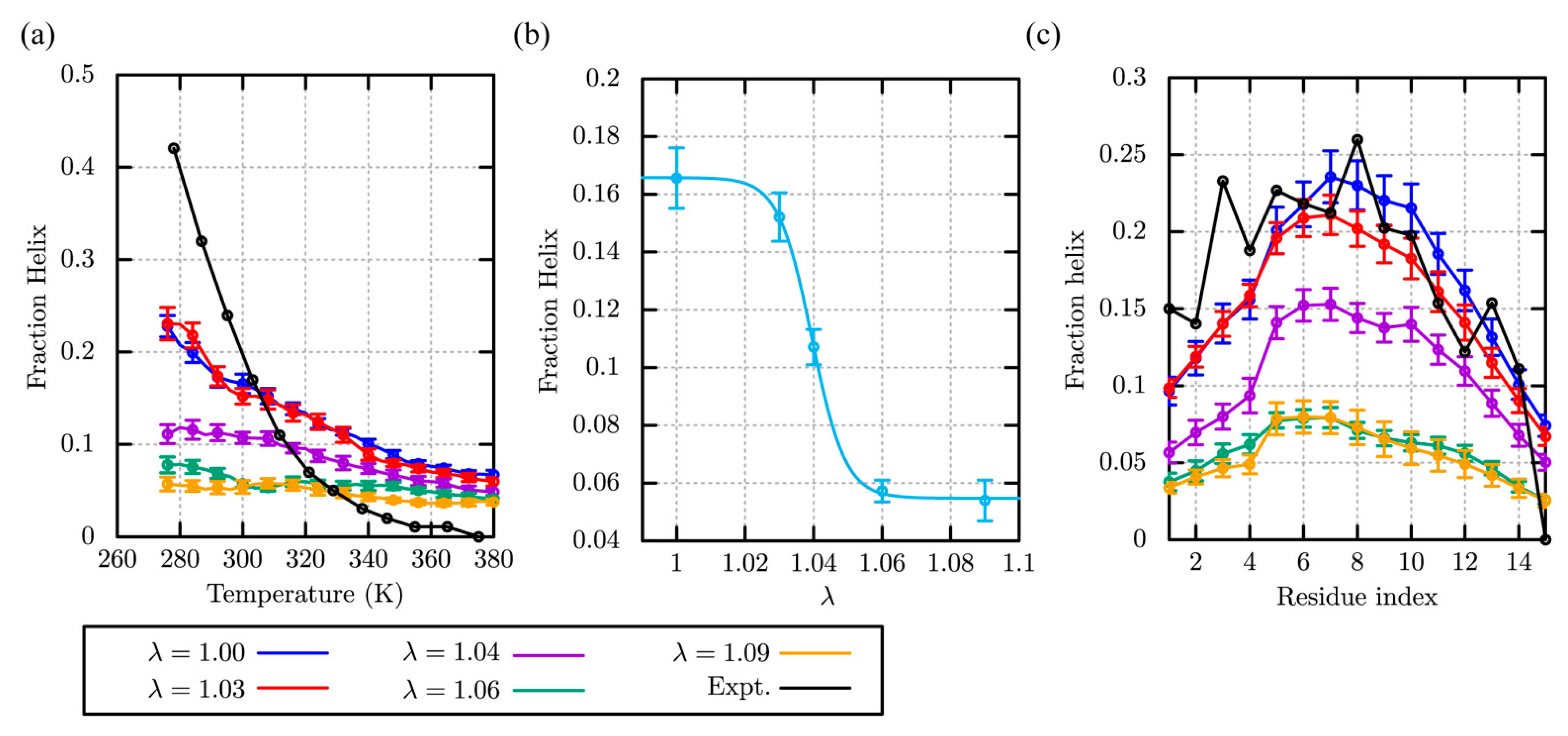
2.1. Folding and Stability of Small Peptides in Solution
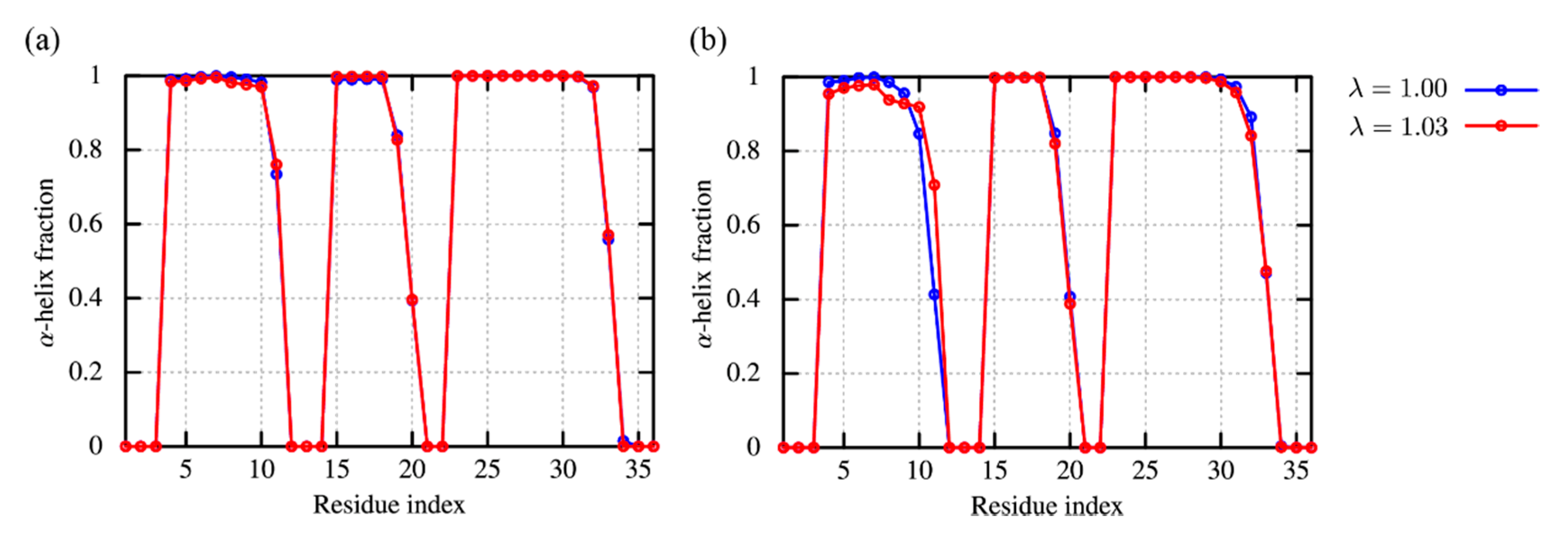
2.1.1. (AAQAA)3
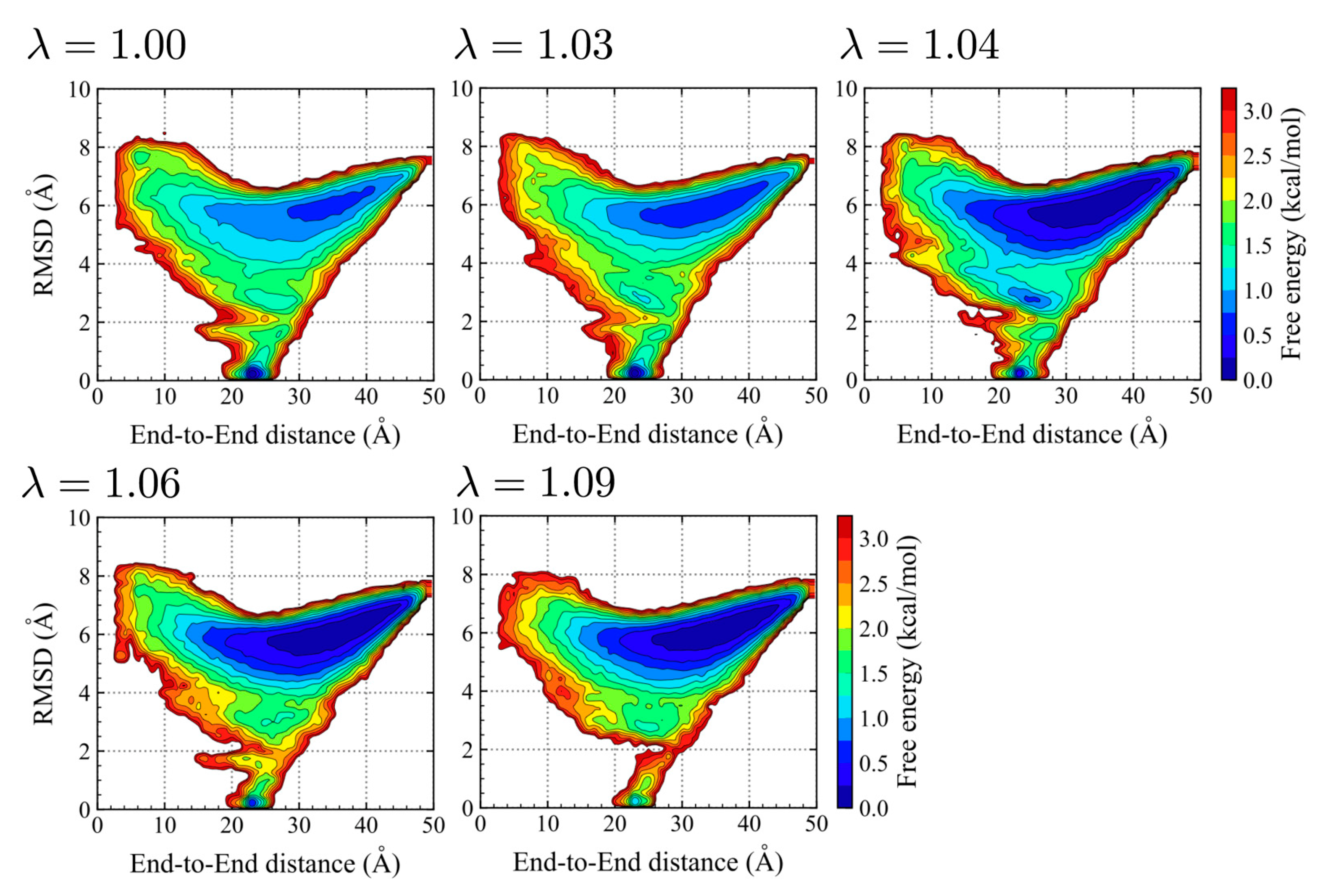
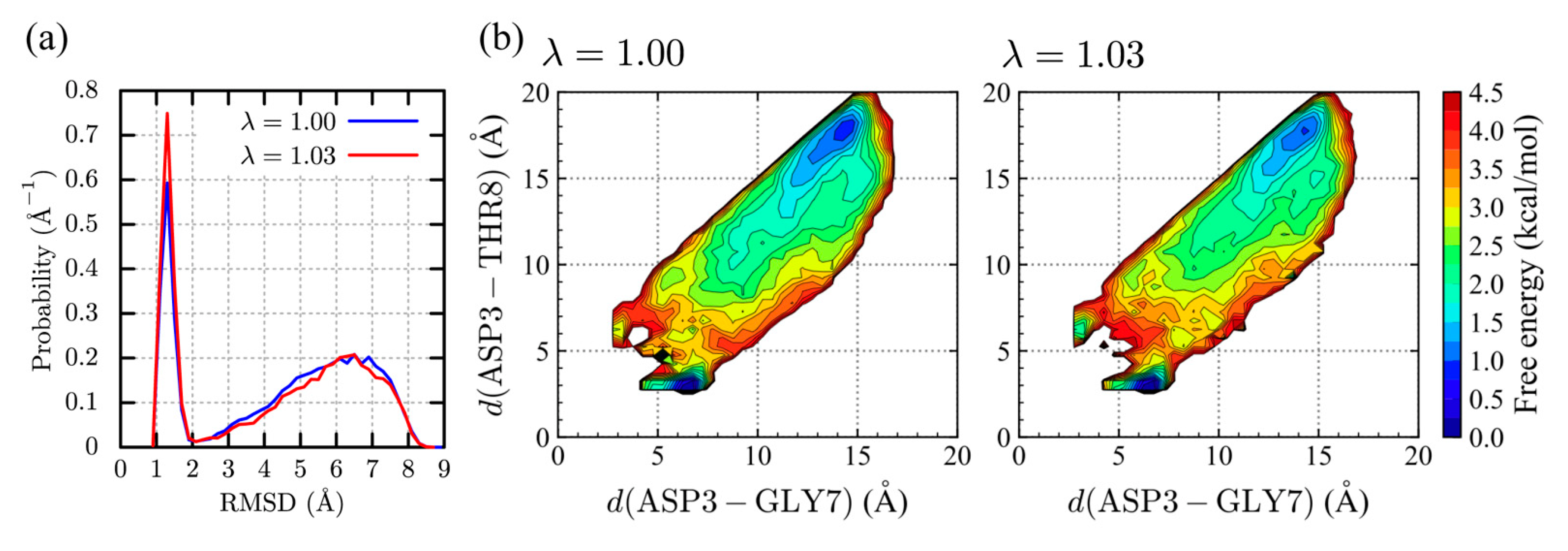
2.1.2. Chignolin
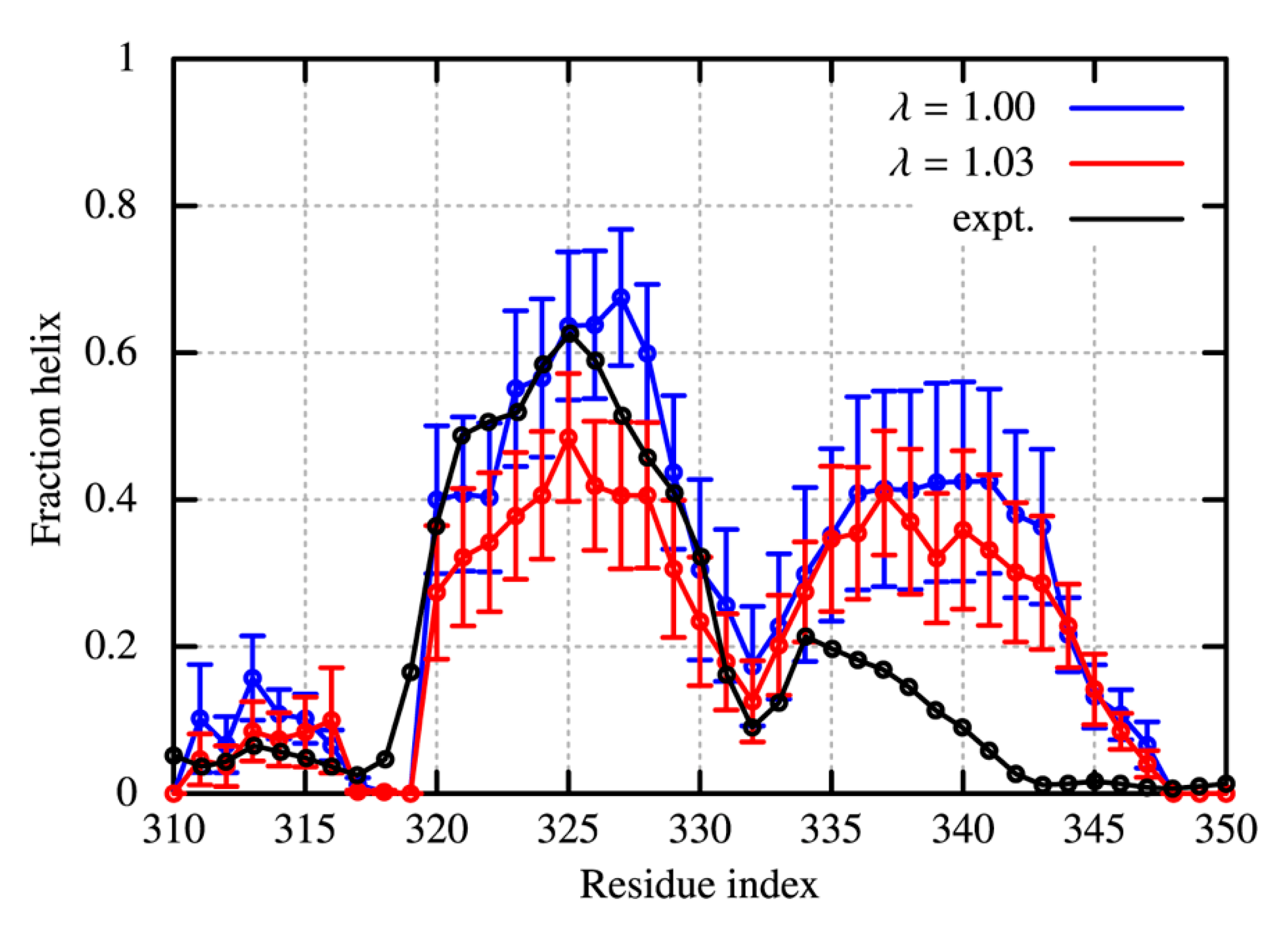
2.1.3. TDP-43 in the CTD
2.2. Dynamics and Stability of Globular Proteins in Solution
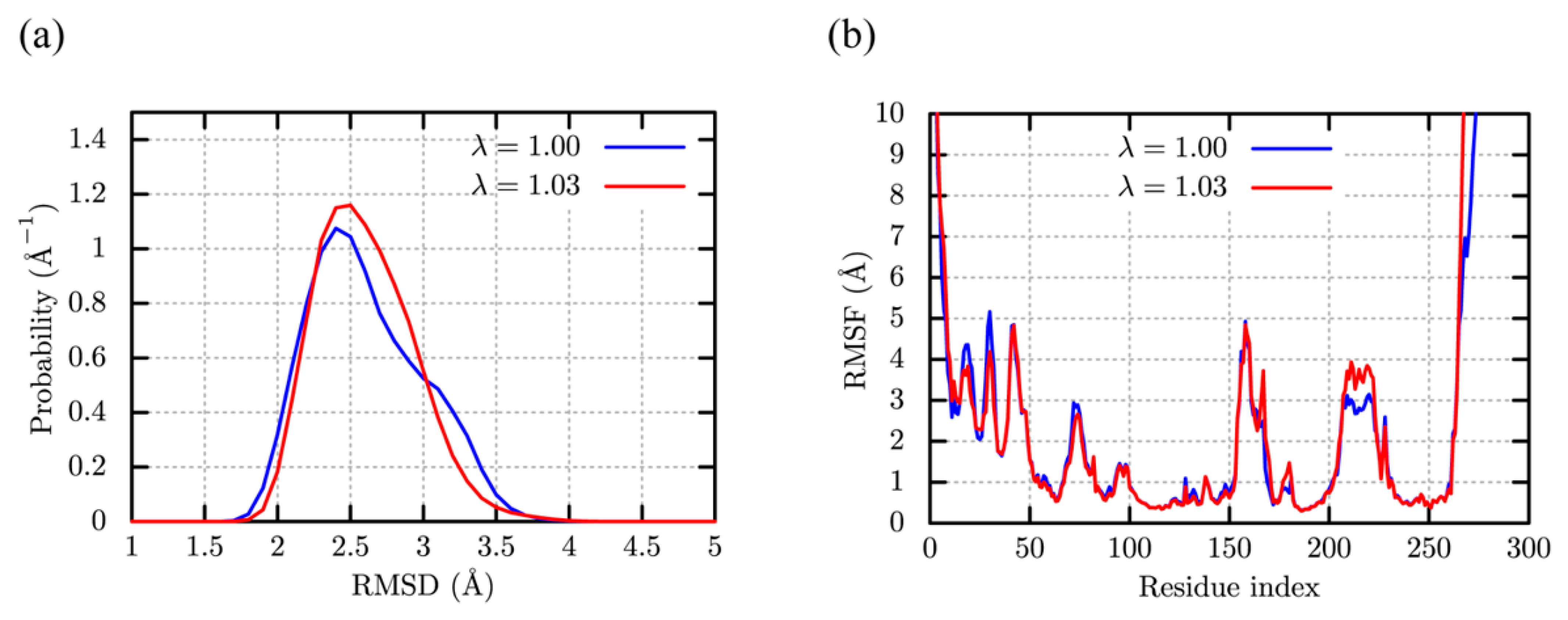
c-Src Kinase
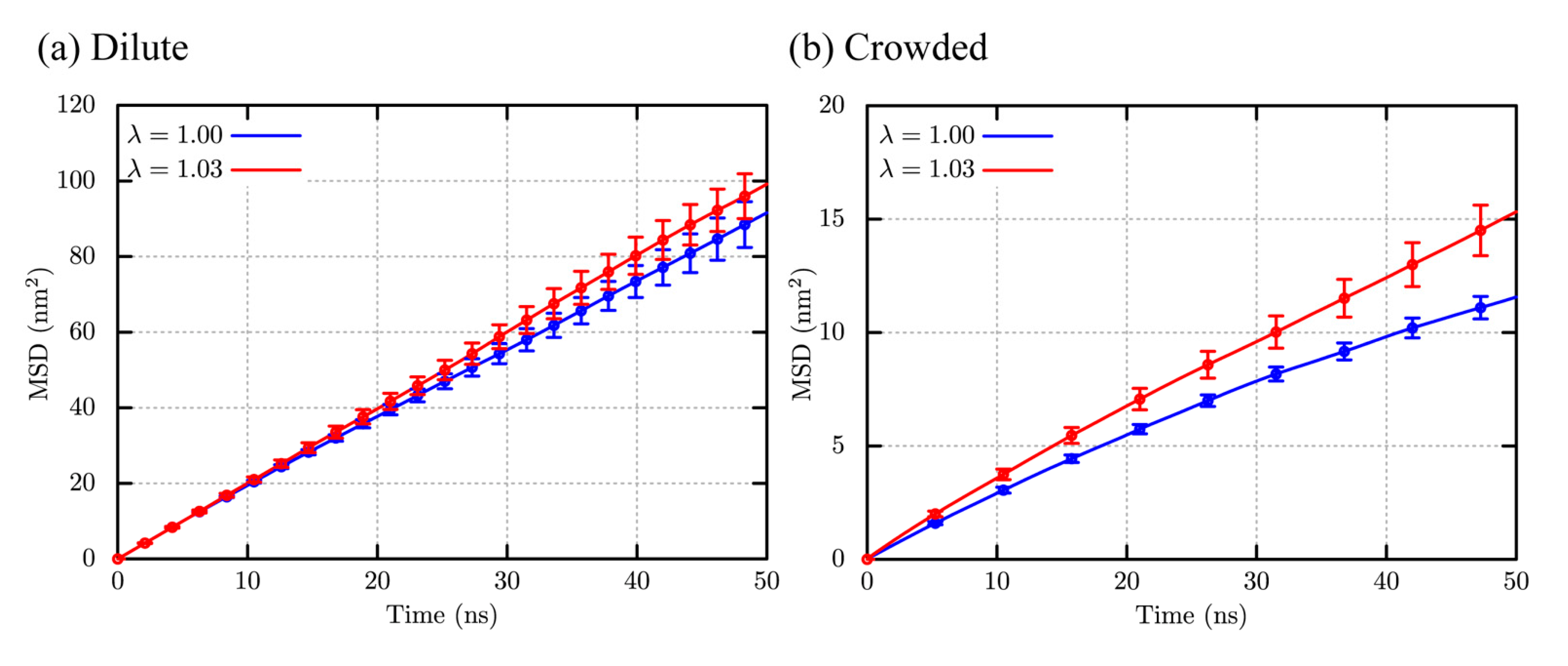
2.3. Structural Stability and Diffusivity of Globular Proteins in the Crowded Solutions
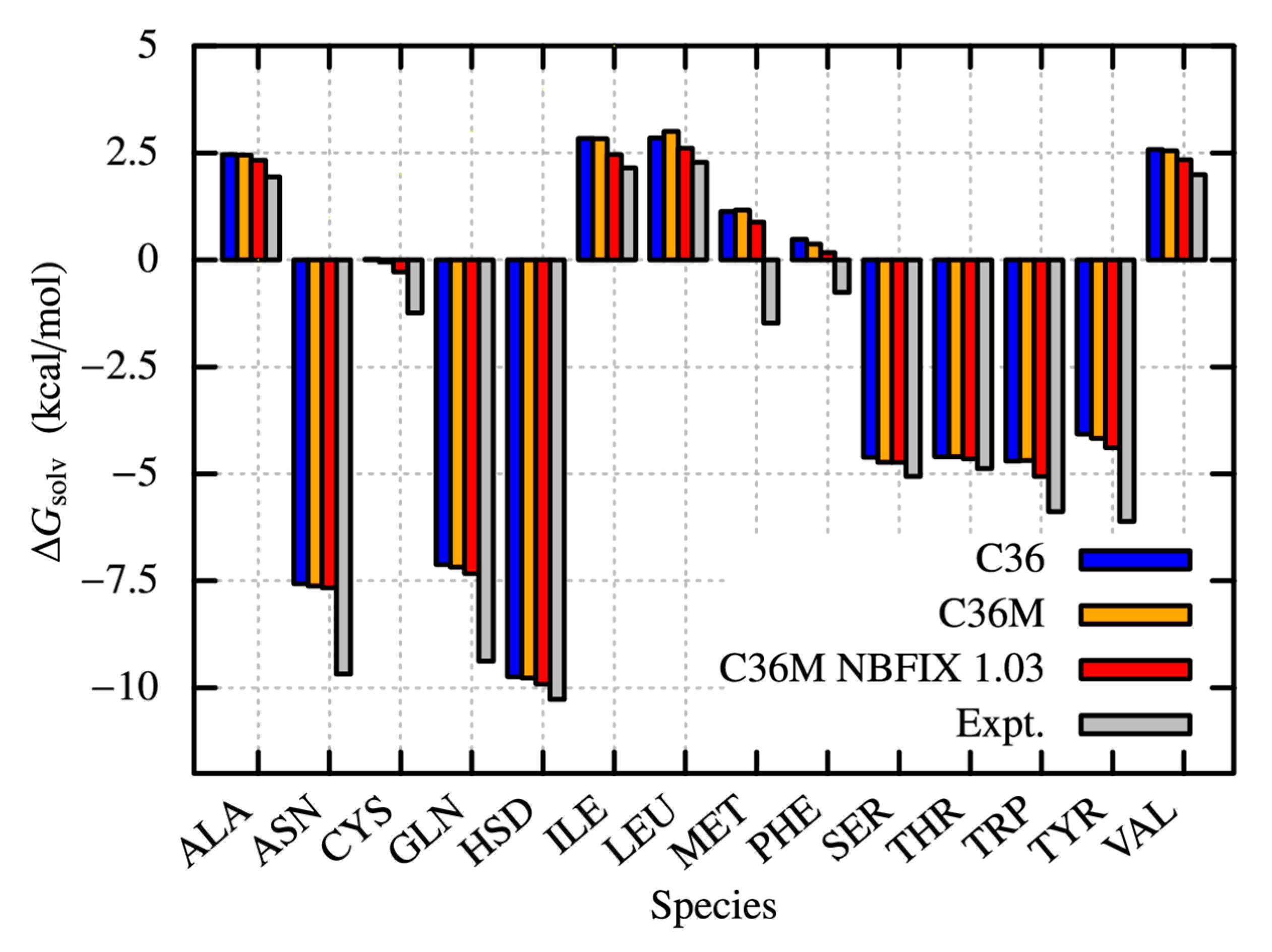
2.4. Solvation Free Energies of Amino Acid Analogues
3. Discussion and Conclusions
4. Materials and Methods
4.1. (AAQAA)3
4.2. Chignolin
4.3. TDP-43
4.4. c-Src Kinase
4.5. Villin Crowding Solution
4.6. Amino-Acid Analogues
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Brini, E.; Simmerling, C.; Dill, K. Protein storytelling through physics. Science 2020, 370, eaaz3041. [Google Scholar] [CrossRef]
- Schlick, T.; Portillo-Ledesma, S. Biomolecular modeling thrives in the age of technology. Nat. Comput. Sci. 2021, 1, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Shaw, D.E.; Maragakis, P.; Lindorff-Larsen, K.; Piana, S.; Dror, R.O.; Eastwood, M.P.; Bank, J.A.; Jumper, J.M.; Salmon, J.K.; Shan, Y.; et al. Atomic-level characterization of the structural dynamics of proteins. Science 2010, 330, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. Biomolecular simulation: A computational microscope for molecular biology. Annu. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef] [PubMed]
- Munro, J.B.; Altman, R.B.; Tung, C.S.; Cate, J.H.; Sanbonmatsu, K.Y.; Blanchard, S.C. Spontaneous formation of the unlocked state of the ribosome is a multistep process. Proc. Natl. Acad. Sci. USA 2010, 107, 709–714. [Google Scholar] [CrossRef]
- Song, X.; Jensen, M.O.; Jogini, V.; Stein, R.A.; Lee, C.H.; McHaourab, H.S.; Shaw, D.E.; Gouaux, E. Mechanism of NMDA receptor channel block by MK-801 and memantine. Nature 2018, 556, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Jung, J.; Kobayashi, C.; Dokainish, H.M.; Re, S.; Sugita, Y. Elucidation of interactions regulating conformational stability and dynamics of SARS-CoV-2 S-protein. Biophys. J. 2021, 120, 1060–1071. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, C.; Matsunaga, Y.; Jung, J.; Sugita, Y. Structural and energetic analysis of metastable intermediate states in the E1P-E2P transition of Ca(2+)-ATPase. Proc. Natl. Acad. Sci. USA 2021, 118, e2105507118. [Google Scholar] [CrossRef]
- Man, V.H.; He, X.; Derreumaux, P.; Ji, B.; Xie, X.Q.; Nguyen, P.H.; Wang, J. Effects of All-Atom Molecular Mechanics Force Fields on Amyloid Peptide Assembly: The Case of Abeta16-22 Dimer. J. Chem. Theory Comput. 2019, 15, 1440–1452. [Google Scholar] [CrossRef]
- Zerze, G.H.; Zheng, W.; Best, R.B.; Mittal, J. Evolution of All-Atom Protein Force Fields to Improve Local and Global Properties. J. Phys. Chem. Lett. 2019, 10, 2227–2234. [Google Scholar] [CrossRef]
- Zheng, W.; Dignon, G.L.; Jovic, N.; Xu, X.; Regy, R.M.; Fawzi, N.L.; Kim, Y.C.; Best, R.B.; Mittal, J. Molecular Details of Protein Condensates Probed by Microsecond Long Atomistic Simulations. J. Phys. Chem. B 2020, 124, 11671–11679. [Google Scholar] [CrossRef]
- Shea, J.E.; Best, R.B.; Mittal, J. Physics-based computational and theoretical approaches to intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2021, 67, 219–225. [Google Scholar] [CrossRef]
- Dignon, G.L.; Zheng, W.; Kim, Y.C.; Best, R.B.; Mittal, J. Sequence determinants of protein phase behavior from a coarse-grained model. PLoS Comput. Biol. 2018, 14, e1005941. [Google Scholar] [CrossRef]
- Joseph, J.A.; Reinhardt, A.; Aguirre, A.; Chew, P.Y.; Russell, K.O.; Espinosa, J.R.; Garaizar, A.; Collepardo-Guevara, R. Physics-driven coarse-grained model for biomolecular phase separation with near-quantitative accuracy. Nat. Comput. Sci. 2021, 1, 732–743. [Google Scholar] [CrossRef]
- Tan, C.; Jung, J.; Kobayashi, C.; Torre, D.U.; Takada, S.; Sugita, Y. Implementation of residue-level coarse-grained models in GENESIS for large-scale molecular dynamics simulations. PLoS Comput. Biol. 2022, 18, e1009578. [Google Scholar] [CrossRef]
- Dannenhoffer-Lafage, T.; Best, R.B. A Data-Driven Hydrophobicity Scale for Predicting Liquid-Liquid Phase Separation of Proteins. J. Phys. Chem. B 2021, 125, 4046–4056. [Google Scholar] [CrossRef]
- Regy, R.M.; Thompson, J.; Kim, Y.C.; Mittal, J. Improved coarse-grained model for studying sequence dependent phase separation of disordered proteins. Protein Sci. 2021, 30, 1371–1379. [Google Scholar] [CrossRef]
- Kumar, A.; Chakraborty, D.; Mugnai, M.L.; Straub, J.E.; Thirumalai, D. Sequence Determines the Switch in the Fibril Forming Regions in the Low-Complexity FUS Protein and Its Variants. J. Phys. Chem. Let.t 2021, 12, 9026–9032. [Google Scholar] [CrossRef]
- Yu, I.; Mori, T.; Ando, T.; Harada, R.; Jung, J.; Sugita, Y.; Feig, M. Biomolecular interactions modulate macromolecular structure and dynamics in atomistic model of a bacterial cytoplasm. eLife 2016, 5, e19274. [Google Scholar] [CrossRef]
- Feig, M.; Yu, I.; Wang, P.H.; Nawrocki, G.; Sugita, Y. Crowding in Cellular Environments at an Atomistic Level from Computer Simulations. J. Phys. Chem. B 2017, 121, 8009–8025. [Google Scholar] [CrossRef] [Green Version]
- Zimmerman, S.B.; Minton, A.P. Macromolecular crowding: Biochemical, biophysical, and physiological consequences. Annu. Rev. Biophys. Biomol. Struct. 1993, 22, 27–65. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.X.; Rivas, G.; Minton, A.P. Macromolecular crowding and confinement: Biochemical, biophysical, and potential physiological consequences. Annu. Rev. Biophys. 2008, 37, 375–397. [Google Scholar] [CrossRef] [PubMed]
- Miklos, A.C.; Sarkar, M.; Wang, Y.; Pielak, G.J. Protein crowding tunes protein stability. J. Am. Chem. Soc. 2011, 133, 7116–7120. [Google Scholar] [CrossRef] [PubMed]
- Speer, S.L.; Stewart, C.J.; Sapir, L.; Harries, D.; Pielak, G.J. Macromolecular Crowding Is More than Hard-Core Repulsions. Annu. Rev. Biophys. 2022, 51, 267–300. [Google Scholar] [CrossRef]
- Harada, R.; Sugita, Y.; Feig, M. Protein crowding affects hydration structure and dynamics. J. Am. Chem. Soc. 2012, 134, 4842–4849. [Google Scholar] [CrossRef]
- Harada, R.; Tochio, N.; Kigawa, T.; Sugita, Y.; Feig, M. Reduced native state stability in crowded cellular environment due to protein-protein interactions. J. Am. Chem. Soc. 2013, 135, 3696–3701. [Google Scholar] [CrossRef]
- Nawrocki, G.; Wang, P.H.; Yu, I.; Sugita, Y.; Feig, M. Slow-Down in Diffusion in Crowded Protein Solutions Correlates with Transient Cluster Formation. J. Phys. Chem. B 2017, 121, 11072–11084. [Google Scholar] [CrossRef]
- von Bulow, S.; Siggel, M.; Linke, M.; Hummer, G. Dynamic cluster formation determines viscosity and diffusion in dense protein solutions. Proc. Natl. Acad. Sci. USA 2019, 116, 9843–9852. [Google Scholar] [CrossRef]
- Rickard, M.M.; Zhang, Y.; Gruebele, M.; Pogorelov, T.V. In-Cell Protein-Protein Contacts: Transient Interactions in the Crowd. J. Phys. Chem. Lett. 2019, 10, 5667–5673. [Google Scholar] [CrossRef]
- Murthy, A.C.; Dignon, G.L.; Kan, Y.; Zerze, G.H.; Parekh, S.H.; Mittal, J.; Fawzi, N.L. Molecular interactions underlying liquid-liquid phase separation of the FUS low-complexity domain. Nat. Struct. Mol. Biol. 2019, 26, 637–648. [Google Scholar] [CrossRef]
- Conicella, A.E.; Dignon, G.L.; Zerze, G.H.; Schmidt, H.B.; D’Ordine, A.M.; Kim, Y.C.; Rohatgi, R.; Ayala, Y.M.; Mittal, J.; Fawzi, N.L. TDP-43 alpha-helical structure tunes liquid-liquid phase separation and function. Proc. Natl. Acad. Sci. USA 2020, 117, 5883–5894. [Google Scholar] [CrossRef]
- Ball, P. Water is an active matrix of life for cell and molecular biology. Proc. Natl. Acad. Sci. USA 2017, 114, 13327–13335. [Google Scholar] [CrossRef]
- Nerenberg, P.S.; Jo, B.; So, C.; Tripathy, A.; Head-Gordon, T. Optimizing solute-water van der Waals interactions to reproduce solvation free energies. J. Phys. Chem. B 2012, 116, 4524–4534. [Google Scholar] [CrossRef]
- Best, R.B.; Zheng, W.; Mittal, J. Balanced Protein-Water Interactions Improve Properties of Disordered Proteins and Non-Specific Protein Association. J. Chem. Theory Comput. 2014, 10, 5113–5124. [Google Scholar] [CrossRef]
- Tang, W.S.; Fawzi, N.L.; Mittal, J. Refining All-Atom Protein Force Fields for Polar-Rich, Prion-like, Low-Complexity Intrinsically Disordered Proteins. J. Phys. Chem. B 2020, 124, 9505–9512. [Google Scholar] [CrossRef]
- Best, R.B. Atomistic Force Fields for Proteins. Methods Mol. Biol. 2019, 2022, 3–19. [Google Scholar]
- van der Spoel, D. Systematic design of biomolecular force fields. Curr. Opin. Struct. Biol. 2021, 67, 18–24. [Google Scholar] [CrossRef]
- Mackerell, A.D., Jr.; Feig, M.; Brooks, C.L., 3rd. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef]
- Wu, H.N.; Jiang, F.; Wu, Y.D. Significantly Improved Protein Folding Thermodynamics Using a Dispersion-Corrected Water Model and a New Residue-Specific Force Field. J. Phys. Chem. Lett. 2017, 8, 3199–3205. [Google Scholar] [CrossRef]
- Robustelli, P.; Piana, S.; Shaw, D.E. Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. USA 2018, 115, E4758–E4766. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; Mackerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmuller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]
- Piana, S.; Lindorff-Larsen, K.; Shaw, D.E. How robust are protein folding simulations with respect to force field parameterization? Biophys. J. 2011, 100, L47–L49. [Google Scholar] [CrossRef]
- Piana, S.; Klepeis, J.L.; Shaw, D.E. Assessing the accuracy of physical models used in protein-folding simulations: Quantitative evidence from long molecular dynamics simulations. Curr. Opin. Struct. Biol. 2014, 24, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Abascal, J.L.; Vega, C. A general purpose model for the condensed phases of water: TIP4P/2005. J. Chem. Phys. 2005, 123, 234505. [Google Scholar] [CrossRef] [Green Version]
- Thomasen, F.E.; Pesce, F.; Roesgaard, M.A.; Tesei, G.; Lindorff-Larsen, K. Improving Martini 3 for Disordered and Multidomain Proteins. J. Chem. Theory Comput. 2022, 18, 2033–2041. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Best, R.B.; de Sancho, D.; Mittal, J. Residue-specific alpha-helix propensities from molecular simulation. Biophys. J. 2012, 102, 1462–1467. [Google Scholar] [CrossRef]
- Piana, S.; Donchev, A.G.; Robustelli, P.; Shaw, D.E. Water dispersion interactions strongly influence simulated structural properties of disordered protein states. J. Phys. Chem. B 2015, 119, 5113–5123. [Google Scholar] [CrossRef] [PubMed]
- Honda, S.; Yamasaki, K.; Sawada, Y.; Morii, H. 10 residue folded peptide designed by segment statistics. Structure 2004, 12, 1507–1518. [Google Scholar] [CrossRef]
- Cowan-Jacob, S.W.; Fendrich, G.; Manley, P.W.; Jahnke, W.; Fabbro, D.; Liebetanz, J.; Meyer, T. The crystal structure of a c-Src complex in an active conformation suggests possible steps in c-Src activation. Structure 2005, 13, 861–871. [Google Scholar] [CrossRef]
- McKnight, C.J.; Matsudaira, P.T.; Kim, P.S. NMR structure of the 35-residue villin headpiece subdomain. Nat. Struct. Biol. 1997, 4, 180–184. [Google Scholar] [CrossRef]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Miao, Y.; Feher, V.A.; McCammon, J.A. Gaussian Accelerated Molecular Dynamics: Unconstrained Enhanced Sampling and Free Energy Calculation. J. Chem. Theory Comput. 2015, 11, 3584–3595. [Google Scholar] [CrossRef]
- Oshima, H.; Re, S.; Sugita, Y. Replica-Exchange Umbrella Sampling Combined with Gaussian Accelerated Molecular Dynamics for Free-Energy Calculation of Biomolecules. J. Chem. Theory Comput. 2019, 15, 5199–5208. [Google Scholar] [CrossRef]
- Shalongo, W.; Dugad, L.; Stellwagen, E. Distribution of Helicity within the Model Peptide Acetyl(AAQAA)3amide. J. Am. Chem. Soc. 2002, 116, 8288–8293. [Google Scholar] [CrossRef]
- Sugita, Y.; Kitao, A.; Okamoto, Y. Multidimensional replica-exchange method for free-energy calculations. J. Chem. Phys. 2000, 113, 6042–6051. [Google Scholar] [CrossRef]
- Satoh, D.; Shimizu, K.; Nakamura, S.; Terada, T. Folding free-energy landscape of a 10-residue mini-protein, chignolin. FEBS Lett. 2006, 580, 3422–3426. [Google Scholar] [CrossRef]
- Re, S.; Oshima, H.; Kasahara, K.; Kamiya, M.; Sugita, Y. Encounter complexes and hidden poses of kinase-inhibitor binding on the free-energy landscape. Proc. Natl. Acad. Sci. USA 2019, 116, 18404–18409. [Google Scholar] [CrossRef]
- Kasahara, K.; Re, S.; Nawrocki, G.; Oshima, H.; Mishima-Tsumagari, C.; Miyata-Yabuki, Y.; Kukimoto-Niino, M.; Yu, I.; Shirouzu, M.; Feig, M.; et al. Reduced efficacy of a Src kinase inhibitor in crowded protein solution. Nat. Commun. 2021, 12, 4099. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Touw, W.G.; Baakman, C.; Black, J.; te Beek, T.A.; Krieger, E.; Joosten, R.P.; Vriend, G. A series of PDB-related databanks for everyday needs. Nucleic Acids Res. 2015, 43, D364–D368. [Google Scholar] [CrossRef]
- Yeh, I.-C.; Hummer, G. System-Size Dependence of Diffusion Coefficients and Viscosities from Molecular Dynamics Simulations with Periodic Boundary Conditions. J. Phys. Chem. B 2004, 108, 15873–15879. [Google Scholar] [CrossRef]
- Ortega, A.; Amoros, D.; Garcia de la Torre, J. Prediction of hydrodynamic and other solution properties of rigid proteins from atomic- and residue-level models. Biophys. J. 2011, 101, 892–898. [Google Scholar] [CrossRef]
- Feller, S.E.; Pastor, R.W.; Rojnuckarin, A.; Bogusz, S.; Brooks, B.R. Effect of Electrostatic Force Truncation on Interfacial and Transport Properties of Water. J. Phys. Chem. 1996, 100, 17011–17020. [Google Scholar] [CrossRef]
- Korson, L.; Drost-Hansen, W.; Millero, F.J. Viscosity of water at various temperatures. J. Phys. Chem. 1969, 73, 34–39. [Google Scholar] [CrossRef]
- Wolfenden, R.; Andersson, L.; Cullis, P.M.; Southgate, C.C. Affinities of amino acid side chains for solvent water. Biochemistry 1981, 20, 849–855. [Google Scholar] [CrossRef]
- Jung, J.; Mori, T.; Kobayashi, C.; Matsunaga, Y.; Yoda, T.; Feig, M.; Sugita, Y. GENESIS: A hybrid-parallel and multi-scale molecular dynamics simulator with enhanced sampling algorithms for biomolecular and cellular simulations. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2015, 5, 310–323. [Google Scholar] [CrossRef]
- Kobayashi, C.; Jung, J.; Matsunaga, Y.; Mori, T.; Ando, T.; Tamura, K.; Kamiya, M.; Sugita, Y. GENESIS 1.1: A hybrid-parallel molecular dynamics simulator with enhanced sampling algorithms on multiple computational platforms. J. Comput. Chem. 2017, 38, 2193–2206. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Patriksson, A.; van der Spoel, D. A temperature predictor for parallel tempering simulations. Phys. Chem. Chem. Phys. 2008, 10, 2073–2077. [Google Scholar] [CrossRef]
- Tuckerman, M.; Berne, B.J.; Martyna, G.J. Reversible multiple time scale molecular dynamics. J. Chem. Phys. 1992, 97, 1990–2001. [Google Scholar] [CrossRef]
- Jung, J.; Kasahara, K.; Kobayashi, C.; Oshima, H.; Mori, T.; Sugita, Y. Optimized Hydrogen Mass Repartitioning Scheme Combined with Accurate Temperature/Pressure Evaluations for Thermodynamic and Kinetic Properties of Biological Systems. J. Chem. Theory Comput. 2021, 17, 5312–5321. [Google Scholar] [CrossRef]
- Shirts, M.R.; Chodera, J.D. Statistically optimal analysis of samples from multiple equilibrium states. J. Chem. Phys. 2008, 129, 124105. [Google Scholar] [CrossRef]
- Hettne, K.M.; Williams, A.J.; van Mulligen, E.M.; Kleinjans, J.; Tkachenko, V.; Kors, J.A. Automatic vs. manual curation of a multi-source chemical dictionary: The impact on text mining. J. Cheminform. 2010, 2, 3. [Google Scholar] [CrossRef]
- Shirts, M.R.; Pitera, J.W.; Swope, W.C.; Pande, V.S. Extremely precise free energy calculations of amino acid side chain analogs: Comparison of common molecular mechanics force fields for proteins. J. Chem. Phys. 2003, 119, 5740–5761. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DMSD (nm2/ns) | DPBC (nm2/ns) | D (nm2/ns) | D′ (nm2/ns) | |
| Dilute | 0.33 (0.30) | 0.17 (0.17) | 0.50 (0.48) | 0.20 (0.19) |
| Crowded | 0.048 (0.031) | 0.15 (0.15) | 0.20 (0.18) | 0.077 (0.070) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsubara, D.; Kasahara, K.; Dokainish, H.M.; Oshima, H.; Sugita, Y. Modified Protein-Water Interactions in CHARMM36m for Thermodynamics and Kinetics of Proteins in Dilute and Crowded Solutions. Molecules 2022, 27, 5726. https://doi.org/10.3390/molecules27175726
Matsubara D, Kasahara K, Dokainish HM, Oshima H, Sugita Y. Modified Protein-Water Interactions in CHARMM36m for Thermodynamics and Kinetics of Proteins in Dilute and Crowded Solutions. Molecules. 2022; 27(17):5726. https://doi.org/10.3390/molecules27175726
Chicago/Turabian StyleMatsubara, Daiki, Kento Kasahara, Hisham M. Dokainish, Hiraku Oshima, and Yuji Sugita. 2022. "Modified Protein-Water Interactions in CHARMM36m for Thermodynamics and Kinetics of Proteins in Dilute and Crowded Solutions" Molecules 27, no. 17: 5726. https://doi.org/10.3390/molecules27175726