
Can We Merge the Weak and Strong Tetrel Bonds? Electronic Features of Tetrahedral Molecules Interacted with Halide Anions


Abstract
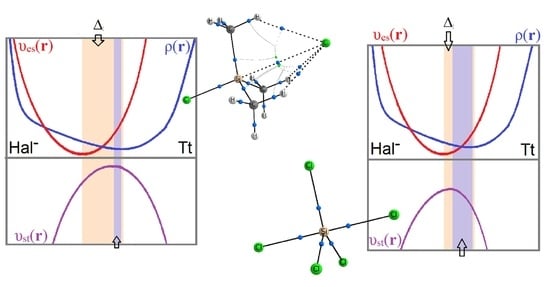
:
1. Introduction
1.1. Tetrel Bonds
1.2. Orbital-Free Quantum Crystallography Approach
- -
- What electronic features of TtBs formed by tetrahedral molecules, where Tt = C, Si, Ge, Sn, Pb, might be made visible?
- -
- How does the behavior of the Pauli potential, and electrostatic and static potentials differ for weak and strong bonds formed by a Tt atom?
- -
- Could the exchange-correlation contribution to the static potential characterize TtB quantitatively?
- -
- Is there a visible change in the properties at the junction of weak and strong TtBs? Can we merge them?
2. Materials and Methods
3. Results and Discussion
3.1. Electron Density Properties

3.2. Electronic Criterion for Weak and Strong Bonds Involving Tt



4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Legon, A.C. Tetrel, Pnictogen and Chalcogen Bonds Identified in the Gas Phase before They Had Names: A Systematic Look at Non-Covalent Interactions. Phys. Chem. Chem. Phys. 2017, 19, 14884–14896. [Google Scholar] [CrossRef] [PubMed]
- Terraneo, G.; Resnati, G. Bonding Matters. Cryst. Growth Des. 2017, 17, 1439–1440. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. A Look at Bonds and Bonding. Struct. Chem. 2019, 30, 1153–1157. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. The Fundamental Nature and Role of the Electrostatic Potential in Atoms and Molecules. Theor. Chem. Acc. 2002, 108, 134–142. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen Bonding: An Electrostatically-Driven Highly Directional Noncovalent Interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the Halogen Bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Aakeroy, C.B.; Bryce, D.L.; Desiraju, G.R.; Frontera, A.; Legon, A.C.; Nicotra, F.; Rissanen, K.; Scheiner, S.; Terraneo, G.; Metrangolo, P.; et al. Definition of the Chalcogen Bond (IUPAC Recommendations 2019). Pure Appl. Chem. 2019, 91, 1889–1892. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Frontera, A. Not Only Hydrogen Bonds: Other Noncovalent Interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef]
- Grabowski, S.J. Tetrel Bond–σ-Hole Bond as a Preliminary Stage of the SN2 Reaction. Phys. Chem. Chem. Phys. 2014, 16, 1824–1834. [Google Scholar] [CrossRef] [PubMed]
- Sethio, D.; Oliveira, V.; Kraka, E. Quantitative Assessment of Tetrel Bonding Utilizing Vibrational Spectroscopy. Molecules 2018, 23, 2763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Scheiner, S. Effects of Halogen, Chalcogen, Pnicogen, and Tetrel Bonds on IR and NMR Spectra. Molecules 2019, 24, 2822. [Google Scholar] [CrossRef] [PubMed]
- Hou, M.; Liu, Z.; Li, Q. The π-Hole Tetrel Bond between X2TO and CO2: Substituent Effects and Its Potential Adsorptivity for CO2. Int. J. Quantum Chem. 2020, 120, e26251. [Google Scholar] [CrossRef]
- Scheiner, S. Steric Crowding in Tetrel Bonds. J. Phys. Chem. A 2018, 122, 2550–2562. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Origins and Properties of the Tetrel Bond. Phys. Chem. Chem. Phys. 2021, 23, 5702–5717. [Google Scholar] [CrossRef]
- Zierkiewicz, W.; Michalczyk, M.; Scheiner, S. Comparison between Tetrel Bonded Complexes Stabilized by σ and π Hole Interactions. Molecules 2018, 23, 1416. [Google Scholar] [CrossRef]
- Grabowski, S.J.; Scheiner, S. Tetrel Bonds with π-Electrons Acting as Lewis Bases—Theoretical Results and Experimental Evidences. Molecules 2018, 23, 1183. [Google Scholar] [CrossRef]
- Scheiner, S. Tetrel Bonding as a Vehicle for Strong and Selective Anion Binding. Molecules 2018, 23, 1147. [Google Scholar] [CrossRef]
- Liu, M.; Li, Q.; Cheng, J.; Li, W.; Li, H.B. Tetrel Bond of Pseudohalide Anions with XH3F (X = C, Si, Ge, and Sn) and Its Role in SN2 Reaction. J. Chem. Phys. 2016, 145, 224310. [Google Scholar] [CrossRef]
- Frontera, A.; Bauzá, A. S⋅⋅⋅Sn Tetrel Bonds in the Activation of Peroxisome Proliferator-Activated Receptors (PPARs) by Organotin Molecules. Chem. A Eur. J. 2018, 24, 16582–16587. [Google Scholar] [CrossRef]
- Daolio, A.; Scilabra, P.; Terraneo, G.; Resnati, G. C(Sp3) Atoms as Tetrel Bond Donors: A Crystallographic Survey. Coord. Chem. Rev. 2020, 413, 213265. [Google Scholar] [CrossRef]
- Scilabra, P.; Kumar, V.; Ursini, M.; Resnati, G. Close Contacts Involving Germanium and Tin in Crystal Structures: Experimental Evidence of Tetrel Bonds. J. Mol. Model. 2018, 24, 37. [Google Scholar] [CrossRef] [PubMed]
- Bartashevich, E.; Matveychuk, Y.; Tsirelson, V. Identification of the Tetrel Bonds between Halide Anions and Carbon Atom of Methyl Groups Using Electronic Criterion. Molecules 2019, 24, 1083. [Google Scholar] [CrossRef] [PubMed]
- Bartashevich, E.; Mukhitdinova, S.; Yushina, I.; Tsirelson, V. Electronic Criterion for Categorizing the Chalcogen and Halogen Bonds: Sulfur-Iodine Interactions in Crystals. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2019, 75, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Seth, S.K.; Frontera, A. Tetrel Bonding Interactions at Work: Impact on Tin and Lead Coordination Compounds. Coord. Chem. Rev. 2019, 384, 107–125. [Google Scholar] [CrossRef]
- Kumar, V.; Rodrigue, C.; Bryce, D.L. Short and Linear Intermolecular Tetrel Bonds to Tin. Cocrystal Engineering with Triphenyltin Chloride. Cryst. Growth Des. 2020, 20, 2027–2034. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules. A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Bader, R.F.W.; Essén, H. The Characterization of Atomic Interactions. J. Chem. Phys. 1998, 80, 1943–1960. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. A Description of the Chemical Bond in Terms of Local Properties of Electron Density and Energy. Croat. Chem. Acta 1984, 57, 1259–1281. [Google Scholar]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen Bond Strengths Revealed by Topological Analyses of Experimentally Observed Electron Densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Mata, I.; Alkorta, I.; Espinosa, E.; Molins, E. Relationships between Interaction Energy, Intermolecular Distance and Electron Density Properties in Hydrogen Bonded Complexes under External Electric Fields. Chem. Phys. Lett. 2011, 507, 185–189. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From Weak to Strong Interactions: A Comprehensive Analysis of the Topological and Energetic Properties of the Electron Density Distribution Involving X–H⋯F–Y Systems. J. Chem. Phys. 2002, 117, 5529. [Google Scholar] [CrossRef]
- Vener, M.V.; Egorova, A.N.; Churakov, A.V.; Tsirelson, V.G. Intermolecular Hydrogen Bond Energies in Crystals Evaluated Using Electron Density Properties: DFT Computations with Periodic Boundary Conditions. J. Comput. Chem. 2012, 33, 2303–2309. [Google Scholar] [CrossRef] [PubMed]
- Bushmarinov, I.S.; Lyssenko, K.A.; Antipin, M.Y. Atomic Energy in the “Atoms in Molecules” Theory and Its Use for Solving Chemical Problems. Russ. Chem. Rev. 2009, 78, 283–302. [Google Scholar] [CrossRef]
- Ananyev, I.V.; Karnoukhova, V.A.; Dmitrienko, A.O.; Lyssenko, K.A. Toward a Rigorous Definition of a Strength of Any Interaction between Bader’s Atomic Basins. J. Phys. Chem. A 2017, 121, 4517–4522. [Google Scholar] [CrossRef] [PubMed]
- Bartashevich, E.V.; Tsirelson, V.G. Interplay between Non-Covalent Interactions in Complexes and Crystals with Halogen Bonds. Russ. Chem. Rev. 2014, 83, 1181–1203. [Google Scholar] [CrossRef]
- Kuznetsov, M.L. Can Halogen Bond Energy Be Reliably Estimated from Electron Density Properties at Bond Critical Point? The Case of the (A)NZ—Y•••X− (X, Y = F, Cl, Br) Interactions. Int. J. Quantum Chem. 2019, 119, e25869. [Google Scholar] [CrossRef]
- Kuznetsov, M.L.; Costa, P.J. Relationships between Interaction Energy and Electron Density Properties for Homo Halogen Bonds of the [(A)NY–X···X–Z(B)m] Type (X = Cl, Br, I). Molecules 2019, 24, 2733. [Google Scholar] [CrossRef]
- Bartashevich, E.V.; Tsirelson, V.G. Atomic Dipole Polarization in Charge-Transfer Complexes with Halogen Bonding. Phys. Chem. Chem. Phys. 2013, 15, 2530–2538. [Google Scholar] [CrossRef]
- Alkorta, I.; Legon, A.C. Nucleophilicities of Lewis Bases b and Electrophilicities of Lewis Acids a Determined from the Dissociation Energies of Complexes B··· A Involving Hydrogen Bonds, Tetrel Bonds, Pnictogen Bonds, Chalcogen Bonds and Halogen Bonds. Molecules 2017, 22, 1786. [Google Scholar] [CrossRef]
- Bartashevich, E.V.; Matveychuk, Y.V.; Mukhitdinova, S.E.; Sobalev, S.A.; Khrenova, M.G.; Tsirelson, V.G. The Common Trends for the Halogen, Chalcogen, and Pnictogen Bonds via Sorting Principles and Local Bonding Properties. Theor. Chem. Acc. 2020, 139, 26. [Google Scholar] [CrossRef]
- Wesolowski, T.A.; Wang, Y.A. Recent Progress in Orbital-Free Density Functional Theory; World Scientific: Singapore, 2013; ISBN 9814436739. [Google Scholar]
- Tsirelson, V.; Stash, A. Orbital-Free Quantum Crystallography: View on Forces in Crystals. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2020, 76, 769–778. [Google Scholar] [CrossRef]
- Tsirelson, V.; Stash, A. Developing Orbital-Free Quantum Crystallography: The Local Potentials and Associated Partial Charge Densities. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2021, 77, 467–477. [Google Scholar] [CrossRef]
- Zhao, D.X.; Gong, L.D.; Yang, Z.Z. The Relations of Bond Length and Force Constant with the Potential Acting on an Electron in a Molecule. J. Phys. Chem. A 2005, 109, 10121–10128. [Google Scholar] [CrossRef]
- Zhao, D.-X.; Yang, Z.-Z. Investigation of the Distinction between van Der Waals Interaction and Chemical Bonding Based on the PAEM-MO Diagram. J. Comput. Chem. 2014, 35, 965–977. [Google Scholar] [CrossRef]
- Bartashevich, E.; Tsirelson, V. A Comparative View on the Potential Acting on an Electron in a Molecule and the Electrostatic Potential through the Typical Halogen Bonds. J. Comput. Chem. 2018, 39, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, I.P.; Mosna, R.A.; Site, L.D. Classical Kinetic Energy, Quantum Fluctuation Terms and Kinetic-Energy Functionals. Theor. Chem. Acc. 2007, 118, 407–415. [Google Scholar] [CrossRef]
- Liu, S. Steric Effect: A Quantitative Description from Density Functional Theory. J. Chem. Phys. 2007, 126, 244103. [Google Scholar] [CrossRef]
- Delle Site, L. Bader’s Interatomic Surface and Bohmian Mechanics. Europhys. Lett. 2002, 57, 20–24. [Google Scholar] [CrossRef]
- March, N.H. Concept of the Pauli Potential in Density Functional Theory. J. Mol. Struct. THEOCHEM 2010, 943, 77–82. [Google Scholar] [CrossRef]
- Shteingolts, S.A.; Stash, A.I.; Tsirelson, V.G.; Fayzullin, R.R. Orbital-Free Quantum Crystallographic View on Noncovalent Bonding: Insights into Hydrogen Bonds, Π⋅⋅⋅π and Reverse Electron Lone Pair⋅⋅⋅π Interactions. Chem. Eur. J. 2021, 27, 7789–7809. [Google Scholar] [CrossRef]
- Levina, E.O.; Khrenova, M.G.; Tsirelson, V.G. The Explicit Role of Electron Exchange in the Hydrogen Bonded Molecular Complexes. J. Comput. Chem. 2021, 42, 870–882. [Google Scholar] [CrossRef]
- Stash, A.I.; Terekhova, E.O.; Ivanov, S.A.; Tsirelson, V.G. X-ray Diffraction Study of the Atomic Interactions, Anharmonic Displacements and Inner-Crystal Field in Orthorhombic KNbO3. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2021, 77, 728–739. [Google Scholar] [CrossRef]
- Bartashevich, E.; Stash, A.; Yushina, I.; Minyaev, M.; Bolshakov, O.; Rakitin, O.; Tsirelson, V. Bonding Features in Appel’s Salt from the Orbital-Free Quantum Crystallographic Perspective. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2021, 77, 478–487. [Google Scholar] [CrossRef]
- Tsirelson, V.G.; Stash, A.I.; Karasiev, V.V.; Liu, S. Pauli Potential and Pauli Charge from Experimental Electron Density. Comput. Theor. Chem. 2013, 1006, 92–99. [Google Scholar] [CrossRef]
- Astakhov, A.A.; Stash, A.I.; Tsirelson, V.G. Improving Approximate Determination of the Noninteracting Electronic Kinetic Energy Density from Electron Density. Int. J. Quantum Chem. 2016, 116, 237–246. [Google Scholar] [CrossRef]
- Gritsenko, O.V.; Mentel, M.; Baerends, E.J. On the Errors of Local Density (LDA) and Generalized Gradient (GGA) Approximations to the Kohn-Sham Potential and Orbital Energies. J. Chem. Phys. 2016, 144, 204114. [Google Scholar] [CrossRef]
- Herring, C. Explicit Estimation of Ground-State Kinetic Energies from Electron Densities. Phys. Rev. A 1986, 34, 2614. [Google Scholar] [CrossRef]
- Bartashevich, E.; Yushina, I.; Kropotina, K.; Muhitdinova, S.; Tsirelson, V. Testing the Tools for Revealing and Characterizing the Iodine-Iodine Halogen Bond in Crystals. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2017, 73, 217–226. [Google Scholar] [CrossRef]
- Bartashevich, E.V.; Yushina, I.D.; Stash, A.I.; Tsirelson, V.G. Halogen Bonding and Other Iodine Interactions in Crystals of Dihydrothiazolo(Oxazino)Quinolinium Oligoiodides from the Electron-Density Viewpoint. Cryst. Growth Des. 2014, 14, 5674–5684. [Google Scholar] [CrossRef]
- Granovsky, A.A. Firefly, Version 8. Available online: http://classic.chem.msu.su/gran/firefly/index.html (accessed on 15 August 2022).
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General Atomic and Molecular Electronic Structure System. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158. [Google Scholar] [CrossRef]
- Barbieri, P.L.; Fantin, P.A.; Jorge, F.E. Gaussian Basis Sets of Triple and Quadruple Zeta Valence Quality for Correlated Wave Functions. Mol. Phys. 2006, 104, 2945–2954. [Google Scholar] [CrossRef]
- MacHado, S.F.; Camiletti, G.G.; Neto, A.C.; Jorge, F.E.; Jorge, R.S. Gaussian Basis Set of Triple Zeta Valence Quality for the Atoms from K to Kr: Application in DFT and CCSD(T) Calculations of Molecular Properties. Mol. Phys. 2009, 107, 1713–1727. [Google Scholar] [CrossRef]
- Campos, C.T.; Jorge, F.E. Triple Zeta Quality Basis Sets for Atoms Rb through Xe: Application in CCSD(T) Atomic and Molecular Property Calculations. Mol. Phys. 2012, 111, 167–173. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- Fradera, X.; Austen, M.A.; Bader, R.F.W. The Lewis Model and Beyond. J. Phys. Chem. A 1999, 103, 304–314. [Google Scholar] [CrossRef]
- Müller, A.M.K. Explicit Approximate Relation between Reduced Two- and One-Particle Density Matrices. Phys. Lett. A 1984, 105, 446–452. [Google Scholar] [CrossRef]
- Buijse, M.A.; Baerends, E.J. An Approximate Exchange-Correlation Hole Density as a Functional of the Natural Orbitals. Mol. Phys. 2009, 100, 401–421. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll (Version 17.11.14). Available online: http://www.aim.tkgristmill.com (accessed on 15 August 2022).
- TIBCO Statistica, v. 13. TIBCO Software Inc.: Palo Alto, CA, USA. Available online: https://www.tibco.com/products/tibco-statistica (accessed on 15 August 2022).
- Chakalov, E.R.; Tupikina, E.Y.; Bartashevich, E.V.; Ivanov, D.M.; Tolstoy, P.M. The Distance between Minima of Electron Density and Electrostatic Potential as a Measure of Halogen Bond Strength. Molecules 2022, 15, 4848. [Google Scholar] [CrossRef]
- Mayer, I. Covalent Bonding: The Role of Exchange Effects. J. Phys. Chem. A 2014, 118, 2543–2546. [Google Scholar] [CrossRef]
- Outeiral, C.; Vincent, M.A.; Martín Pendás, Á.; Popelier, P.L.A. Revitalizing the Concept of Bond Order through Delocalization Measures in Real Space. Chem. Sci. 2018, 9, 5517–5529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsirelson, V.G. Quantum Chemistry. Molecules, Molecular Systems and Solids; BINOM Publishing: Moscow, Russia, 2014; ISBN 978-5-9963-1668-7. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
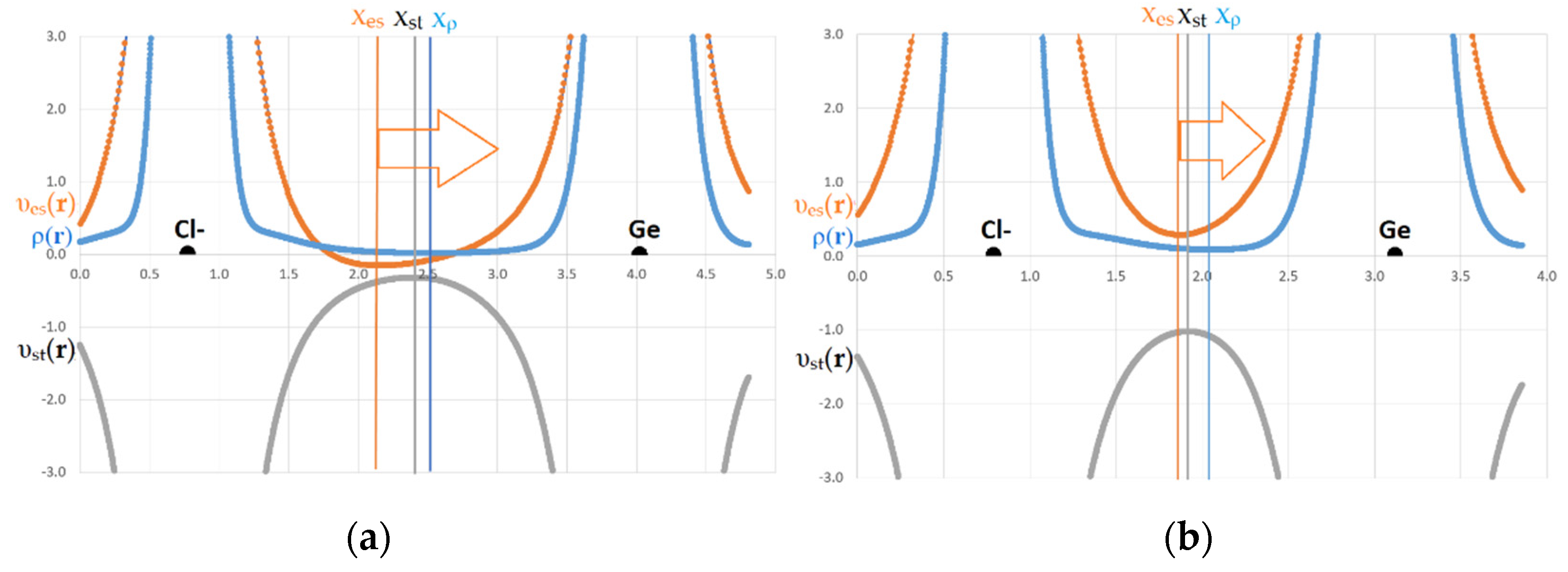{kind=link}
{kind=link}
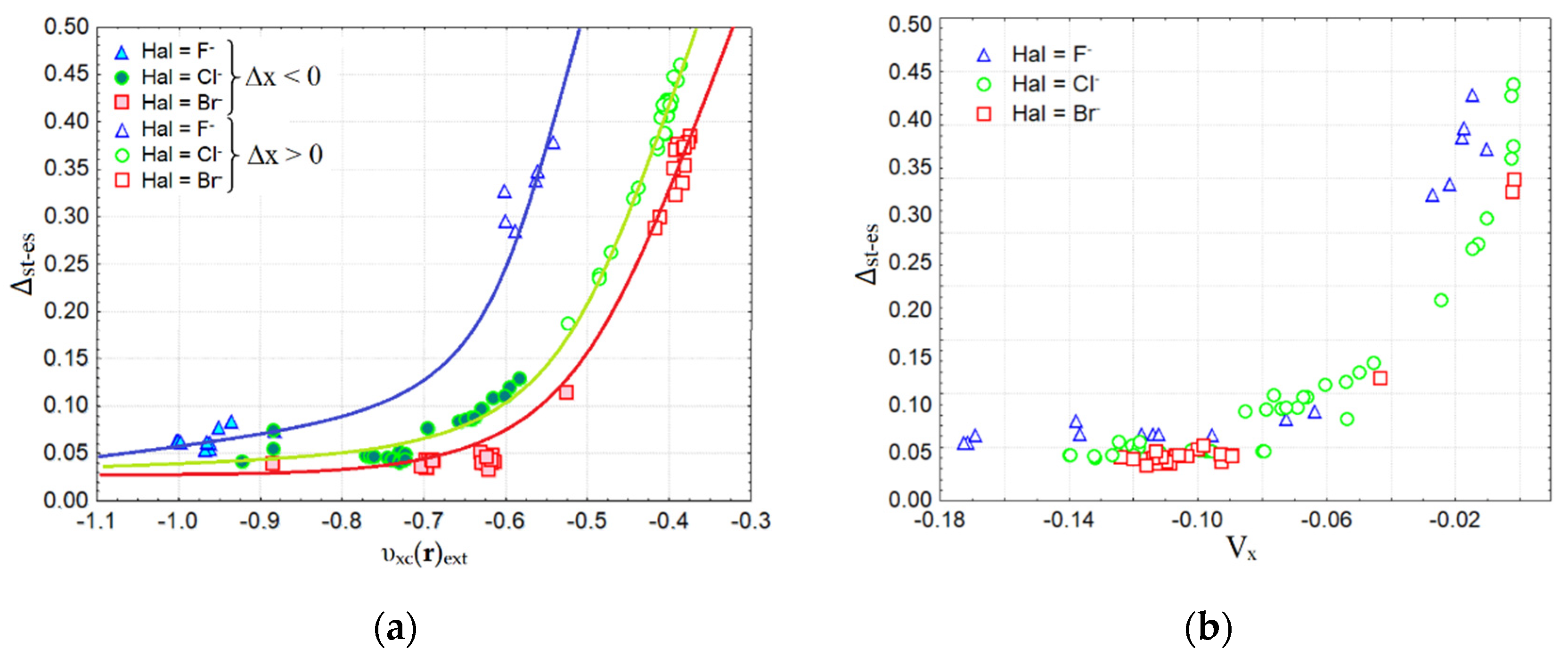{kind=link}
{kind=link}
{kind=link}
| Type of the Set, (Number of Cases) | a0 | a1 | R, Correlation Coefficient |
|---|---|---|---|
| Hal = F–, (19) | 3.515 (3.913) | 4.144 (5.138) | 0.99 (0.99) |
| Hal = Cl–, (56) | 5.550 (3.416) | 6.468 (6.279) | 0.97 (0.96) |
| Hal = Br–, (36) | 6.600 (4.945) | 7.705 (7.824) | 0.97 (0.98) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartashevich, E.V.; Mukhitdinova, S.E.; Klyuev, I.V.; Tsirelson, V.G. Can We Merge the Weak and Strong Tetrel Bonds? Electronic Features of Tetrahedral Molecules Interacted with Halide Anions. Molecules 2022, 27, 5411. https://doi.org/10.3390/molecules27175411
Bartashevich EV, Mukhitdinova SE, Klyuev IV, Tsirelson VG. Can We Merge the Weak and Strong Tetrel Bonds? Electronic Features of Tetrahedral Molecules Interacted with Halide Anions. Molecules. 2022; 27(17):5411. https://doi.org/10.3390/molecules27175411
Chicago/Turabian StyleBartashevich, Ekaterina V., Svetlana E. Mukhitdinova, Iliya V. Klyuev, and Vladimir G. Tsirelson. 2022. "Can We Merge the Weak and Strong Tetrel Bonds? Electronic Features of Tetrahedral Molecules Interacted with Halide Anions" Molecules 27, no. 17: 5411. https://doi.org/10.3390/molecules27175411