
The Chemistry of Green and Roasted Coffee by Selectable 1D/2D Gas Chromatography Mass Spectrometry with Spectral Deconvolution


Abstract
:1. Introduction
2. Results and Discussion
2.1. Coffee Sourcing and Description
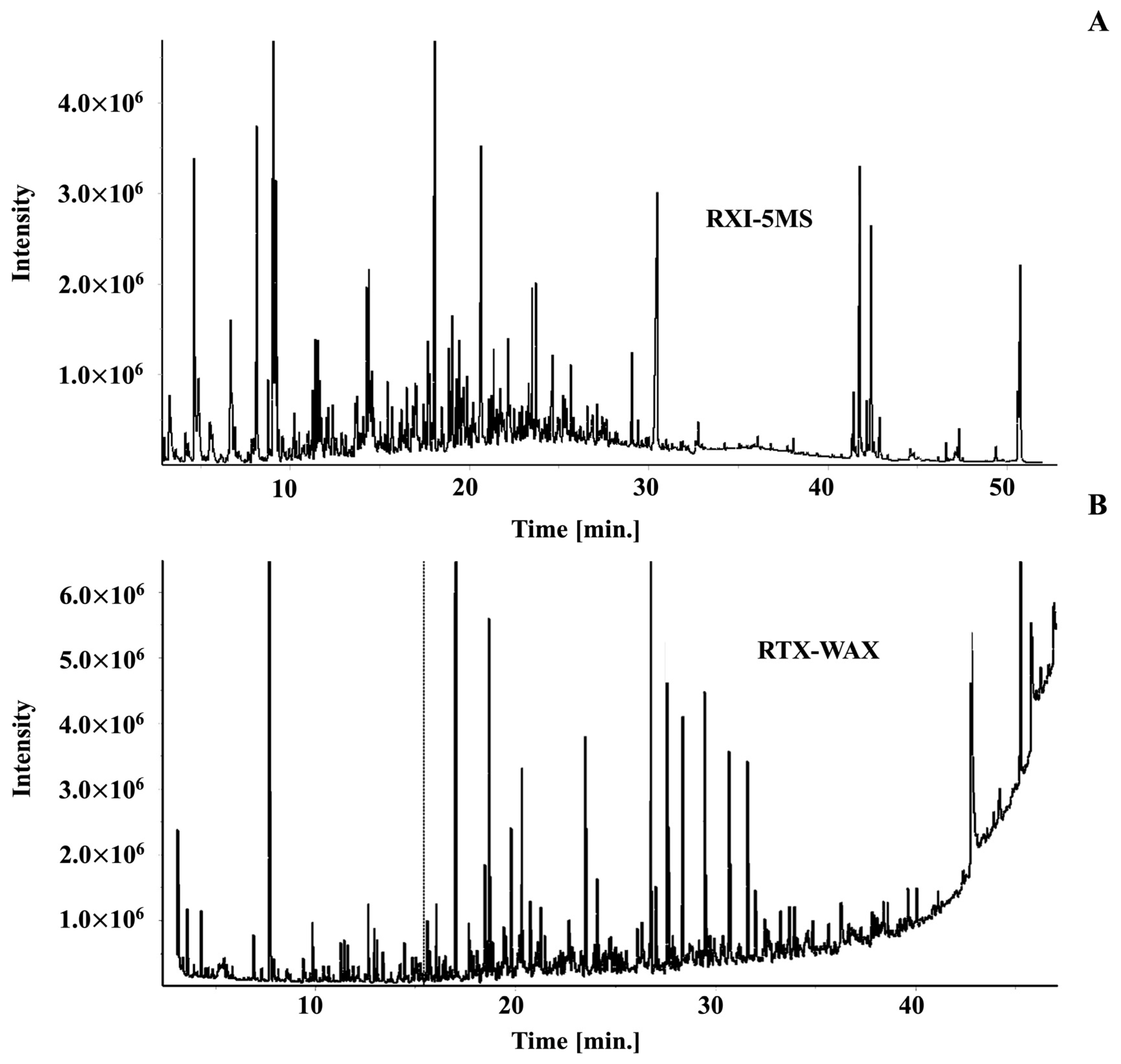
2.2. Building the Initial Target Compound Library
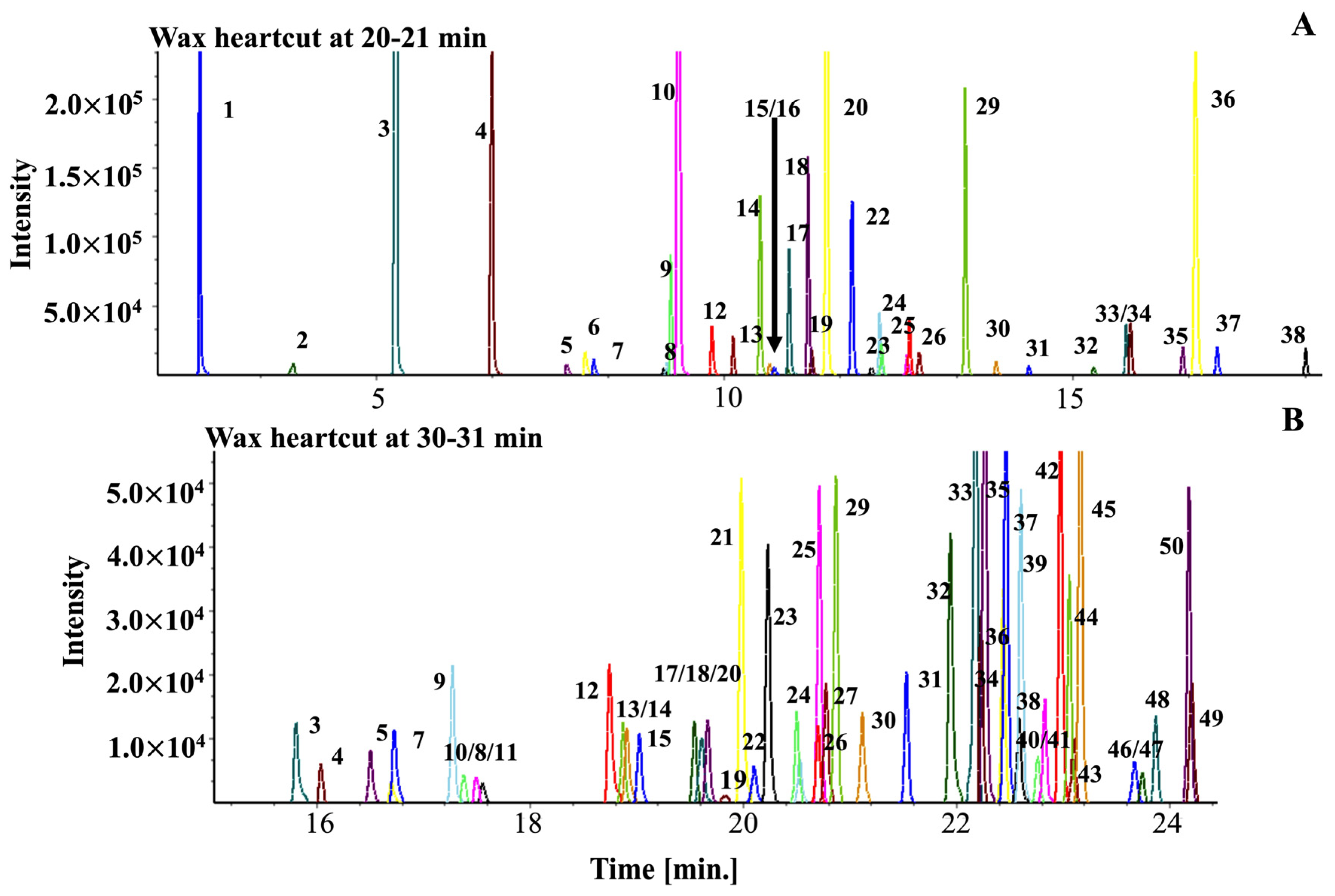
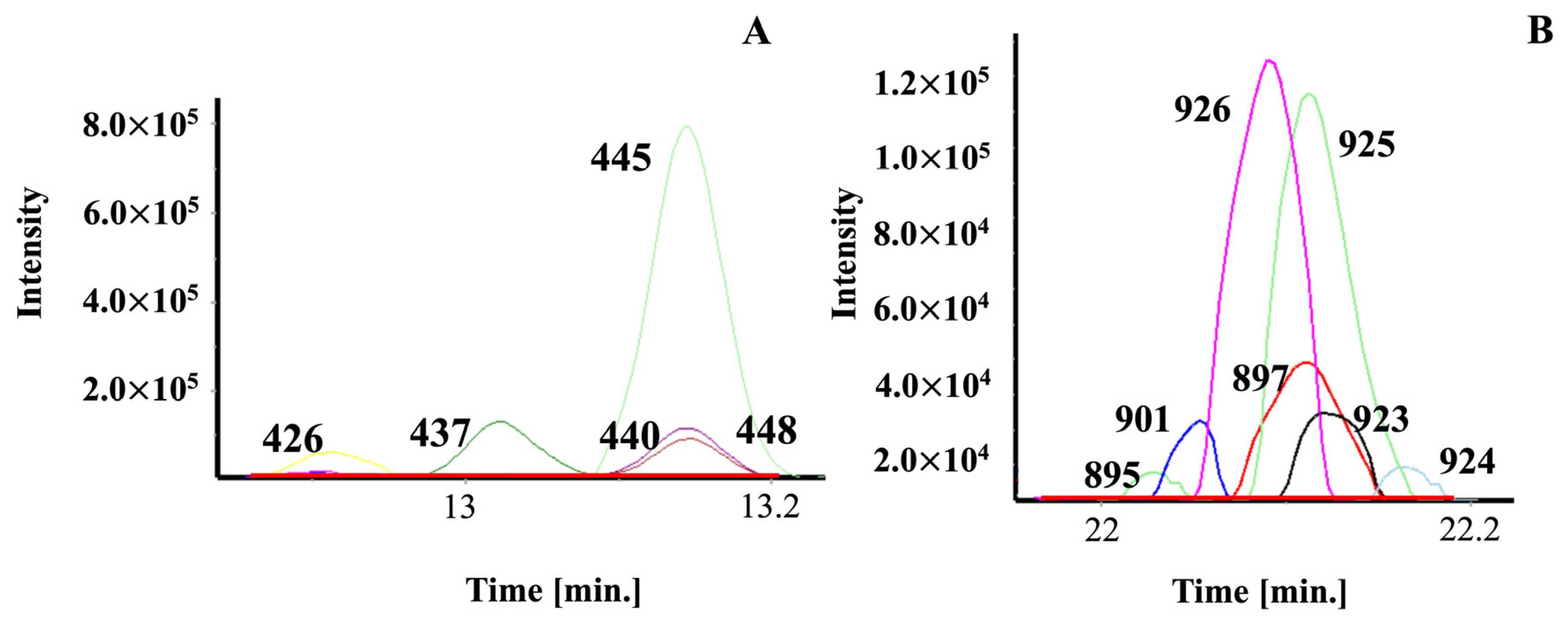
2.3. Applying the Coffee Library
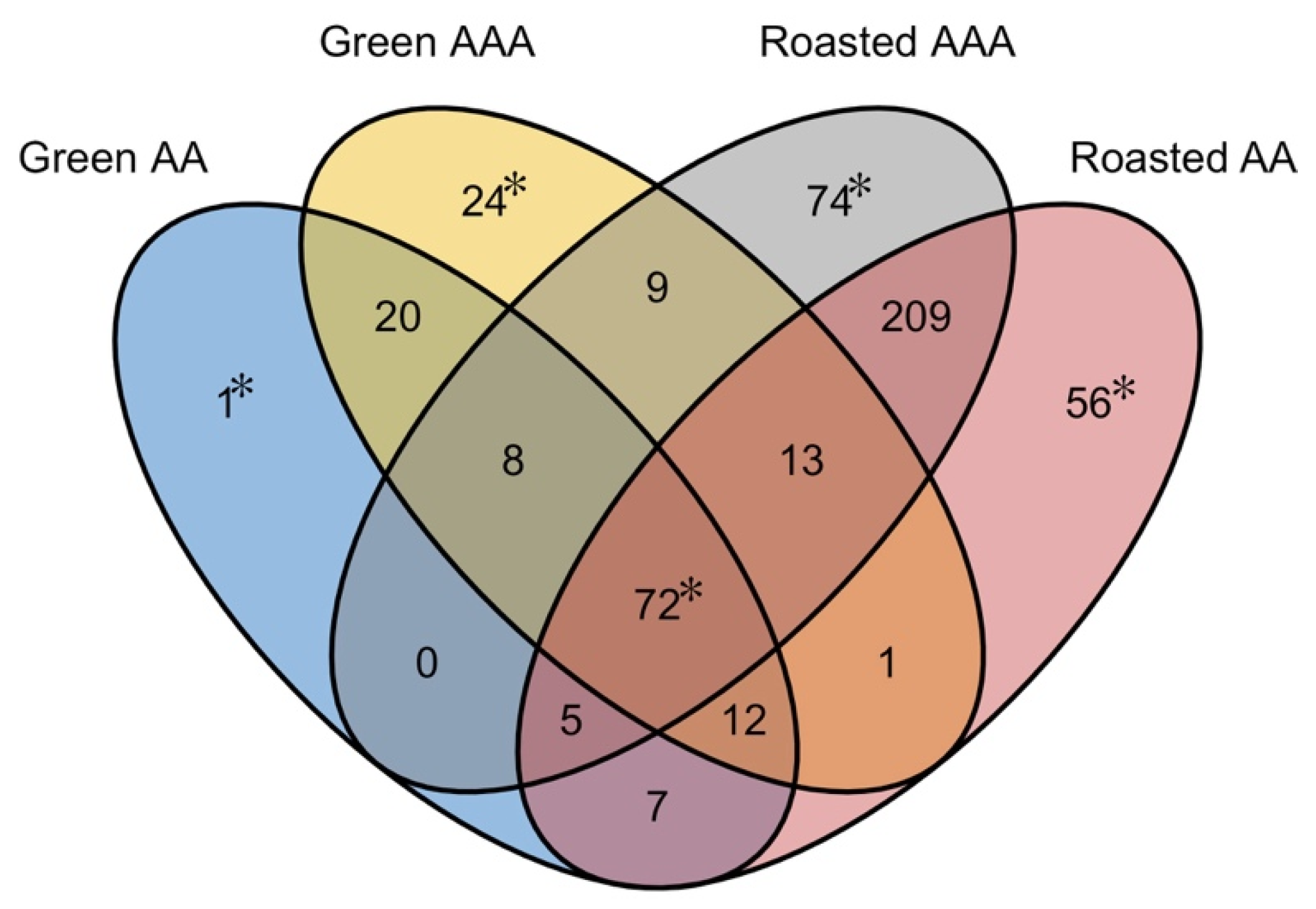
2.4. Comparison of Green and Roasted Coffees
2.5. Comparison of Green Coffees

{kind=link}
{kind=link}
{kind=link}
{kind=link}
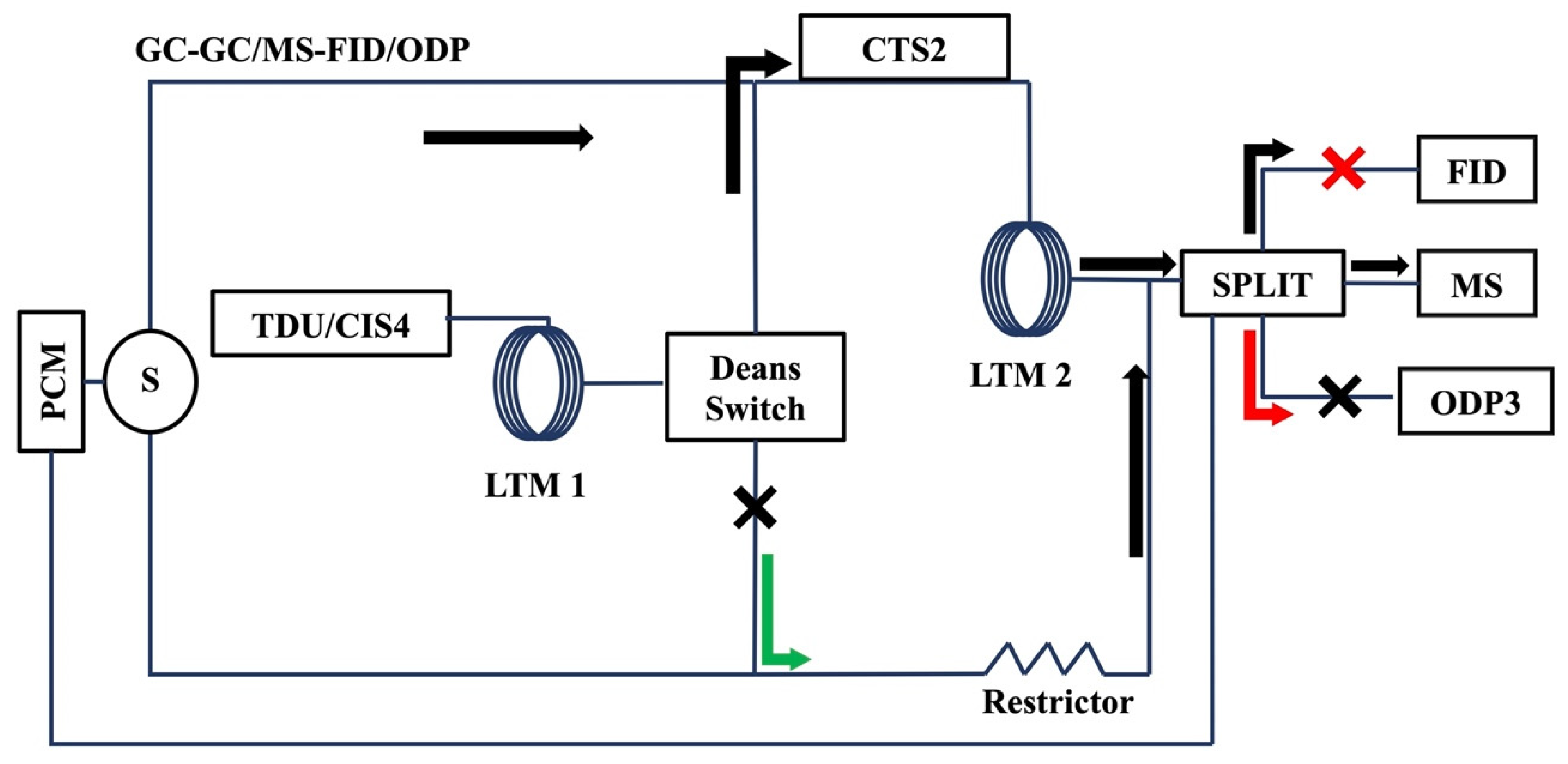{kind=link}
| Compound | Green AAA | Green AA | Roast AAA | Roast AA | Retention Time | Lit Odor |
|---|---|---|---|---|---|---|
| Unique to Green AAA | ||||||
| 171 | 40,806 | 0 | 0 | 0 | 19.05 | nr |
| 3-methyl-1-butanol | 19,745 | 0 | 0 | 0 | 4.28 | fermented |
| 3-hexen-2-one | 16,409 | 0 | 0 | 0 | 6.80 | nr |
| methyl benzoate | 12,172 | 0 | 0 | 0 | 14.23 | phenolic |
| 2-methyl-cyclopentanone | 12,079 | 0 | 0 | 0 | 6.80 | nr |
| 2-ethyl furan | 11,877 | 0 | 0 | 0 | 3.78 | chemical |
| 2,4-dimethyl-1,3-pentadiene, | 10,381 | 0 | 0 | 0 | 3.78 | caramel |
| methyl 3-(3,5-di-tert-butyl-4-hydroxyphenyl)propionate | 10,130 | 0 | 0 | 0 | 36.71 | nr |
| 643 | 9657 | 0 | 0 | 0 | 28.22 | |
| 6-methyl-5-hepten-2-one | 9540 | 0 | 0 | 0 | 10.70 | citrus, green |
| 217 | 9156 | 0 | 0 | 0 | 17.38 | |
| nerol oxide | 8261 | 0 | 0 | 0 | 16.57 | green |
| 175 | 8223 | 0 | 0 | 0 | 13.73 | |
| 3,5-dimethyl-2-isobutylpyrazine | 8083 | 0 | 0 | 0 | 17.58 | cocoa |
| 265 | 7659 | 0 | 0 | 0 | 19.85 | |
| 431 | 7345 | 0 | 0 | 0 | 25.35 | |
| 4-methylpyrrolo [1,2-a]pyrazine | 7323 | 0 | 0 | 0 | 21.47 | nr |
| 1-phenyl-2-butanone | 7244 | 0 | 0 | 0 | 18.35 | earthy |
| cyclopentanone | 7172 | 0 | 0 | 0 | 5.26 | nr |
| 457 | 6984 | 0 | 0 | 0 | 26.68 | |
| 568 | 6590 | 0 | 0 | 0 | 31.47 | |
| cis-linalool oxide (furan) | 5701 | 0 | 0 | 0 | 13.51 | earthy, floral |
| 46 | 5528 | 0 | 0 | 0 | 10.59 | |
| ortho-hydroxybiphenyl | 5516 | 0 | 0 | 0 | 26.52 | nr |
| Unique to Green AA | ||||||
| 450 | 0 | 24,258 | 0 | 0 | 12.00 |
2.6. Comparison of Roasted AAA to Roasted AA
| Compound | Green AAA | Green AA | Roast AAA | Roast AA | Retention Time | Lit Odor |
|---|---|---|---|---|---|---|
| Unique to Roast AAA | ||||||
| 4-(2-methylprop-2-enoyloxy)butyl 2-methylprop-2-enoate | 0 | 0 | 1,872,381 | 0 | 27.19 | nr |
| 2,3,5-trimethylpyrazine | 0 | 0 | 795,151 | 0 | 11.16 | nutty |
| 1-[1-(furan-2-ylmethyl)pyrrol-2-yl]ethanone | 0 | 0 | 694,986 | 0 | 25.52 | nr |
| furan-2-ylmethyl pentanoate | 0 | 0 | 194,721 | 0 | 18.19 | nr |
| 719 | 0 | 0 | 191,413 | 0 | 13.26 | |
| 3-methylpentane-2,4-dione | 0 | 0 | 182,840 | 0 | 14.56 | nr |
| 5-ethyl-2,3-dimethylpyrazine | 0 | 0 | 142,250 | 0 | 13.98 | burnt |
| (E)-3-(furan-2-yl)-2-methylprop-2-enal | 0 | 0 | 128,245 | 0 | 17.29 | spicy |
| 678 | 0 | 0 | 123,536 | 0 | 31.43 | |
| 3-phenyl-2-propenal | 0 | 0 | 87,809 | 0 | 19.47 | spicy |
| 694 | 0 | 0 | 82,049 | 0 | 33.61 | |
| 1-(4-methoxyphenyl)-propan-1-one | 0 | 0 | 80,471 | 0 | 23.51 | musty |
| pentane-2,3-dione | 0 | 0 | 74,227 | 0 | 3.70 | buttery |
| 2,3-dihydro-1-benzofuran | 0 | 0 | 73,915 | 0 | 18.19 | phenolic |
| 472 | 0 | 0 | 68,503 | 0 | 24.85 | |
| 503 | 0 | 0 | 67,741 | 0 | 24.62 | |
| 99 | 0 | 0 | 64,820 | 0 | 9.71 | |
| 473 | 0 | 0 | 64,012 | 0 | 25.16 | |
| 732 | 0 | 0 | 62,312 | 0 | 17.30 | |
| 406 | 0 | 0 | 57,488 | 0 | 23.40 | |
| 390 | 0 | 0 | 52,419 | 0 | 19.96 | |
| 305 | 0 | 0 | 48,447 | 0 | 19.08 | |
| 536 | 0 | 0 | 46,637 | 0 | 26.24 | |
| 344 | 0 | 0 | 42,780 | 0 | 20.71 | |
| 2-pyridinemethanol | 0 | 0 | 42,327 | 0 | 20.18 | nr |
| 2-furanacetaldehyde-α-propyl | 0 | 0 | 37,037 | 0 | 16.58 | nr |
| furfuryl formate | 0 | 0 | 36,063 | 0 | 8.24 | nr |
| 252 | 0 | 0 | 36,031 | 0 | 17.51 | |
| 321 | 0 | 0 | 34,425 | 0 | 18.49 | |
| terpineol | 0 | 0 | 33,957 | 0 | 17.28 | citrus, woody |
| 2-acetyl-4-methylpyridine | 0 | 0 | 33,281 | 0 | 14.61 | nr |
| 427 | 0 | 0 | 31,760 | 0 | 24.89 | |
| 395 | 0 | 0 | 29,745 | 0 | 24.01 | |
| 417 | 0 | 0 | 28,972 | 0 | 25.79 | |
| hexane-2,3-dione | 0 | 0 | 27,926 | 0 | 5.20 | buttery |
| 118 | 0 | 0 | 27,593 | 0 | 16.23 | |
| 4-ethenyl-2,6-dimethoxyphenol | 0 | 0 | 27,338 | 0 | 27.80 | burnt |
| 2-ethoxyaniline | 0 | 0 | 26,009 | 0 | 16.49 | nr |
| 616 | 0 | 0 | 24,918 | 0 | 30.13 | |
| 2-acetyl-3-ethylpyrizine | 0 | 0 | 24,790 | 0 | 16.52 | nutty |
| 579 | 0 | 0 | 24,744 | 0 | 25.62 | |
| 258 | 0 | 0 | 24,703 | 0 | 20.21 | |
| 2-methylthiolan-3-one | 0 | 0 | 24,697 | 0 | 10.73 | sulfurous |
| 2-cyclohexene-1,4-dione | 0 | 0 | 22,742 | 0 | 12.13 | nr |
| 73 | 0 | 0 | 21,729 | 0 | 8.29 | |
| 652 | 0 | 0 | 20,165 | 0 | 28.20 | |
| 3-methylbut-2-enyl acetate | 0 | 0 | 19,106 | 0 | 8.77 | fruity |
| 218 | 0 | 0 | 18,875 | 0 | 17.43 | |
| 674 | 0 | 0 | 18,580 | 0 | 29.94 | |
| 333 | 0 | 0 | 18,410 | 0 | 21.44 | |
| 334 | 0 | 0 | 17,704 | 0 | 21.51 | |
| 313 | 0 | 0 | 17,266 | 0 | 20.93 | |
| 206 | 0 | 0 | 16,915 | 0 | 15.48 | |
| 691 | 0 | 0 | 16,395 | 0 | 32.39 | |
| (E)-but-2-enal | 0 | 0 | 16,165 | 0 | 4.43 | nr |
| 371 | 0 | 0 | 15,208 | 0 | 20.35 | |
| 436 | 0 | 0 | 15,106 | 0 | 21.32 | |
| 131 | 0 | 0 | 14,928 | 0 | 14.71 | |
| 1-phenyl-propan-1-one | 0 | 0 | 14,205 | 0 | 16.48 | floral, fruity |
| 250 | 0 | 0 | 14,062 | 0 | 16.47 | |
| 730 | 0 | 0 | 13,064 | 0 | 15.32 | |
| 214 | 0 | 0 | 12,678 | 0 | 17.24 | |
| 204 | 0 | 0 | 12,331 | 0 | 15.32 | |
| 3-methylthiophene | 0 | 0 | 11,185 | 0 | 5.04 | fatty |
| 48 | 0 | 0 | 10,644 | 0 | 12.99 | |
| 351 | 0 | 0 | 10,465 | 0 | 21.54 | |
| 40 | 0 | 0 | 9174 | 0 | 6.77 | |
| 6-tridecyloxan-2-one | 0 | 0 | 8075 | 0 | 40.72 | fatty |
| 37 | 0 | 0 | 7152 | 0 | 9.54 | |
| 3-methylbut-2-en-1-ol | 0 | 0 | 6839 | 0 | 5.07 | sweet fruit |
| 1,3,5-trimethylbenzene | 0 | 0 | 6117 | 0 | 10.88 | nr |
| 3-methylbut-3-en-1-ol | 0 | 0 | 5895 | 0 | 4.26 | nr |
| 27 | 0 | 0 | 5870 | 0 | 9.20 | |
| 725 | 0 | 0 | 5115 | 0 | 6.82 | |
| Unique to Roast AA | ||||||
| 1-(furan-2-ylmethyl)pyrrole | 0 | 0 | 0 | 938,004 | 16.40 | vegetable |
| phenylacetonitrile | 0 | 0 | 0 | 316,086 | 29.51 | nr |
| 5H-furan-2-one | 0 | 0 | 0 | 286,753 | 14.63 | buttery |
| 3-methylfuran | 0 | 0 | 0 | 258,698 | 2.97 | nr |
| 2-(furan-2-ylmethyl)furan | 0 | 0 | 0 | 220,455 | 13.87 | roasted |
| 5-(formylfuran-2-yl)methyl acetate | 0 | 0 | 0 | 192,403 | 19.99 | nr |
| 2-(furan-2-ylmethyl)-5-methylfuran | 0 | 0 | 0 | 168,726 | 16.32 | nr |
| 2-oxopropyl acetate | 0 | 0 | 0 | 130,636 | 7.08 | fruity, buttery, dairy |
| ethyl 3-methylbutanoate | 0 | 0 | 0 | 123,878 | 28.04 | fruity |
| 5-methyl-2(5H)-furanone | 0 | 0 | 0 | 99,098 | 13.48 | nr |
| 6-(5-methyl-furan-2-yl)-hexan-2-one | 0 | 0 | 0 | 73,787 | 18.38 | nr |
| 2-(4-aminophenyl) acetonitrile | 0 | 0 | 0 | 72,461 | 17.02 | nr |
| 4-morpholin-4-yl aniline | 0 | 0 | 0 | 68,907 | 30.07 | nr |
| 2-propylpyrazine | 0 | 0 | 0 | 68,501 | 11.37 | brothy, sulfury, smoky, beany |
| 5-(furan-2-ylmethyl)-5-methylfuran-2-one | 0 | 0 | 0 | 68,166 | 22.16 | nr |
| methylpyridine-3-carboxylate | 0 | 0 | 0 | 67,852 | 15.69 | herbal, tobacco |
| 1-(2-hydroxy-5-methylphenyl)ethanone | 0 | 0 | 0 | 60,350 | 17.94 | floral |
| 2-methyl-furan-3-carboxylic acid n′-acetyl-hydrazide | 0 | 0 | 0 | 55,314 | 22.98 | nr |
| 4-methyl-2H-quinolin-5-one | 0 | 0 | 0 | 47,832 | 26.54 | nr |
| 1H-pyrrolo [2,3-b]pyridine | 0 | 0 | 0 | 43,278 | 17.04 | nr |
| 2,5-dimethylfuran | 0 | 0 | 0 | 42,981 | 3.82 | meaty |
| 1-thiophen-3-ylethanone | 0 | 0 | 0 | 39,659 | 13.48 | nr |
| thiophene-3-carbaldehyde | 0 | 0 | 0 | 34,872 | 10.65 | nr |
| trans-isoeugenol | 0 | 0 | 0 | 33,906 | 23.72 | spicy |
| 2-acetylcyclohexan-1-one | 0 | 0 | 0 | 30,995 | 16.09 | nr |
| 1-(5-methylthiophen-2-yl)ethanone | 0 | 0 | 0 | 30,609 | 17.06 | floral |
| nonanoic acid | 0 | 0 | 0 | 30,342 | 19.64 | waxy |
| 1-(4-hydroxyphenyl)propan-1-one | 0 | 0 | 0 | 27,581 | 18.74 | nr |
| 2-methyl-5,6,7,8-tetrahydroquinoxaline | 0 | 0 | 0 | 26,835 | 19.60 | animal |
| 6-methyl-2H-1-benzopyran-2-one | 0 | 0 | 0 | 26,168 | 26.49 | coconut |
| 5,6,7,8-tetrahydroquinoxaline | 0 | 0 | 0 | 25,822 | 17.16 | nr |
| 4-nitrophenyl pentanoate | 0 | 0 | 0 | 24,919 | 17.48 | nr |
| 6-nonyloxan-2-one | 0 | 0 | 0 | 23,085 | 34.22 | waxy |
| 2,5-dimethyl-3-(2-methylprpyl) pyrazine | 0 | 0 | 0 | 22,126 | 17.23 | nr |
| 1-thiophen-2-ylethanone | 0 | 0 | 0 | 20,122 | 13.25 | onion |
| 2-methyl-5-[(5-methylfuran-2-yl)methyl]furan | 0 | 0 | 0 | 19,517 | 19.09 | nr |
| 1,3-thiazole | 0 | 0 | 0 | 19,465 | 4.49 | green, nutty, tomato |
| cis-dehydroxy linalool oxide | 0 | 0 | 0 | 18,958 | 10.85 | floral green |
| (3E)-3,7-dimethylocta-1,3,6-triene | 0 | 0 | 0 | 18,666 | 12.09 | nr |
| 2,3,5-trimethyl-6-prop-2-enyl pyrazine | 0 | 0 | 0 | 18,344 | 20.39 | nr |
| 7-methyl-3-methylene-octa-1,6-diene | 0 | 0 | 0 | 18,224 | 10.34 | spicy, wood |
| 3-ethylpyridine | 0 | 0 | 0 | 17,528 | 18.05 | tobacco |
| 3-methylpyridine | 0 | 0 | 0 | 15,821 | 7.32 | green |
| 1-pyridin-4-ylethanone | 0 | 0 | 0 | 15,585 | 13.60 | burnt |
| 1-(5-methylfuran-2-yl)butan-1-one | 0 | 0 | 0 | 15,300 | 17.41 | nr |
| 2-[(4-ethylphenoxy)methyl]oxirane | 0 | 0 | 0 | 15,066 | 20.55 | nr |
| 1-(4-methylthiophen-3-yl)ethanone | 0 | 0 | 0 | 13,022 | 15.35 | nr |
| 1-ethyl-4-methoxy-9H-pyrido [3,4-b]indole | 0 | 0 | 0 | 12,922 | 31.55 | nr |
| 2,6,6-trimethyl-2-cyclohexene-1,4-dione | 0 | 0 | 0 | 12,523 | 15.01 | musty, citrus |
| 1-(4H-pyridin-1-yl)ethanone | 0 | 0 | 0 | 12,011 | 11.22 | nr |
| 1-(4-methylthiophen-2-yl)ethanone | 0 | 0 | 0 | 11,558 | 15.29 | nutty |
| 2-methylcyclopent-2-en-1-one | 0 | 0 | 0 | 10,775 | 8.01 | nr |
| naphthalene | 0 | 0 | 0 | 9395 | 17.02 | mothballs |
| propanoic acid | 0 | 0 | 0 | 8969 | 4.32 | nr |
| 1-methyl-4-(propan-2-y)lbenzene | 0 | 0 | 0 | 7475 | 11.88 | spicy, citrus |
| 2,4-dimethyl-1,3-thiazole | 0 | 0 | 0 | 6090 | 7.46 | briney, sulfury, burnt, rubber, medicine |
2.7. Compounds that Persisted through Roasting
| Compound | Green AAA | Green AA | Roast AAA | Roast AA | Retention Time | Lit Odor |
|---|---|---|---|---|---|---|
| hexadecanoic acid | 1,108,940 | 6200 | 233,280 | 59,944 | 37.27 | waxy |
| toluene | 830,552 | 107,867 | 183,937 | 76,509 | 4.83 | nr |
| pyridine | 501,494 | 13,372 | 1,495,635 | 452,851 | 4.48 | resinous, roasted, burnt |
| benzophenone | 376,506 | 20,060 | 296,941 | 40,875 | 29.49 | balsamic |
| 1-(furan-2-yl)ethan-1-one | 269,574 | 103,844 | 684,350 | 128,217 | 8.42 | fruity sweet, caramel |
| nonanal | 231,092 | 5426 | 314,988 | 26,032 | 14.54 | waxy, rose, orange |
| decanal | 144,208 | 36,319 | 193,021 | 20,118 | 17.70 | aldehydic |
| 734 | 131,831 | 31,844 | 181,302 | 162,176 | 22.10 | |
| 2-methyl-1-(1,1-dimethylethyl)-2-methyl-1,3-propanediyl ester | 127,289 | 4785 | 210,279 | 39,256 | 28.59 | nr |
| 2-ethenyl-6-methylpyrazine | 111,984 | 4900 | 76,380 | 105,157 | 11.64 | roasted, potato |
| furan-2-carbaldehyde | 97,096 | 5747 | 1,026,548 | 67,410 | 6.36 | woody, almond, baked bread |
| 5-methylfuran-2-carbaldehyde | 93,410 | 7790 | 2,726,476 | 35,266 | 10.02 | caramel-like, bready, coffee-like |
| 378 | 90,465 | 51,972 | 830,442 | 89,822 | 22.12 | |
| 2-ethenyl-5-methylpyrazine | 72,870 | 5200 | 51,693 | 68,303 | 11.71 | rubber, smoky, chemical, greasy, onion |
| 449 | 69,464 | 16,976 | 126,788 | 53,076 | 26.66 | |
| 2,4-ditert-butylphenol | 67,202 | 24,294 | 53,059 | 39,125 | 26.44 | nr |
| furan-2-ylmethyl acetate | 64,927 | 17,522 | 2,093,291 | 5186 | 11.00 | fruity sweet, banana-like |
| octanal | 64,004 | 64,330 | 69,829 | 38,039 | 11.21 | waxy, citrus, green, fatty |
| 2-ethylpyrazine | 60,417 | 761,061 | 535,929 | 761,061 | 8.50 | nutty |
| 2-methylpyrazine | 47,786 | 8794 | 734,866 | 246,899 | 6.08 | nutty |
| (E,E)-2,4-heptadien-6-ynal | 44,850 | 44,963 | 108,827 | 114,800 | 9.89 | nr |
| 2-ethyl-1-hexnol | 39,167 | 45,579 | 39,044 | 88,651 | 12.07 | citrus, floral |
| 420 | 34,364 | 6842 | 89,710 | 4916 | 26.82 | |
| 506 | 33,761 | 4750 | 218,646 | 193,545 | 25.37 | |
| 4-ethenyl-1,2-dimethoxybenzene | 33,129 | 4965 | 648,094 | 596,689 | 22.49 | floral, green |
| phenol | 31,362 | 4808 | 67,790 | 571,972 | 10.74 | medicinal, tar, phenolic |
| linalool | 30,870 | 11,402 | 78,965 | 98,374 | 14.40 | fruity, floral |
| 1,3-benzothiazole | 30,124 | 41,072 | 33,267 | 72,051 | 18.31 | meaty |
| 1-phenylethan-1-one | 30,092 | 130,549 | 48,599 | 1,207,900 | 13.31 | floral |
| 2,2′-(oxydimethylene)difuran | 26,876 | 58,142 | 597,198 | 131,155 | 20.61 | coffee, nutty, earthy |
| 331 | 26,456 | 10,222 | 319,210 | 151,125 | 19.41 | |
| 514 | 26,220 | 11,153 | 561,508 | 166,993 | 27.00 | |
| 1,3-xylene | 24,927 | 14,683 | 33,280 | 16,454 | 7.19 | nr |
| 1H-indole | 24,822 | 7635 | 258,353 | 30,262 | 20.41 | fecal |
| 4-ethyl-2-methoxyphenol | 18,664 | 4659 | 475,344 | 131,434 | 19.94 | balsamic, clove, phenolic, woody, smoke, |
| 2-(2-propenyl)furan | 18,134 | 18,360 | 8514 | 18,360 | 6.96 | nr |
| 2-phenylacetaldehyde | 17,357 | 15,093 | 82,885 | 530,637 | 12.56 | green, floral, honey, cocoa |
| 619 | 16,717 | 9832 | 180,995 | 104,992 | 31.06 | |
| styrene | 16,637 | 14,947 | 22,648 | 14,979 | 7.78 | nr |
| 751 | 16,394 | 4650 | 89,238 | 56,635 | 41.24 | |
| 1-(1-methyl-1H-pyrrol-2-yl)-ethanone | 15,794 | 4263 | 253,898 | 62,326 | 13.58 | musty |
| p-methylacetophenone | 14,685 | 67,536 | 139,498 | 802,460 | 17.08 | na |
| 471 | 14,261 | 5482 | 442,113 | 5482 | 24.69 | |
| 489 | 13,635 | 6069 | 142,306 | 34,194 | 26.40 | |
| 382 | 12,689 | 24,900 | 90,967 | 223,435 | 23.03 | |
| 277 | 11,900 | 8253 | 498,094 | 500,537 | 17.10 | |
| 1-(furan-2-yl)propan-1-one | 11,781 | 9982 | 281,461 | 296,132 | 11.42 | fruity |
| 681 | 11,748 | 14,780 | 464,226 | 646,418 | 33.22 | |
| 3-pheny pyridine | 11,322 | 33,697 | 72,022 | 70,303 | 25.13 | nr |
| heptan-2-one | 11,163 | 24,213 | 21,113 | 44,952 | 7.81 | cheesy |
| 1-methyl-4-(prop-1-en-2-yl)cyclohex-1-ene | 10,476 | 9440 | 19,822 | 21,809 | 12.01 | citrus |
| 1,2-xylene | 10,334 | 20,449 | 17,638 | 31,242 | 7.83 | nr |
| 5-methyl-6,7-dihydro-5H-cyclopenta[b]pyrazine | 9832 | 12,867 | 86,032 | 127,990 | 15.71 | nr |
| 3,4-dimethyl-1H-pyrrole-2-carboxaldehyde | 9744 | 17,959 | 163,351 | 1,435,806 | 16.27 | nr |
| 132 | 9723 | 6481 | 15,159 | 18,572 | 13.42 | |
| methyl salicylate | 9104 | 9255 | 75,020 | 98,271 | 17.39 | wintergreen |
| 198 | 8682 | 13,196 | 46,484 | 48,733 | 13.71 | |
| furan-2-ylmethyl propanoate | 8673 | 4994 | 296,194 | 270,827 | 14.00 | nr |
| 490 | 8517 | 9901 | 336,296 | 462,642 | 27.02 | |
| (6E,8E)-megastigma-4,6,8-trien-3-one | 8268 | 13,727 | 71,780 | 479,280 | 29.44 | nr |
| dodecanal | 7960 | 10,237 | 18,759 | 167,465 | 23.61 | aldehydic |
| 1-(4-hydroxyphenyl)-2-methylpropan-1-one | 7565 | 5897 | 130,450 | 108,157 | 22.50 | nr |
| 3,7-dimethyl-6,7-dihydro-5H-cyclopentapyrazine | 7277 | 9821 | 49,083 | 91,792 | 18.24 | nr |
| 2-[(methyldithio) methyl]-furan | 7261 | 15,630 | 123,674 | 128,211 | 17.92 | sulfury |
| 1-(pyridin-2-yl)ethanone | 7225 | 6667 | 24,502 | 20,747 | 12.23 | popcorn |
| 1-(2-hydroxyphenyl)ethanone | 7223 | 36,783 | 65,120 | 63,927 | 16.40 | phenolic |
| 2-methoxyphenol | 7107 | 17,135 | 124,736 | 204,167 | 14.08 | phenolic, woody |
| 6-methoxy-2-methylquinoline | 7060 | 6876 | 39,934 | 76,719 | 24.52 | nr |
| 15 | 6361 | 12,780 | 7646 | 12,870 | 3.32 | |
| 2-phenylbut-2-enal | 4983 | 58,614 | 135,175 | 61,768 | 19.78 | musty, green |
| 2-methylpyridine | 4886 | 4923 | 8723 | 9524 | 6.04 | sweat |
| 733 | 4012 | 8892 | 132,975 | 479,806 | 18.86 |
2.8. Relevance of GC-GC/MS for Natural Product Profiling
3. Materials and Methods
3.1. Workflow
3.1.1. Database Building
3.1.2. Target Compound Identification
- is the reduced ion intensity relative to the main ion, at scan ;
- is the absolute i-th confirming ion intensity at scan ;
- is the expected relative ion abundance ratio for the i-th ion;
- is the absolute abundance of the main ion.
3.2. Coffee Roasting
3.3. Coffee Extraction
3.4. Chemical Standards
3.5. Selectable 1D/2D GC/MS
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wilton, N.M.; Wise, S.A.; Robbat, A., Jr. Errors in alkylated polycyclic aromatic hydrocarbon and sulfur heterocycle concentrations caused by currently employed standardized methods. Anal. Chim. Acta 2017, 977, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Robbat, A., Jr.; Kfoury, N.; Baydakov, E.; Gankin, Y. Optimizing targeted/untargeted metabolomics by automating gas chromatography/mass spectrometry workflows. J. Chromatogr. A 2017, 1505, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Kowalsick, A.; Kfoury, N.; Robbat, A., Jr.; Ahmed, S.; Orians, C.; Griffin, T.; Cash, S.B.; Stepp, J.R. Metabolite profiling of Camellia sinensis by automated sequential, multidimensional gas chromatography/mass spectrometry reveals strong monsoon effects on tea constituents. J. Chromatogr. A 2014, 1370, 230–239. [Google Scholar] [CrossRef]
- Robbat, A., Jr.; Kowalsick, A.; Howell, J. Tracking juniper berry content in oils and distillates by spectral deconvolution of gas chromatography/mass spectrometry data. J. Chromatogr. A 2011, 1218, 5531–5541. [Google Scholar] [CrossRef] [PubMed]
- Zeigler, C.; Schantz, M.; Wise, S.; Robbat, A., Jr. Mass Spectra and Retention Indexes for Polycyclic Aromatic Sulfur Heterocycles and Some Alkylated Analogs. Polycycl. Aromat. Compd. 2012, 32, 154–176. [Google Scholar] [CrossRef]
- Zeigler, C.; Wilton, N.; Robbat, A., Jr. Toward the Accurate Analysis of C-1-C-4 Polycyclic Aromatic Sulfur Heterocycles. Anal. Chem. 2012, 84, 2245–2252. [Google Scholar] [CrossRef]
- Zeigler, C.D.; Robbat, A., Jr. Comprehensive Profiling of Coal Tar and Crude Oil to Obtain Mass Spectra and Retention Indices for Alkylated PAH Shows Why Current Methods Err. Environ. Sci. Technol. 2012, 46, 3935–3942. [Google Scholar] [CrossRef]
- Morimoto, J.; Rosso, M.C.; Kfoury, N.; Bicchi, C.; Cordero, C.; Robbat, A., Jr. Untargeted/Targeted 2D Gas Chromatography/Mass Spectrometry Detection of the Total Volatile Tea Metabolome. Molecules 2019, 24, 3757. [Google Scholar] [CrossRef] [Green Version]
- Stilo, F.; Tredici, G.; Bicchi, C.; Robbat, A., Jr.; Morimoto, J.; Cordero, C. Climate and Processing Effects on Tea (Camellia sinensis L. Kuntze) Metabolome: Accurate Profiling and Fingerprinting by Comprehensive Two-Dimensional Gas Chromatography/Time-of-Flight Mass Spectrometry. Molecules 2020, 25, 2447. [Google Scholar] [CrossRef]
- Stilo, F.; Bicchi, C.; Robbat, A.; Reichenbach, S.E.; Cordero, C. Untargeted approaches in food-omics: The potential of comprehensive two-dimensional gas chromatography/mass spectrometry. TrAC Trends Anal. Chem. 2021, 135, 116162. [Google Scholar] [CrossRef]
- Kfoury, N.; Baydakov, E.; Gankin, Y.; Robbat, A., Jr. Differentiation of key biomarkers in tea infusions using a target/nontarget gas chromatography/mass spectrometry workflow. Food Res. Int. 2018, 113, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Kfoury, N.; Morimoto, J.; Kern, A.; Scott, E.R.; Orians, C.M.; Ahmed, S.; Griffin, T.; Cash, S.B.; Stepp, J.R.; Xue, D.; et al. Striking changes in tea metabolites due to elevational effects. Food Chem. 2018, 264, 334–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kfoury, N.; Scott, E.R.; Orians, C.M.; Ahmed, S.; Cash, S.B.; Griffin, T.; Matyas, C.; Stepp, J.R.; Han, W.; Xue, D.; et al. Plant-Climate Interaction Effects: Changes in the Relative Distribution and Concentration of the Volatile Tea Leaf Metabolome in 2014–2016. Front. Plant. Sci. 2019, 10, 1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebanov, L.; Ghiasvand, A.; Paull, B. Data handling and data analysis in metabolomic studies of essential oils using GC-MS. J. Chromatogr. A 2021, 1640, 461896. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Ma, A.; Meng, X.; Shen, G.-a.; Qi, X. QPMASS: A parallel peak alignment and quantification software for the analysis of large-scale gas chromatography-mass spectrometry (GC-MS)-based metabolomics datasets. J. Chromatogr. A 2020, 1620, 460999. [Google Scholar] [CrossRef]
- Spicer, R.; Salek, R.M.; Moreno, P.; Canueto, D.; Steinbeck, C. Navigating freely-available software tools for metabolomics analysis. Metabolomics 2017, 13, 106. [Google Scholar] [CrossRef] [Green Version]
- Misra, B.B. New tools and resources in metabolomics: 2016–2017. Electrophoresis 2018, 39, 909–923. [Google Scholar] [CrossRef]
- Barcaru, A.; Mol, H.G.J.; Tienstra, M.; Vivo-Truyols, G. Bayesian approach to peak deconvolution and library search for high resolution gas chromatography–Mass spectrometry. Anal. Chim. Acta 2017, 983, 76–90. [Google Scholar] [CrossRef]
- Shekari, N.; Vosough, M.; Heidar, K.T. Chromatographic fingerprinting through chemometric techniques for herbal slimming pills: A way of adulterant identification. Forensic Sci. Int. 2018, 286, 213–222. [Google Scholar] [CrossRef]
- Sichilongo, K.; Padiso, T.; Turner, Q. AMDIS-Metab R data manipulation for the geographical and floral differentiation of selected honeys from Zambia and Botswana based on volatile chemical compositions using SPME-GC-MS. Eur. Food Res. Technol. 2020, 246, 1679–1690. [Google Scholar] [CrossRef]
- Zhang, Y.-M.; Zhang, Y.-Y.; Zhang, Q.; Lv, Y.; Sun, T.; Han, L.; Bai, C.-C.; Yu, Y.-J. Automatic peak detection coupled with multivariate curve resolution-alternating least squares for peak resolution in gas chromatography-mass spectrometry. J. Chromatogr. A 2019, 1601, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Chua, C.K.; Lu, B.; Lv, Y.; Gu, X.Y.; Di Thng, A.; Zhang, H.J. An optimized band-target entropy minimization for mass spectral reconstruction of severely co-eluting and trace-level components. Anal. Bioanal. Chem. 2018, 410, 6549–6560. [Google Scholar] [CrossRef] [PubMed]
- Chua, C.K.; Lv, Y.; Zhao, W.; Ren, Y.; Zhang, H.J. Improving annotation of known-unknowns with accurately reconstructed mass spectra. Int. J. Mass Spectrom. 2020, 451, 116321. [Google Scholar] [CrossRef]
- Lu, B.; Lv, Y.; Du, F.L.; Chua, C.K.; Zhang, H.J. Lower limit of detection achieved by raw band–target entropy minimization (rBTEM) for trace and coeluted gas chromatography-mass spectrometry components. Anal. Lett. 2019, 52, 1579–1589. [Google Scholar] [CrossRef]
- Cramer, J.A.; Hammond, M.H.; Loegel, T.N.; Morris, R.E. Evolving window factor analysis-multivariate curve resolution with automated library matching for enhanced peak deconvolution in gas chromatography-mass spectrometry fuel data. J. Chromatogr. A 2018, 1581, 125–134. [Google Scholar] [CrossRef]
- Dabrowski, L. Evaluation of a Simplified Method for GC/MS Qualitative Analysis of Polycyclic Aromatic Hydrocarbons, Polychlorinated Biphenyls, and Organic Pesticides Using PARADISe Computer Program. Molecules 2020, 25, 3727. [Google Scholar] [CrossRef]
- Johnsen, L.G.; Skou, P.B.; Khakimov, B.; Bro, R. Gas chromatography − mass spectrometry data processing made easy. J. Chromatogr. A 2017, 1503, 57–64. [Google Scholar] [CrossRef]
- Song, J.-J.; Wang, X.; Wang, Y.-Y.; Zhang, Y.-Y.; Yu, Y.-J. High-throughput identification of volatile and semi-volatile organic compounds in archaeological samples by gas chromatography–mass spectrometry combined with advanced chemometrics methodology. Microchem. J. 2020, 158, 1695. [Google Scholar] [CrossRef]
- Zhang, Y.-Y.; Zhang, Q.; Zhang, Y.-M.; Wang, W.-W.; Zhang, L.; Yu, Y.-J.; Bai, C.-C.; Guo, J.-Z.; Fu, H.-Y.; She, Y. A comprehensive automatic data analysis strategy for gas chromatography-mass spectrometry based untargeted metabolomics. J. Chromatogr. A 2020, 1616, 469787. [Google Scholar] [CrossRef]
- Haiduc, A.; Zanetti, F.; Zhao, X.; Schlage, W.K.; Scherer, M.; Pluym, N.; Schlenger, P.; Ivanov, N.V.; Majeed, S.; Hoeng, J.; et al. Analysis of chemical deposits on tooth enamel exposed to total particulate matter from cigarette smoke and tobacco heating system 2.2 aerosol by novel GC–MS deconvolution procedures. J. Chromatogr. B-Anal. Technol. Biomed. Life Sci. 2020, 1152, 122228. [Google Scholar] [CrossRef]
- Liang, D.; Liu, Q.; Zhou, K.; Jia, W.; Xie, G.; Chen, T. IP4M: An integrated platform for mass spectrometry-based metabolomics data mining. BMC Bioinform. 2020, 21, 444. [Google Scholar] [CrossRef] [PubMed]
- Aksenov, A.A.; Laponogov, I.; Zhang, Z.; Doran, S.L.F.; Belluomo, I.; Veselkov, D.; Bittremieux, W.; Nothias, L.F.; Nothias-Esposito, M.; Maloney, K.N.; et al. Auto-deconvolution and molecular networking of gas chromatography–mass spectrometry data. Nat. Biotechnol. 2020, 39, 169. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Almenara, X.; Guijas, C.; Billings, E.; Montenegro-Burke, J.R.; Uritboonthai, W.; Aisporna, A.E.; Chen, E.; Benton, H.P.; Siuzdak, G. The METLIN small molecule dataset for machine learning-based retention time prediction. Nat. Commun. 2019, 10, 5811. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, X.R. Peak alignment of gas chromatography–mass spectrometry data with deep learning. J. Chromatogr. A 2019, 1604, 460476. [Google Scholar] [CrossRef] [Green Version]
- Risum, A.B.; Bro, R. Using deep learning to evaluate peaks in chromatographic data. Talanta 2019, 204, 255–260. [Google Scholar] [CrossRef]
- Robbat, A., Jr.; Kowalsick, A. Using Automated Sequential Two-dimensional Gas Chromatography/Mass Spectrometry to produce a Library of Essential Oil Compounds and Track their presence in Gin, Based on Spectral Deconvolution Software. In Flavor, Frangrance, and Odor Analysis; Marsili, R., Ed.; CRC Press: Boca Raton, FL, USA, 2011; pp. 183–206. [Google Scholar]
- Sunarharum, W.B.; Williams, D.J.; Smyth, H.E. Complexity of coffee flavor: A compositional and sensory perspective. Food Res. Int. 2014, 62, 315–325. [Google Scholar] [CrossRef]
- Toci, A.T.; Farah, A. Volatile compounds as potential defective coffee beans’ markers. Food Chem. 2008, 108, 1133–1141. [Google Scholar] [CrossRef]
- Holscher, W.; Steinhart, H. Aroma compounds in green coffee. In Developments in Food Science; Charalambous, G., Ed.; Elsevier: Amsterdam, The Netherlands, 1995; Volume 37, pp. 785–803. [Google Scholar]
- Bonnländer, B.; Cappuccio, R.; Liverani, F.S.; Winterhalter, P. Analysis of enantiomeric linalool ratio in green and roasted coffee. Flavour Fragr. J. 2006, 21, 637–641. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frost, S.C.; Walker, P.; Orians, C.M.; Robbat, A., Jr. The Chemistry of Green and Roasted Coffee by Selectable 1D/2D Gas Chromatography Mass Spectrometry with Spectral Deconvolution. Molecules 2022, 27, 5328. https://doi.org/10.3390/molecules27165328
Frost SC, Walker P, Orians CM, Robbat A Jr. The Chemistry of Green and Roasted Coffee by Selectable 1D/2D Gas Chromatography Mass Spectrometry with Spectral Deconvolution. Molecules. 2022; 27(16):5328. https://doi.org/10.3390/molecules27165328
Chicago/Turabian StyleFrost, Scott C., Paige Walker, Colin M. Orians, and Albert Robbat, Jr. 2022. "The Chemistry of Green and Roasted Coffee by Selectable 1D/2D Gas Chromatography Mass Spectrometry with Spectral Deconvolution" Molecules 27, no. 16: 5328. https://doi.org/10.3390/molecules27165328