
Molecular Basis of Inhibitory Mechanism of Naltrexone and Its Metabolites through Structural and Energetic Analyses


Abstract
:1. Introduction
2. Results
2.1. Docking Results
2.2. Stability of the Systems
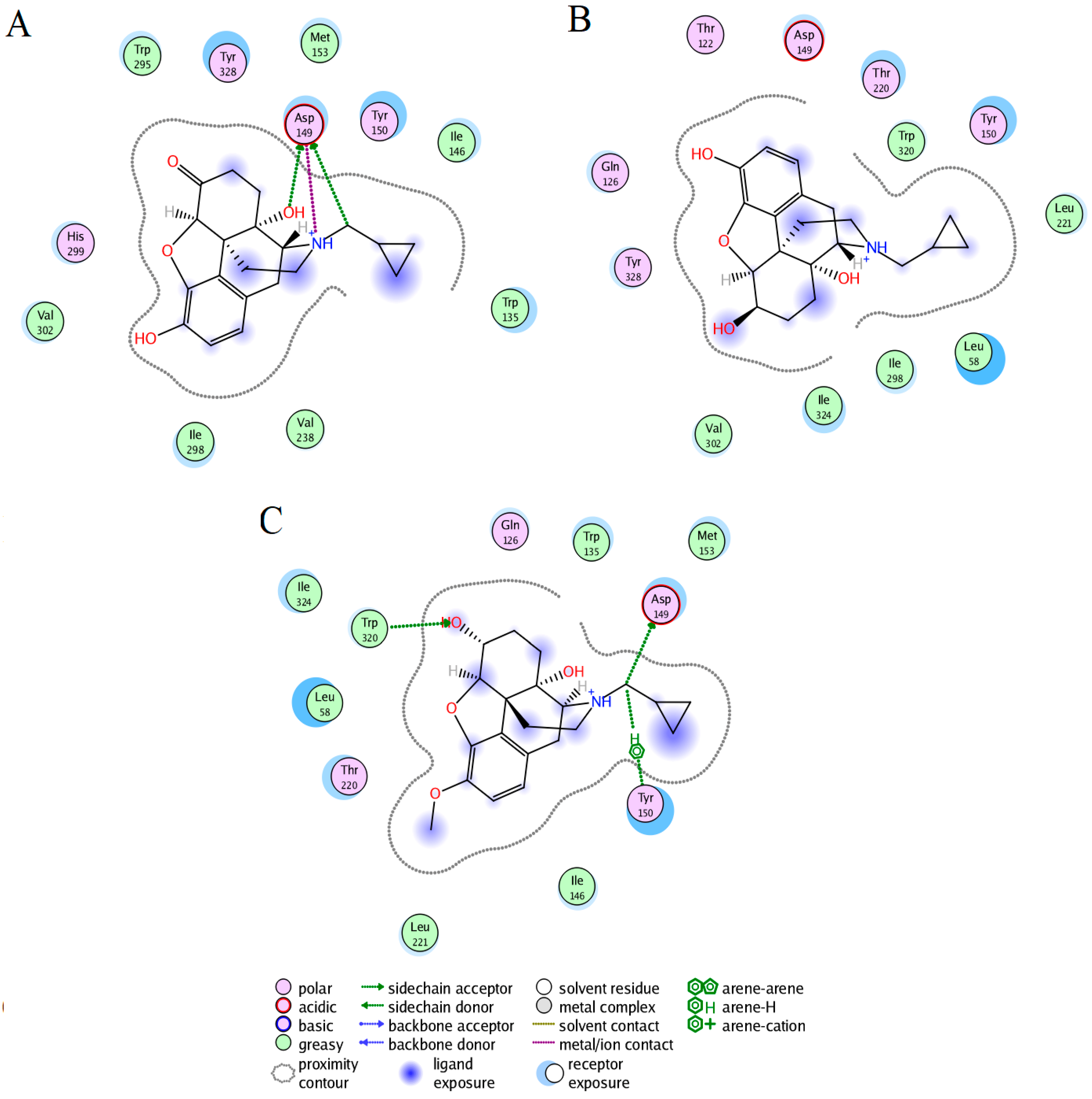
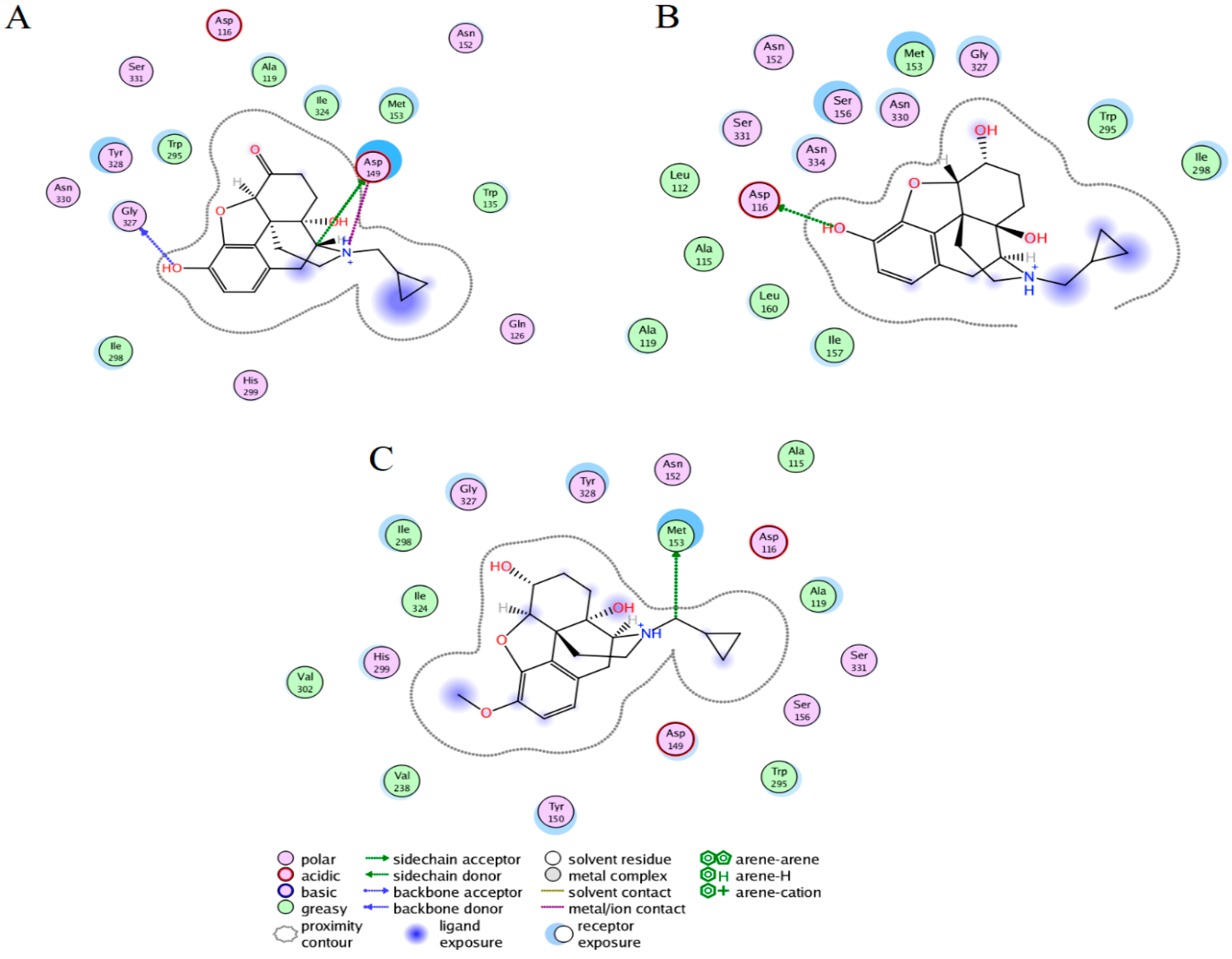
2.3. Ligand Interactions on the hMOR–Ligand Complex through MD Simulations
2.4. Binding Free Energy Calculations
2.5. Per-Residue Free Energy Decomposition for the hMOR–Naltrexone Complex
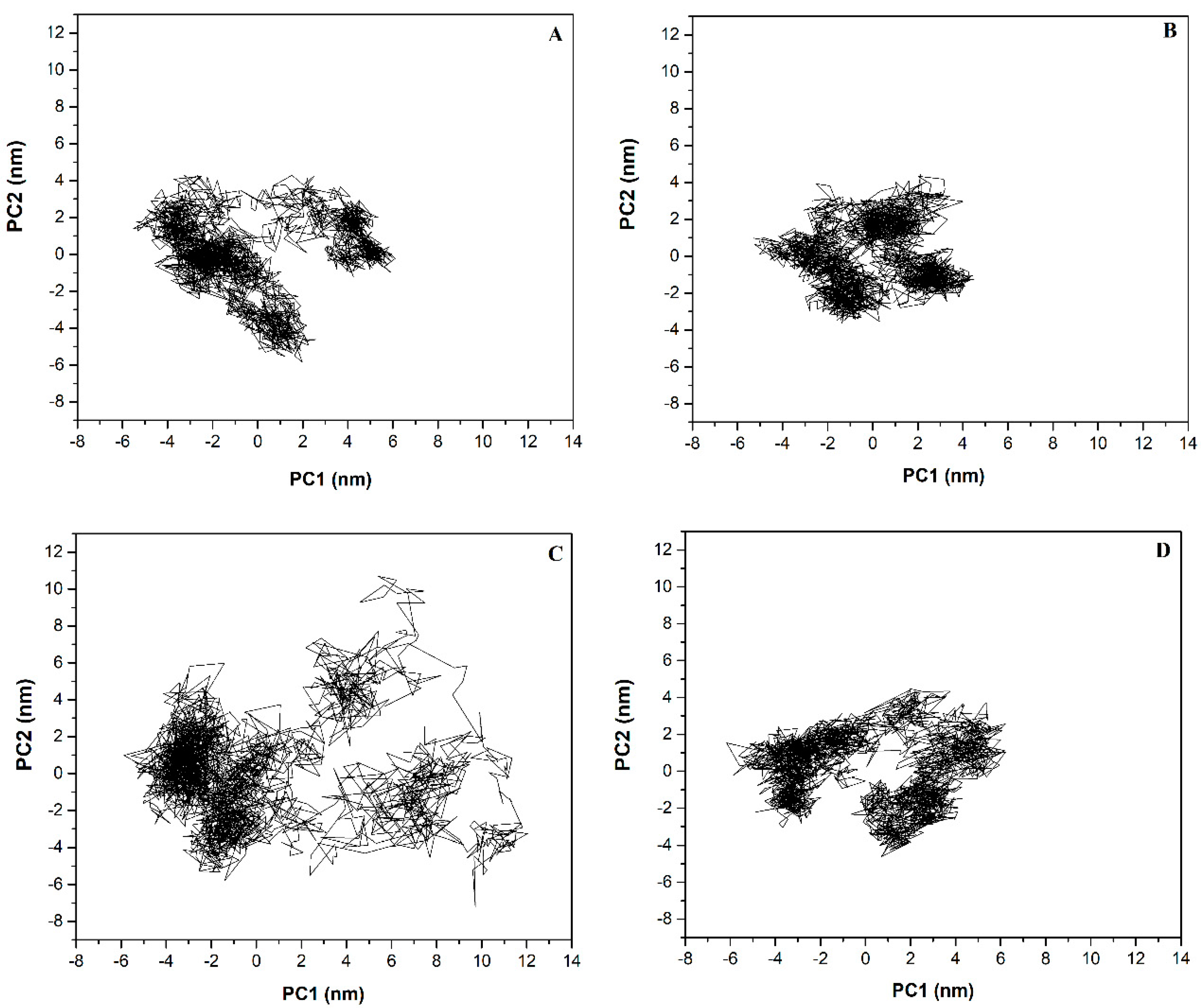
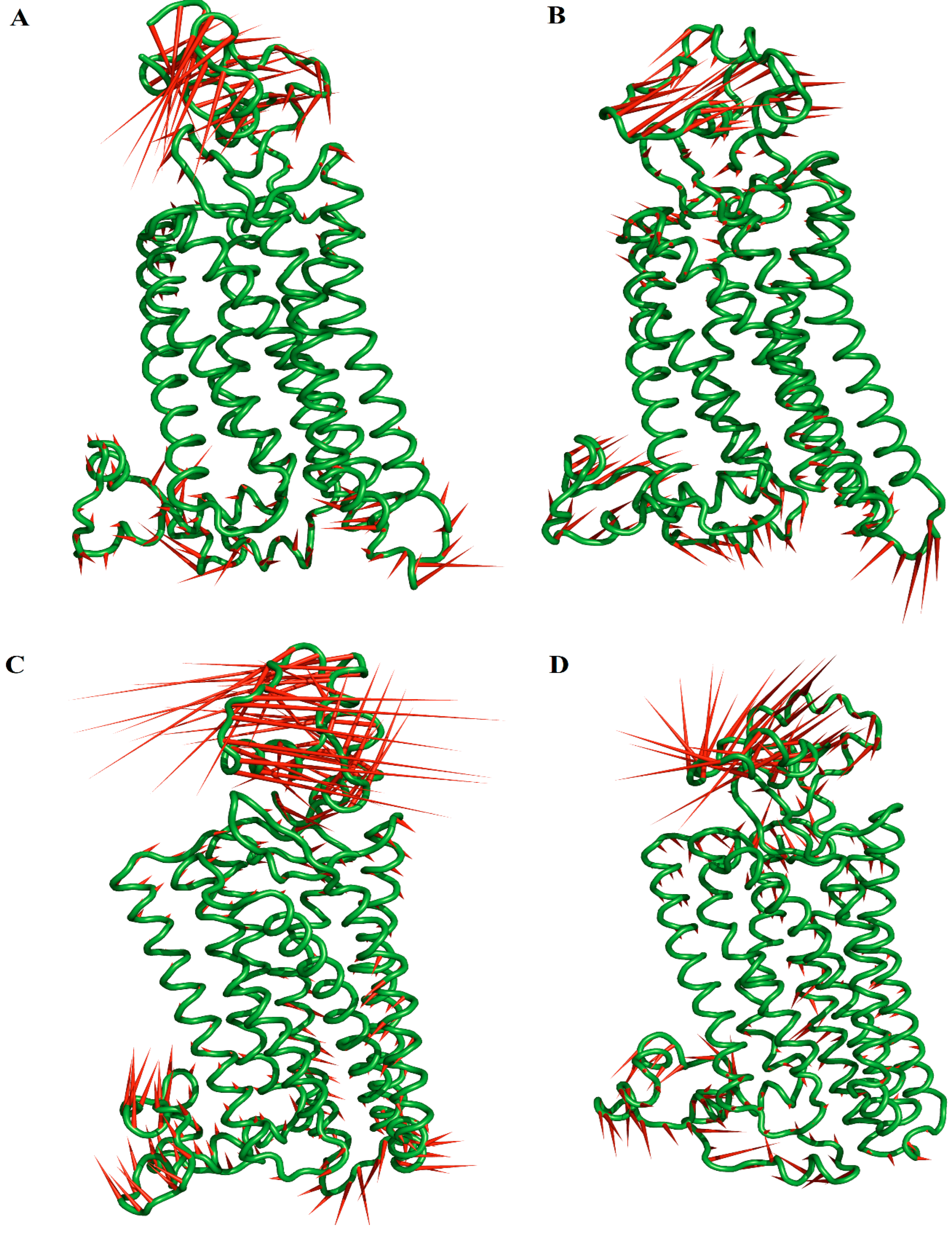
2.6. Principal Component (PC) Analysis
3. Discussion
4. Material and Methods
4.1. Structural Modeling
4.2. Docking Studies
4.3. Anchoring of the Receptor–Ligand Complex onto the Membrane
4.4. MD Simulations
4.5. MD Trajectory Analysis
4.6. Binding Free Energy
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Litten, R.Z.; Allen, J.P. Advances in development of medications for alcoholism treatment. Psychopharmacology 1998, 139, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Volpicelli, J.R.; Alterman, A.I.; Hayashida, M.; O’Brien, C.P. Naltrexone in the treatment of alcohol dependence. Arch. Gen. Psychiatry 1992, 49, 876–879. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, S.S. Opioid antagonists in the treatment of alcohol dependence: Clinical efficacy and presentation of relapse. Alcohol Alcohol. 1996, 31, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Anton, R.F.; O’Malley, S.S.; Ciraulo, D.C.; Cisler, R.A.; Couper, D.; Donovan, D.M.; Gastfriend, D.R.; Hosking, J.D.; Johnson, B.A.; LoCastro, J.S.; et al. Combined pharmacotherapies and behavioral interventions for alcohol dependence: The COMBINE Study: A Randomized Controlled Trial. JAMA 2006, 295, 2003–2017. [Google Scholar] [CrossRef] [Green Version]
- Renault, P.F. Treatment of heroin-dependent persons with antagonists: Current status. Bull. Narc. 1978, 30, 21–29. [Google Scholar]
- Krupitsky, E.; Zvartau, E.; Masalov, D.; Tsoy, M.; Burakov, A.; Egorova, V.; Didenko, T.; Romanova, T.; Ivanova, E.; Bespalov, A.; et al. Naltrexone with or without fluoxetine for preventing relapse to heroin addiction in St. Petersburg, Russia. J. Subst. Abus. Treat. 2006, 31, 319–328. [Google Scholar] [CrossRef]
- Crabtree, B.L. Review of naltrexone, a long-acting opiate antagonist. Clin. Pharm. 1984, 3, 273–280. [Google Scholar]
- O’Malley, S.; Jaffe, A.J.; Chang, G.; Schottenfeld, R.S.; Meyer, R.E.; Rounsaville, B. Naltrexone and coping skills therapy for alcohol dependence. Arch. Gen. Psychiatry 1992, 49, 881–887. [Google Scholar] [CrossRef]
- Volpicelli, J.R.; Rhines, K.C.; Rhines, J.S.; Volpicelli, L.A.; Alterman, A.I.; O’Brien, C.P. Naltrexone and alcohol dependence: Role of subject compliance. Arch. Gen. Psychiatry 1997, 54, 737–742. [Google Scholar] [CrossRef]
- Verebey, K.; Mulé, S.J. Naltrexone pharmacology, pharmacokinetics, and metabolism: Current status. J. Drug Alcohol Abus. 1975, 2, 357–363. [Google Scholar] [CrossRef]
- Schmidt, W.K.; Tam, S.W.; Shotzberger, G.S.; Smith, D.H., Jr.; Clark, R.; Bernier, V.G. Nalbuphine. Drug Alcohol Depend. 1985, 14, 339–362. [Google Scholar] [CrossRef]
- Okada, T.; Le Trong, I.; Fox, B.A.; Behnke, C.A.; Stenkamp, R.E.; Palczewski, K. X-Ray diffraction analysis of three-dimensional crystals of bovine rhodopsin obtained from mixed micelles. J. Struct. Biol. 2000, 130, 73–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palczewski, K.; Kumasaka, T.; Hori, T.; Behnke, C.A.; Motoshima, H.; Fox, B.A.; Le Trong, I.; Teller, D.C.; Okada, T.; Stenkamp, R.E.; et al. Crystal Structure of Rhodopsin: A G Protein-Coupled Receptor. Science 2000, 289, 739–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teller, D.C.; Okada, T.; Behnke, C.A.; Palczewski, K.; Stenkamp, R.E. Advances in Determination of a High-Resolution Three-Dimensional Structure of Rhodopsin, a Model of G-Protein-Coupled Receptors (GPCRs). Biochemistry 2001, 40, 7761–7772. [Google Scholar] [CrossRef] [Green Version]
- Okada, T.; Fujiyoshi, Y.; Silow, M.; Navarro, J.; Landau, E.M.; Shichida, Y. Functional role of internal water molecules in rhodopsin revealed by x-ray crystallography. Proc. Natl. Acad. Sci. USA 2002, 99, 5982–5987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elling, C.E.; Thirstrup, K.; Nielsen, S.M.; Hjorth, S.A.; Schwartz, T.W. Metal-ion sites as structural and functional probes of helix-helix interactions in 7TM receptors. Ann. N. Y. Acad. Sci. 1997, 814, 142–151. [Google Scholar] [CrossRef]
- Mizobe, T.; Maze, M.; Lam, V.; Suryanarayana, S.; Kobilka, B.K. Arrangement of transmembrane domains in adrenergic receptors. Similarity to bacteriorhodopsin. J. Biol. Chem. 1996, 271, 2387–2389. [Google Scholar] [CrossRef] [Green Version]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Carroll, F.I.; Dolle, R.E. The discovery and development of the N-substituted trans-3,4-dimethyl-4-(3′-hydroxyphenyl)piperidine class of pure opioid receptor antagonists. ChemMedChem 2014, 9, 1638–1654. [Google Scholar] [CrossRef]
- Yuan, Y.; Zaidi, S.; Elbegdorj, O.; Aschenbach, L.C.K.; Li, G.; Stevens, D.L.; Scoggins, K.L.; Dewey, W.L.; Selley, D.E.; Zhang, Y. Design, Synthesis, and Biological Evaluation of 14-Heteroaromatic-Substituted Naltrexone Derivatives: Pharmacological Profile Switch from Mu Opioid Receptor Selectivity to Mu/Kappa Opioid Receptor Dual Selectivity. J. Med. Chem. 2013, 56, 9156–9169. [Google Scholar] [CrossRef] [Green Version]
- Shang, Y.; LeRouzic, V.; Schneider, S.; Bisignano, P.; Pasternak, G.W.; Filizola, M. Mechanistic insights into the allosteric modulation of opioid receptors by sodium ions. Biochemistry 2014, 53, 5140–5149. [Google Scholar] [CrossRef] [PubMed]
- Livingston, K.E.; Traynor, J.R. Disruption of the Na+ ion binding site as a mechanism for positive allosteric modulation of the mu-opioid receptor. Proc. Natl. Acad. Sci. USA 2014, 111, 18369–18374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Manglik, A.; Venkatakrishnan, A.J.; Laeremans, T.; Feinberg, E.N.; Sanborn, A.L.; Kato, H.E.; Livingston, K.E.; Thorsen, T.S.; Kling, R.C.; et al. Structural insights into μ-opioid receptor activation. Nature 2015, 524, 315–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, S.G.; Choi, H.-J.; Fung, J.J.; Pardon, E.; Casarosa, P.; Chae, P.S.; Devree, B.T.; Rosenbaum, D.M.; Thian, F.S.; Kobilka, T.S.; et al. Structure of a nanobody-stabilized active state of the β(2) adrenoceptor. Nature 2011, 469, 175–180. [Google Scholar] [CrossRef] [Green Version]
- Ring, A.M.; Manglik, A.; Kruse, A.C.; Enos, M.D.; Weis, W.I.; Garcia, K.C.; Kobilka, B.K. Adrenaline-activated structure of β2- adrenoceptor stabilized by an engineered nanobody. Nature 2013, 502, 575–579. [Google Scholar] [CrossRef] [Green Version]
- Kruse, A.C.; Ring, A.M.; Manglik, A.; Hu, J.; Hu, K.; Eitel, K.; Hübner, H.; Pardon, E.; Valant, C.; Sexton, P.M.; et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 2013, 504, 101–106. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, A.; Martinez-Rosell, G.; Provasi, D.; de Fabritiis, G.; Filizola, M. Dynamic and kinetic elements of μ-opioid receptor functional selectivity. Sci. Rep. 2017, 7, 11255. [Google Scholar] [CrossRef]
- Manglik, A.; Lin, H.; Aryal, D.K.; McCorvy, J.D.; Dengler, D.; Corder, G.; Levit, A.; Kling, R.C.; Bernat, V.; Hübner, H.; et al. Structurebased discovery of opioid analgesics with reduced side effects. Nature 2016, 537, 185–190. [Google Scholar] [CrossRef] [Green Version]
- de Waal, P.W.; Shi, J.; You, E.; Wang, X.; Melcher, K.; Jiang, Y.; Xu, H.E.; Dickson, B.M. Molecular mechanisms of fentanyl mediated β-arrestin biased signaling. PLoS Comput. Biol. 2020, 16, e1007394. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, P.F.J.; Jarończyk, M.; Dobrowolski, J.C.; Sadlej, J. Molecular dynamics of fentanyl bound to μ-opioid receptor. J. Mol. Model. 2019, 25, 144. [Google Scholar] [CrossRef] [Green Version]
- Koehl, A.; Hu, H.; Maeda, S.; Zhang, Y.; Qu, Q.; Paggi, J.M.; Latorraca, N.R.; Hilger, D.; Dawson, R.; Matile, H.; et al. Structure of the μ-opioid receptor-Gi protein complex. Nature 2018, 558, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Chatterjie, N.; Inturrisi, C.E.; Dayton, H.B.; Blumberg, H. Stereospecific synthesis of the 6beta-hydroxy metabolites of naltrexone and naloxone. J. Med. Chem. 1975, 18, 490–492. [Google Scholar] [CrossRef] [PubMed]
- Huq, F. Molecular Modelling Analysis of the Metabolism of Naltrexone. J. Pharmacol. Toxicol. 2006, 1, 354–361. [Google Scholar] [CrossRef] [Green Version]
- Wolf, M.G.; Hoefling, M.; Aponte-Santamaría, C.; Grubmüller, H.; Groenhof, G. g_membed: Efficient insertion of a membrane protein into an equilibrated lipid bilayer with minimal perturbation. J. Comput. Chem. 2010, 31, 2169–2174. [Google Scholar] [CrossRef] [Green Version]
- Ricarte, A.; Dalton, J.A.R.; Giraldo, J. Structural Assessment of Agonist Efficacy in the μ-Opioid Receptor: Morphine and Fentanyl Elicit Different Activation Patterns. J. Chem. Inf. Model. 2021, 61, 1251–1274. [Google Scholar] [CrossRef]
- Liao, S.; Tan, K.; Floyd, C.; Bong, D.; Pino, M.J.; Wu, C. Probing biased activation of mu-opioid receptor by the biased agonist PZM21 using all atom molecular dynamics simulation. Life Sci. 2021, 269, 119026. [Google Scholar] [CrossRef]
- Sena, D.M.; Cong, X.; Giorgetti, A. Ligand based conformational space studies of the μ-opioid receptor. Biochim. Biophys. Acta Gen. Subj. 2020, 1865, 129838. [Google Scholar] [CrossRef]
- Li, J.-G.; Chen, C.; Yin, J.; Rice, K.; Zhang, Y.; Matecka, D.; de Riel, J.; Desjarlais, R.L.; Liu-Chen, L.-Y. Asp147 in the third transmembrane helix of the rat μ opioid receptor forms ion-pairing with morphine and naltrexone. Life Sci. 1999, 65, 175–185. [Google Scholar] [CrossRef]
- Dalton, J.A.R.; Lans, I.; Giraldo, J. Quantifying conformational changes in GPCRs: Glimpse of a common functional mechanism. BMC Bioinform. 2015, 16, 124. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Sanz, A.; Pardo, L.; Liu-Chen, L.Y. Activation of the mu opioid receptor involves conformational rearrangements of multiple transmembrane domains. Biochemistry 2008, 47, 10576–10586. [Google Scholar] [CrossRef]
- Befort, K.; Tabbara, L.; Bausch, S.; Chavkin, C.; Evans, C.; Kieffer, B. The conserved aspartate residue in the third putative transmembrane domain of the delta-opioid receptor is not the anionic counterpart for cationic opiate binding but is a constituent of the receptor binding site. Mol. Pharmacol. 1996, 49, 216–223. [Google Scholar] [PubMed]
- Mansour, A.; Taylor, L.P.; Fine, J.L.; Thompson, R.C.; Hoversten, M.T.; Mosberg, H.I.; Watson, S.J.; Akil, H. Key residues defining the mu-opioid receptor binding pocket: A site-directed mutagenesis study. J. Neurochem. 1997, 68, 344–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using Modeller. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Chen, V.B.; Arendall, W.B., III; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, M.A.; Robb, J.R.; Cheeseman, G.; Scalmani, V.; Barone, B.; Mennucci, G.A.; Petersson, H.; et al. Gaussian 09; Revision, D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Lomize, M.A.; Lomize, A.L.; Pogozheva, I.D.; Mosberg, H.I. OPM: Orientations of proteins in membranes database. Bioinformatics 2006, 22, 623–625. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Im, W. Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS ONE 2007, 2, e880. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Lim, J.B.; Klauda, J.B.; Im, W. CHARMM-GUI Membrane Builder for Mixed Bilayers and Its Application to Yeast Membranes. Biophys. J. 2009, 97, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickson, C.J.; Madej, B.D.; Skjevik, A.A.; Betz, R.M.; Teigen, K.; Gould, I.R.; Walker, R.C. Lipid14: The amber lipid force field. J. Chem. Theory Comput. 2014, 10, 865–879. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- van Gunsteren, W.F.; Berendsen, H.J.C. Algorithms for macromoleculardynamics and constraint dynamics. Mol. Phys. 1977, 34, 1311–1327. [Google Scholar] [CrossRef]
- Amadei, A.; Linssen, A.B.M.; Berendsen, H.J.C. Essential dynamics of proteins. Proteins Struct. Funct. Bioinform. 1993, 17, 412–425. [Google Scholar] [CrossRef]
- DeLano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific: Palo Alto, CA, USA, 2002. [Google Scholar]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Feig, M.; Onufriev, A.; Lee, M.S.; Im, W.; Case, D.A.; Brooks, C.L. Performance Comparison of Generalized Born and Poisson Methods in the Calculation of Electrostatic Solvation Energies for Protein Structures. J. Comput. Chem. 2004, 25, 265–284. [Google Scholar] [CrossRef]
- Bello, M. Binding mechanism of kinase inhibitors to EGFR and T790M, L858R and L858R/T790M mutants through structural and energetic analysis. Int. J. Biol. Macromol. 2018, 118, 1948–1962. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Systems | ΔEMM | ΔGsolv-GBSA | ΔGMMGBSA |
|---|---|---|---|
| hMOR–naltrexone | −71.36 ± 7.12 | 37.97 ± 6.11 | −33.39 ± 2.68 |
| hMOR–NTXOL | −39.43 ±11.8 | 8.76 ± 1.0 | −30.67 ± 2.95 |
| hMOR–HMNTXOL | −49.99 ± 8.5 | 20.36 ± 8.7 | 29.63 ± 2.62 |
| Residue | hMOR–Naltrexone | hMOR–NTXOL | hMOR–HMNTXOL |
|---|---|---|---|
| A115 | −1.173 | ||
| A119 | −1.128 | ||
| M153 | −1.780 | −1.551 | −3.050 |
| I157 | −1.016 | ||
| W295 | −1.572 | −1.176 | |
| I298 | −1.184 | −1.725 | |
| I324 | −1.216 | −1.382 | |
| G327 | −1.547 | ||
| Y328 | −2.774 | −1.522 | |
| N330 | −1.566 | ||
| N334 | −1.942 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bello, M. Molecular Basis of Inhibitory Mechanism of Naltrexone and Its Metabolites through Structural and Energetic Analyses. Molecules 2022, 27, 4919. https://doi.org/10.3390/molecules27154919
Bello M. Molecular Basis of Inhibitory Mechanism of Naltrexone and Its Metabolites through Structural and Energetic Analyses. Molecules. 2022; 27(15):4919. https://doi.org/10.3390/molecules27154919
Chicago/Turabian StyleBello, Martiniano. 2022. "Molecular Basis of Inhibitory Mechanism of Naltrexone and Its Metabolites through Structural and Energetic Analyses" Molecules 27, no. 15: 4919. https://doi.org/10.3390/molecules27154919