
Stability and Existence of Noncanonical I-motif DNA Structures in Computer Simulations Based on Atomistic and Coarse-Grained Force Fields


Abstract
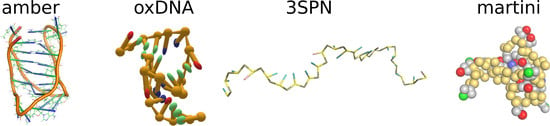
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
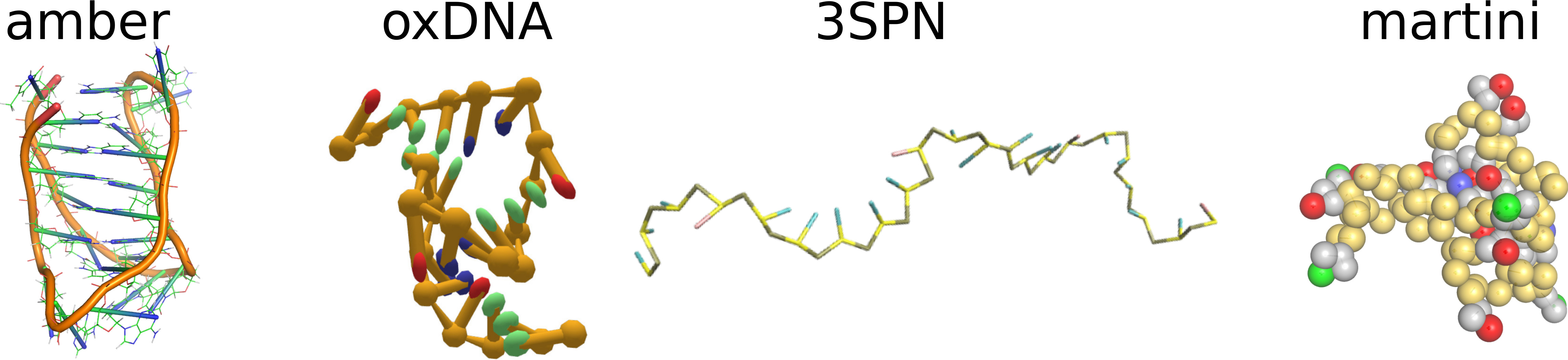
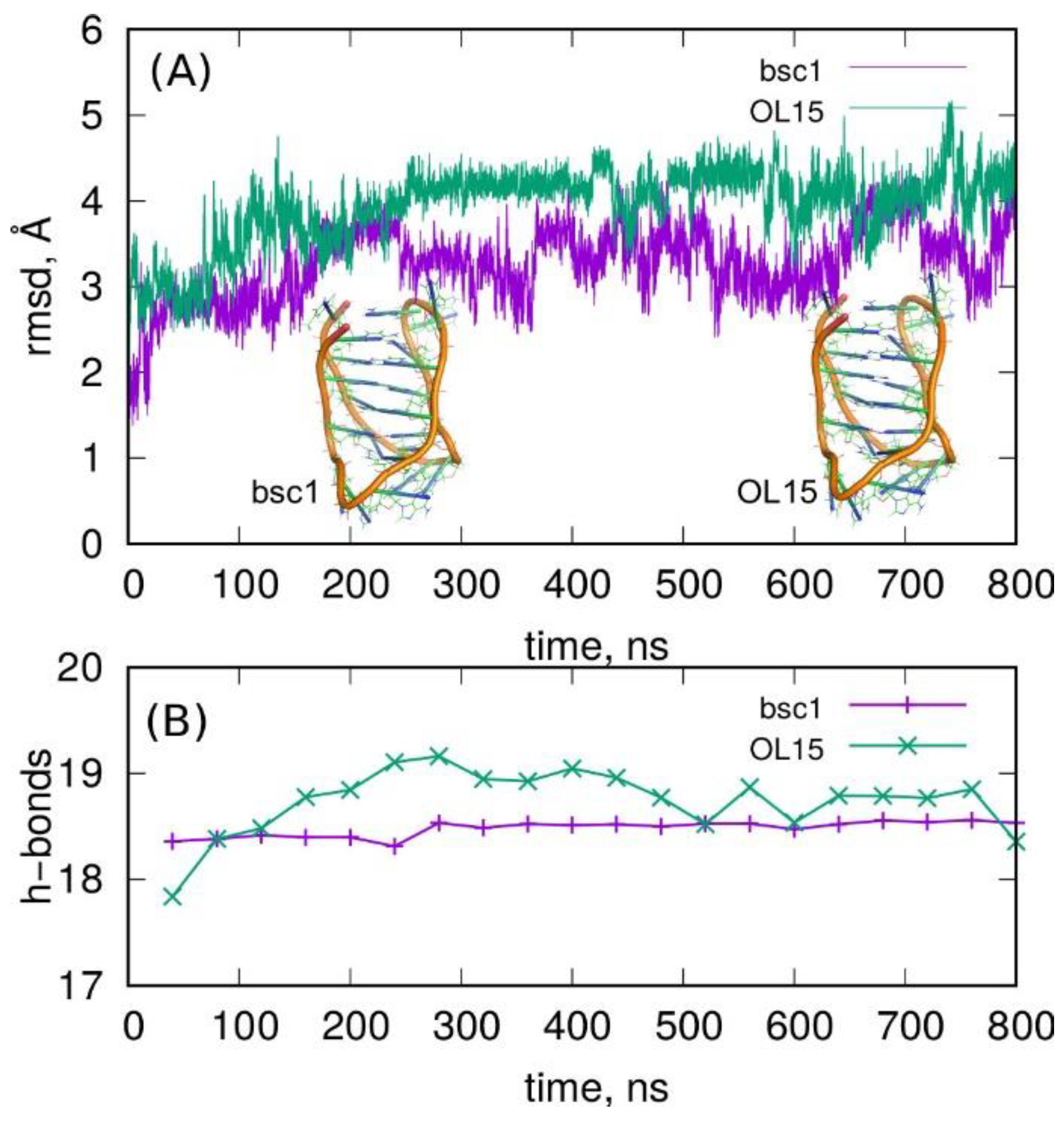
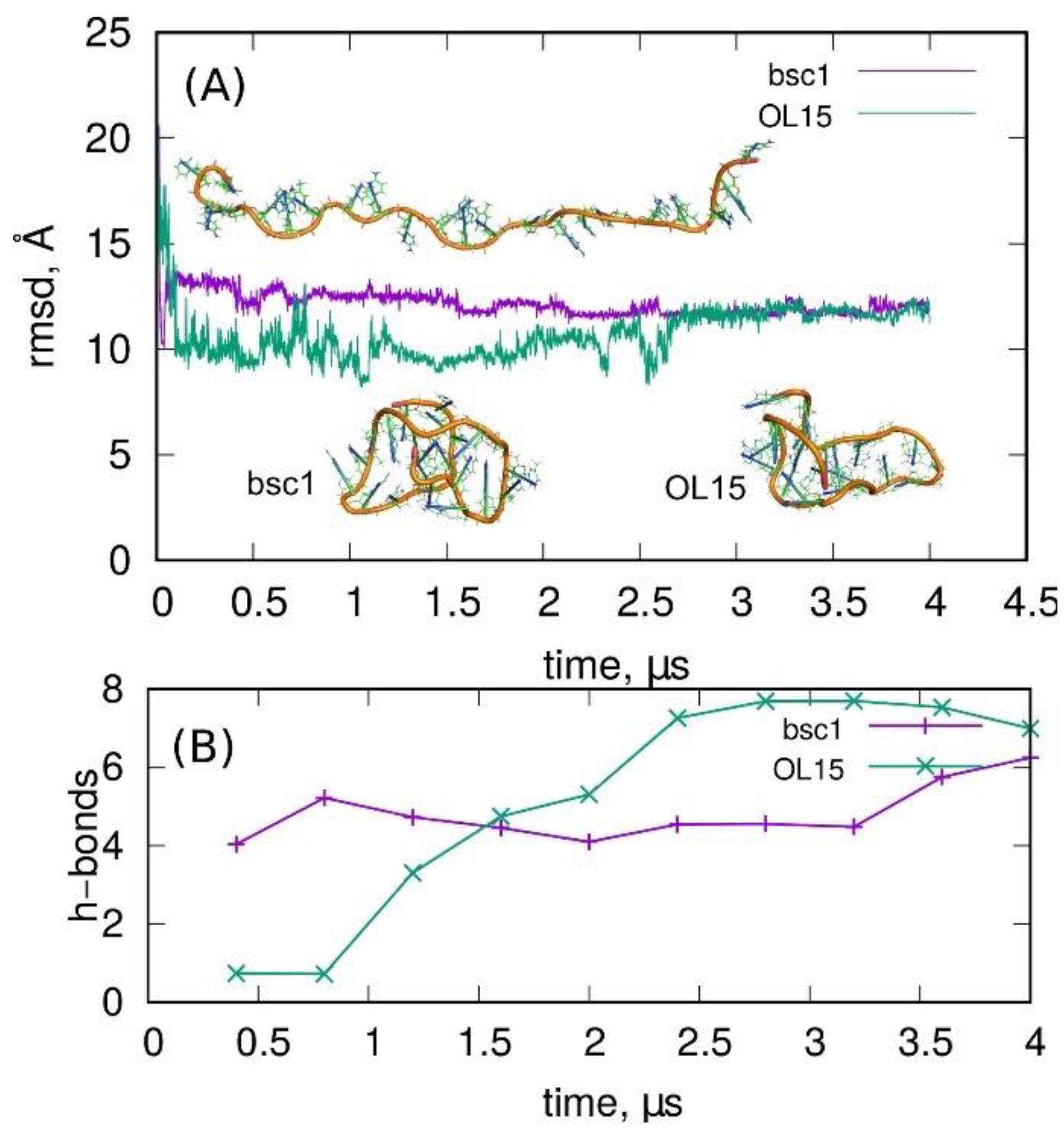
2.1. Stability of I-Motif in the Amber Atomistic Force Field Representations
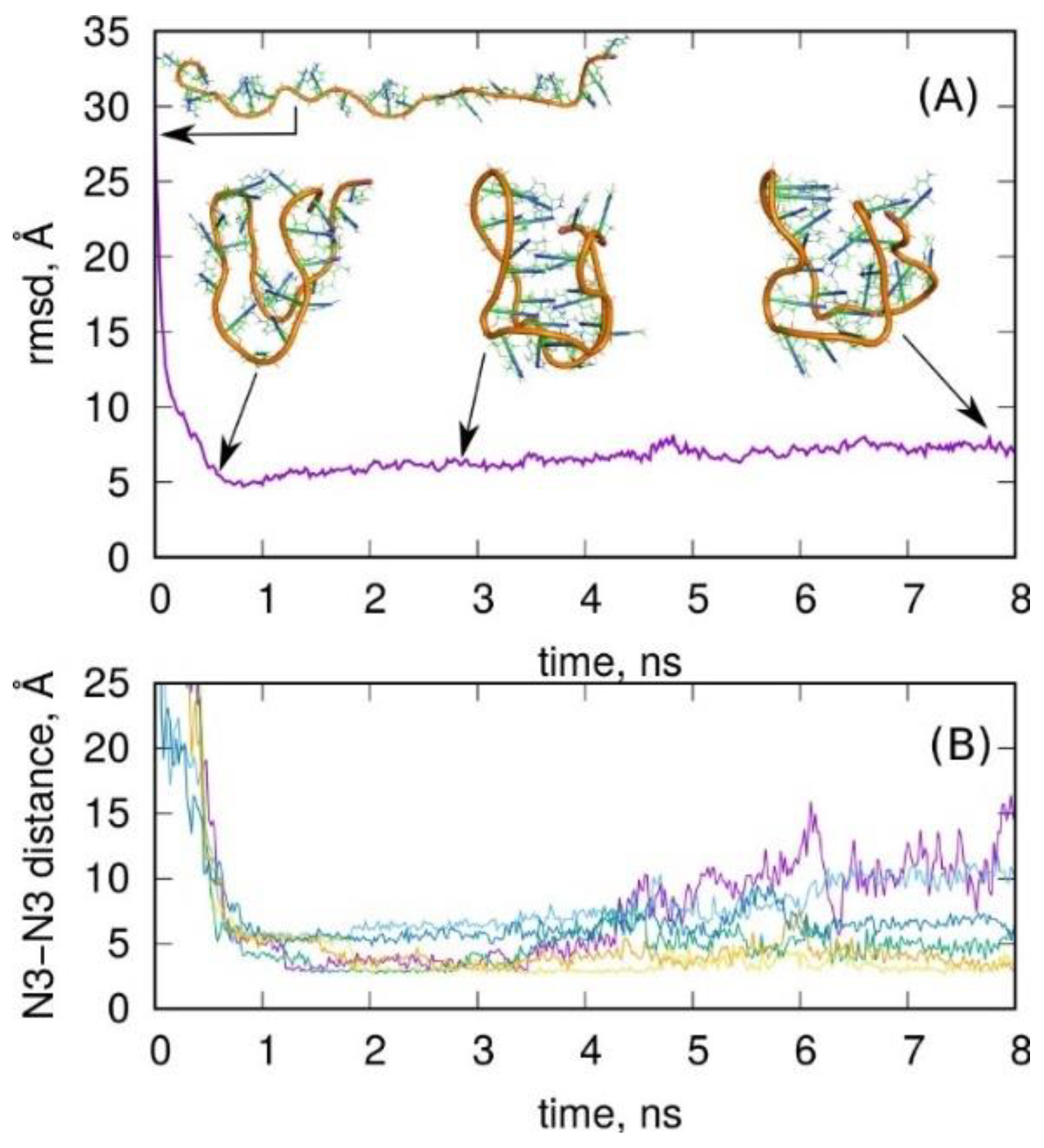
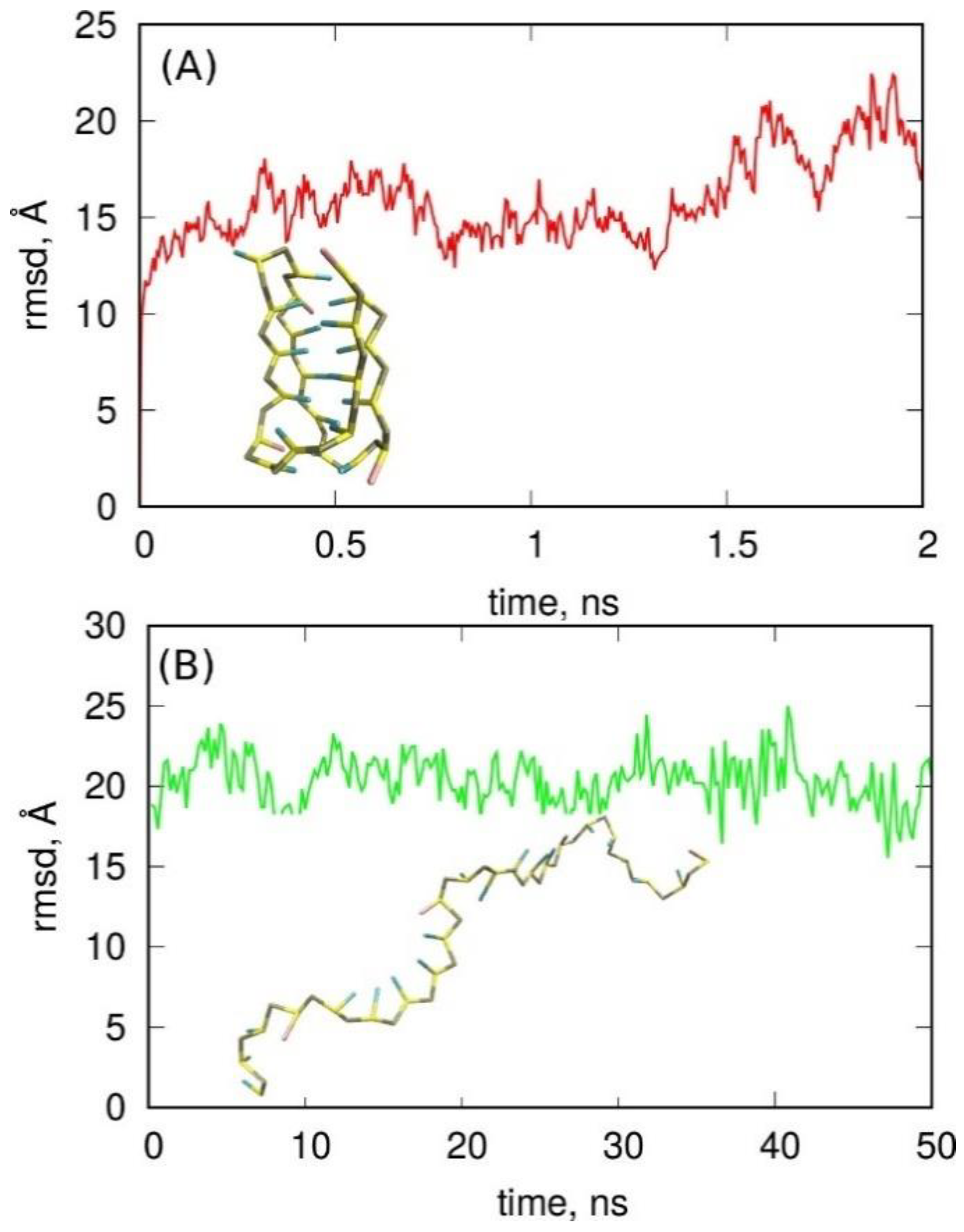
2.2. Folding of Cytosine-Rich Sequence into I-Motif
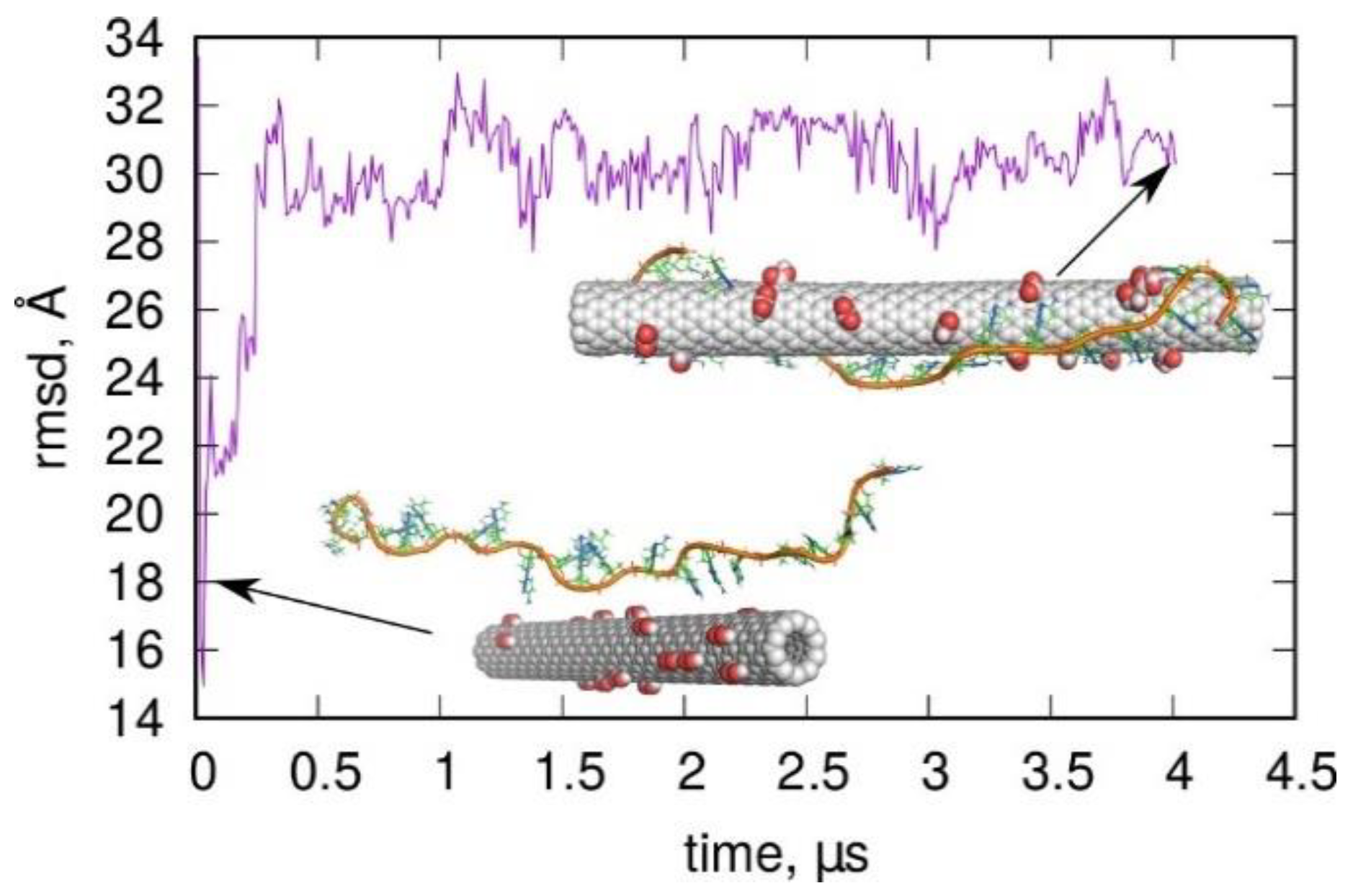
2.3. Folding of Cytosine-Rich Sequence in the Presence of a Carbon Nanotube
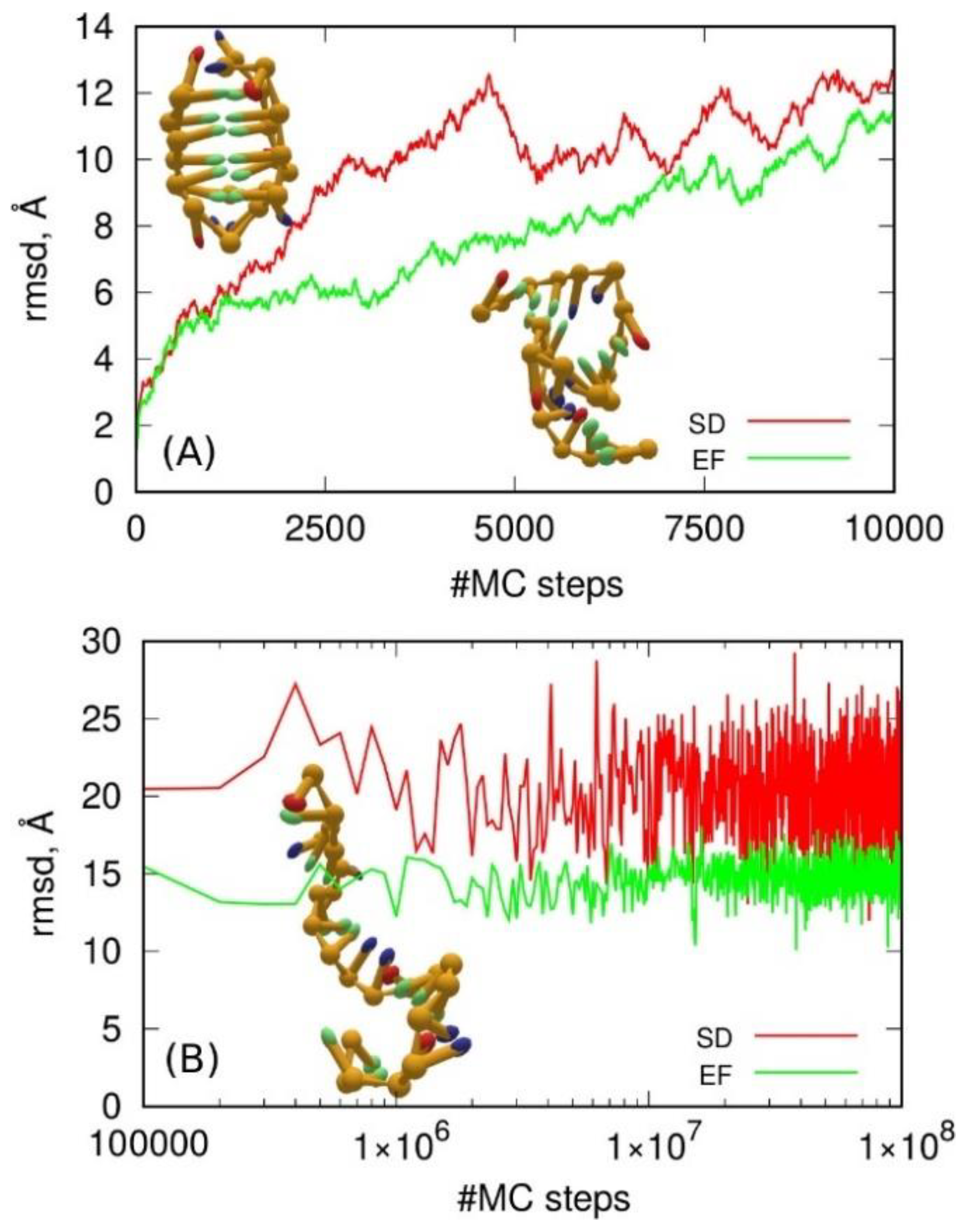
2.4. Coarse-Grained Force Fields for DNA
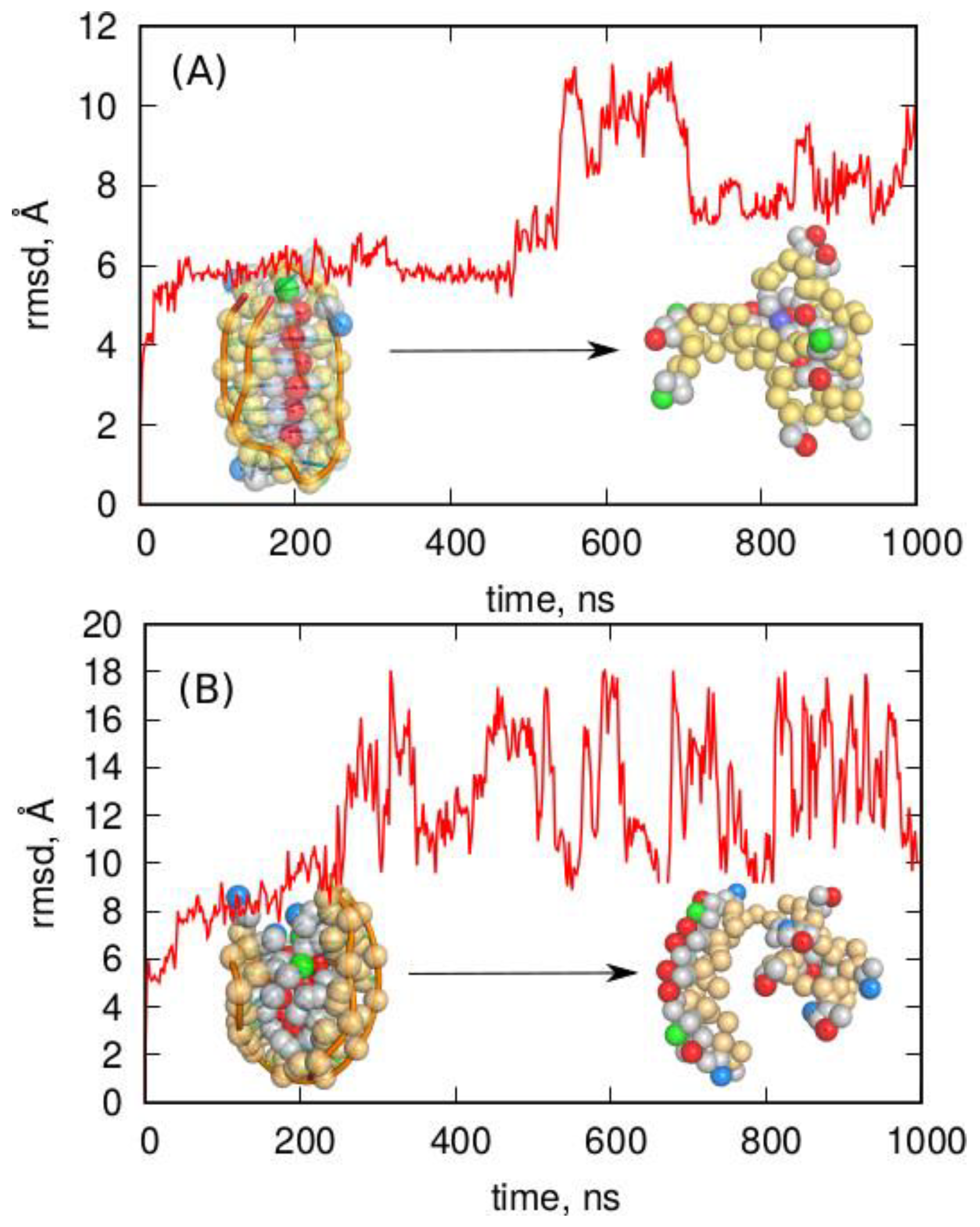
2.4.1. oxDNA Model
2.4.2. 3SPN Model
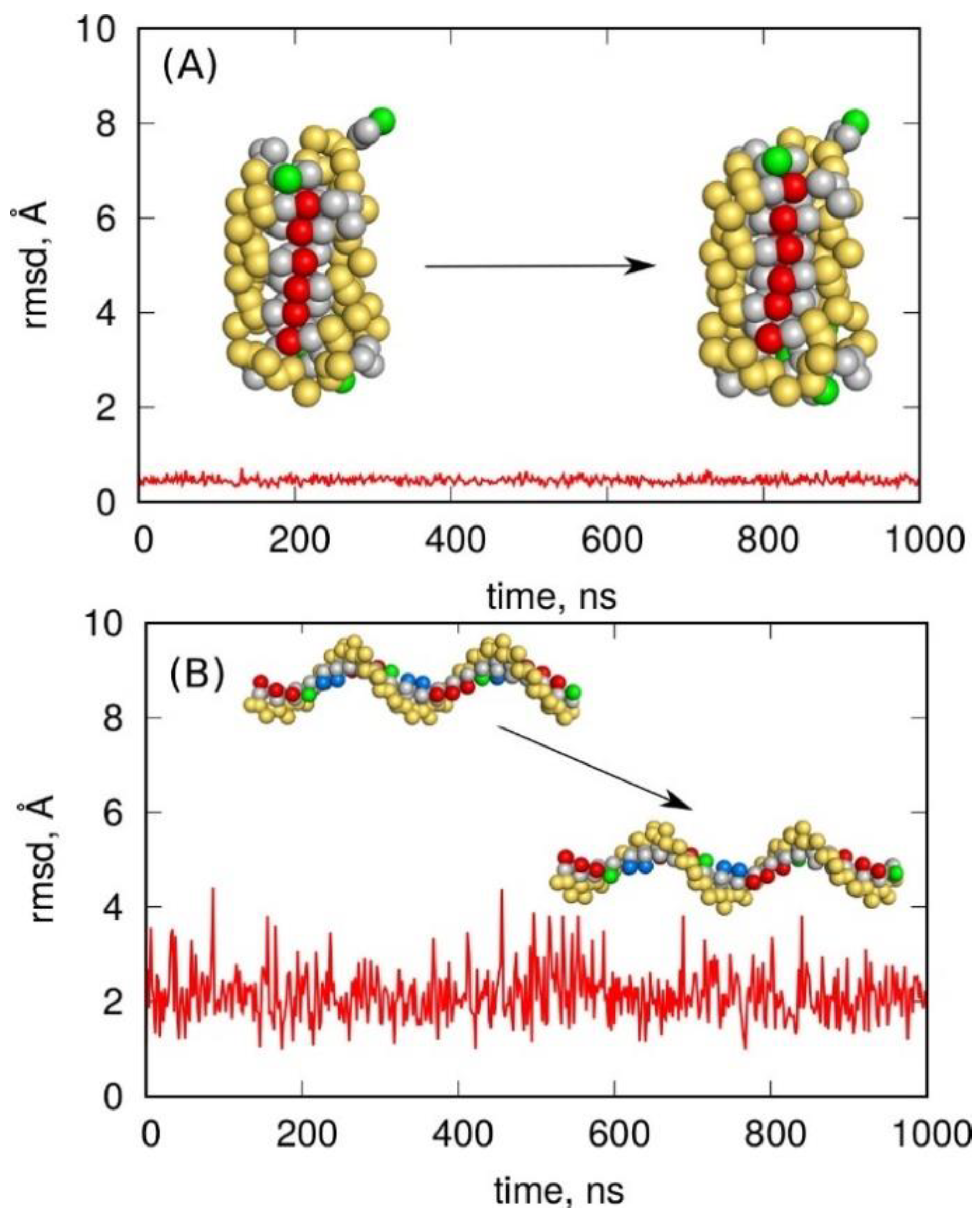
2.4.3. Martini-DNA Model
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Huppert, J.L.; Balasubramanian, S. G-Quadruplexes in Promoters throughout the Human Genome. Nucleic Acids Res. 2007, 35, 406–413. [Google Scholar] [CrossRef]
- Dembska, A. The Analytical and Biomedical Potential of Cytosine-Rich Oligonucleotides: A Review. Anal. Chim. Acta 2016, 930, 12. [Google Scholar] [CrossRef]
- Zeraati, M.; Langley, D.B.; Schofield, P.; Moye, A.L.; Rouet, R.; Hughes, W.E.; Bryan, T.M.; Dinger, M.E.; Christ, D. I-Motif DNA Structures Are Formed in the Nuclei of Human Cells. Nat. Chem. 2018, 10, 631–637. [Google Scholar] [CrossRef]
- Phan, A.T.; Mergny, J.-L. Human Telomeric DNA: G-Quadruplex, i-Motif and Watson–Crick Double Helix. Nucleic Acids Res. 2002, 30, 4618–4625. [Google Scholar] [CrossRef]
- Russo Krauss, I.; Ramaswamy, S.; Neidle, S.; Haider, S.; Parkinson, G.N. Structural Insights into the Quadruplex–Duplex 3′ Interface Formed from a Telomeric Repeat: A Potential Molecular Target. J. Am. Chem. Soc. 2016, 138, 1226–1233. [Google Scholar] [CrossRef] [Green Version]
- Balasubramanian, S.; Neidle, S. G-Quadruplex Nucleic Acids as Therapeutic Targets. Curr. Opin. Chem. Biol. 2009, 13, 345–353. [Google Scholar] [CrossRef] [Green Version]
- Burge, S.; Parkinson, G.N.; Hazel, P.; Todd, A.K.; Neidle, S. Quadruplex DNA: Sequence, Topology and Structure. Nucleic Acids Res. 2006, 34, 5402–5415. [Google Scholar] [CrossRef] [Green Version]
- Gehring, K.; Leroy, J.-L.; Guéron, M. A Tetrameric DNA Structure with Protonated Cytosine-Cytosine Base Pairs. Nature 1993, 363, 561–565. [Google Scholar] [CrossRef]
- Day, H.A.; Pavlou, P.; Waller, Z.A.E. I-Motif DNA: Structure, Stability and Targeting with Ligands. Bioorganic Med. Chem. 2014, 22, 4407–4418. [Google Scholar] [CrossRef]
- Chen, Y.; Qu, K.; Zhao, C.; Wu, L.; Ren, J.; Wang, J.; Qu, X. Insights into the Biomedical Effects of Carboxylated Single-Wall Carbon Nanotubes on Telomerase and Telomeres. Nat. Commun. 2012, 3, 1074. [Google Scholar] [CrossRef]
- De Cian, A.; Lacroix, L.; Douarre, C.; Temime-Smaali, N.; Trentesaux, C.; Riou, J.-F.; Mergny, J.-L. Targeting Telomeres and Telomerase. Biochimie 2008, 90, 131–155. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Peng, Y.; Ren, J.; Qu, X. Carboxyl-Modified Single-Walled Carbon Nanotubes Selectively Induce Human Telomeric i-Motif Formation. Proc. Natl. Acad. Sci. USA 2006, 103, 19658–19663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolski, P.; Nieszporek, K.; Panczyk, T. Cytosine-Rich DNA Fragments Covalently Bound to Carbon Nanotube as Factors Triggering Doxorubicin Release at Acidic PH. A Molecular Dynamics Study. Int. J. Mol. Sci. 2021, 22, 8466. [Google Scholar] [CrossRef]
- Wolski, P.; Nieszporek, K.; Panczyk, T. Carbon Nanotubes and Short Cytosine-Rich Telomeric DNA Oligomeres as Platforms for Controlled Release of Doxorubicin—A Molecular Dynamics Study. Int. J. Mol. Sci. 2020, 21, 3619. [Google Scholar] [CrossRef] [PubMed]
- Smiatek, J.; Chen, C.; Liu, D.; Heuer, A. Stable Conformations of a Single Stranded Deprotonated DNA I-Motif. J. Phys. Chem. B 2011, 115, 13788–13795. [Google Scholar] [CrossRef]
- Panczyk, T.; Wojton, P.; Wolski, P. Mechanism of Unfolding and Relative Stabilities of G-Quadruplex and I-Motif Noncanonical DNA Structures Analyzed in Biased Molecular Dynamics Simulations. Biophys. Chem. 2019, 250, 106173. [Google Scholar] [CrossRef]
- Bian, Y.; Ren, W.; Song, F.; Yu, J.; Wang, J. Exploration of the Folding Dynamics of Human Telomeric G-Quadruplex with a Hybrid Atomistic Structure-Based Model. J. Chem. Phys. 2018, 148, 204107. [Google Scholar] [CrossRef]
- Cheatham, T.E. Simulation and Modeling of Nucleic Acid Structure, Dynamics and Interactions. Curr. Opin. Struct. Biol. 2004, 14, 360–367. [Google Scholar] [CrossRef]
- Pérez, A.; Marchán, I.; Svozil, D.; Sponer, J.; Cheatham, T.E.; Laughton, C.A.; Orozco, M. Refinement of the AMBER Force Field for Nucleic Acids: Improving the Description of α/γ Conformers. Biophys. J. 2007, 92, 3817–3829. [Google Scholar] [CrossRef] [Green Version]
- Krepl, M.; Zgarbová, M.; Stadlbauer, P.; Otyepka, M.; Banáš, P.; Koča, J.; Cheatham, T.E.; Jurečka, P.; Šponer, J. Reference Simulations of Noncanonical Nucleic Acids with Different χ Variants of the AMBER Force Field: Quadruplex DNA, Quadruplex RNA, and Z-DNA. J. Chem. Theory Comput. 2012, 8, 2506–2520. [Google Scholar] [CrossRef] [Green Version]
- Galindo-Murillo, R.; Robertson, J.C.; Zgarbová, M.; Šponer, J.; Otyepka, M.; Jurečka, P.; Cheatham, T.E. Assessing the Current State of Amber Force Field Modifications for DNA. J. Chem. Theory Comput. 2016, 12, 4114–4127. [Google Scholar] [CrossRef]
- Ivani, I.; Dans, P.D.; Noy, A.; Pérez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goñi, R.; Balaceanu, A.; et al. Parmbsc1: A Refined Force Field for DNA Simulations. Nat. Methods 2015, 3, 55–58. [Google Scholar] [CrossRef] [Green Version]
- Martínez, L. Introducing the Levinthal’s Protein Folding Paradox and Its Solution. J. Chem. Educ. 2014, 91, 1918–1923. [Google Scholar] [CrossRef]
- Ellis, R.J. Molecular Chaperones: Assisting Assembly in Addition to Folding. Trends Biochem. Sci. 2006, 31, 395–401. [Google Scholar] [CrossRef]
- Chen, C.; Li, M.; Xing, Y.; Li, Y.; Joedecke, C.-C.; Jin, J.; Yang, Z.; Liu, D. Study of PH-Induced Folding and Unfolding Kinetics of the DNA i-Motif by Stopped-Flow Circular Dichroism. Langmuir 2012, 28, 17743–17748. [Google Scholar] [CrossRef]
- Ouldridge, T.E.; Louis, A.A.; Doye, J.P.K. Structural, Mechanical, and Thermodynamic Properties of a Coarse-Grained DNA Model. J. Chem. Phys. 2011, 134, 085101. [Google Scholar] [CrossRef] [Green Version]
- Snodin, B.E.K.; Randisi, F.; Mosayebi, M.; Šulc, P.; Schreck, J.S.; Romano, F.; Ouldridge, T.E.; Tsukanov, R.; Nir, E.; Louis, A.A.; et al. Introducing Improved Structural Properties and Salt Dependence into a Coarse-Grained Model of DNA. J. Chem. Phys. 2015, 142, 234901. [Google Scholar] [CrossRef] [Green Version]
- Knotts, T.A.; Rathore, N.; Schwartz, D.C.; de Pablo, J.J. A Coarse Grain Model for DNA. J. Chem. Phys. 2007, 126, 084901. [Google Scholar] [CrossRef]
- Freeman, G.S.; Hinckley, D.M.; de Pablo, J.J. A Coarse-Grain Three-Site-per-Nucleotide Model for DNA with Explicit Ions. J. Chem. Phys. 2011, 135, 165104. [Google Scholar] [CrossRef] [Green Version]
- Uusitalo, J.J.; Ingólfsson, H.I.; Akhshi, P.; Tieleman, D.P.; Marrink, S.J. Martini Coarse-Grained Force Field: Extension to DNA. J. Chem. Theory Comput. 2015, 11, 3932–3945. [Google Scholar] [CrossRef]
- Panczyk, T.; Wolski, P. Molecular Dynamics Analysis of Stabilities of the Telomeric Watson-Crick Duplex and the Associated i-Motif as a Function of PH and Temperature. Biophys. Chem. 2018, 237, 22–30. [Google Scholar] [CrossRef]
- Wolski, P.; Wojton, P.; Nieszporek, K.; Panczyk, T. Interaction of Human Telomeric I-Motif DNA with Single-Walled Carbon Nanotubes: Insights from Molecular Dynamics Simulations. J. Phys. Chem. B 2019, 123, 10343–10353. [Google Scholar] [CrossRef] [PubMed]
- Šponer, J.; Bussi, G.; Stadlbauer, P.; Kührová, P.; Banáš, P.; Islam, B.; Haider, S.; Neidle, S.; Otyepka, M. Folding of Guanine Quadruplex Molecules–Funnel-like Mechanism or Kinetic Partitioning? An Overview from MD Simulation Studies. Biochim. Et Biophys. Acta (BBA)—Gen. Subj. 2017, 1861, 1246–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.; Kim, S.; Tachikawa, T.; Fujitsuka, M.; Majima, T. PH-Induced Intramolecular Folding Dynamics of i-Motif DNA. J. Am. Chem. Soc. 2011, 133, 16146–16153. [Google Scholar] [CrossRef] [PubMed]
- Panczyk, T.; Wojton, P.; Wolski, P. Molecular Dynamics Study of the Interaction of Carbon Nanotubes with Telomeric DNA Fragment Containing Noncanonical G-Quadruplex and i-Motif Forms. Int. J. Mol. Sci. 2020, 21, 1925. [Google Scholar] [CrossRef] [Green Version]
- Schreck, J.S.; Ouldridge, T.E.; Romano, F.; Louis, A.A.; Doye, J.P.K. Characterizing the Bending and Flexibility Induced by Bulges in DNA Duplexes. J. Chem. Phys. 2015, 142, 165101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietz, H.; Douglas, S.M.; Shih, W.M. Folding DNA into Twisted and Curved Nanoscale Shapes. Science 2009, 325, 725–730. [Google Scholar] [CrossRef] [Green Version]
- Suma, A.; Poppleton, E.; Matthies, M.; Šulc, P.; Romano, F.; Louis, A.A.; Doye, J.P.K.; Micheletti, C.; Rovigatti, L. TacoxDNA: A User-friendly Web Server for Simulations of Complex DNA Structures, from Single Strands to Origami. J. Comput. Chem. 2019, 40, 2586–2595. [Google Scholar] [CrossRef]
- Poppleton, E.; Bohlin, J.; Matthies, M.; Sharma, S.; Zhang, F.; Šulc, P. Design, Optimization and Analysis of Large DNA and RNA Nanostructures through Interactive Visualization, Editing and Molecular Simulation. Nucleic Acids Res. 2020, 48, e72. [Google Scholar] [CrossRef]
- Sambriski, E.J.; Schwartz, D.C.; de Pablo, J.J. A Mesoscale Model of DNA and Its Renaturation. Biophys. J. 2009, 96, 1675–1690. [Google Scholar] [CrossRef] [Green Version]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 19. [Google Scholar] [CrossRef] [Green Version]
- Liwo, A.; Czaplewski, C.; Sieradzan, A.K.; Lipska, A.G.; Samsonov, S.A.; Murarka, R.K. Theory and Practice of Coarse-Grained Molecular Dynamics of Biologically Important Systems. Biomolecules 2021, 11, 1347. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The MARTINI Force Field: Coarse Grained Model for Biomolecular Simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolski, P.; Nieszporek, K.; Panczyk, T. G-Quadruplex and I-Motif Structures within the Telomeric DNA Duplex. A Molecular Dynamics Analysis of Protonation States as Factors Affecting Their Stability. J. Phys. Chem. B 2019, 123, 468–479. [Google Scholar] [CrossRef]
- Panczyk, T.; Camp, P.J. Lorentz Forces Induced by a Static Magnetic Field Have Negligible Effects on Results from Classical Molecular Dynamics Simulations of Aqueous Solutions. J. Mol. Liq. 2021, 330, 115701. [Google Scholar] [CrossRef]
- Dai, J.; Chen, D.; Jones, R.A.; Hurley, L.H.; Yang, D. NMR Solution Structure of the Major G-Quadruplex Structure Formed in the Human BCL2 Promoter Region. Nucleic Acids Res. 2006, 34, 5133–5144. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A Message-Passing Parallel Molecular Dynamics Implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Periole, X.; Cavalli, M.; Marrink, S.-J.; Ceruso, M.A. Combining an Elastic Network with a Coarse-Grained Molecular Force Field: Structure, Dynamics, and Intermolecular Recognition. J. Chem. Theory Comput. 2009, 5, 2531–2543. [Google Scholar] [CrossRef] [Green Version]
- Phan, A.T.; Guéron, M.; Leroy, J.-L. The Solution Structure and Internal Motions of a Fragment of the Cytidine-Rich Strand of the Human Telomere 1 1Edited by I. Tinoco. J. Mol. Biol. 2000, 299, 123–144. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber Biomolecular Simulation Programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser InterfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Dupradeau, F.-Y.; Pigache, A.; Zaffran, T.; Savineau, C.; Lelong, R.; Grivel, N.; Lelong, D.; Rosanski, W.; Cieplak, P. The R.E.D. Tools: Advances in RESP and ESP Charge Derivation and Force Field Library Building. Phys. Chem. Chem. Phys. 2010, 12, 7821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Fu, A.; Xue, X.; Guo, F.; Huai, W.; Chu, T.; Wang, Z. Density Functional Theory Prediction of p K a for Carboxylated Single-Wall Carbon Nanotubes and Graphene. Chem. Phys. 2017, 490, 47–54. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panczyk, T.; Nieszporek, K.; Wolski, P. Stability and Existence of Noncanonical I-motif DNA Structures in Computer Simulations Based on Atomistic and Coarse-Grained Force Fields. Molecules 2022, 27, 4915. https://doi.org/10.3390/molecules27154915
Panczyk T, Nieszporek K, Wolski P. Stability and Existence of Noncanonical I-motif DNA Structures in Computer Simulations Based on Atomistic and Coarse-Grained Force Fields. Molecules. 2022; 27(15):4915. https://doi.org/10.3390/molecules27154915
Chicago/Turabian StylePanczyk, Tomasz, Krzysztof Nieszporek, and Pawel Wolski. 2022. "Stability and Existence of Noncanonical I-motif DNA Structures in Computer Simulations Based on Atomistic and Coarse-Grained Force Fields" Molecules 27, no. 15: 4915. https://doi.org/10.3390/molecules27154915