
Improving the Enzymatic Cascade of Reactions for the Reduction of CO2 to CH3OH in Water: From Enzymes Immobilization Strategies to Cofactor Regeneration and Cofactor Suppression



Abstract
:1. Introduction
- Make enzymes more robust, i.e., methods of immobilization that also facilitate enzymes recovery and recycling;
- Increase the availability of CO2 in solution either by adding specific enzymes that drive the hydration/dehydration process or by using materials capable of capturing CO2;
- In situ and non-in situ methods that allow selective regeneration of the cofactor or even reduce or eliminate the cofactor itself, using artificial cofactors or adopting a direct electron transfer.
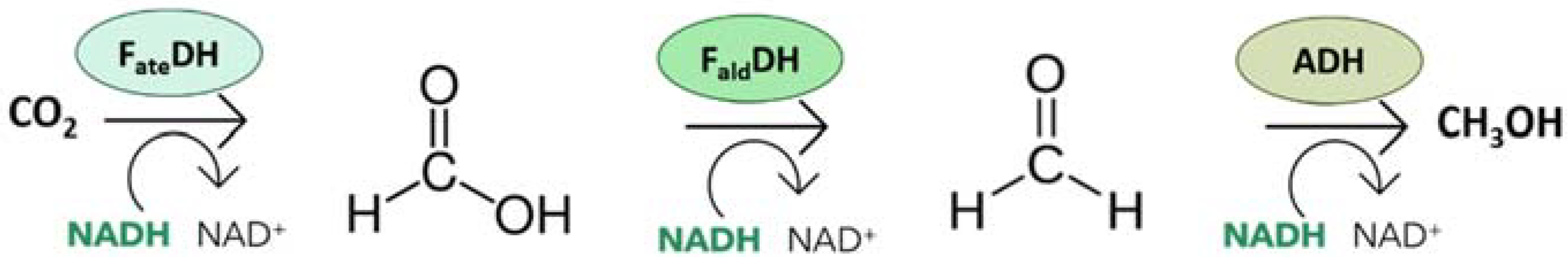
2. The Dehydrogenases
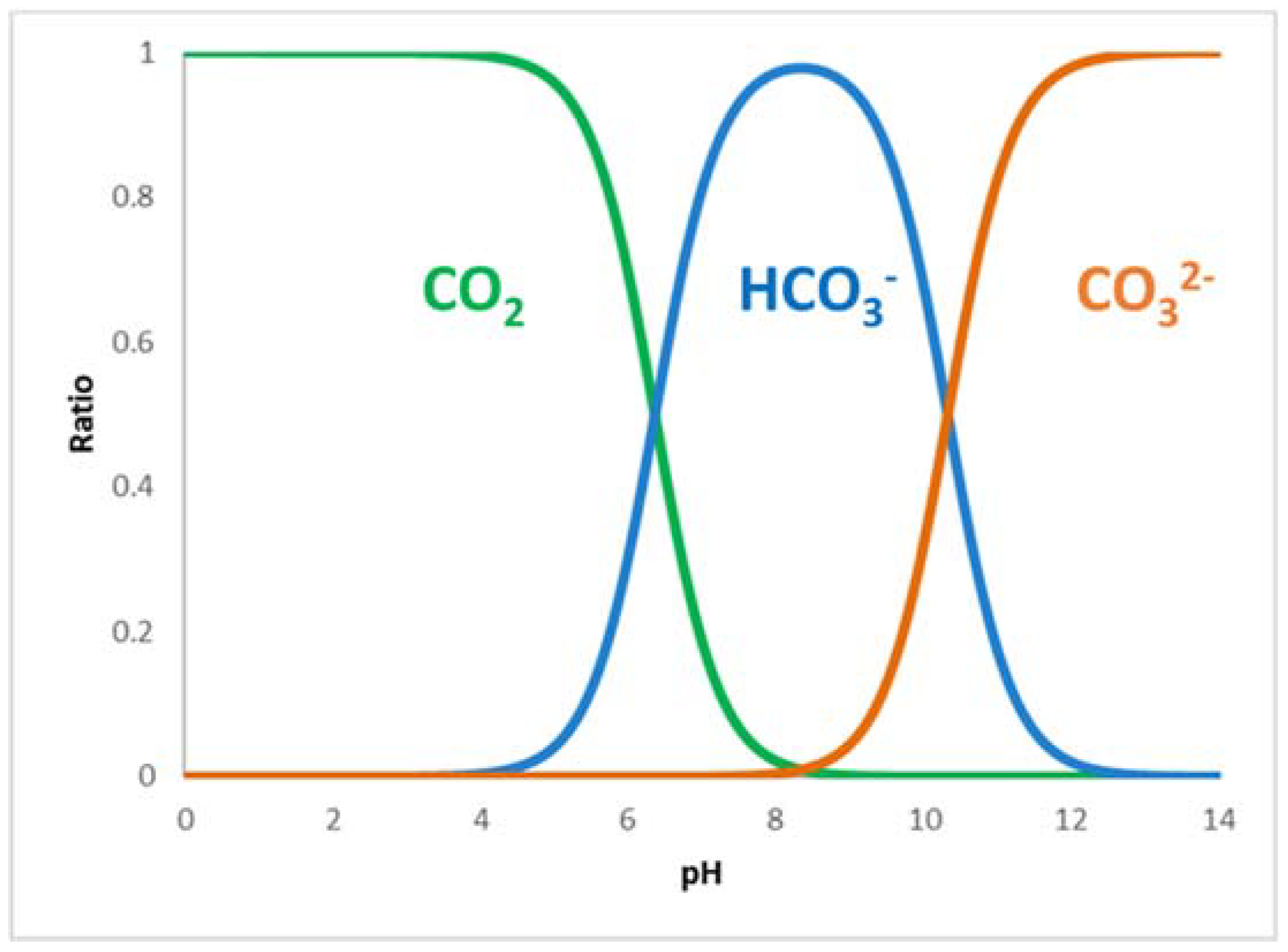
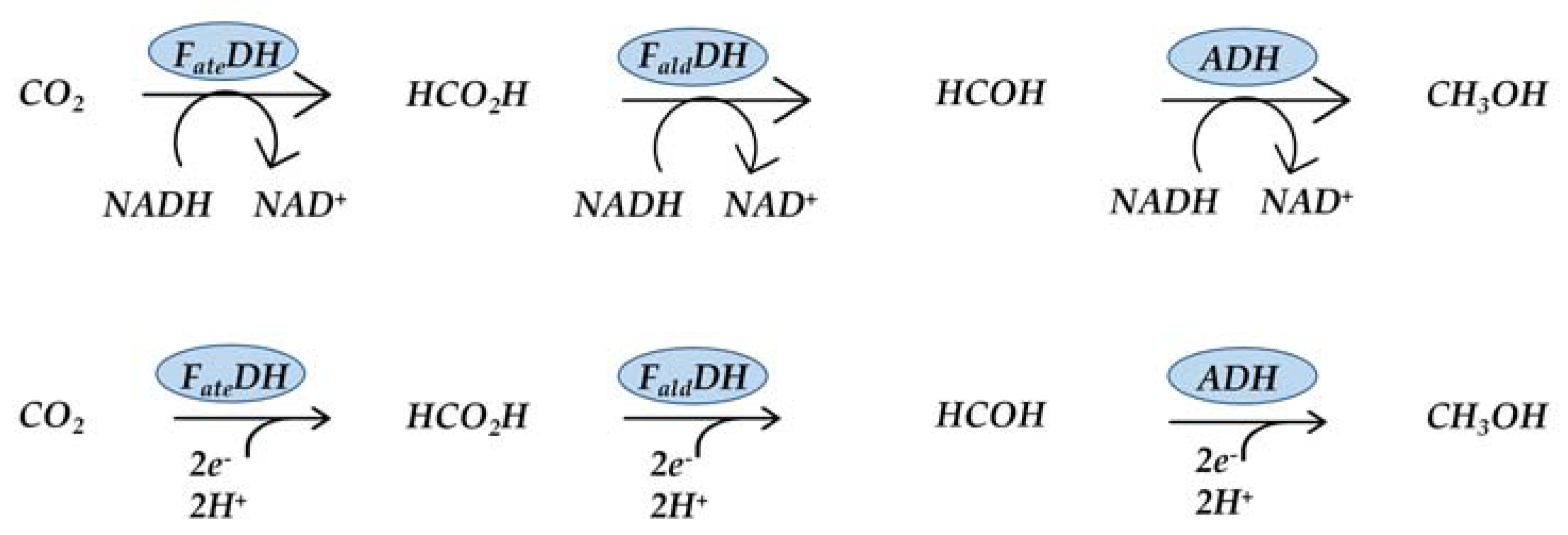
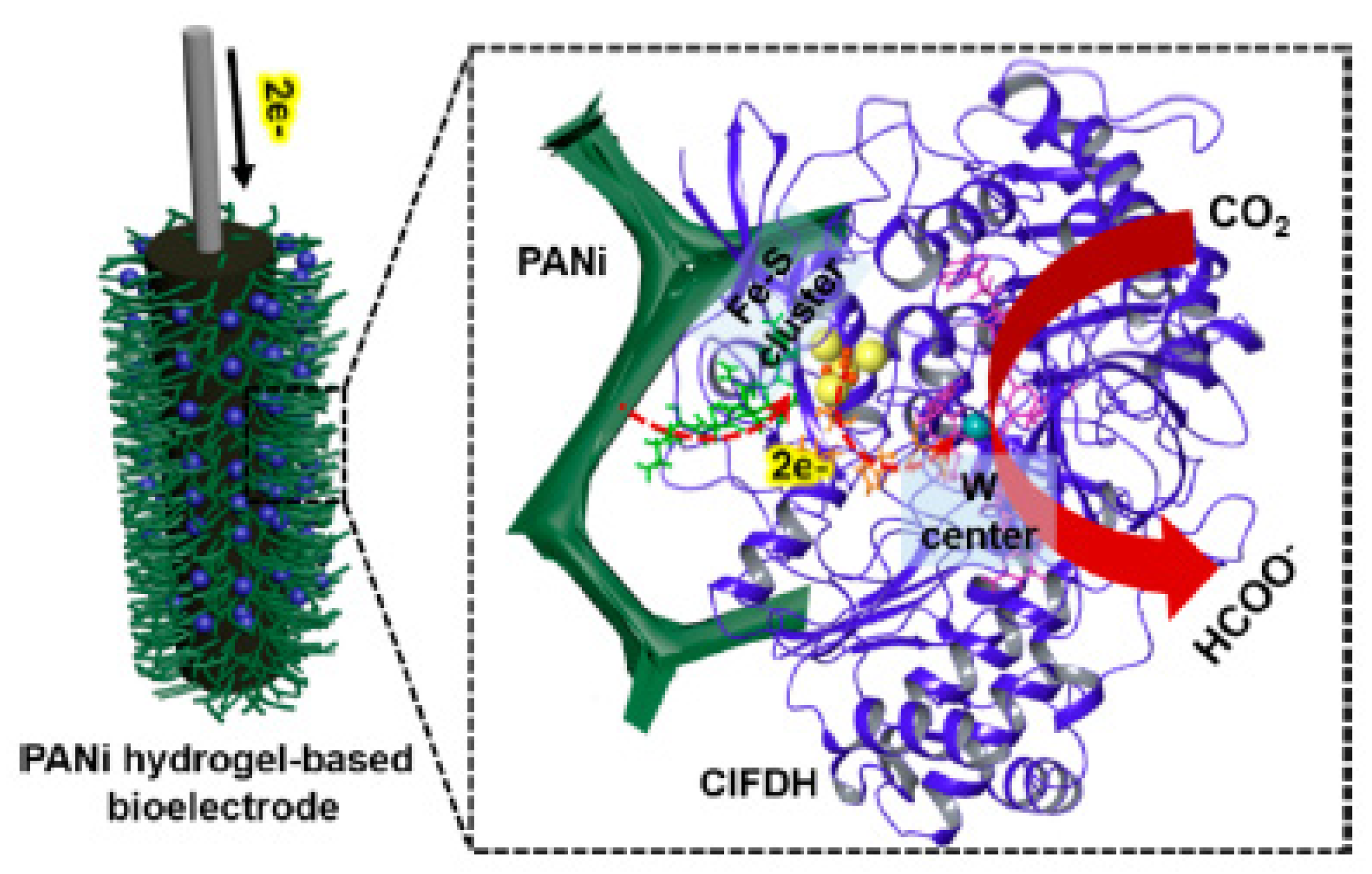
2.1. Formate Dehydrogenase
2.2. Formaldehyde Dehydrogenase
2.3. Alcohol Dehydrogenase
3. Study of Reaction Conditions
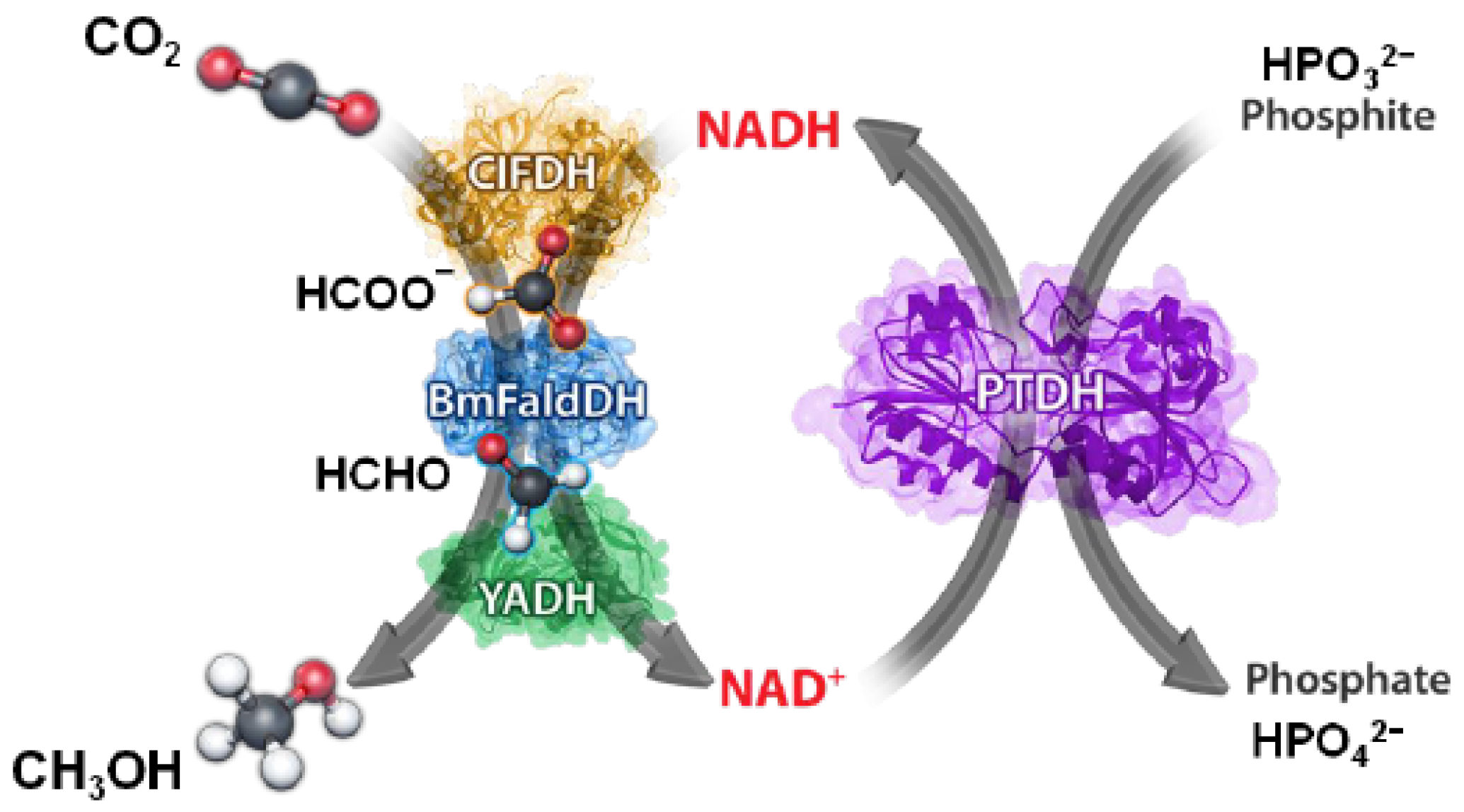
3.1. Enzymes Immobilization and Co-Immobilization

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Immobilization Matrix | Immobilized Enzymes | Note/Key Outcome | Ref. |
|---|---|---|---|
| SiO2 sol–gel | FateDH, FaldDH, ADH | Yield-free enzymes = 10–20% Yield-immobilized enzymes = 40–90% | [45] |
| SiO2 sol–gel | FateDH, FaldDH, ADH | Yield-free enzymes = 98.1% Yield-immobilized enzymes = 92.1% | [46] |
| ALG-SiO2, hybrid gel | FateDH, FaldDH, ADH | Yield-free enzymes = 98.8% Yield-immobilized enzymes = 98.1% | [47] |
| PS NPs | FateDH, FaldDH, ADH | Yield-free enzymes = 12%. Yield-immobilized enzymes = 127% (80% initial activity retained after 11 cycles). Enzymatic regeneration with GDH | [48] |
| Capsules-in-bead scaffold | FateDH, FaldDH, ADH | Immobilized enzymes were more active than free enzymes when a free cofactor was presented. | [49] |
| Titania–protamine particles | FateDH, FaldDH, ADH | Yield-free enzymes = 5–10%. Yield immobilized enzymes = 35–60% (50% initial activity retained after 10 cycles) | [50] |
| ALG-SiO2, hybrid gel | FateDH, FaldDH, ADH | Yield-immobilized enzymes = 100% (external reg.) Yield-immobilized enzymes = 80% (with in situ reg.) Chemical regeneration with SDT | [8] |
| Phospholipids–silica nanocapsules | FateDH, FaldDH, ADH | Free enzymes = 0.06 mmol MeOH/genzyme. Immobilized enzymes = 0.88 mmol MeOH/genzyme Free with PtDH = 0.16 mmol MeOH/genzyme Immobilized enzyme with PtDH = 4.30 mmol MeOH/genzyme. Enzymatic regeneration with PtDH | [18] |
| Hybrid microcapsules | FateDH, FaldDH, ADH | Yield free enzymes = 35.5%. Yield immobilized enzymes = 71.6% (52.6% initial activity retained after 9 cycles) | [21] |
| Flat-sheet polymeric membranes | FateDH, FaldDH, ADH | Free enzymes: [MeOH] = 0.5 mM Co-immobilized enzymes: [MeOH] = 0.6 mM mMSeq-immobilized enzymes: [MeOH] = 0.7 mM Enzymatic regeneration with GDH and glutamate | [29] |
| CF electrode with alginate matrix | FateDH, FaldDH, ADH | Electrochemical CO2 reduction to methanol around 0.15 ppm. Faradaic efficiencies of around 10%. No NADH but direct electron transfer | [51] |
| PS nanofibrous membrane | FateDH | Free enzymes: [Formate] = 0.6 mM Immobilized enzymes: [Formate] = 0.3 mM (53% initial activity retained after 8 cycles) Electrochemical regeneration | [34] |
| Magnetic NPs | FateDH, FaldDH, ADH | Stepwise scheme led to only a 2.3% yield of methanol per NADH; batch system under CO2 pressure, the combination of the four immobilized enzymes increased the methanol yield by 64-fold | [52] |
| ZIF-8 entrapped in PVDF microporous asymmetric membrane | FateDH, FaldDH, ADH | Immobilized enzymes without membrane (EMS) = 5 µmol. Immobilized enzymes with membrane (ECMS) = 6 µmol. Disord. Immobilized enzymes with membrane (DEMM) = 7 µmol. Ord. Immobilized enzymes + NADH without membrane (OEMM) = 13 µmol. Ord. Immobilized enzymes + NADH with membrane (OECMM) = 14 µmol. Over 50 % of their original productivity was retained after 12 h of use | [53] |
| Titania NPs | ADH | The results revealed that immobilization of enzymes led to higher catalytic. The activity of ADH from 30% to more than 80% of its initial activity after 30 days of storage at 4 °C. (84% initial activity retained after 10 cycles) | [54] |
| MOF, NU-1006 | FateDH | Immobilized Enzyme + cofactor Rh: [Formic acid] = 144 mM. Photochemical regeneration with Rh complex | [55] |
| Zeolite particles | FateDH | Yield imm. Enzyme = 34–37% | [56] |
| MOF, ZIF-8 | FateDH | Compared with the free multienzyme system, formate yield was increased by 4.6-fold. Co-immobilized with CA and enzymatic regeneration with GDH | [57] |
| Graphene + CF electrode with alginate matrix | FateDH, FaldDH, ADH | Electrochemical CO2 reduction to methanol around 20 ppm. Faradaic efficiencies of around 12%. No NADH but direct electron transfer | [58] |
| MOF, ZIF-8 | FateDH, FaldDH, ADH | Free enzymes: [MeOH] = 0.061 mM. Immobilized enzymes: [MeOH] = 0.320 mM. Immobilized enzymes + NADH regeneration: [MeOH] = 0.742 mM. Electrochemical regeneration with Rh complex-grafted electrode | [59] |
| MCF | FateDH, FaldDH, ADH | Catalytic activity-free enzyme systems = 0.3 mmol MeOH/genzyme min. Catalytic activity immobilized enzymes systems = 1.35 mmol MeOH/genzyme min | [60] |
| Gold and graphite electrodes | FateDH | Electrochemical CO2 reduction imm. enzyme: [Formate] = 3.7 µM. Faradaic efficiencies of around 100% No NADH but direct electron transfer | [61] |
3.2. Cofactor Regeneration
3.2.1. Enzymatic Regeneration of the Cofactor
3.2.2. Chemical Regeneration of the Cofactor
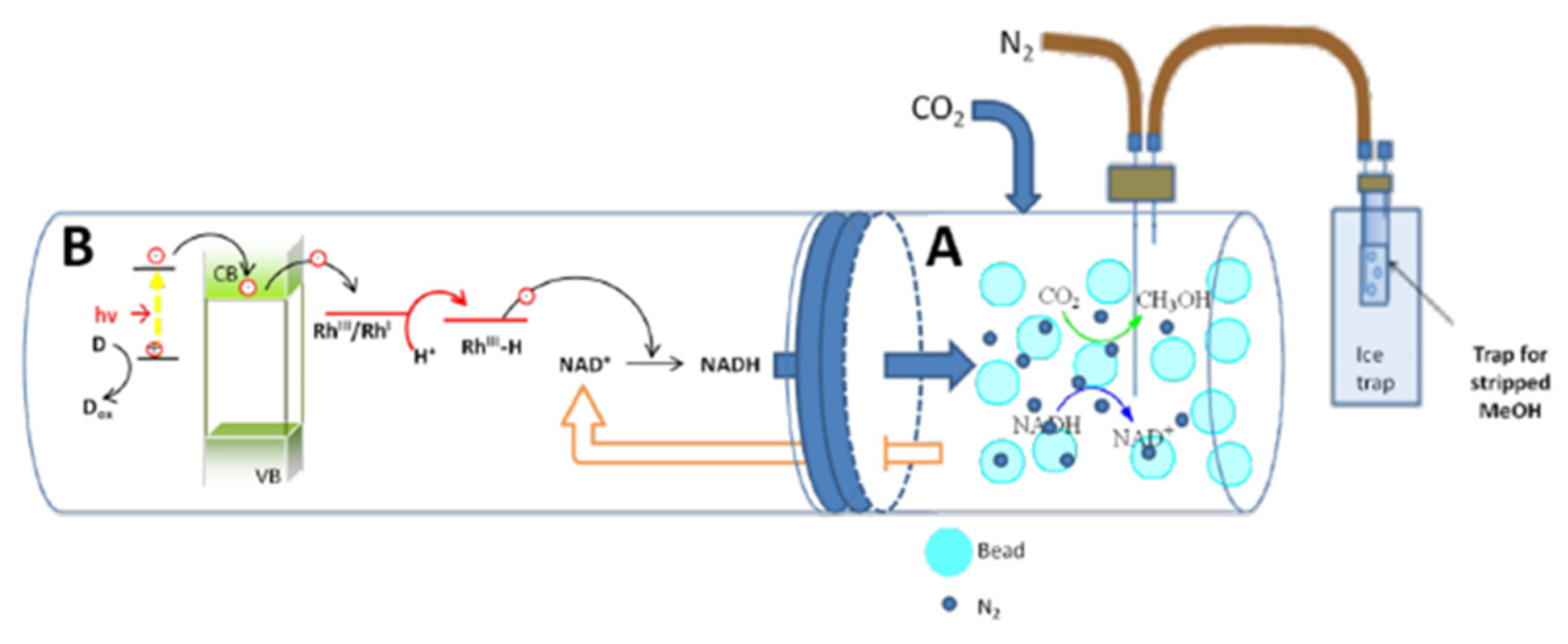
3.2.3. Photochemical Regeneration
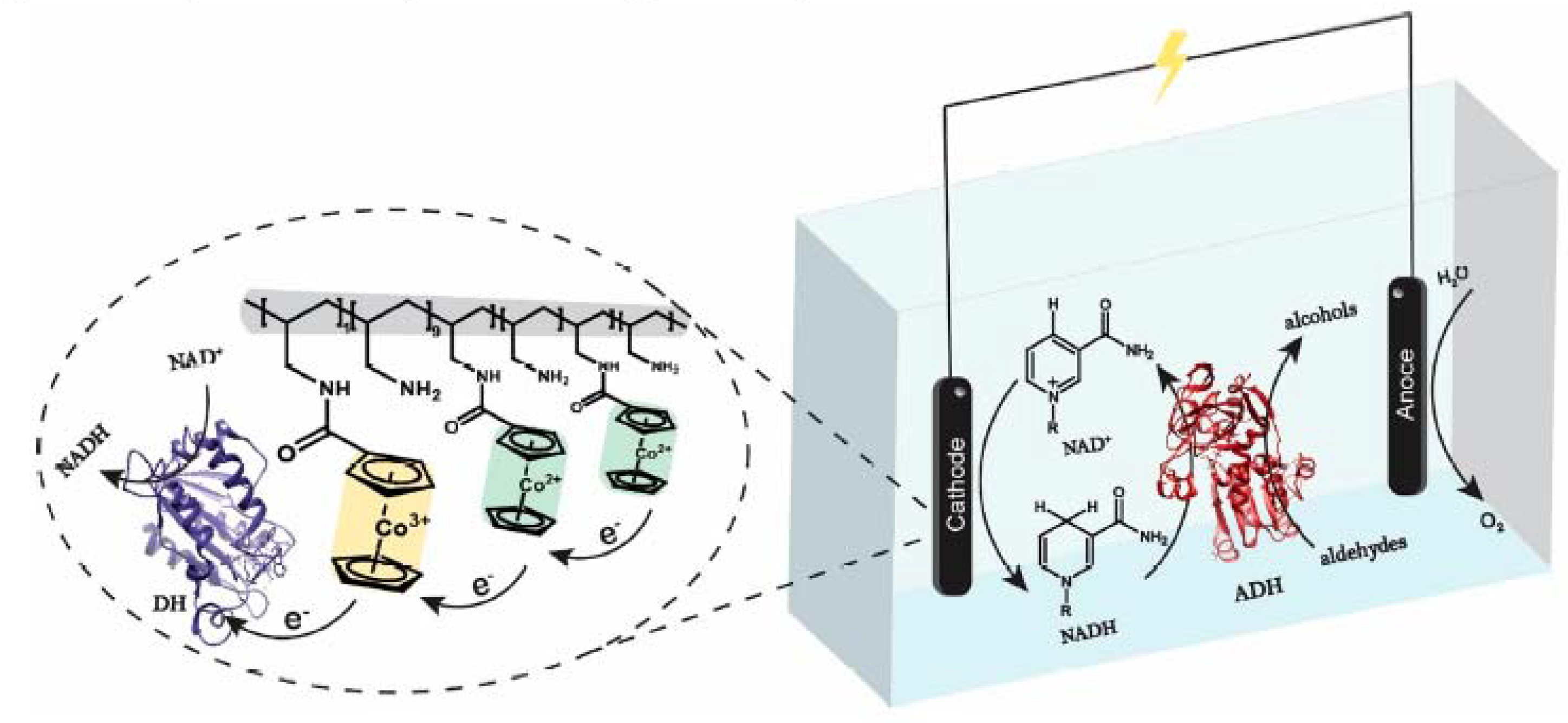
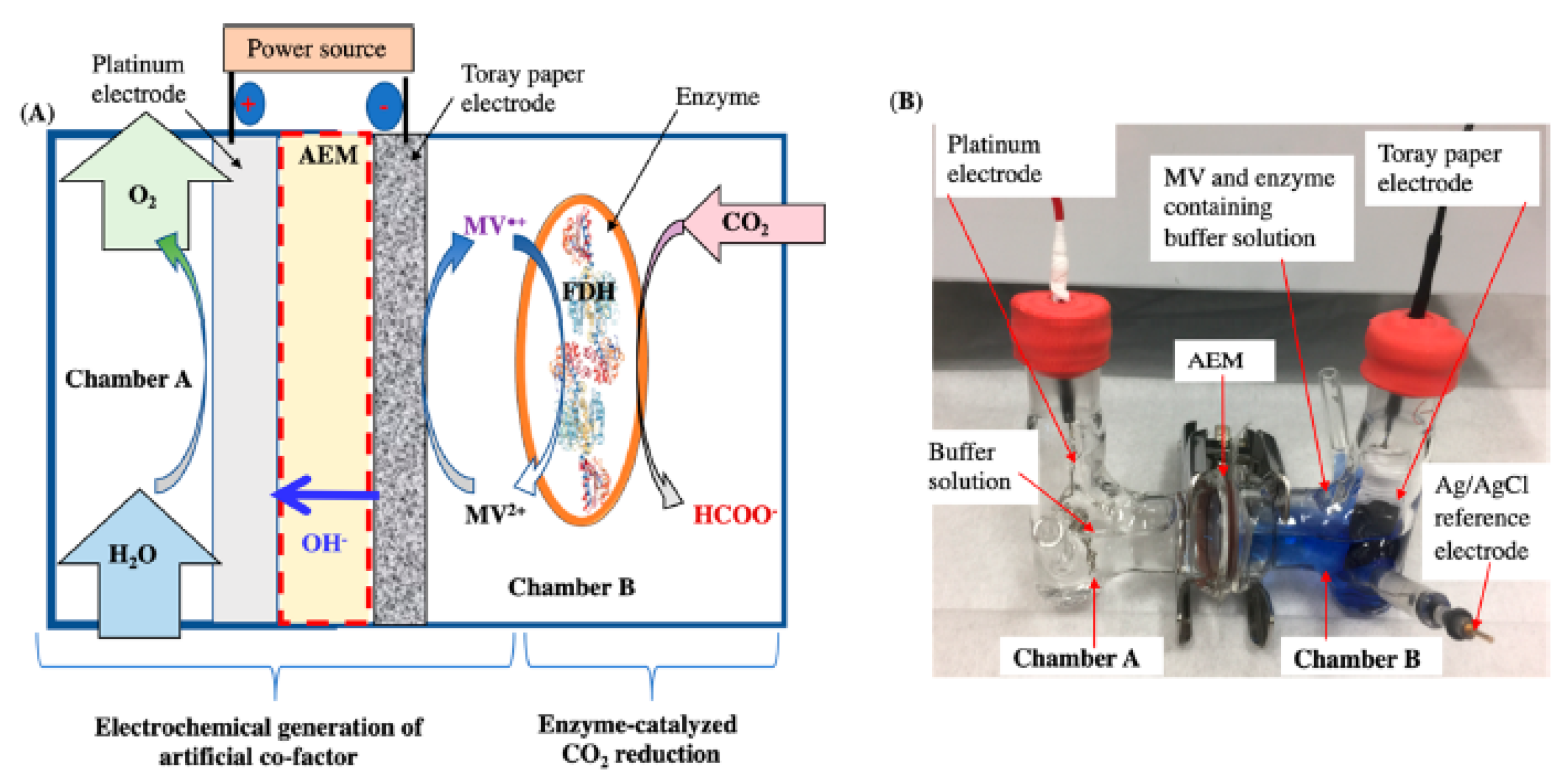
3.2.4. Electrochemical Regeneration of the Cofactor
| Regeneration Method | Type of Regenerator | Yield/Key Outcome | Ref. |
|---|---|---|---|
| Enzymatic regeneration | GDH | YMeOH reached 127% | [48] |
| Enzymatic regeneration | GDH | YMeOH reached up to 95.3% | [72] |
| Enzymatic regeneration | PTDH or GlyDH | PTDH is 4 times more active than GlyDH, [CH3OH] increases from 0.1 mM without PTDH to 0.9 mM with PTDH | [18] |
| Enzymatic regeneration | PTDH | The multienzymatic cascade reaction, along with PTDH, yielded 3.28 mM methanol | [64] |
| Enzymatic regeneration | GCDH | Yield of methanol reached 100% after coupling GCDH regeneration | [68] |
| Enzymatic regeneration | GCDH-XDH | XDH for NADH regeneration was found to be more efficient than GCDH producing at least 8 mM CH3OH yield | [65] |
| Enzymatic regeneration | GDH | Yield of methanol was increased 64-folds compared to the reaction without a regeneration system | [52] |
| Enzymatic regeneration | GDH | Formate yield was increased 4.6-fold compared to the reaction with free enzymes | [57] |
| Photochemical regeneration | Carbon-containing TiO2/H2/[Cp*Rh(bpy)(H2O)]2+ | NADH conversion reaches 94.29% in the presence of H2 as an electron’s donor | [73] |
| Photochemical regeneration | P-doped TiO2 nanoparticles/H2O/[Cp*Rh(bpy)(H2O)]2+ | If P to Ti molar ratio is 6%, TiO2 nanoparticle can photo catalytically reproduce 34.6% NADH under visible light | [74] |
| Photochemical regeneration | Cobaloxime/TEOA /eosin | NADH conversion reaches a yield of 36% | [75] |
| Photochemical regeneration | CCG-IP/TEOA/[Cp*Rh(bpy)(H2O)]2+ | NADH conversion reaches a yield of 38.99% (first cycle) and 36.81% (third cycle) | [76] |
| Photochemical regeneration | CrF5(H2O)]2−@TiO2/Water-Glycerol/[Cp*Rh(bpy)H2O]Cl2 | NADH conversion reaches the maximum yield (very close to 100%) | [67] |
| Photochemical regeneration | TiO2/EDTA/[Cp*Rh(bpy)(H2O)]2+ | In the presence of 1.5 mg/mL TiO2, the NADH yield reached approximately 90% after 30 min of irradiation | [62] |
| Photochemical regeneration | ATCN-DSCN/TEOA/[Cp*Rh(bpy)H2O]2+ | NADH yield of ~74% | [77] |
| Photochemical regeneration | Ionic porphyrin (ZnTPyPBr)/TEOA/[Cp*Rh(bpy)(H2O)]2+ | Yield of NADH increase by 17.9% after 1 h, a seven-fold increase in methanol concentration | [68] |
| Photochemical regeneration | TiO2/H2O/[Cp*Rh(bpy)(H2O)]2+ | Yield of NADH conversion 45.54% (after 2 h) | [78] |
| Electrochemical regeneration | carbon nanofibers cathode | Yield ~ 99% pure 1,4-NADH | [79] |
| Electrochemical regeneration | Cu nanorods on glassy carbon | 1,4-NADH conversion yield reaches 67%/with electron mediator [Cp*Rh(bpy)Cl]Cl complex reaches almost 100% | [69] |
| Electrochemical regeneration | Ni NP-MWCNT cathode | Yield ~ 98% pure 1,4-NADH | [80] |
| Electrochemical regeneration | Cu foam electrode | NADH conversion yield reaches 93–99% 1,4-NADH (active isomer): 75–79% | [34] |
| Electrochemical regeneration | DH/Cc-PAA biocathode | Bioactive 1,4-NADH yield: 97–100% Faradaic efficiencies: 78–99% | [70] |
| Electrochemical regeneration | Rh modified electrode | NADH conversion yield reaches more than 90% in 20 min | [81] |
| Electrochemical regeneration | CuNPS on carbon felt electrode | NADH regeneration yield achieves a maximum of 92.1% | [82] |
| Electrochemical regeneration | Rh complex-grafted electrode | Yield NADH ~ 80% 1,4-NADH reaches almost 100% | [59] |
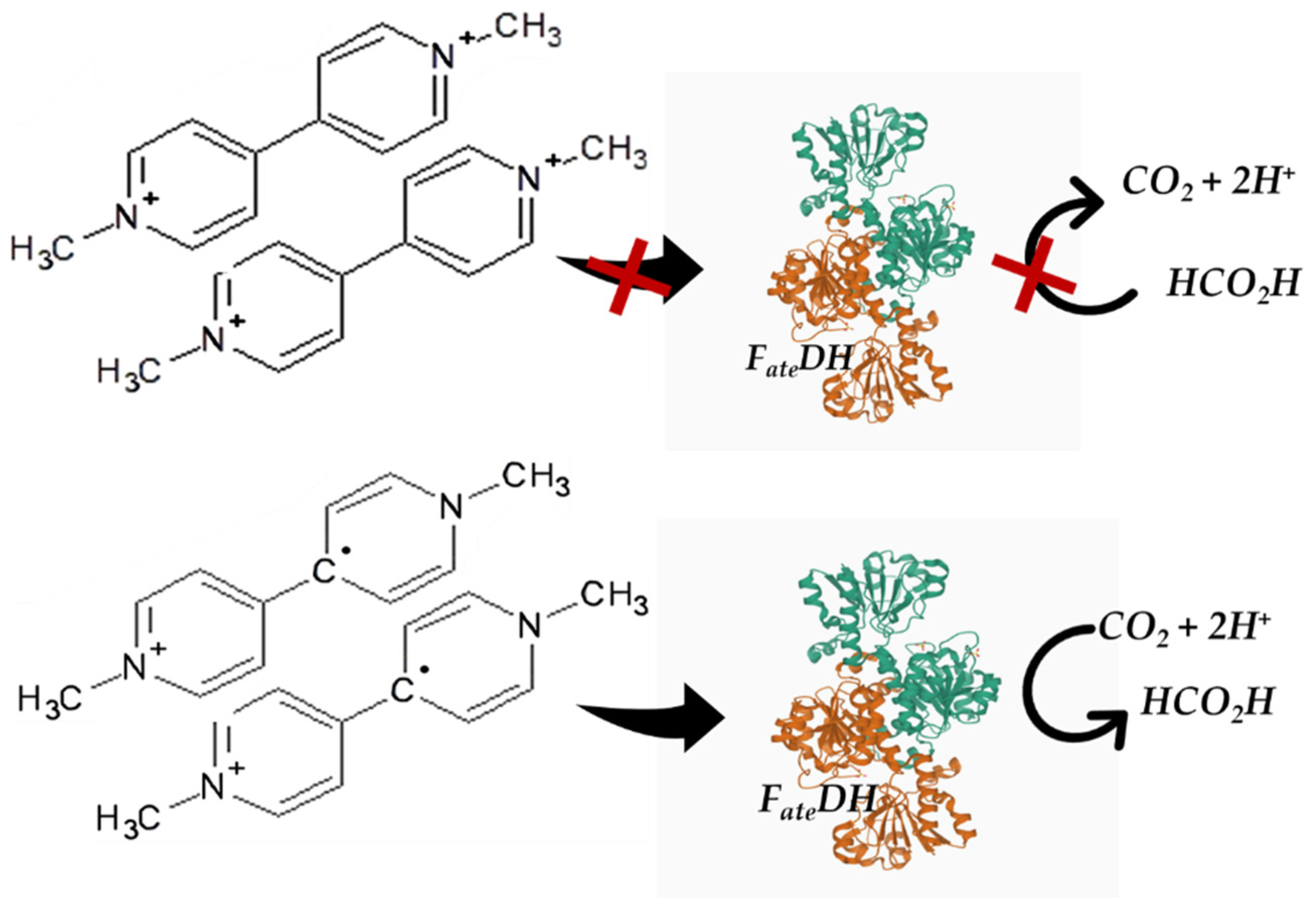
3.3. Cofactor Substitution
3.4. Cofactor Free Use of the Cascade f Reactions
3.5. Coupling of Immobilization and Regeneration Methods: The Results
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Portner, H.O.; Roberts, D.C.; Adams, H.; Adler, C.; Aldunce, P.; Ali, E.; Begum, R.A.; Betts, R.; Kerr, R.B.; Biesbroek, R.; et al. Climate Change 2022: Impacts, Adaptation, and Vulnerability; Contribution of Working Group II to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change; IPCC: Geneva, Switzerland, 2022. [Google Scholar]
- UNFCCC Sites and Platforms. Available online: https://unfccc.int/documents/460952 (accessed on 8 March 2022).
- Aresta, M.; Karimi, I.; Kawi, S. (Eds.) An Economy Based on Carbon Dioxide and Water, 1st ed.; Springer International Publishing: Berlin, Germany, 2019; ISBN 978-3-030-15867-5. [Google Scholar]
- Aresta, M.; Dibenedetto, A. The Carbon Dioxide Revolution; Springer International Publishing: Berlin, Germany, 2021. [Google Scholar] [CrossRef]
- Gao, W.; Liang, S.; Wang, R.; Jiang, Q.; Zhang, Y.; Zheng, Q.; Xie, B.; Toe, C.Y.; Zhu, X.; Wang, J.; et al. Industrial Carbon Dioxide Capture and Utilization: State of the Art and Future Challenges. Chem. Soc. Rev. 2020, 49, 8584–8686. [Google Scholar] [CrossRef] [PubMed]
- Aresta, M.; Dibenedetto, A. Utilisation of CO2 as a Chemical Feedstock: Opportunities and Challenges. Dalton Trans. 2007, 28, 2975–2992. [Google Scholar] [CrossRef] [PubMed]
- Aresta, M.; Dibenedetto, A.; Angelini, A. Catalysis for the Valorization of Exhaust Carbon: From CO2 to Chemicals, Materials, and Fuels. Technological Use of CO2. Chem. Rev. 2014, 114, 1709–1742. [Google Scholar] [CrossRef] [PubMed]
- Dibenedetto, A.; Stufano, P.; Macyk, W.; Baran, T.; Fragale, C.; Costa, M.; Aresta, M.; Dibenedetto, A.; Macyk, W. Hybrid Technologies for an Enhanced Carbon Recycling Based on the Enzymatic Reduction of CO2 to Methanol in Water: Chemical and Photochemical NADH Regeneration. ChemSusChem 2012, 5, 373–378. [Google Scholar] [CrossRef]
- Schlager, S.; Dibenedetto, A.; Aresta, M.; Apaydin, D.H.; Dumitru, L.M.; Neugebauer, H.; Sariciftci, N.S. Biocatalytic and Bioelectrocatalytic Approaches for the Reduction of Carbon Dioxide Using Enzymes. Energy Technol. 2017, 5, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Aresta, M.; Dibenedetto, A.; Quaranta, E. State of the Art and Perspectives in Catalytic Processes for CO2 Conversion into Chemicals and Fuels: The Distinctive Contribution of Chemical Catalysis and Biotechnology. J. Catal. 2016, 343, 2–45. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A.; Pastore, C. Biotechnology to Develop Innovative Syntheses Using CO2. Environ. Chem. Lett. 2005, 3, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Marcolongo, D.M.S.; Aresta, M.; Dibenedetto, A. Stepping toward the Carbon Circular Economy (CCE): Integration of Solar Chemistry and Biosystems for an Effective CO2 Conversion into Added Value Chemicals and Fuels. In Advances in Inorganic Chemistry; Academic Press: Cambridge, MA, USA, 2021; Volume 78, pp. 289–351. ISBN 9780323851152. [Google Scholar]
- Marpani, F.; Pinelo, M.; Meyer, A.S. Enzymatic Conversion of CO2 to CH3OH via Reverse Dehydrogenase Cascade Biocatalysis: Quantitative Comparison of Efficiencies of Immobilized Enzyme Systems. Biochem. Eng. J. 2017, 127, 217–228. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Jiang, Y.; Jiang, Z.; Wang, X.; Wang, X.; Zhang, S.; Han, P.; Yang, C. Enzymatic Conversion of Carbon Dioxide. Chem. Soc. Rev. 2015, 44, 5981–6000. [Google Scholar] [CrossRef]
- Sultana, S.; Chandra Sahoo, P.; Martha, S.; Parida, K. A Review of Harvesting Clean Fuels from Enzymatic CO2 Reduction. RSC Adv. 2016, 6, 44170–44194. [Google Scholar] [CrossRef]
- Calzadiaz-Ramirez, L.; Meyer, A.S. Formate Dehydrogenases for CO2 Utilization. Curr. Opin. Biotechnol. 2022, 73, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Gakhar, L.; Wickersham, K.; Francis, K.; Vardi-Kilshtain, A.; Major, D.T.; Cheatum, C.M.; Kohen, A. Structural and Kinetic Studies of Formate Dehydrogenase from Candida Boidinii. Biochemistry 2016, 55, 2760–2771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cazelles, R.; Drone, J.; Fajula, F.; Ersen, O.; Moldovan, S.; Galarneau, A. Reduction of CO2 to Methanol by a Polyenzymatic System Encapsulated in Phospholipids–Silica Nanocapsules. New J. Chem. 2013, 37, 3721–3730. [Google Scholar] [CrossRef]
- Garrels, R.; Charles, L.C. Solutions, Minerals, and Equilibria, 1st ed.; Harper & Row: New York, NY, USA, 1965; ISBN 0877353336. [Google Scholar]
- Sato, R.; Amao, Y. Can Formate Dehydrogenase from: Candida Boidinii Catalytically Reduce Carbon Dioxide, Bicarbonate, or Carbonate to Formate? New J. Chem. 2020, 44, 11922–11926. [Google Scholar] [CrossRef]
- Wang, X.; Li, Z.; Shi, J.; Wu, H.; Jiang, Z.; Zhang, W.; Song, X.; Ai, Q. Bioinspired Approach to Multienzyme Cascade System Construction for Efficient Carbon Dioxide Reduction. ACS Catal. 2014, 4, 962–972. [Google Scholar] [CrossRef]
- Nielsen, C.F.; Lange, L.; Meyer, A.S. Classification and Enzyme Kinetics of Formate Dehydrogenases for Biomanufacturing via CO2 Utilization. Biotechnol. Adv. 2019, 37, 107408. [Google Scholar] [CrossRef]
- Maia, L.B.; Fonseca, L.; Moura, I.; Moura, J.J.G. Reduction of Carbon Dioxide by a Molybdenum-Containing Formate Dehydrogenase: A Kinetic and Mechanistic Study. J. Am. Chem. Soc. 2016, 138, 8834–8846. [Google Scholar] [CrossRef]
- Choe, H.; Joo, J.C.; Cho, D.H.; Kim, M.H.; Lee, S.H.; Jung, K.D.; Kim, Y.H. Efficient CO2-Reducing Activity of NAD-Dependent Formate Dehydrogenase from Thiobacillus sp. KNK65MA for Formate Production from CO2 Gas. PLoS ONE 2014, 9, e103111. [Google Scholar] [CrossRef]
- Altaş, N.; Aslan, A.S.; Karataş, E.; Chronopoulou, E.; Labrou, N.E.; Binay, B. Heterologous Production of Extreme Alkaline Thermostable NAD+-Dependent Formate Dehydrogenase with Wide-Range PH Activity from Myceliophthora Thermophila. Process Biochem. 2017, 61, 110–118. [Google Scholar] [CrossRef]
- Alissandratos, A.; Kim, H.K.; Matthews, H.; Hennessy, J.E.; Philbrook, A.; Easton, C.J. Clostridium Carboxidivorans Strain P7T Recombinant Formate Dehydrogenase Catalyzes Reduction of CO2 to Formate. Appl. Environ. Microbiol. 2013, 79, 741–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabavi Zadeh, P.S.; Zezzi Do Valle Gomes, M.; Åkerman, B.; Palmqvist, A.E.C. Förster Resonance Energy Transfer Study of the Improved Biocatalytic Conversion of CO2 to Formaldehyde by Coimmobilization of Enzymes in Siliceous Mesostructured Cellular Foams. ACS Catal. 2018, 8, 7251–7260. [Google Scholar] [CrossRef]
- Zezzi do Valle Gomes, M.; Palmqvist, A.E.C. Immobilization of Formaldehyde Dehydrogenase in Tailored Siliceous Mesostructured Cellular Foams and Evaluation of Its Activity for Conversion of Formate to Formaldehyde. Colloids Surf. B Biointerfaces 2018, 163, 41–46. [Google Scholar] [CrossRef]
- Luo, J.; Meyer, A.S.; Mateiu, R.V.; Pinelo, M. Cascade Catalysis in Membranes with Enzyme Immobilization for Multi-Enzymatic Conversion of CO2 to Methanol. New Biotechnol. 2015, 32, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Zezzi Do Valle Gomes, M.; Palmqvist, A.E.C. Influence of Operating Conditions and Immobilization on Activity of Alcohol Dehydrogenase for the Conversion of Formaldehyde to Methanol. New J. Chem. 2017, 41, 11391–11397. [Google Scholar] [CrossRef]
- Zeuner, B.; Ma, N.; Berendt, K.; Meyer, A.S.; Andric, P.; Jørgensen, J.H.; Pinelo, M. Immobilization of Alcohol Dehydrogenase on Ceramic Silicon Carbide Membranes for Enzymatic CH3OH Production. J. Chem. Technol. Biotechnol. 2018, 93, 2952–2961. [Google Scholar] [CrossRef]
- Rover, L.; Fernandes, J.C.B.; Neto, G.D.O.; Kubota, L.T.; Katekawa, E.; Serrano, S.H.P. Study of NADH Stability Using Ultraviolet-Visible Spectrophotometric Analysis and Factorial Design. Anal. Biochem. 1998, 260, 50–55. [Google Scholar] [CrossRef]
- Schmakel, C.O.; Santhanam, K.S.V.; Elving, P.J. Nicotinamide Adenine Dinucleotide (NAD+) and Related Compounds. Electrochemical Redox Pattern and Allied Chemical Behavior. J. Am. Chem. Soc. 1975, 97, 5083–5092. [Google Scholar] [CrossRef]
- Barin, R.; Biria, D.; Rashid-Nadimi, S.; Asadollahi, M.A. Enzymatic CO2 Reduction to Formate by Formate Dehydrogenase from Candida Boidinii Coupling with Direct Electrochemical Regeneration of NADH. J. CO2 Util. 2018, 28, 117–125. [Google Scholar] [CrossRef]
- Kim, S.; Kim, M.K.; Lee, S.H.; Yoon, S.; Jung, K.D. Conversion of CO2 to Formate in an Electroenzymatic Cell Using Candida Boidinii Formate Dehydrogenase. J. Mol. Catal. B Enzym. 2014, 102, 9–15. [Google Scholar] [CrossRef]
- Olson, B.J.S.C.; Skavdahl, M.; Ramberg, H.; Osterman, J.C.; Markwell, J. Formate Dehydrogenase in Arabidopsis Thaliana: Characterization and Possible Targeting to the Chloroplast. Plant Sci. 2000, 159, 205–212. [Google Scholar] [CrossRef]
- Wang, Y.; Li, M.; Zhao, Z.; Liu, W. Effect of Carbonic Anhydrase on Enzymatic Conversion of CO2 to Formic Acid and Optimization of Reaction Conditions. J. Mol. Catal. B Enzym. 2015, 116, 89–94. [Google Scholar] [CrossRef]
- Liu, W.; Hou, Y.; Hou, B.; Zhao, Z. Enzyme-Catalyzed Sequential Reduction of Carbon Dioxide to Formaldehyde. Chin. J. Chem. Eng. 2014, 22, 1328–1332. [Google Scholar] [CrossRef]
- Cavalcante, F.T.T.; Cavalcante, A.L.G.; de Sousa, I.G.; Neto, F.S.; Dos Santos, J.C.S. Current Status and Future Perspectives of Supports and Protocols for Enzyme Immobilization. Catal 2021, 11, 1222. [Google Scholar] [CrossRef]
- Rodrigues, R.C.; Berenguer-Murcia, Á.; Carballares, D.; Morellon-Sterling, R.; Fernandez-Lafuente, R. Stabilization of Enzymes via Immobilization: Multipoint Covalent Attachment and Other Stabilization Strategies. Biotechnol. Adv. 2021, 52, 107821. [Google Scholar] [CrossRef]
- Arana-Peña, S.; Carballares, D.; Morellon-Sterlling, R.; Berenguer-Murcia, Á.; Alcántara, A.R.; Rodrigues, R.C.; Fernandez-Lafuente, R. Enzyme Co-Immobilization: Always the Biocatalyst Designers’ Choice…or Not? Biotechnol. Adv. 2021, 51, 107584. [Google Scholar] [CrossRef]
- Liu, D.M.; Dong, C. Recent Advances in Nano-Carrier Immobilized Enzymes and Their Applications. Process Biochem. 2020, 92, 464–475. [Google Scholar] [CrossRef]
- Schmid-Dannert, C.; López-Gallego, F. Advances and Opportunities for the Design of Self-Sufficient and Spatially Organized Cell-Free Biocatalytic Systems. Curr. Opin. Chem. Biol. 2019, 49, 97–104. [Google Scholar] [CrossRef]
- López-Gallego, F.; Jackson, E.; Betancor, L. Heterogeneous Systems Biocatalysis: The Path to the Fabrication of Self-Sufficient Artificial Metabolic Cells. Chem. – A Eur. J. 2017, 23, 17841–17849. [Google Scholar] [CrossRef]
- Obert, R.; Dave, B.C. Enzymatic Conversion of Carbon Dioxide to Methanol: Enhanced Methanol Production in Silica Sol-Gel Matrices. J. Am. Chem. Soc. 1999, 121, 12192–12193. [Google Scholar] [CrossRef]
- Jiang, Z.; Wu, H.; Xu, S.; Huang, S. Enzymatic Conversion of Carbon Dioxide to Methanol by Dehydrogenases Encapsulated in Sol-Gel Matrix. ACS Div. Fuel Chem. Prepr. 2002, 47, 306. [Google Scholar] [CrossRef]
- Xu, S.W.; Lu, Y.; Li, J.; Jiang, Z.Y.; Wu, H. Efficient Conversion of CO2 to Methanol Catalyzed by Three Dehydrogenases Co-Encapsulated in an Alginate−Silica (ALG−SiO2) Hybrid Gel. Ind. Eng. Chem. Res. 2006, 45, 4567–4573. [Google Scholar] [CrossRef]
- El-Zahab, B.; Donnelly, D.; Wang, P. Particle-Tethered NADH for Production of Methanol from CO2 Catalyzed by Coimmobilized Enzymes. Biotechnol. Bioeng. 2008, 99, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Sun, Q.; Zhang, L.; Jiang, Z. Capsules-in-Bead Scaffold: A Rational Architecture for Spatially Separated Multienzyme Cascade System. J. Mater. Chem. 2009, 19, 9068–9074. [Google Scholar] [CrossRef]
- Sun, Q.; Jiang, Y.; Jiang, Z.; Zhang, L.; Sun, X.; Li, J. Green and Efficient Conversion of CO2 to Methanol by Biomimetic Coimmobilization of Three Dehydrogenases in Protamine-Templated Titania. Ind. Eng. Chem. Res. 2009, 48, 4210–4215. [Google Scholar] [CrossRef]
- Schlager, S.; Dumitru, L.M.; Haberbauer, M.; Fuchsbauer, A.; Neugebauer, H.; Hiemetsberger, D.; Wagner, A.; Portenkirchner, E.; Sariciftci, N.S. Electrochemical Reduction of Carbon Dioxide to Methanol by Direct Injection of Electrons into Immobilized Enzymes on a Modified Electrode. ChemSusChem 2016, 9, 631–635. [Google Scholar] [CrossRef] [Green Version]
- Marques Netto, C.G.C.; Andrade, L.H.; Toma, H.E. Carbon Dioxide/Methanol Conversion Cycle Based on Cascade Enzymatic Reactions Supported on Superparamagnetic Nanoparticles. An. Acad. Bras. Cienc. 2017, 90, 593–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, D.; Ao, S.; Deng, H.; Wang, M.; Qin, C.; Zhang, J.; Jia, Y.; Ye, P.; Ni, H. Ordered Coimmobilization of a Multienzyme Cascade System with a Metal Organic Framework in a Membrane: Reduction of CO2 to Methanol. ACS Appl. Mater. Interfaces 2019, 11, 33581–33588. [Google Scholar] [CrossRef]
- Ghannadi, S.; Abdizadeh, H.; Miroliaei, M.; Saboury, A.A. Immobilization of Alcohol Dehydrogenase on Titania Nanoparticles to Enhance Enzyme Stability and Remove Substrate Inhibition in the Reaction of Formaldehyde to Methanol. Ind. Eng. Chem. Res. 2019, 58, 9844–9854. [Google Scholar] [CrossRef]
- Chen, Y.; Li, P.; Zhou, J.; Buru, C.T.; Aorević, L.; Li, P.; Zhang, X.; Cetin, M.M.; Stoddart, J.F.; Stupp, S.I.; et al. Integration of Enzymes and Photosensitizers in a Hierarchical Mesoporous Metal-Organic Framework for Light-Driven CO2 Reduction. J. Am. Chem. Soc. 2020, 142, 1768–1773. [Google Scholar] [CrossRef]
- Pietricola, G.; Ottone, C.; Fino, D.; Tommasi, T. Enzymatic Reduction of CO2 to Formic Acid Using FDH Immobilized on Natural Zeolite. J. CO2 Util. 2020, 42, 101343. [Google Scholar] [CrossRef]
- Ren, S.; Wang, Z.; Bilal, M.; Feng, Y.; Jiang, Y.; Jia, S.; Cui, J. Co-Immobilization Multienzyme Nanoreactor with Co-Factor Regeneration for Conversion of CO2. Int. J. Biol. Macromol. 2020, 155, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Seelajaroen, H.; Bakandritsos, A.; Otyepka, M.; Zbořil, R.; Sariciftci, N.S. Immobilized Enzymes on Graphene as Nanobiocatalyst. ACS Appl. Mater. Interfaces 2020, 12, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, J.; Ji, M.; Liu, Y.; Wang, N.; Zhang, X.; Zhang, S.; Ji, X. Encapsulation of Multiple Enzymes in a Metal-Organic Framework with Enhanced Electro-Enzymatic Reduction of CO2 to Methanol. Green Chem. 2021, 23, 2362–2371. [Google Scholar] [CrossRef]
- Zezzi Do Valle Gomes, M.; Masdeu, G.; Eiring, P.; Kuhlemann, A.; Sauer, M.; Åkerman, B.; Palmqvist, A.E.C. Improved Biocatalytic Cascade Conversion of CO2 to Methanol by Enzymes Co-Immobilized in Tailored Siliceous Mesostructured Cellular Foams. Catal. Sci. Technol. 2021, 11, 6952–6959. [Google Scholar] [CrossRef]
- Alvarez-Malmagro, J.; Oliveira, A.R.; Gutiérrez-Sánchez, C.; Villajos, B.; Pereira, I.A.C.; Vélez, M.; Pita, M.; De Lacey, A.L. Bioelectrocatalytic Activity of W-Formate Dehydrogenase Covalently Immobilized on Functionalized Gold and Graphite Electrodes. ACS Appl. Mater. Interfaces 2021, 13, 11891–11900. [Google Scholar] [CrossRef]
- Wang, X.; Saba, T.; Yiu, H.H.P.; Howe, R.F.; Anderson, J.A.; Shi, J. Cofactor NAD(P)H Regeneration Inspired by Heterogeneous Pathways. Chem 2017, 2, 621–654. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, X.; Ji, X.; Bueno, C.; Zhang, Z.; Zhang, X.; Ji, X. Developing and Regenerating Cofactors for Sustainable Enzymatic CO2 Conversion. Processes 2022, 10, 230. [Google Scholar] [CrossRef]
- Singh, R.K.; Singh, R.; Sivakumar, D.; Kondaveeti, S.; Kim, T.; Li, J.; Sung, B.H.; Cho, B.K.; Kim, D.R.; Kim, S.C.; et al. Insights into Cell-Free Conversion of CO2 to Chemicals by a Multienzyme Cascade Reaction. ACS Catal. 2018, 8, 11085–11093. [Google Scholar] [CrossRef]
- Marpani, F.; Sárossy, Z.; Pinelo, M.; Meyer, A.S. Kinetics Based Reaction Optimization of Enzyme Catalyzed Reduction of Formaldehyde to Methanol with Synchronous Cofactor Regeneration. Biotechnol. Bioeng. 2017, 114, 2762–2770. [Google Scholar] [CrossRef] [PubMed]
- Aresta, M.; Dibenedetto, A. Integration of Solar Chemistry and Biotechnology for Building-up an Effective Man-Made C-Cycle That May Complement the Natural C-Cycle. Int. J. Plant Biol. 2020, 8, 1117. [Google Scholar]
- Aresta, M.; Dibenedetto, A.; Baran, T.; Angelini, A.; Łabuz, P.; Macyk, W. An Integrated Photocatalytic/Enzymatic System for the Reduction of CO2 to Methanol in Bioglycerol-Water. Beilstein J. Org. Chem. 2014, 10, 2556–2565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Tong, J.; Meng, X.; Cai, Y.; Ma, S.; Huo, F.; Luo, J.; Xu, B.H.; Zhang, S.; Pinelo, M. Development of an Ionic Porphyrin-Based Platform as a Biomimetic Light-Harvesting Agent for High-Performance Photoenzymatic Synthesis of Methanol from CO2. ACS Sustain. Chem. Eng. 2021, 9, 11503–11511. [Google Scholar] [CrossRef]
- Kim, S.H.; Chung, G.Y.; Kim, S.H.; Vinothkumar, G.; Yoon, S.H.; Jung, K.D. Electrochemical NADH Regeneration and Electroenzymatic CO2 Reduction on Cu Nanorods/Glassy Carbon Electrode Prepared by Cyclic Deposition. Electrochim. Acta 2016, 210, 837–845. [Google Scholar] [CrossRef]
- Yuan, M.; Kummer, M.J.; Milton, R.D.; Quah, T.; Minteer, S.D. Efficient NADH Regeneration by a Redox Polymer-Immobilized Enzymatic System. ACS Catal. 2019, 9, 5486–5495. [Google Scholar] [CrossRef]
- Immanuel, S.; Sivasubramanian, R.; Gul, R.; Dar, M.A. Recent Progress and Perspectives on Electrochemical Regeneration of Reduced Nicotinamide Adenine Dinucleotide (NADH). Chem. Asian J. 2020, 15, 4256–4270. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Su, Z.; Wang, P.; Ma, G.; Zhang, S. Tethering of Nicotinamide Adenine Dinucleotide inside Hollow Nanofibers for High-Yield Synthesis of Methanol from Carbon Dioxide Catalyzed by Coencapsulated Multienzymes. ACS Nano 2015, 9, 4600–4610. [Google Scholar] [CrossRef]
- Jiang, Z.; Lü, C.; Wu, H. Photoregeneration of NADH Using Carbon-Containing TiO2. Ind. Eng. Chem. Res. 2005, 44, 4165–4170. [Google Scholar] [CrossRef]
- Shi, Q.; Yang, D.; Jiang, Z.; Li, J. Visible-Light Photocatalytic Regeneration of NADH Using P-Doped TiO2 Nanoparticles. J. Mol. Catal. B Enzym. 2006, 43, 44–48. [Google Scholar] [CrossRef]
- Kim, J.A.; Kim, S.; Lee, J.; Baeg, J.O.; Kim, J. Photochemical Production of NADH Using Cobaloxime Catalysts and Visible-Light Energy. Inorg. Chem. 2012, 51, 8057–8063. [Google Scholar] [CrossRef]
- Yadav, R.K.; Oh, G.H.; Park, N.J.; Kumar, A.; Kong, K.J.; Baeg, J.O. Highly Selective Solar-Driven Methanol from CO2 by a Photocatalyst/Biocatalyst Integrated System. J. Am. Chem. Soc. 2014, 136, 16728–16731. [Google Scholar] [CrossRef]
- Meng, J.; Tian, Y.; Li, C.; Lin, X.; Wang, Z.; Sun, L.; Zhou, Y.; Li, J.; Yang, N.; Zong, Y.; et al. A Thiophene-Modified Doubleshell Hollow g-C3N4 Nanosphere Boosts NADH Regeneration via Synergistic Enhancement of Charge Excitation and Separation. Catal. Sci. Technol. 2019, 9, 1911–1921. [Google Scholar] [CrossRef]
- Guo, M.; Gu, F.; Meng, L.; Liao, Q.; Meng, Z.; Liu, W. Synthesis of Formaldehyde from CO2 Catalyzed by the Coupled Photo-Enzyme System. Sep. Purif. Technol. 2022, 286, 120480. [Google Scholar] [CrossRef]
- Ali, I.; Gill, A.; Omanovic, S. Direct Electrochemical Regeneration of the Enzymatic Cofactor 1,4-NADH Employing Nano-Patterned Glassy Carbon/Pt and Glassy Carbon/Ni Electrodes. Chem. Eng. J. 2012, 188, 173–180. [Google Scholar] [CrossRef]
- Ali, I.; Ullah, N.; McArthur, M.A.; Coulombe, S.; Omanovic, S. Direct Electrochemical Regeneration of Enzymatic Cofactor 1,4-NADH on a Cathode Composed of Multi-Walled Carbon Nanotubes Decorated with Nickel Nanoparticles. Can. J. Chem. Eng. 2018, 96, 68–73. [Google Scholar] [CrossRef]
- Chen, Y.; Li, P.; Noh, H.; Kung, C.-W.; Buru, C.T.; Wang, X.; Zhang, X.; Omar Farha, K.; Chen, Y.; Noh, H.; et al. Stabilization of Formate Dehydrogenase in a Metal–Organic Framework for Bioelectrocatalytic Reduction of CO2. Angew. Chem. Int. Ed. 2019, 58, 7682–7686. [Google Scholar] [CrossRef]
- Song, H.; Ma, C.; Liu, P.; You, C.; Lin, J.; Zhu, Z. A Hybrid CO2 Electroreduction System Mediated by Enzyme-Cofactor Conjugates Coupled with Cu Nanoparticle-Catalyzed Cofactor Regeneration. J. CO2 Util. 2019, 34, 568–575. [Google Scholar] [CrossRef]
- Paul, C.E.; Arends, I.W.C.E.; Hollmann, F. Is Simpler Better? Synthetic Nicotinamide Cofactor Analogues for Redox Chemistry. ACS Catal. 2014, 4, 788–797. [Google Scholar] [CrossRef]
- Jayathilake, B.S.; Bhattacharya, S.; Vaidehi, N.; Narayanan, S.R. Efficient and Selective Electrochemically Driven Enzyme-Catalyzed Reduction of Carbon Dioxide to Formate Using Formate Dehydrogenase and an Artificial Cofactor. Acc. Chem. Res. 2019, 52, 676–685. [Google Scholar] [CrossRef]
- Amao, Y.; Ikeyama, S. Discovery of the Reduced Form of Methylviologen Activating Formate Dehydrogenase in the Catalytic Conversion of Carbon Dioxide to Formic Acid. Chem. Lett. 2015, 44, 1182–1184. [Google Scholar] [CrossRef]
- Amao, Y.; Kataoka, R. Methanol Production from CO2 with the Hybrid System of Biocatalyst and Organo-Photocatalyst. Catal. Today 2018, 307, 243–247. [Google Scholar] [CrossRef]
- Zhang, Z.; Vasiliu, T.; Li, F.; Laaksonen, A.; Mocci, F.; Ji, X. Electrochemically Driven Efficient Enzymatic Conversion of CO2 to Formic Acid with Artificial Cofactors. J. CO2 Util. 2021, 52, 101679. [Google Scholar] [CrossRef]
- Ikeyama, S.; Amao, Y. An Artificial Co-Enzyme Based on the Viologen Skeleton for Highly Efficient CO2 Reduction to Formic Acid with Formate Dehydrogenase. ChemCatChem 2017, 9, 833–838. [Google Scholar] [CrossRef]
- Miyaji, A.; Amao, Y. Artificial Co-Enzyme Based on Carbamoyl-Modified Viologen Derivative Cation Radical for Formate Dehydrogenase in the Catalytic CO2 Reduction to Formate. New J. Chem. 2020, 44, 18803–18812. [Google Scholar] [CrossRef]
- Miyaji, A.; Amao, Y. Visible-Light Driven Reduction of CO2 to Formate by a Water-Soluble Zinc Porphyrin and Formate Dehydrogenase System with Electron-Mediated Amino and Carbamoyl Group-Modified Viologen. New J. Chem. 2021, 45, 5780–5790. [Google Scholar] [CrossRef]
- Ikeyama, S.; Katagiri, T.; Amao, Y. The Improvement of Formic Acid Production from CO2 with Visible-Light Energy and Formate Dehydrogenase by the Function of the Viologen Derivative with Carbamoylmethyl Group as an Electron Carrier. J. Photochem. Photobiol. A Chem. 2018, 358, 362–367. [Google Scholar] [CrossRef]
- Ikeyama, S.; Amao, Y. A Novel Electron Carrier Molecule Based on a Viologen Derivative for Visible Light-Driven CO2 Reduction to Formic Acid with the System of Zinc Porphyrin and Formate Dehydrogenase. Sustain. Energy Fuels 2017, 1, 1730–1733. [Google Scholar] [CrossRef]
- Amao, Y.; Abe, R.; Shiotani, S. Effect of Chemical Structure of Bipyridinium Salts as Electron Carrier on the Visible-Light Induced Conversion of CO2 to Formic Acid with the System Consisting of Water-Soluble Zinc Porphyrin and Formate Dehydrogenase. J. Photochem. Photobiol. A Chem. 2015, 313, 149–153. [Google Scholar] [CrossRef] [Green Version]
- Ikeyama, S.; Amao, Y. The Effect of the Functional Ionic Group of the Viologen Derivative on Visible-Light Driven CO2 Reduction to Formic Acid with the System Consisting of Water-Soluble Zinc Porphyrin and Formate Dehydrogenase. Photochem. Photobiol. Sci. 2018, 17, 60–68. [Google Scholar] [CrossRef]
- Ishibashi, T.; Ikeyama, S.; Ito, M.; Ikeda, S.; Amao, Y. Light-Driven CO2 Reduction to Formic Acid with a Hybrid System of Biocatalyst and Semiconductor Based Photocatalyst. Chem. Lett. 2018, 47, 1505–1508. [Google Scholar] [CrossRef] [Green Version]
- Kuk, S.K.; Gopinath, K.; Singh, R.K.; Kim, T.D.; Lee, Y.; Choi, W.S.; Lee, J.K.; Park, C.B. NADH-Free Electroenzymatic Reduction of CO2 by Conductive Hydrogel-Conjugated Formate Dehydrogenase. ACS Catal. 2019, 9, 5584–5589. [Google Scholar] [CrossRef]









| Enzyme | Reaction | Km | Ref. |
|---|---|---|---|
| FateDH | CO2 → HCO2− | 30–50 mM | [18] |
| HCO2− → CO2 | 0.5 mM | [18] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Spiridione, C.; Aresta, M.; Dibenedetto, A. Improving the Enzymatic Cascade of Reactions for the Reduction of CO2 to CH3OH in Water: From Enzymes Immobilization Strategies to Cofactor Regeneration and Cofactor Suppression. Molecules 2022, 27, 4913. https://doi.org/10.3390/molecules27154913
Di Spiridione C, Aresta M, Dibenedetto A. Improving the Enzymatic Cascade of Reactions for the Reduction of CO2 to CH3OH in Water: From Enzymes Immobilization Strategies to Cofactor Regeneration and Cofactor Suppression. Molecules. 2022; 27(15):4913. https://doi.org/10.3390/molecules27154913
Chicago/Turabian StyleDi Spiridione, Carmela, Michele Aresta, and Angela Dibenedetto. 2022. "Improving the Enzymatic Cascade of Reactions for the Reduction of CO2 to CH3OH in Water: From Enzymes Immobilization Strategies to Cofactor Regeneration and Cofactor Suppression" Molecules 27, no. 15: 4913. https://doi.org/10.3390/molecules27154913