
Potent Antibiotic Lemonomycin: A Glimpse of Its Discovery, Origin, and Chemical Synthesis


{kind=link}
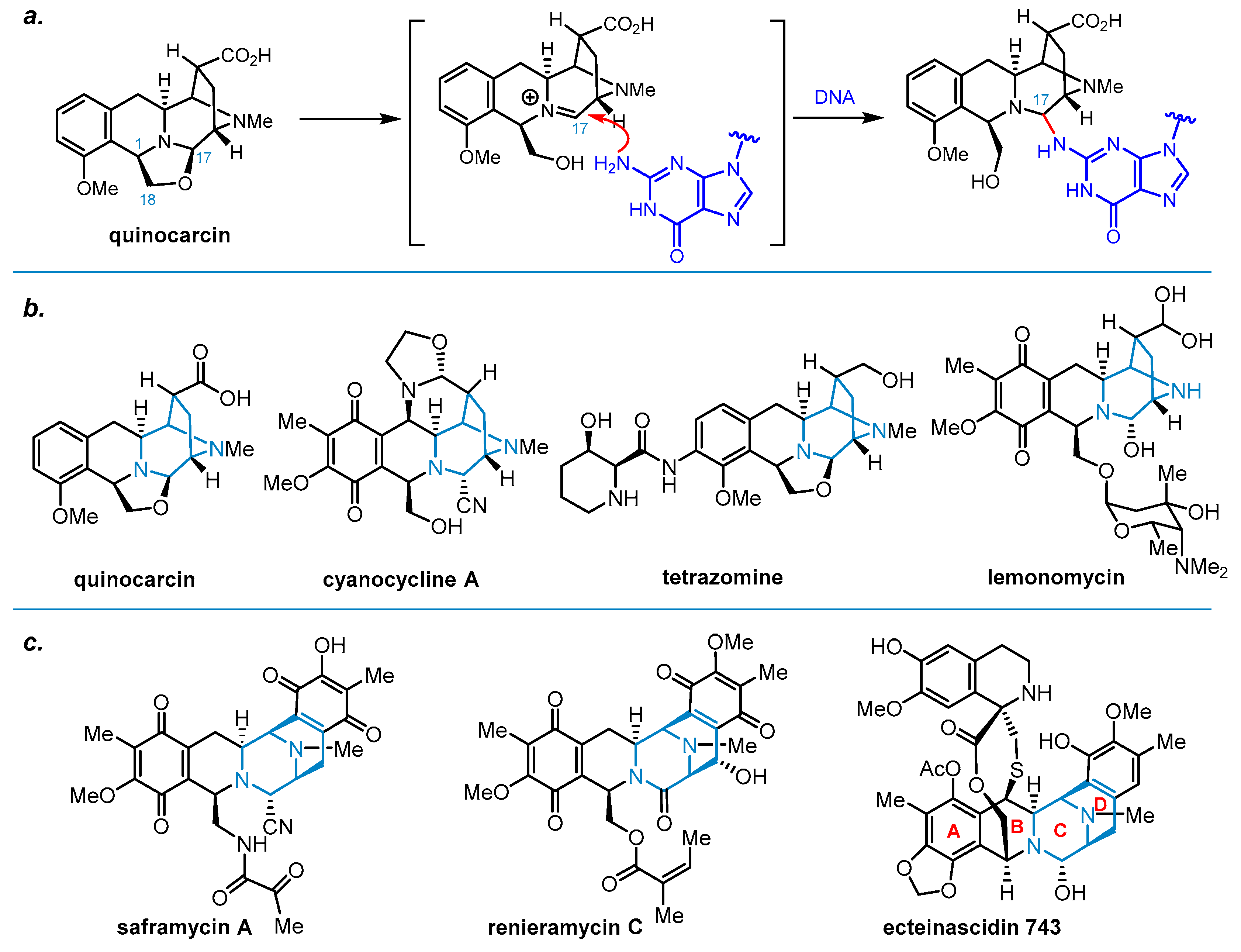{kind=link}
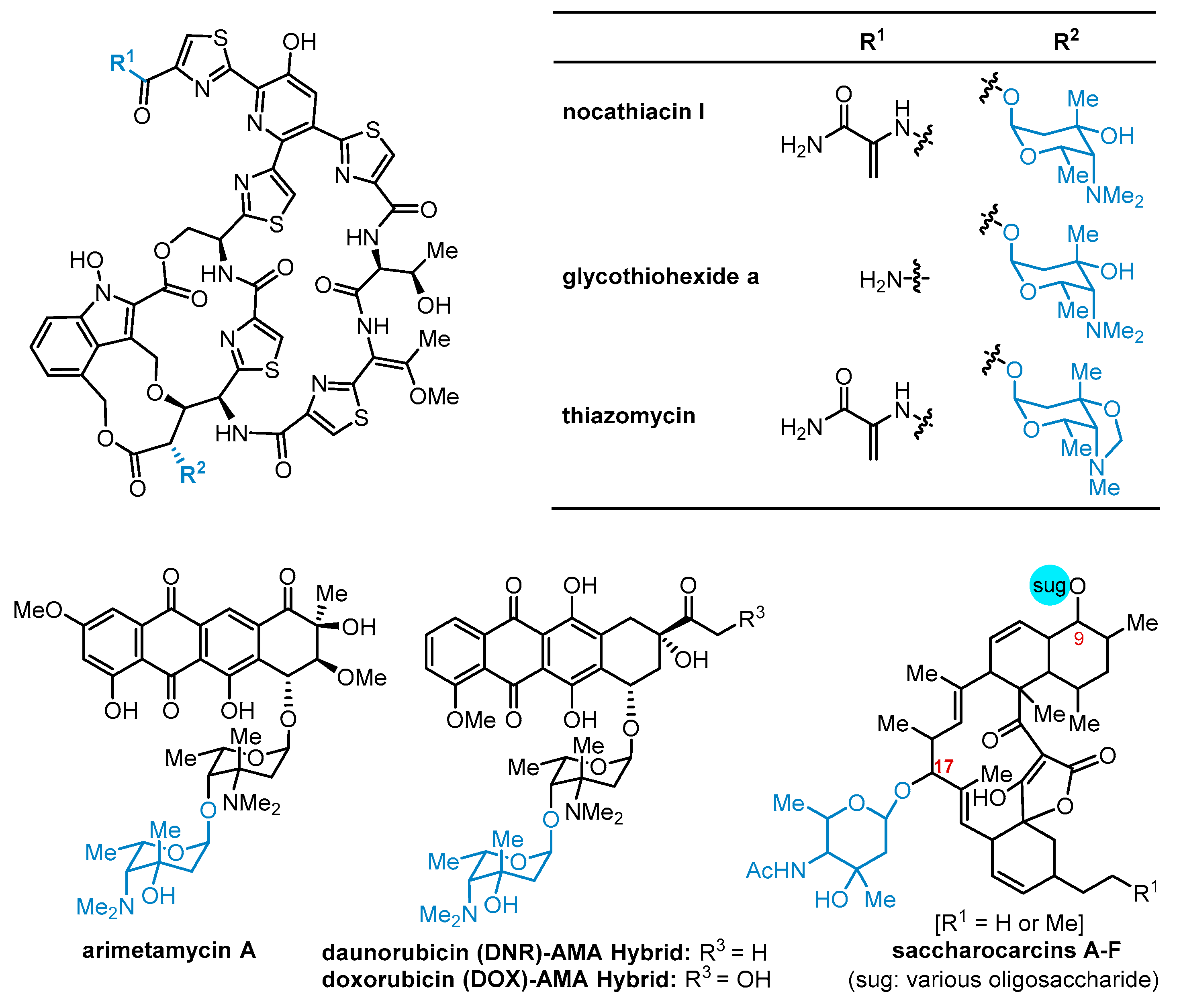{kind=link}
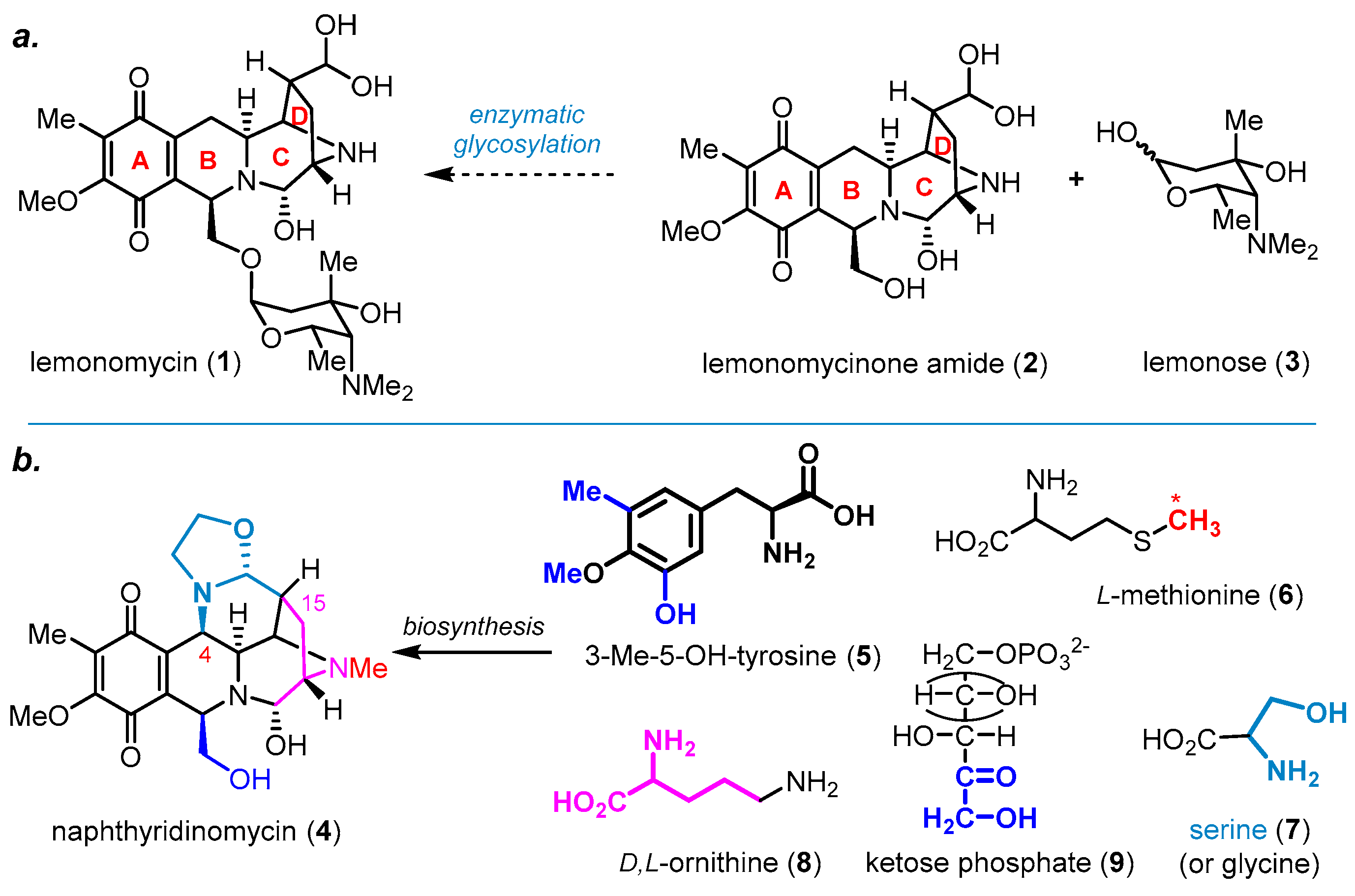{kind=link}
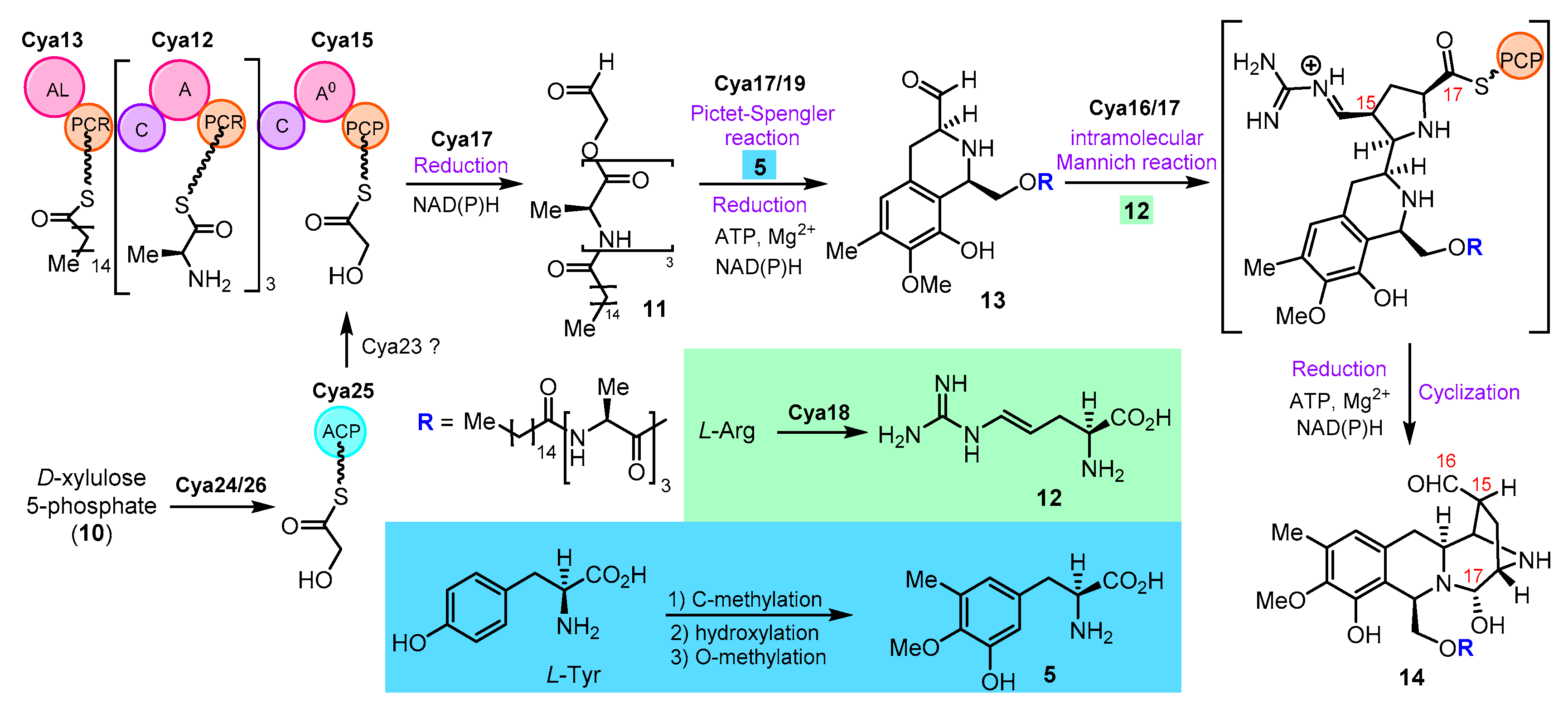{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Isolation and Structural Elucidation
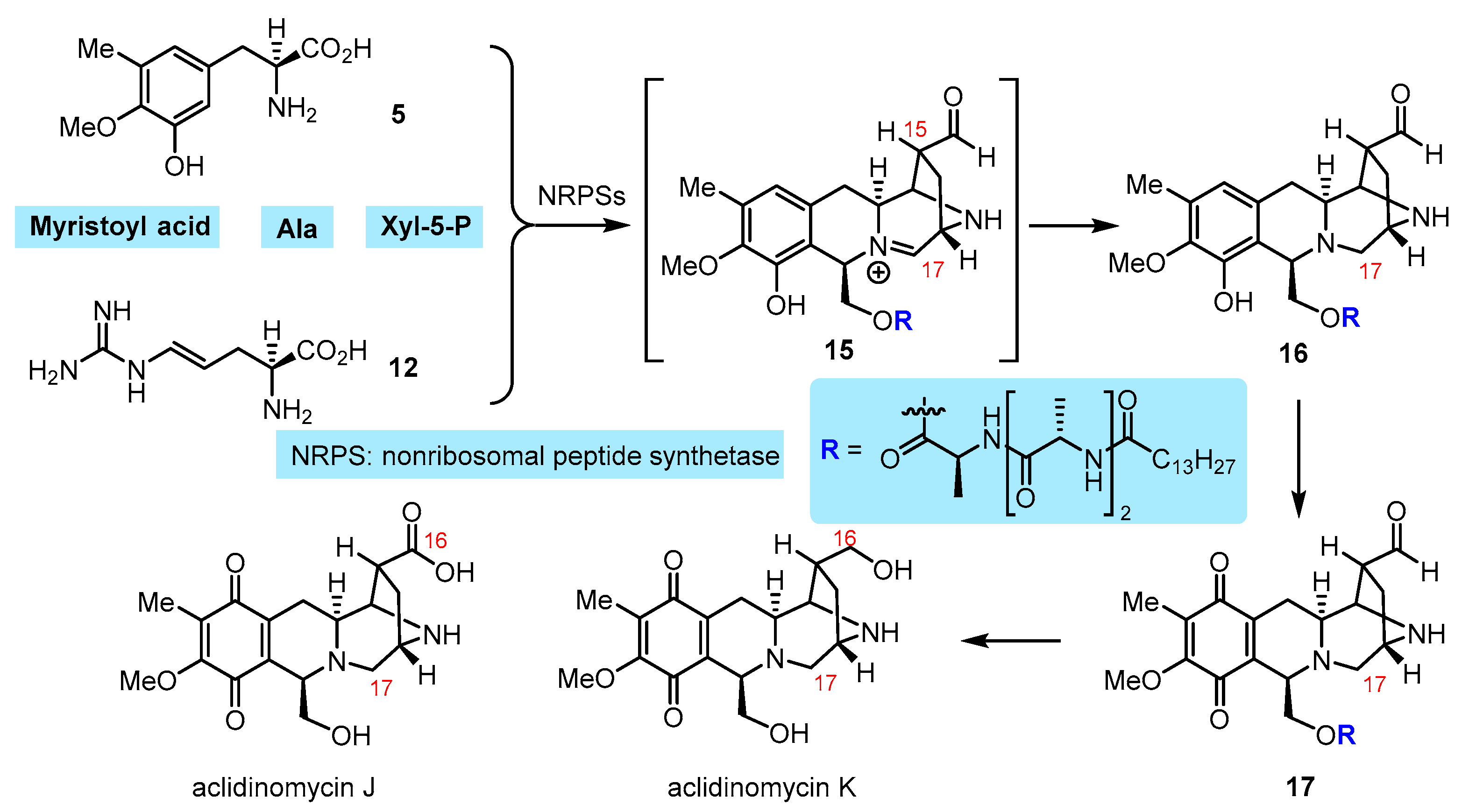
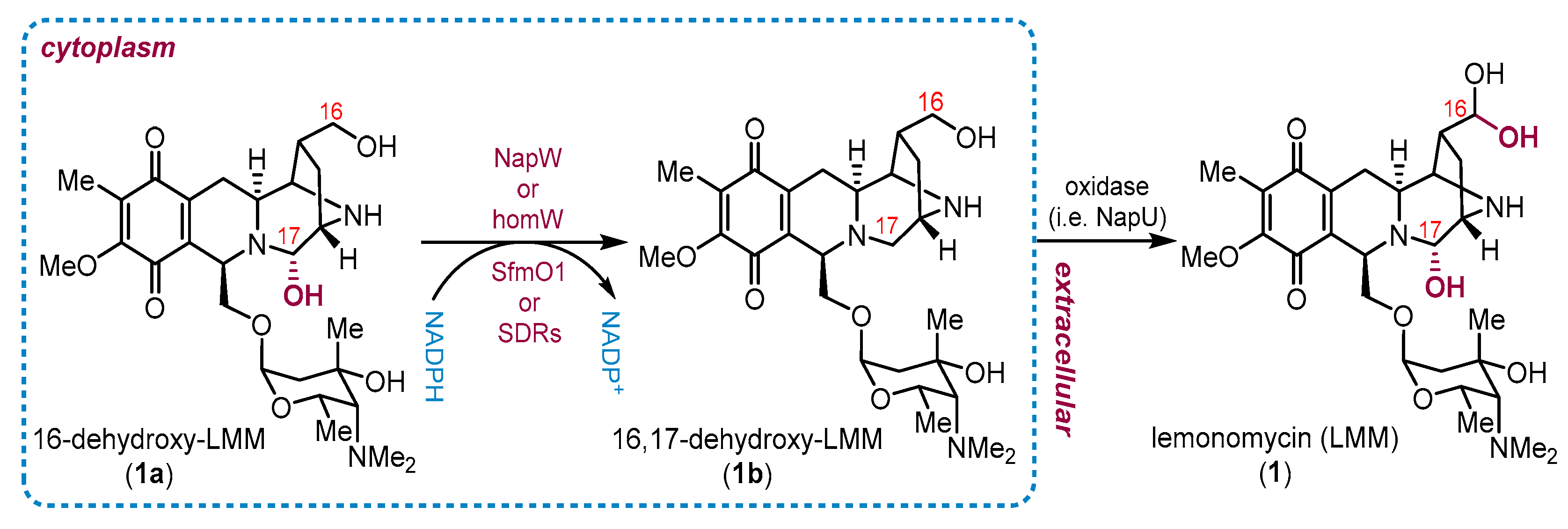
2. Current Status of Biosynthesis
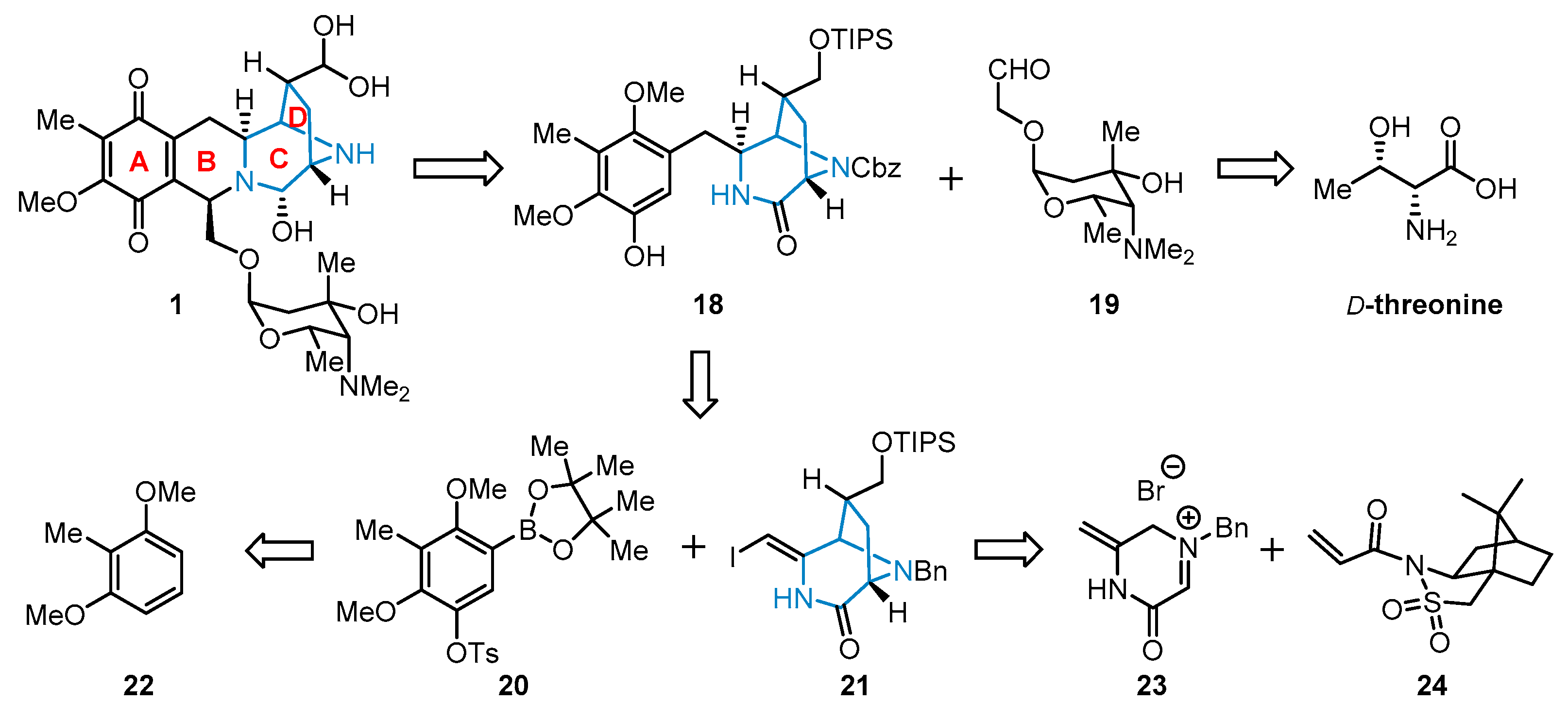
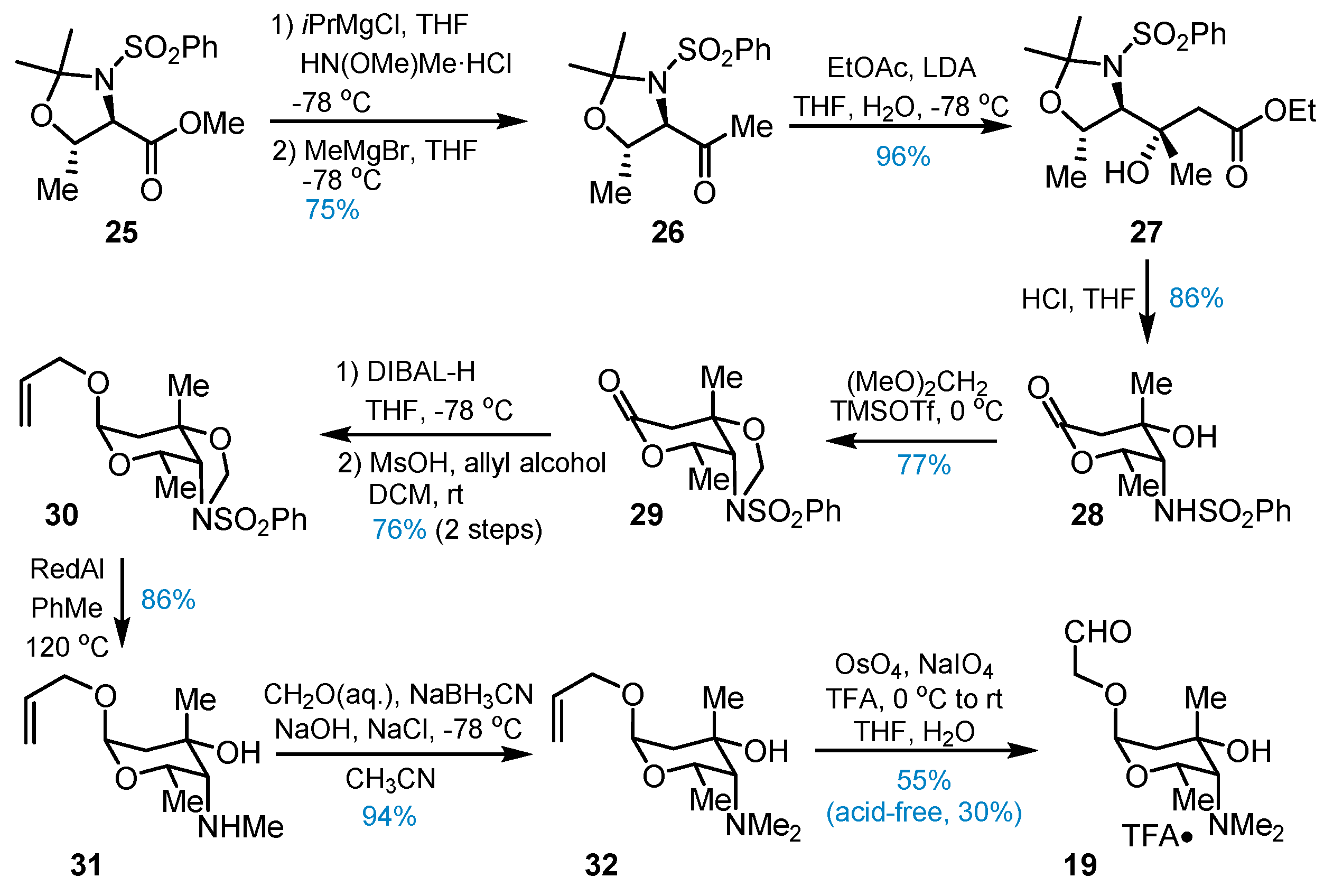
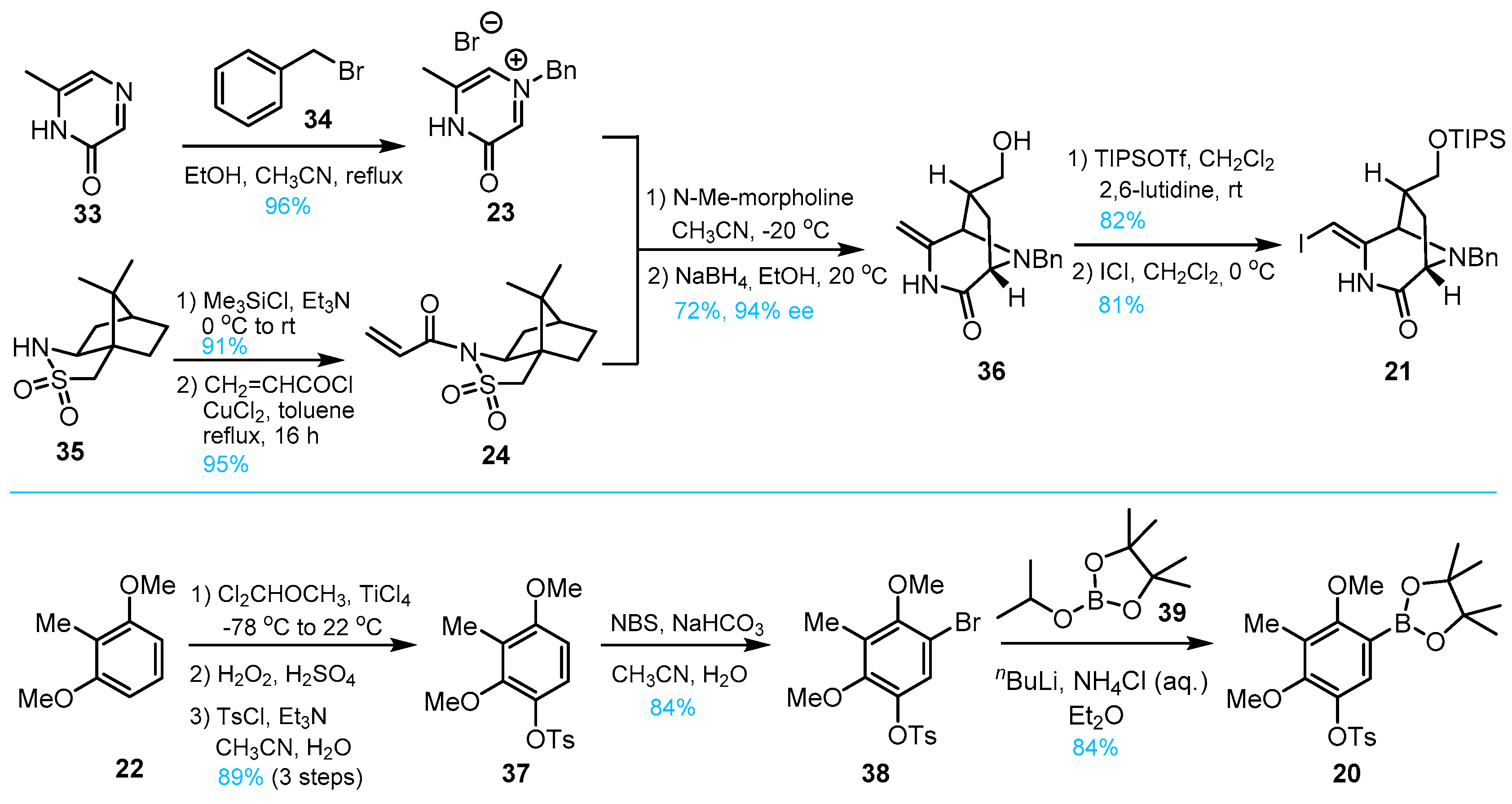
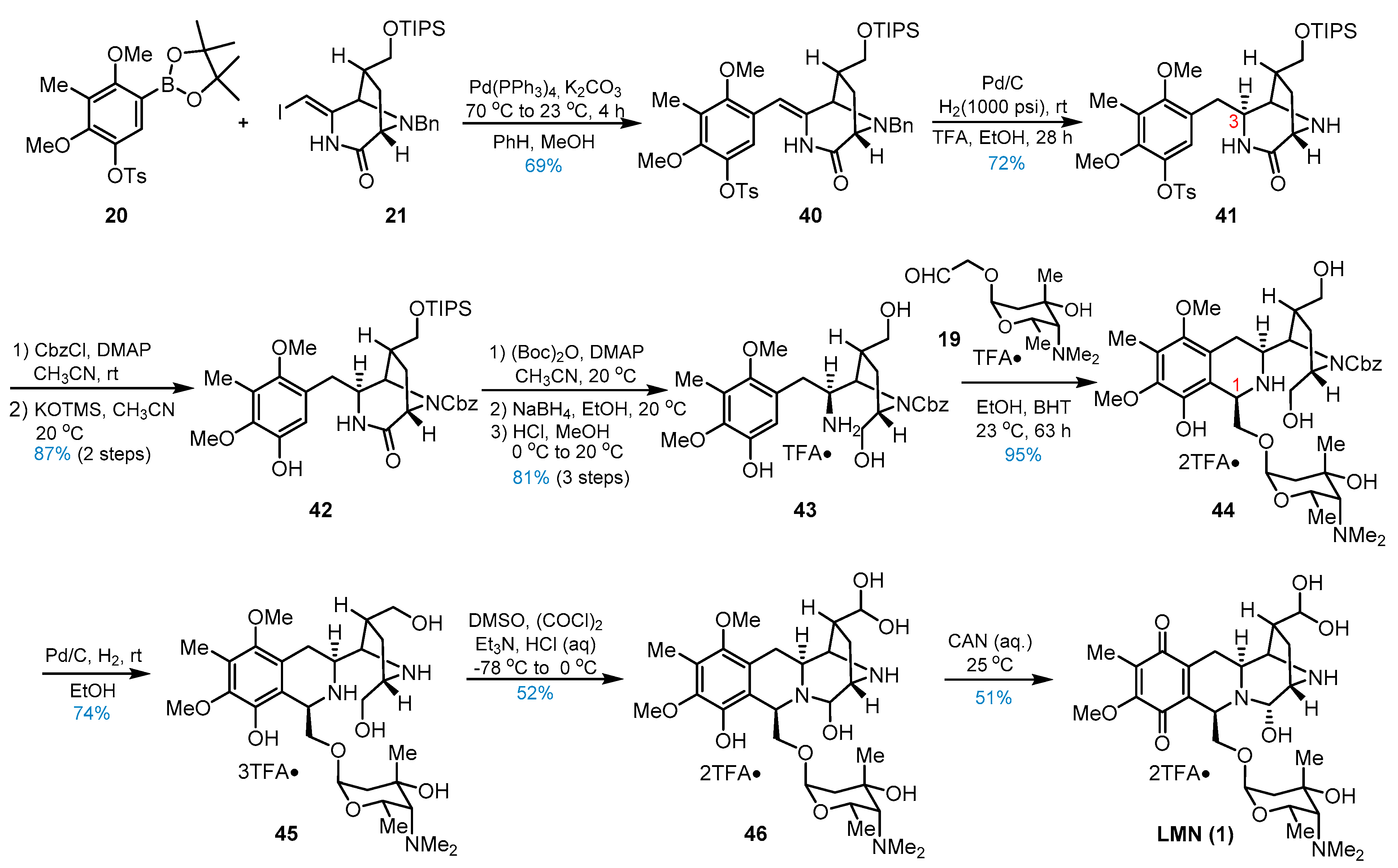
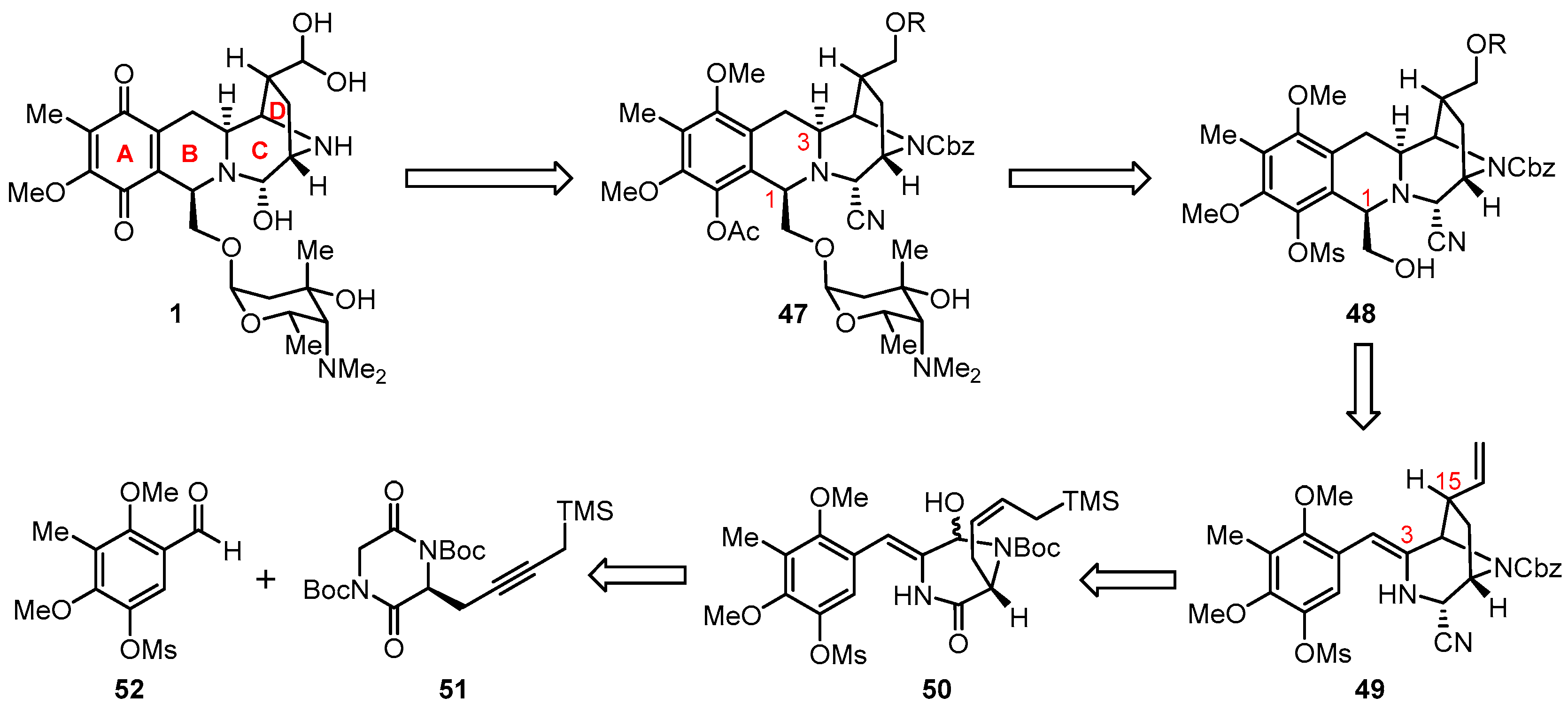
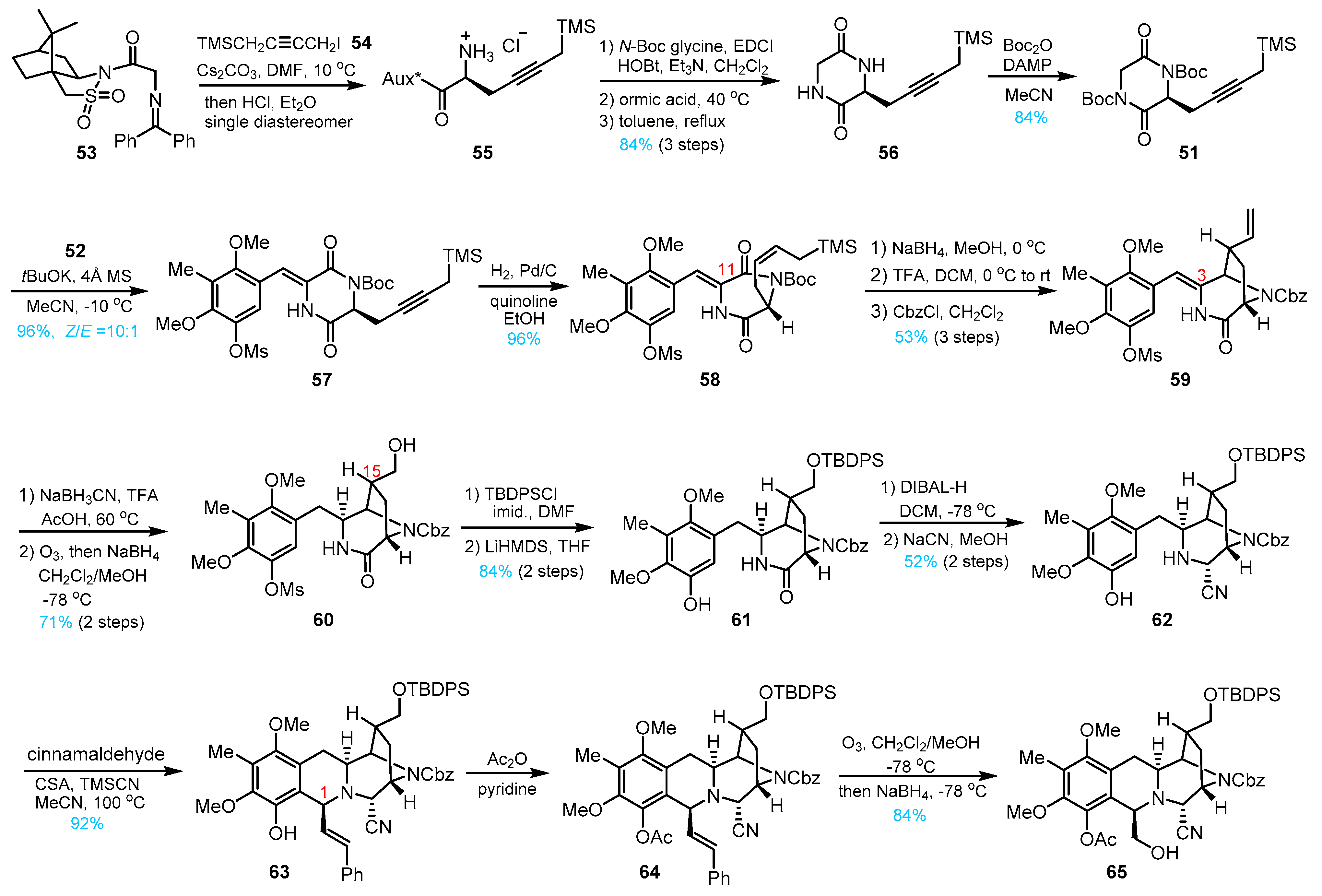
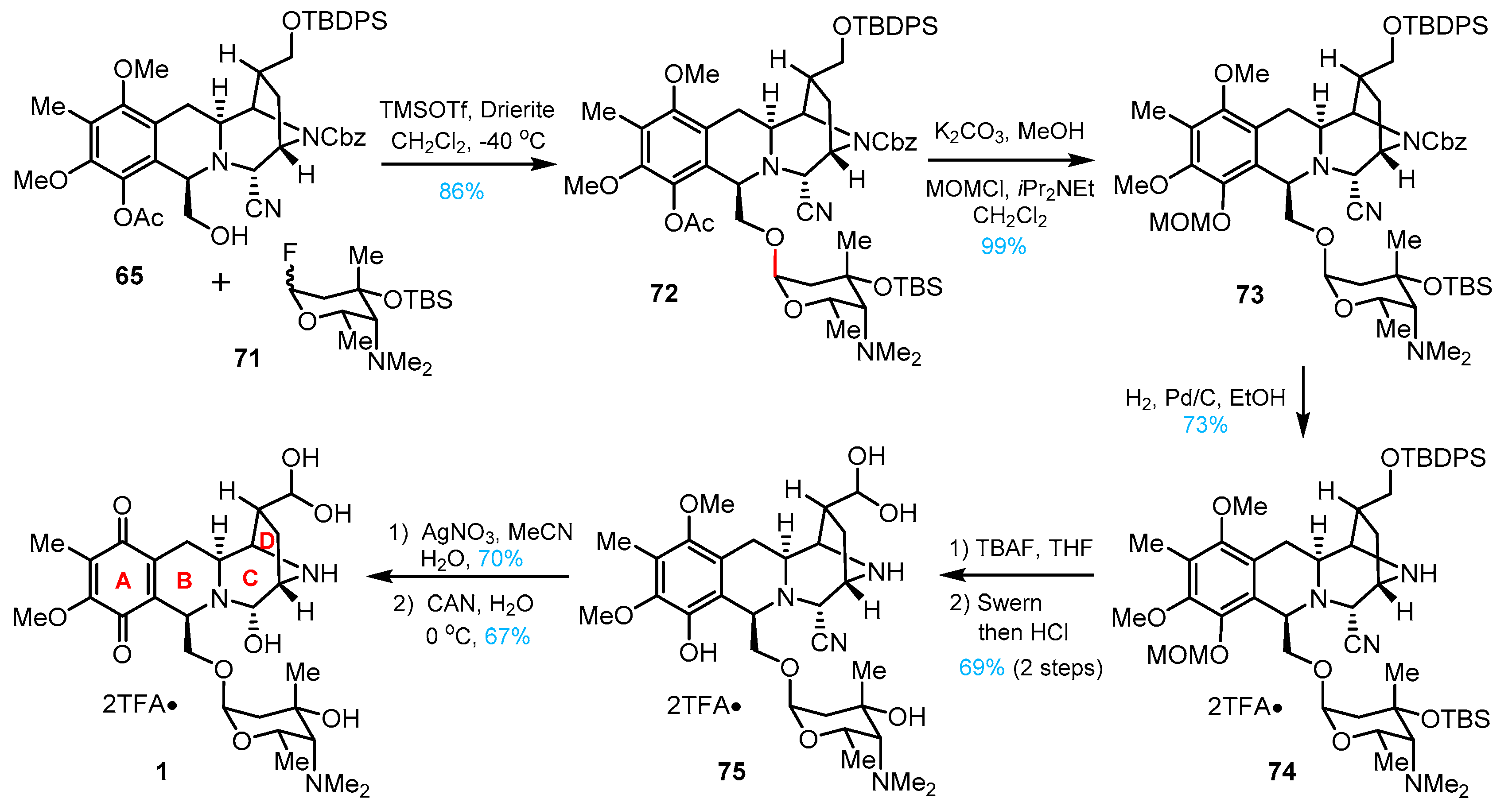
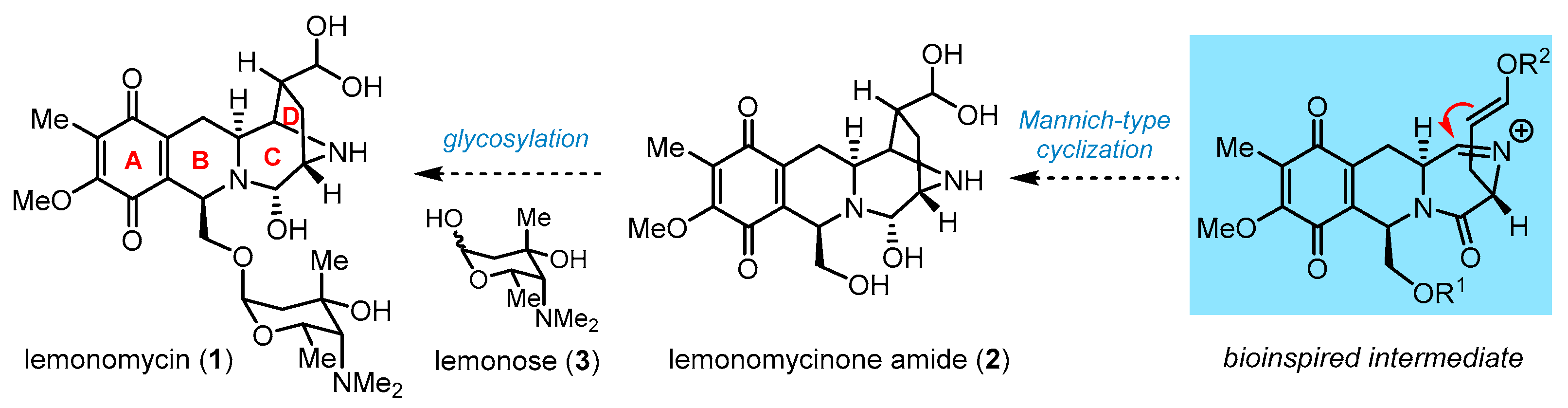
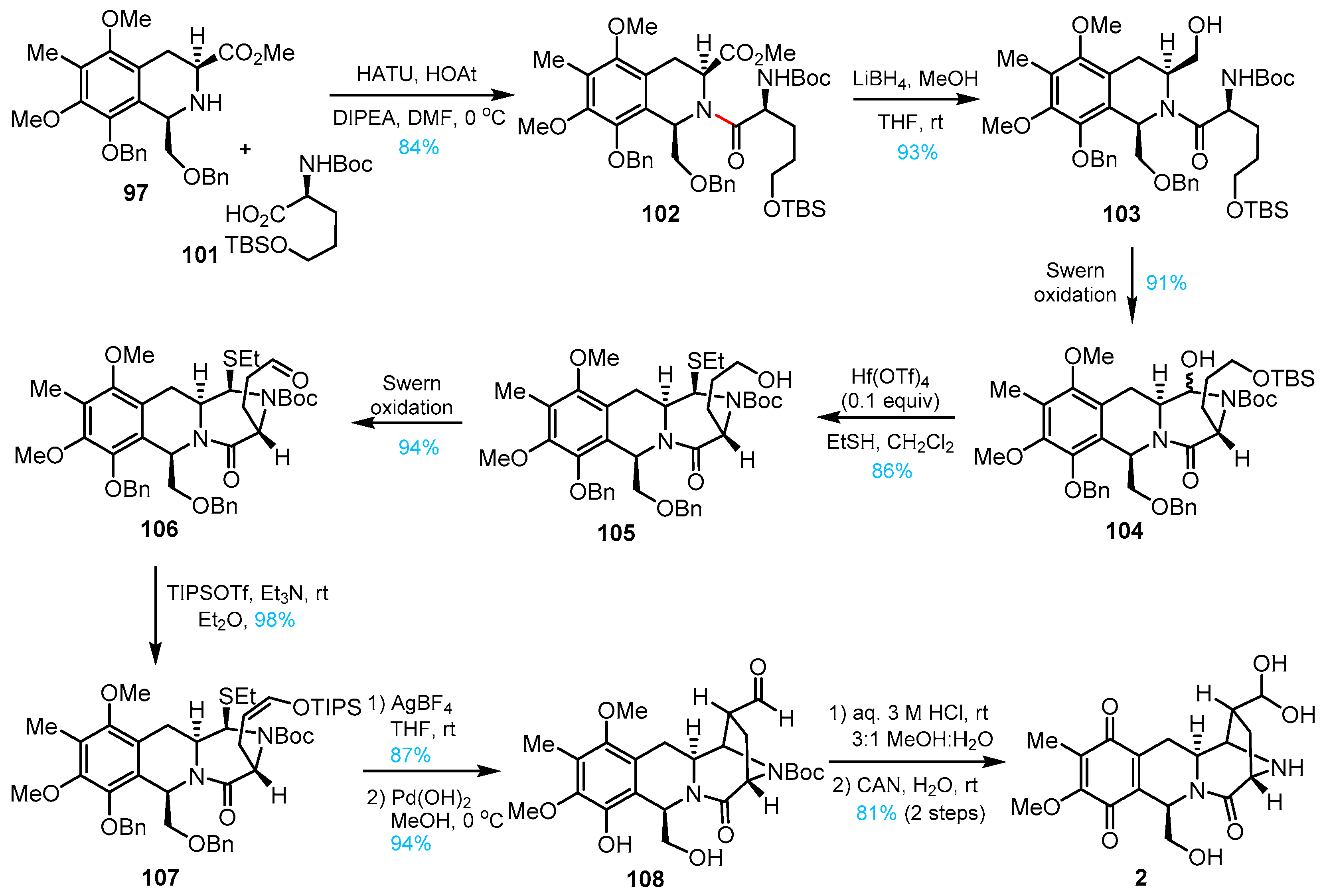
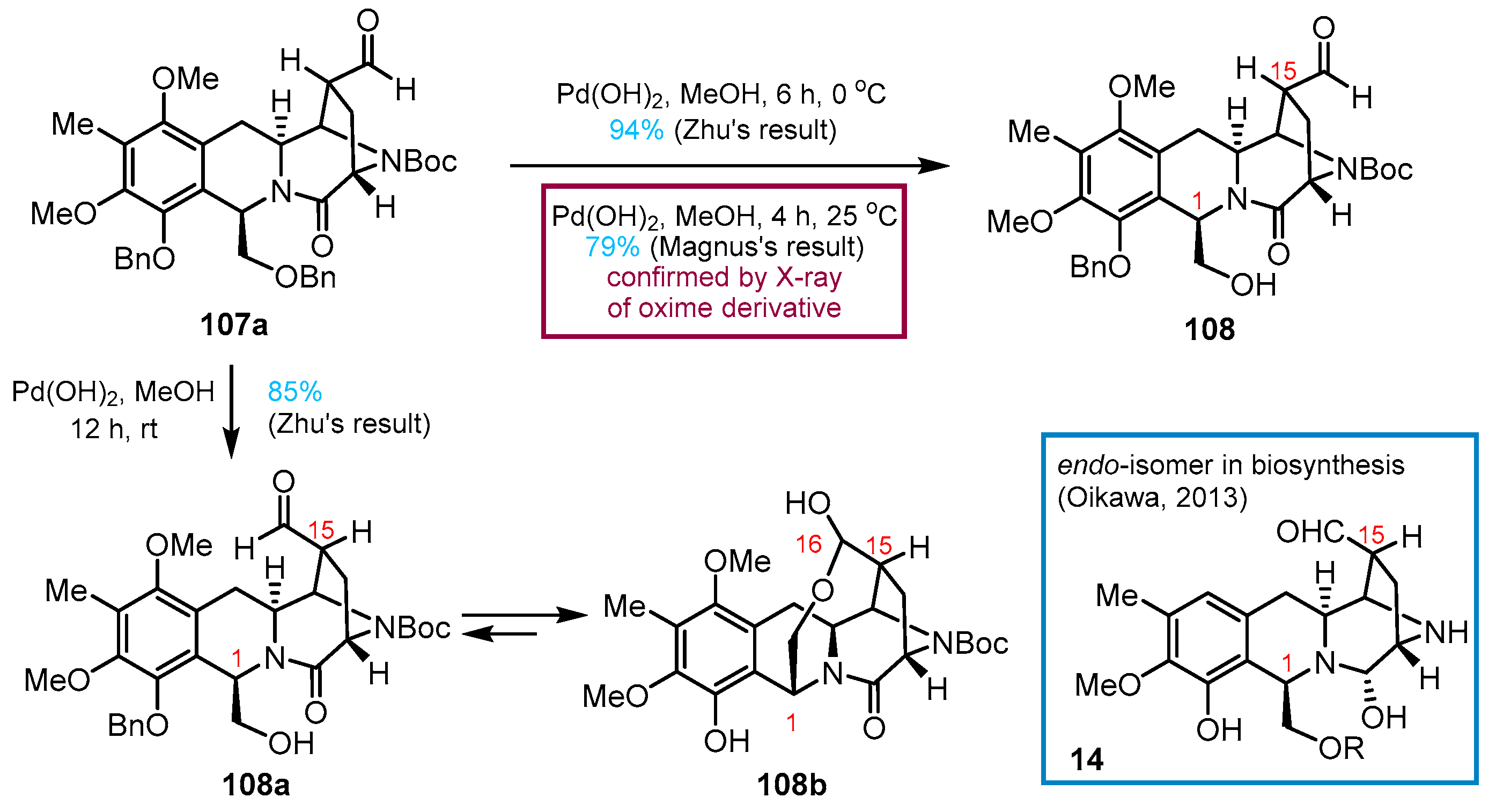
3. Total Synthesis of Lemonomycin
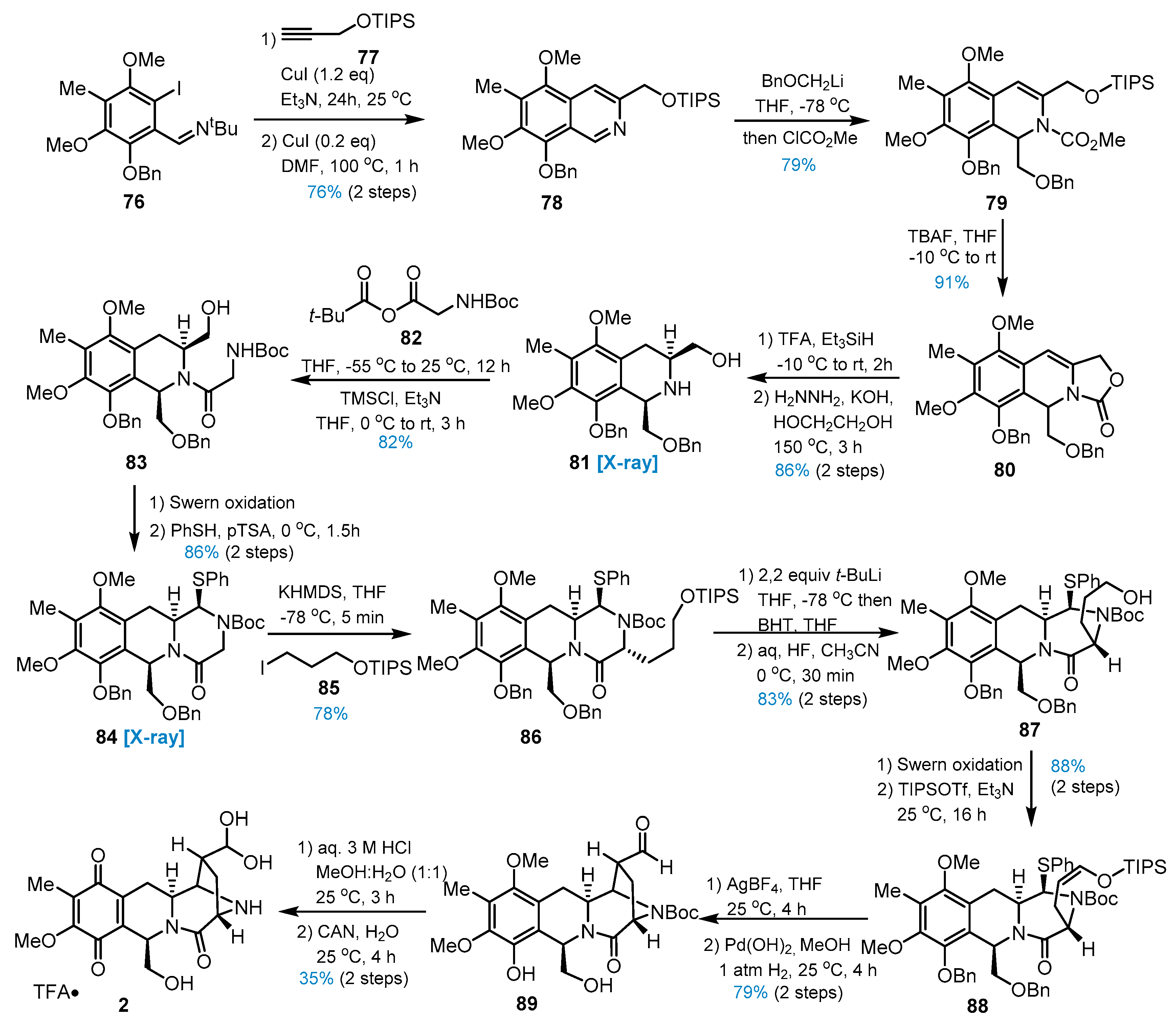
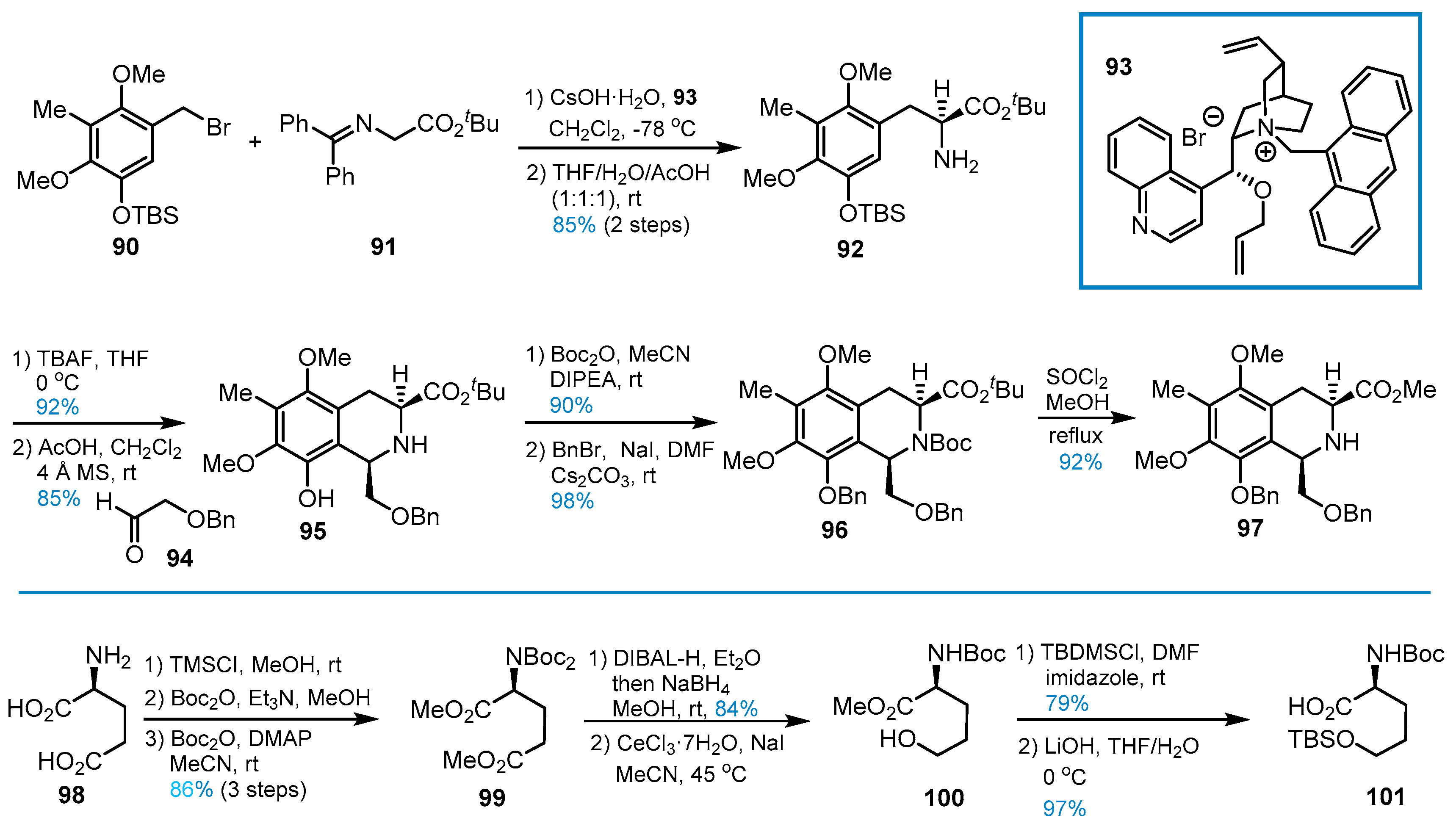
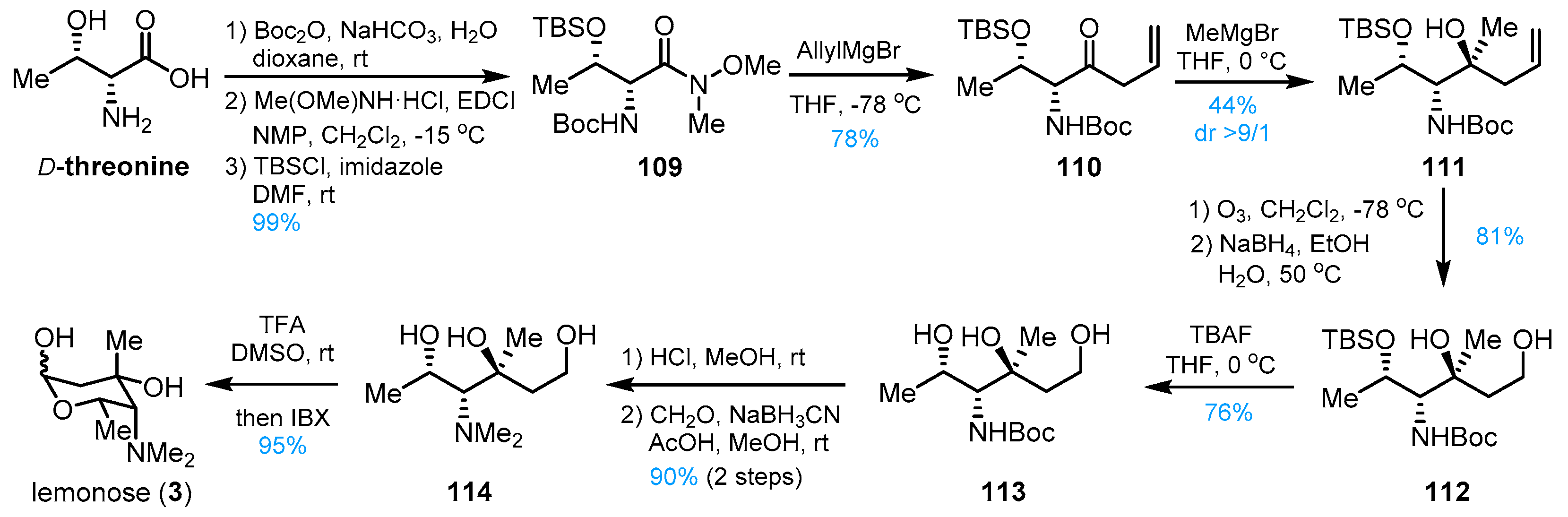
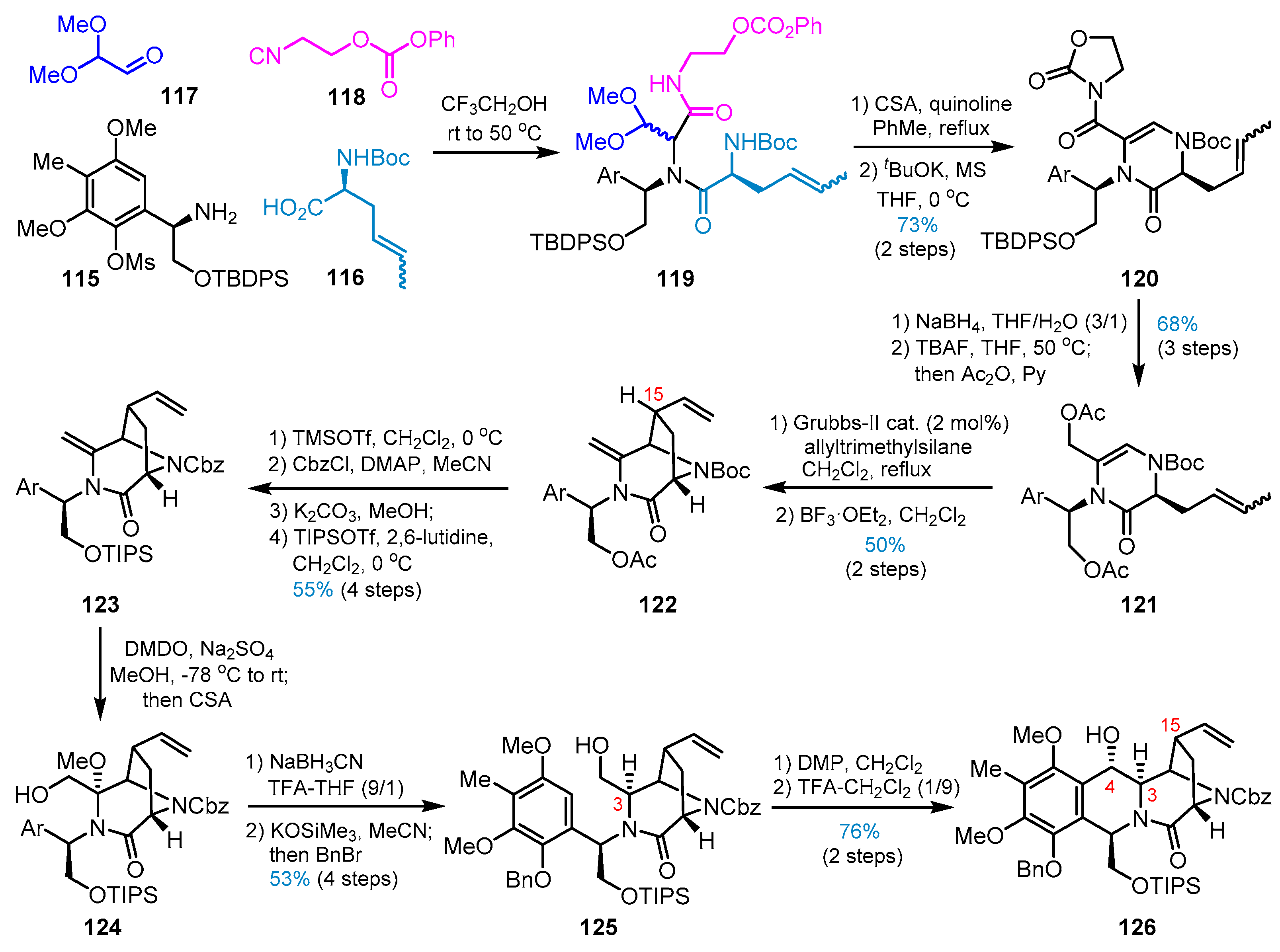
4. Other Studies toward Lemonomycin
5. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations; Government of the United Kingdom: London, UK, 2016.
- Butler, M.S.; Paterson, D.L. Antibiotics in the clinical pipeline in October 2019. J. Antibiot. 2020, 73, 329–364. [Google Scholar] [CrossRef]
- Whaley, H.A.; Patterson, E.L.; Dann, M.; Shay, A.J.; Porter, J.N. Isolation and characterization of Lemonomycin, a new antibiotic. Antimicrob. Agents Chemother. 1964, 8, 83–86. [Google Scholar]
- Ashley, E.; Stoltz, B. A Dipolar cycloaddition strategy for the synthesis of 3,8-diazabicyclo [3.2.1]octane tetrahydroisoquinoline antitumor antibiotics: The total synthesis of (-)-lemonomycin. Strateg. Tactics Org. Synth. 2012, 8, 351–373. [Google Scholar]
- He, H.Y.; Shen, B.; Carter, G.T. Structural elucidation of lemonomycin, a potent antibiotic from Streptomyces candidus. Tetrahedron Lett. 2000, 41, 2067–2071. [Google Scholar] [CrossRef]
- Scott, J.D.; Williams, R.M. Chemistry and biology of the tetrahydroisoquinoline antitumor amtibiotics. Chem. Rev. 2002, 102, 1669–1730. [Google Scholar] [CrossRef]
- Siengalewicz, P.; Rinner, U.; Mulzer, j. Recent progress in the total synthesis of naphthyridinomycin and lemonomycin tetrahydroisoquinoline antitumor antibiotics (TAAs). Chem. Soc. Rev. 2008, 37, 2676–2690. [Google Scholar] [CrossRef]
- Hill, G.C.; Wunz, T.P.; Remers, W.A. Computer simulation of the binding of quinocarcin to DNA. Prediction of mode of action and absolute configuration. J. Comput. Aided Mol. Des. 1988, 2, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Sygusch, J.; Brisse, F.; Hanessian, S.; Kluepfel, D. The molecular structure of naphthyridinomycin—A broad spectrum antibiotic. Tetrahedron Lett. 1974, 15, 4021–4023. [Google Scholar] [CrossRef]
- Kluepfel, D.; Baker, H.A.; Piattoni, G.; Sehgal, S.N.; Sidorowicz, A.; Singh, K.; Vézina, C. Naphthyridinomycin, a new broad-spectrum antibiotic. J. Antibiot. 1975, 28, 497–502. [Google Scholar] [CrossRef]
- Zmijewski, M.; Mikolajczak, M., Jr.; Viswanatha, V.; Hruby, V.J. Biosynthesis of the antitumor antibiotic naphthyridinomycin. J. Am. Chem. Soc. 1982, 104, 4969–4971. [Google Scholar] [CrossRef]
- Zmijewski, M.; Palaniswamy, V.A., Jr.; Gould, S.J. Naphthyridinomycin biosynthesis. The involvement of ornithine and the origin of the oxazolidine nitrogen. J. Chem. Soc. Chem. Commun. 1985, 1261–1262. [Google Scholar] [CrossRef]
- Palaniswamy, V.A.; Gould, S.J. The incorporation of 3’-methyltyrosine and 5’-methyl DOPA into naphthyridinomycin. J. Am. Chem. Soc. 1986, 108, 5651–5652. [Google Scholar] [CrossRef]
- Peng, C.; Pu, J.Y.; Song, L.Q.; Jian, X.H.; Tang, M.C.; Tang, G.L. Hijacking a hydroxyethyl unit from a central metabolic ketose into a nonribosomal peptide assembly line. Proc. Natl. Acad. Sci. USA 2012, 109, 8540–8545. [Google Scholar] [CrossRef] [Green Version]
- Pu, J.Y.; Peng, C.; Tang, M.C.; Zhang, Y.; Guo, J.P.; Song, L.Q.; Hua, Q.; Tang, G.L. Naphthyridinomycin biosynthesis revealing the use of leader peptide to guide nonribosomal peptide assembly. Org. Lett. 2013, 15, 3674–3677. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wen, W.-H.; Pu, J.-Y.; Tang, M.-C.; Zhang, L.; Peng, C.; Xu, Y.; Tang, G.-L. Extracellularly oxidative activation and inactivation of matured prodrug for cryptic self-resistance in naphthyridinomycin biosynthesis. Proc. Natl Acad. Sci. USA 2018, 115, 11232–11237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thibodeaux, C.J.; Melançon, C.E., III; Liu, H.W. Natural-product sugar biosynthesis and enzymatic glycodiversification. Angew. Chem. Int. Ed. 2008, 47, 9814–9859. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Phillips, G.N., Jr.; Thorson, J.S. The structural biology of enzymes involved in natural product glycosylation. Nat. Prod. Rep. 2012, 29, 1201–1237. [Google Scholar] [CrossRef] [Green Version]
- Hiratsuka, T.; Koketsu, K.; Minami, A.; Kaneko, S.; Yamazaki, C.; Watanabe, K.; Oguri, H.; Oikawa, H. Core assembly mechanism of quinocarcin/SF-1739: Bimodular complex nonribosomal peptide synthetases for sequential Mannich-type reactions. Chem. Biol. 2013, 20, 1523–1535. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Song, Y.; Tang, M.; Li, M.; Deng, J.; Wong, N.; Ju, J. Genome-directed discovery of tetrahydroisoquinolines from deep-sea derived Streptomyces niveus SCSIO 3406. J. Org. Chem. 2021, 86, 11107–11116. [Google Scholar] [CrossRef]
- Wen, W.-H.; Zhang, Y.; Zhang, Y.-Y.; Yu, Q.; Jiang, C.-C.; Tang, M.-C.; Pu, J.-Y.; Wu, L.; Zhao, Y.-L.; Shi, T.; et al. Reductive inactivation of the hemiaminal pharmacophore for resistance against tetrahydroisoquinoline antibiotics. Nat. Commun. 2021, 12, 7085. [Google Scholar] [CrossRef]
- Almabruk, K.H.; Dinh, L.K.; Philmus, B. Self-resistance of natural product producers: Past, present, and future focusing on self-resistant protein variants. ACS Chem. Biol. 2018, 13, 1426–1437. [Google Scholar] [CrossRef] [PubMed]
- Ashley, E.R.; Cruz, E.G.; Stoltz, B.M. The total synthesis of (-)-lemonomycin. J. Am. Chem. Soc. 2003, 125, 15000–15001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiss, M.; Russell-Maynard, J.; Joule, A. 1,3-Dipolar cycloadditions to oxidopyraziniums. Tetrahedron Lett. 1987, 28, 2187–2190. [Google Scholar] [CrossRef]
- Allway, P.A.; Sutherland, J.K.; Joule, J.A. A synthesis of (±)-7-methoxycarbonyl-2-(3-methoxyphenylmethylidene)-8-methyl-3,8-diazabicyclo[3.2.1] Octan-4-one (1b) using dipolar cycloaddition to a 3-oxidopyrazinium. Tetrahedron Lett. 1990, 31, 4781–4782. [Google Scholar] [CrossRef]
- Yates, N.D.; Peters, D.A.; Allway, P.A.; Beddeoes, R.L.; Scopes, D.I.C.; Joule, J.A. 1,3-Dipolar cycloadditions to oxidopyraziniums. Heterocycles 1995, 40, 331–347. [Google Scholar]
- Reetz, M.T.; Schmitz, A. Stereoselective Grignard-type reaction of chiral N,N-dibenzylamino ketones. Tetrahedron Lett. 1999, 40, 2737–2740. [Google Scholar] [CrossRef]
- Reetz, M.T. Synthesis and diastereoselective reactions of N,N-dibenzylamino aldehydes and related compounds. Chem. Rev. 1999, 99, 1121–1162. [Google Scholar] [CrossRef]
- Pappo, R.; Allen, D.S., Jr.; Lemieux, R.U.; Johnson, W.S. Osmium tetroxide-catalyzed periodate oxidation of olefinic bonds. J. Org. Chem. 1956, 21, 478–479. [Google Scholar] [CrossRef]
- Thom, C.; Kocieński, P. A practical method for N-acylation of bornane-2,10-sultam and 2-oxazolidinones. Synthesis 1992, 6, 582–586. [Google Scholar] [CrossRef]
- Yoshida, A.; Akaiwa, M.; Asakawa, T.; Hamashima, Y.; Yokoshima, S.; Fukuyama, T.; Kan, T. Total synthesis of (-)-lemonomycin. Chem. Eur. J. 2012, 18, 11192–11195. [Google Scholar] [CrossRef]
- Fukuyama, T.; Nunes, J.J. Stereocontrolled total synthesis of (±)-quinocarcin. J. Am. Chem. Soc. 1988, 110, 5196–5198. [Google Scholar] [CrossRef]
- Fukuyama, T.; Yang, L.; Ajeck, K.L.; Sachleben, R.A. Total synthesis of (±)-saframycin A. J. Am. Chem. Soc. 1990, 112, 3712–3713. [Google Scholar] [CrossRef]
- Mori, K.; Rikimaru, K.; Kan, T.; Fukuyama, T. Synthetic studies on (+)-naphthyridinomycin: Stereoselective synthesis of the tetracyclic core framework. Org. Lett. 2004, 6, 3095–3097. [Google Scholar] [CrossRef] [PubMed]
- Majetich, G.; Lowery, D.; Khetani, V.; Song, J.; Hull, K.; Ringold, C. Intramolecular additions of allylsilanes to conjugated dienones. Direct stereoselective syntheses of (±)-neolemnanyl acetate and (±)-neolemnane. J. Org. Chem. 1991, 56, 3973–3988. [Google Scholar] [CrossRef]
- Gallina, C.; Liberatori, A. Condensation of 1,4-diacetylpiperazine-2,5-dione with aldehydes. Tetrahedron 1974, 30, 667–673. [Google Scholar] [CrossRef]
- Ritter, T.; Stanek, K.; Larrosa, I.; Carreira, E.M. Mild cleavage of aryl mesylates: Methanesulfonate as potent protecting group for phenols. Org. Lett. 2004, 6, 1513–1514. [Google Scholar] [CrossRef]
- Mukaiyama, T. Explorations into new reaction chemistry. Angew. Chem. Int. Ed. 2004, 43, 5590–5614. [Google Scholar] [CrossRef]
- Magnus, P.; Matthews, K.S. Synthesis of the tetrahydroisoquinoline alkaloid (±)-renieramycin G and a (±)-lemonomycinone analogue from a common intermediate. J. Am. Chem. Soc. 2005, 127, 12476–12477. [Google Scholar] [CrossRef]
- Magnus, P.; Matthews, K.S. A divergent strategy for synthesis of the tetrahydroisoquinoline alkaloids renieramycin G and a lemonomycin analog. Tetrahedron 2012, 68, 6343–6360. [Google Scholar] [CrossRef]
- Magnus, P.; Matthews, K.S.; Lynch, V. New strategy for the synthesis of tetrahydroisoquinoline alkaloids. Org. Lett. 2003, 5, 2181–2184. [Google Scholar] [CrossRef]
- Castro, C.E.; Havlin, R.; Honwad, V.K.; Malte, A.M.; Moje, S.W. Copper(I) substitutions. Scope and mechanism of cuprous acetylide substitutions. J. Am. Chem. Soc. 1969, 91, 6464–6470. [Google Scholar] [CrossRef]
- Kaufman, T.S. Convenient one-pot synthesis of primary α-alkoxystannanes. Synlett 1997, 12, 1377–1378. [Google Scholar] [CrossRef]
- Yu, C.; Hu, L. A facile synthesis of cyclic enecarbamates using Dess–Martin periodinane. Tetrahedron Lett. 2001, 42, 5167–5170. [Google Scholar] [CrossRef]
- Pearlman, W.M. Noble metal hydroxides on carbon nonpyrophoric dry catalysts. Tetrahedron Lett. 1967, 8, 1663–1664. [Google Scholar] [CrossRef]
- Wu, Y.; Bernadat, G.; Masson, G.; Couturier, C.; Schlama, T.; Zhu, J. Synthetic studies on (-)-lemonomycin: An efficient asymmetric synthesis of lemonomycinone amide. J. Org. Chem. 2009, 74, 2046–2052. [Google Scholar] [CrossRef]
- Corey, E.J.; Xu, F.; Noe, M.C. A rational approach to catalytic enantioselective enolate alkylation using a structurally rigidified and defined chiral quaternary ammonium salt under phase transfer conditions. J. Am. Chem. Soc. 1997, 119, 12414–12415. [Google Scholar] [CrossRef]
- Lygo, B.; Wainwright, P.G. A new class of asymmetric phase-transfer catalysts derived from Cinchona alkaloids–Application in the enantioselective synthesis of α-amino acids. Tetrahedron Lett. 1997, 38, 8595–8598. [Google Scholar] [CrossRef]
- Lygo, B.; Andrews, B.I. Asymmetric phase-transfer catalysis utilizing chiral quaternary ammonium salts: Asymmetric alkylation of glycine imines. Acc. Chem. Res. 2004, 37, 518–525. [Google Scholar] [CrossRef]
- Kokotos, G.; Padrón, J.M.; Martín, T.; Gibbons, W.A.; Martin, V.S. A general approach to the asymmetric synthesis of unsaturated lipidic α-amino acids. The first synthesis of α-aminoarachidonic acid. J. Org. Chem. 1998, 63, 3741–3744. [Google Scholar] [CrossRef]
- Wu, Y.; Zhu, J.P. Hafnium trifluoromethanesulfonate (hafnium triflate) as a highly efficient catalyst for chemoselective thioacetalization and transthioacetalization of carbonyl compounds. J. Org. Chem. 2008, 73, 9522–9524. [Google Scholar] [CrossRef]
- Bernadat, G.; George, N.; Couturier, C.; Masson, G.; Schlama, T.; Zhu, J. Asymmetric synthesis of 2,4,6-trideoxy-4-(dimethylamino)-3-C-methyl-L-lyxohexopyranose (lemonose). Synlett 2011, 4, 576–578. [Google Scholar]
- Frigerio, M.; Santagostino, M.; Sputore, S.; Palmisano, G. Oxidation of alcohols with o-iodoxybenzoic acid in DMSO: A new insight into an old hypervalent iodine reagent. J. Org. Chem. 1995, 60, 7272–7276. [Google Scholar] [CrossRef]
- Rikimaru, K.; Mori, K.; Kan, T.; Fukuyama, T. Synthetic studies on (–)-lemonomycin: Stereocontrolled construction of the 3,8-diazabicyclo[3.2.1] skeleton. Chem. Commun. 2005, 3, 394–396. [Google Scholar] [CrossRef]
- Siengalewicz, P.; Brecker, L.; Mulzer, J. Stereocontrolled synthesis of the tetracyclic core framework of (-)-lemonomycin. Synlett 2008, 16, 2443–2446. [Google Scholar]
- Jiménez-Somarribas, A.; Williams, R.M. Synthetic studies on lemonomycin: Construction of the tetracyclic core. Tetrahedron 2013, 69, 7505–7512. [Google Scholar] [CrossRef] [Green Version]
- Liao, X.W.; Liu, W.; Dong, W.F.; Guan, B.H.; Chen, S.Z.; Liu, Z.Z. Total synthesis of (−)-renieramycin G from l-tyrosine. Tetrahedron 2009, 65, 5709–5715. [Google Scholar] [CrossRef]
- Kren, V.; Rezanka, T. Sweet antibiotics-the role of glycosidic residues in antibiotic and antitumor activity and their randomization. FEMS Microbiol. Rev. 2008, 32, 858–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Constantine, K.L.; Mueller, L.; Huang, S.; Abid, S.; Lam, K.S.; Li, W.; Leet, J.E. Conformation and absolute configuration of nocathiacin I determined by NMR spectroscopy and chiral capillary electrophoresis. J. Am. Chem. Soc. 2002, 124, 7284–7285. [Google Scholar] [CrossRef]
- Zhang, C.; Herath, K.; Jayasuriya, H.; Ondeyka, J.G.; Zink, D.L.; Occi, J.; Birdsall, G.; Venugopal, J.; Ushio, M.; Burgess, B.; et al. Thiazomycins, thiazolyl peptide antibiotics from Amycolatopsis fastidiosa. J. Nat. Prod. 2009, 72, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Northcote, P.T.; Siegel, M.; Borders, D.B.; Lee, M.D. Glycothiohexide α, a novel antibiotic produced by “Sebekia” sp., LL-14E605. III. Structural elucidation. J. Antibiot. 1994, 47, 901–908. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.S.; Brady, S.F. Arimetamycin A: Improving clinically relevant families of natural products through sequence-guided screening of soil metagenomes. Angew. Chem. Int. Ed. 2013, 52, 11063–11067. [Google Scholar] [CrossRef] [Green Version]
- Huseman, E.D.; Byl, J.A.W.; Chapp, S.M.; Schley, N.D.; Osheroff, N.; Townsend, S.D. Synthesis and cytotoxic evaluation of arimetamycin A and its daunorubicin and doxorubicin hybrids. ACS Cent. Sci. 2021, 7, 1327–1337. [Google Scholar] [CrossRef]
- Hegde, V.R.; Patel, M.G.; Das, P.R.; Pramanik, B.; Puar, M.S. A family of novel macrocyclic lactones, the saccharocarcins produced by Saccharothrix aerocolonigenes subsp. antihiotica. II. Physico-chemical properties and structure determination. J. Antibiot. 1997, 50, 126–134. [Google Scholar] [CrossRef] [Green Version]
- Pfrengle, F.H.-U. Reissig, Amino sugars and their mimetics via 1,2-oxazines. Chem. Soc. Rev. 2010, 39, 549–557. [Google Scholar] [CrossRef]
- Ding, F.; Cai, S.; William, R.; Liu, X.-W. Pathways leading to 3-amino- and 3-nitro-2,3-dideoxy sugars: Strategies and synthesis. RSC Adv. 2013, 3, 13594. [Google Scholar] [CrossRef]
- Skarbek, K.; Milewska, M.J. Biosynthetic and synthetic access to amino sugars. Carbohydr. Res. 2016, 434, 44–71. [Google Scholar] [CrossRef] [PubMed]
- Behera, A.; Kulkarni, S.S. Chemical synthesis of rare, deoxy-amino sugars containing bacterial glycoconjugates as potential vaccine candidates. Molecules 2018, 23, 1997. [Google Scholar] [CrossRef]
- Huseman, E.D.; Townsend, S.D. De novo synthesis of an L-lemonose thioglycoside donor from D-Threonine. Tetrahedron Lett. 2021, 73, 153097. [Google Scholar] [CrossRef]
- Gloster, T.M. Exploitation of carbohydrate processing enzymes in biocatalysis. Curr. Opin. Chem. Biol. 2020, 55, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Kim, H.U.; Chae, T.U.; Cho, J.S.; Kim, J.W.; Shin, J.H.; Kim, D.I.; Ko, Y.-S.; Jang, W.D.; Jang, Y.-S. A comprehensive metabolic map for production of bio-based chemicals. Nat. Catal. 2019, 2, 18–33. [Google Scholar] [CrossRef]
























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tao, S.; Wang, Y.; Hong, R.; Huang, S.-H. Potent Antibiotic Lemonomycin: A Glimpse of Its Discovery, Origin, and Chemical Synthesis. Molecules 2022, 27, 4324. https://doi.org/10.3390/molecules27134324
Tao S, Wang Y, Hong R, Huang S-H. Potent Antibiotic Lemonomycin: A Glimpse of Its Discovery, Origin, and Chemical Synthesis. Molecules. 2022; 27(13):4324. https://doi.org/10.3390/molecules27134324
Chicago/Turabian StyleTao, Shunan, Yang Wang, Ran Hong, and Sha-Hua Huang. 2022. "Potent Antibiotic Lemonomycin: A Glimpse of Its Discovery, Origin, and Chemical Synthesis" Molecules 27, no. 13: 4324. https://doi.org/10.3390/molecules27134324