
Anti-Inflammatory and Anti-Rheumatic Potential of Selective Plant Compounds by Targeting TLR-4/AP-1 Signaling: A Comprehensive Molecular Docking and Simulation Approaches

 , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Methods
2.1. Protein Selection
2.2. Ligand Database Preparation
2.3. Docking Protocol
2.4. Pharmacokinetics Parameters Assessment
2.5. Toxicokinetic Parameter Assessment
2.6. Drug-Likeness Analysis
2.7. MD Simulation
2.8. Analysis of MD Simulation and Calculation of Secondary Structure Content
2.9. Statistical Analysis
3. Results
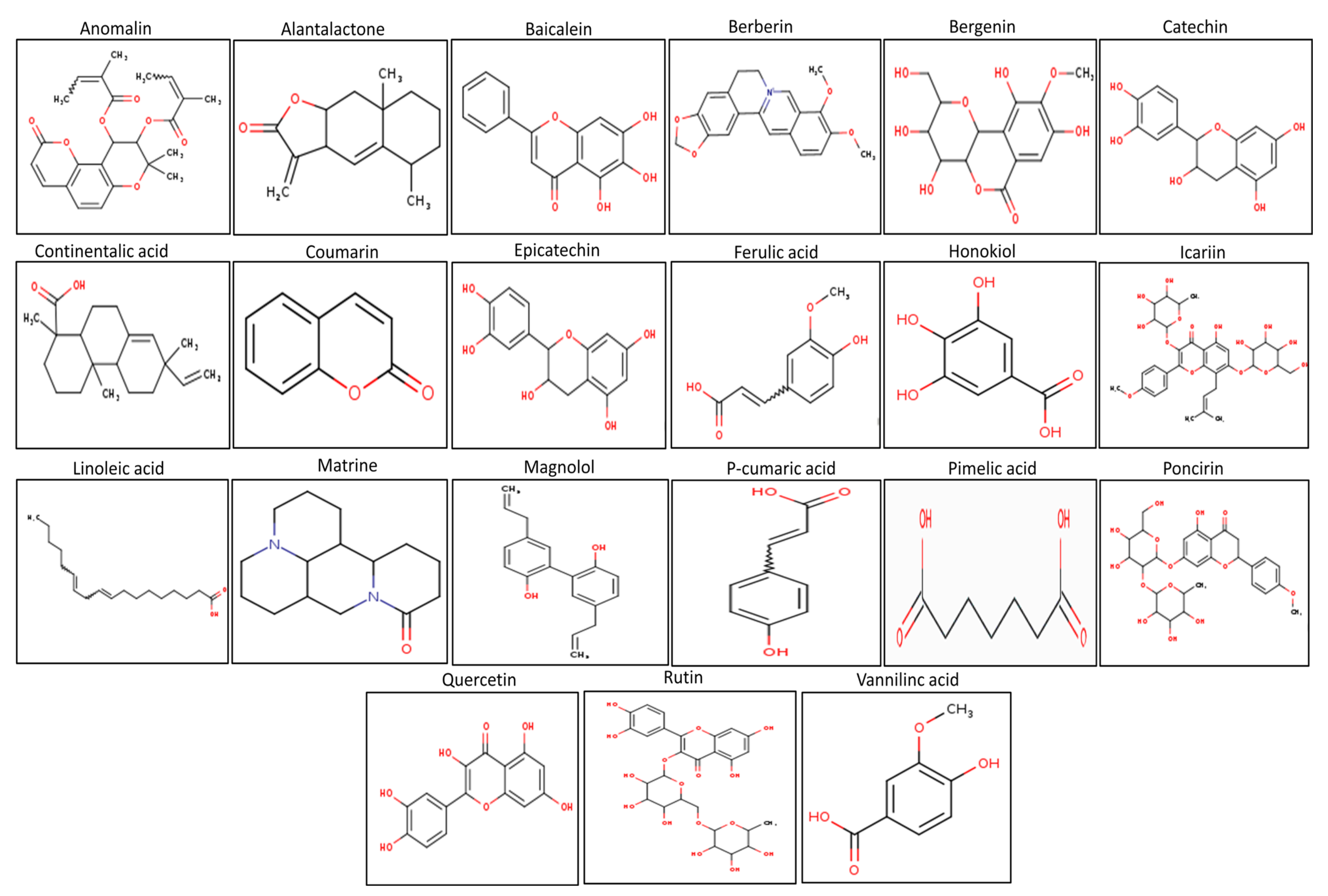
3.1. The Physico-Chemical Analysis of Selected Compounds
3.2. Toxicokinetic Analysis of Selected Natural Compounds
3.3. Pharmacokinetic Behavior
3.4. Drug-Likeness Behavior of the Studied Compounds
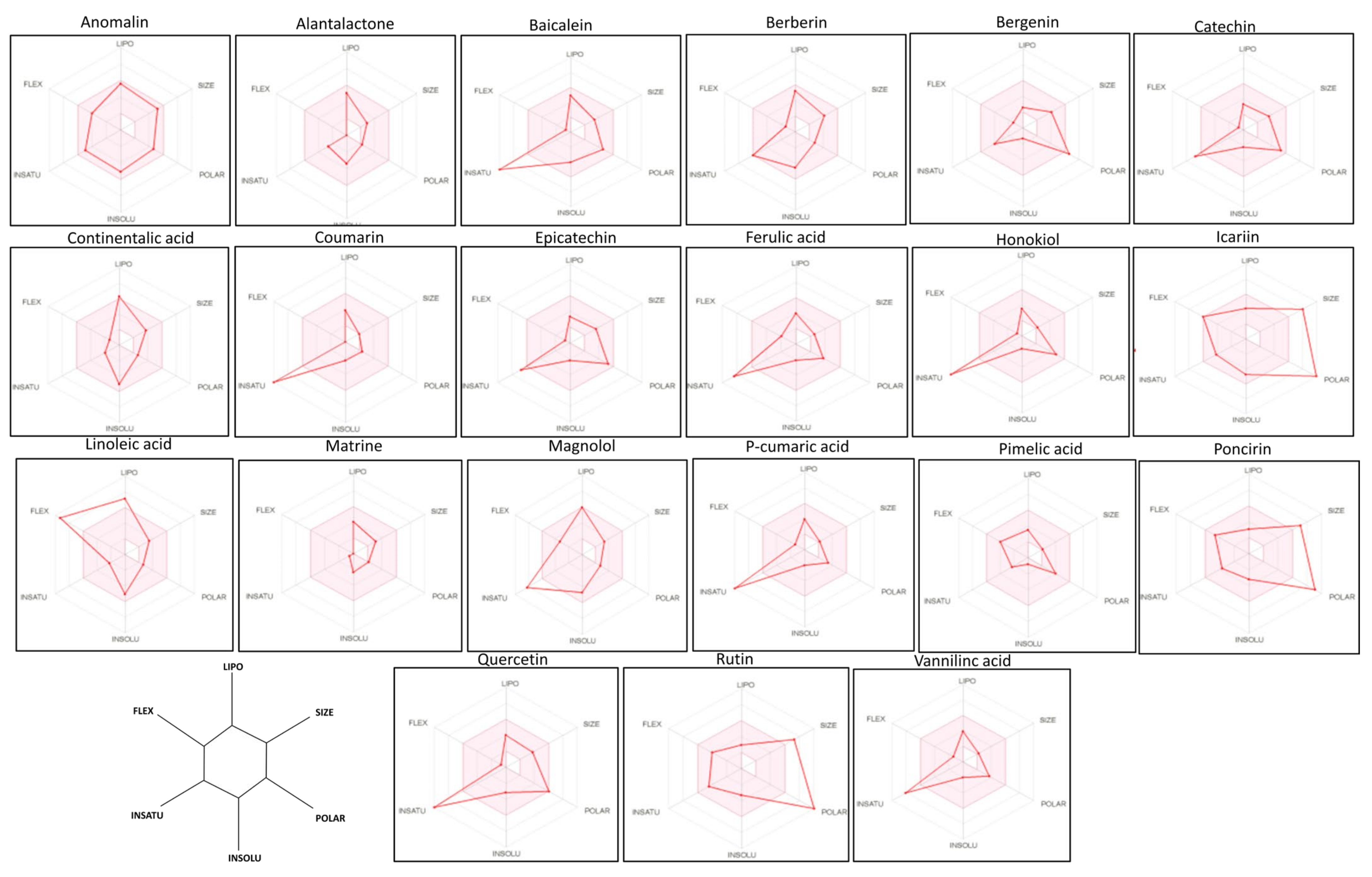
3.5. Bioavailability Radar Analysis of the Drugs
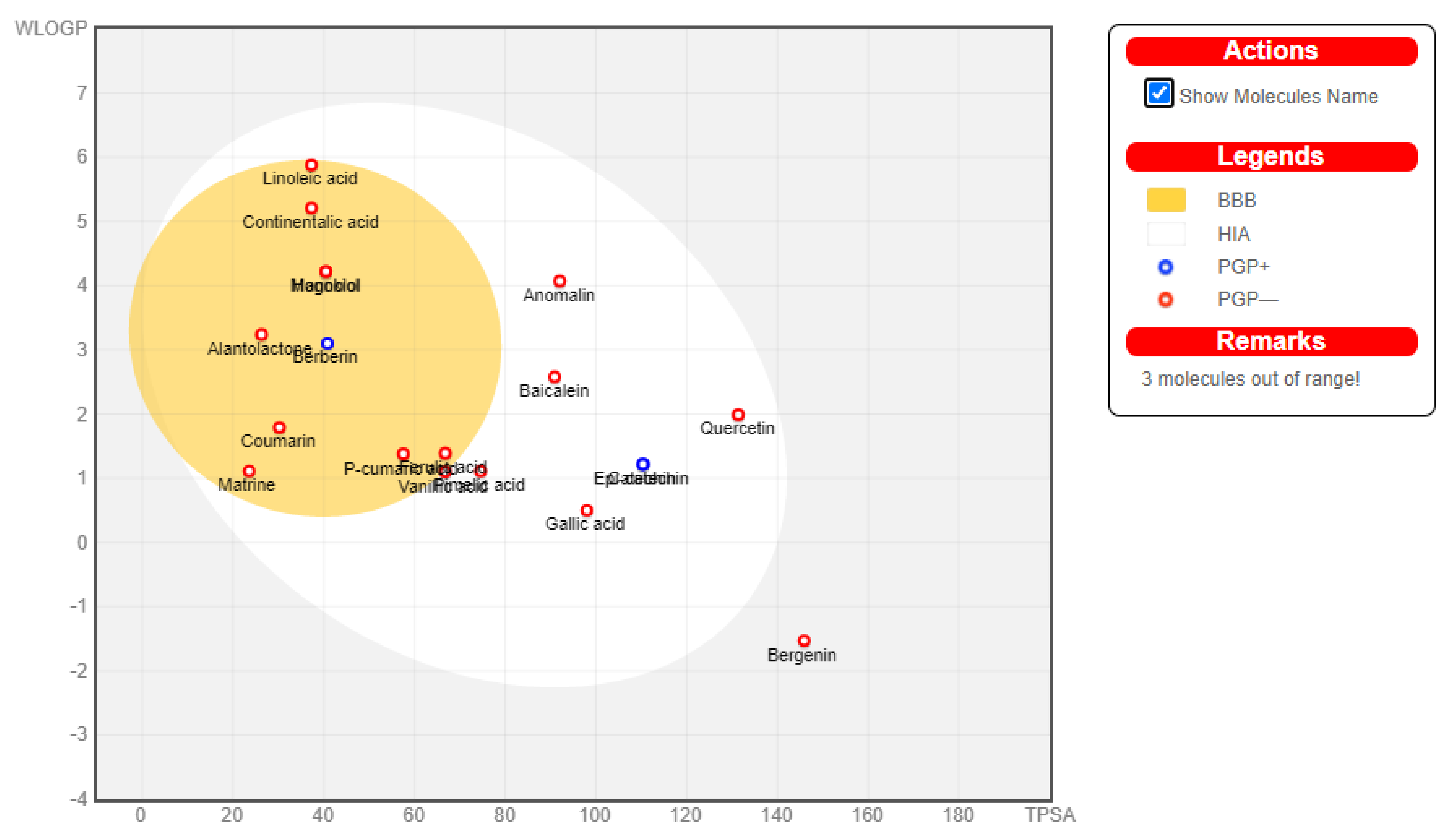
3.6. Boiled Egg Analysis
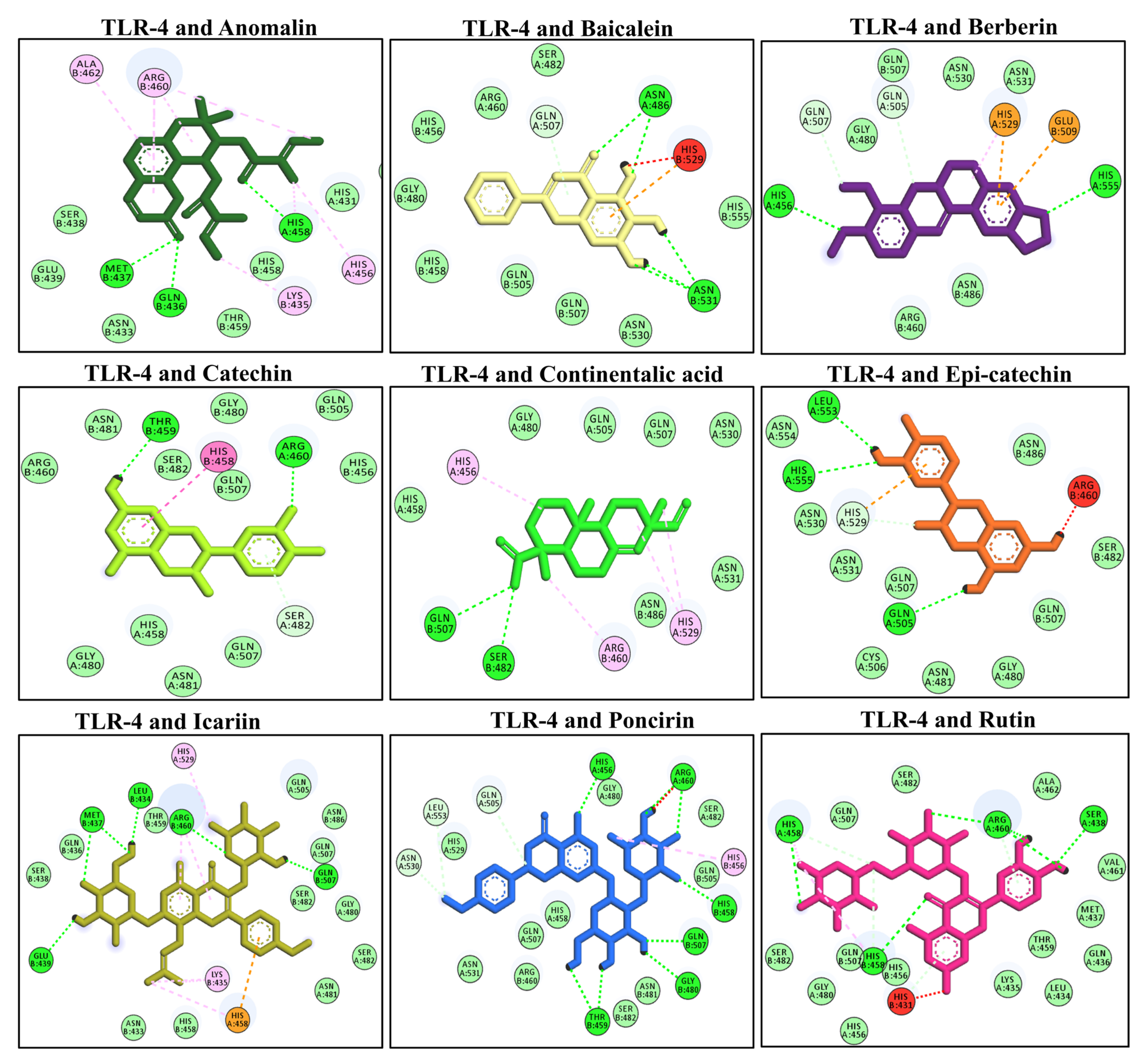
3.7. Molecular Docking of Selected Compounds with TLR-4
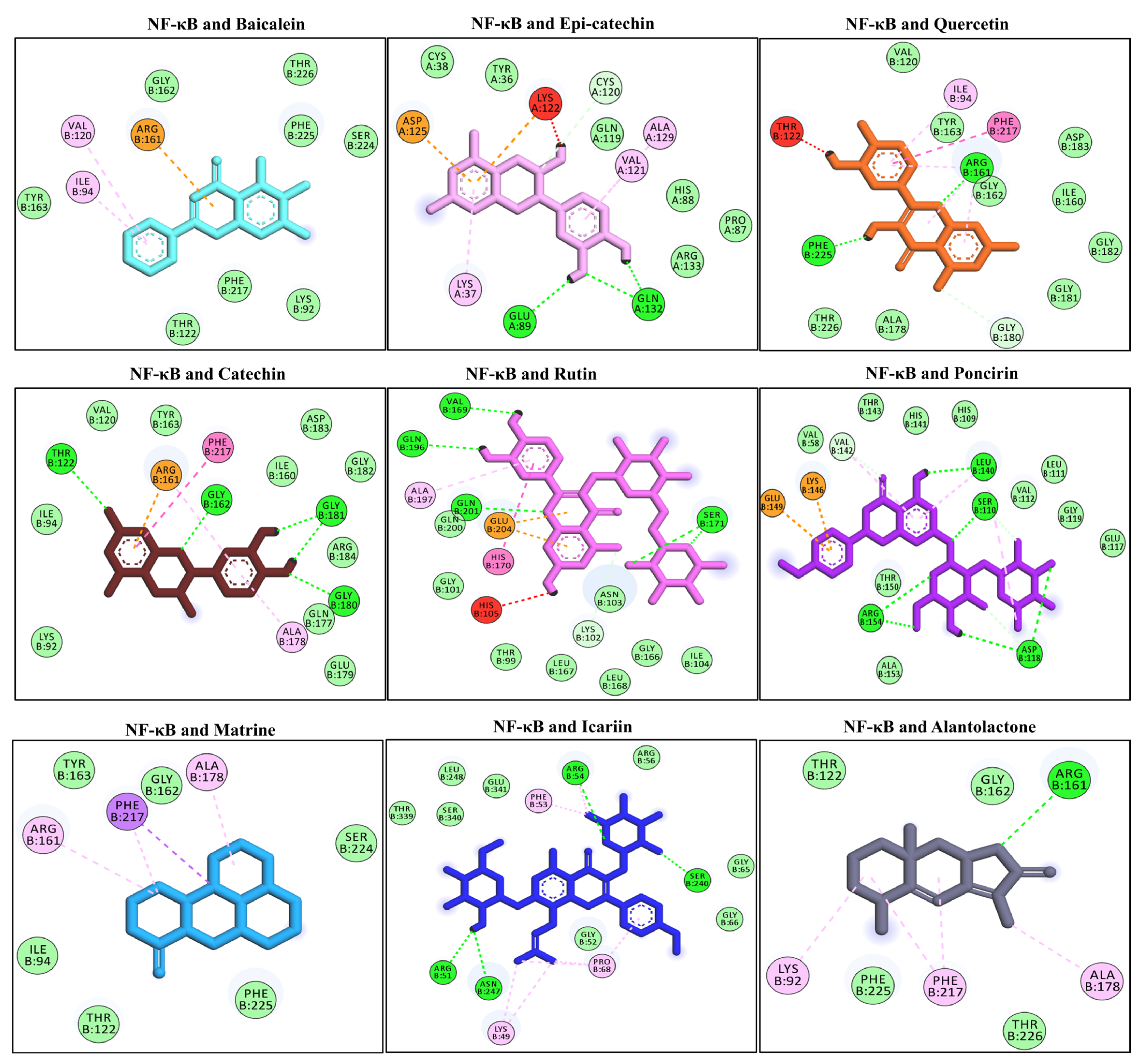
3.8. Docking Interaction with the NF-κB
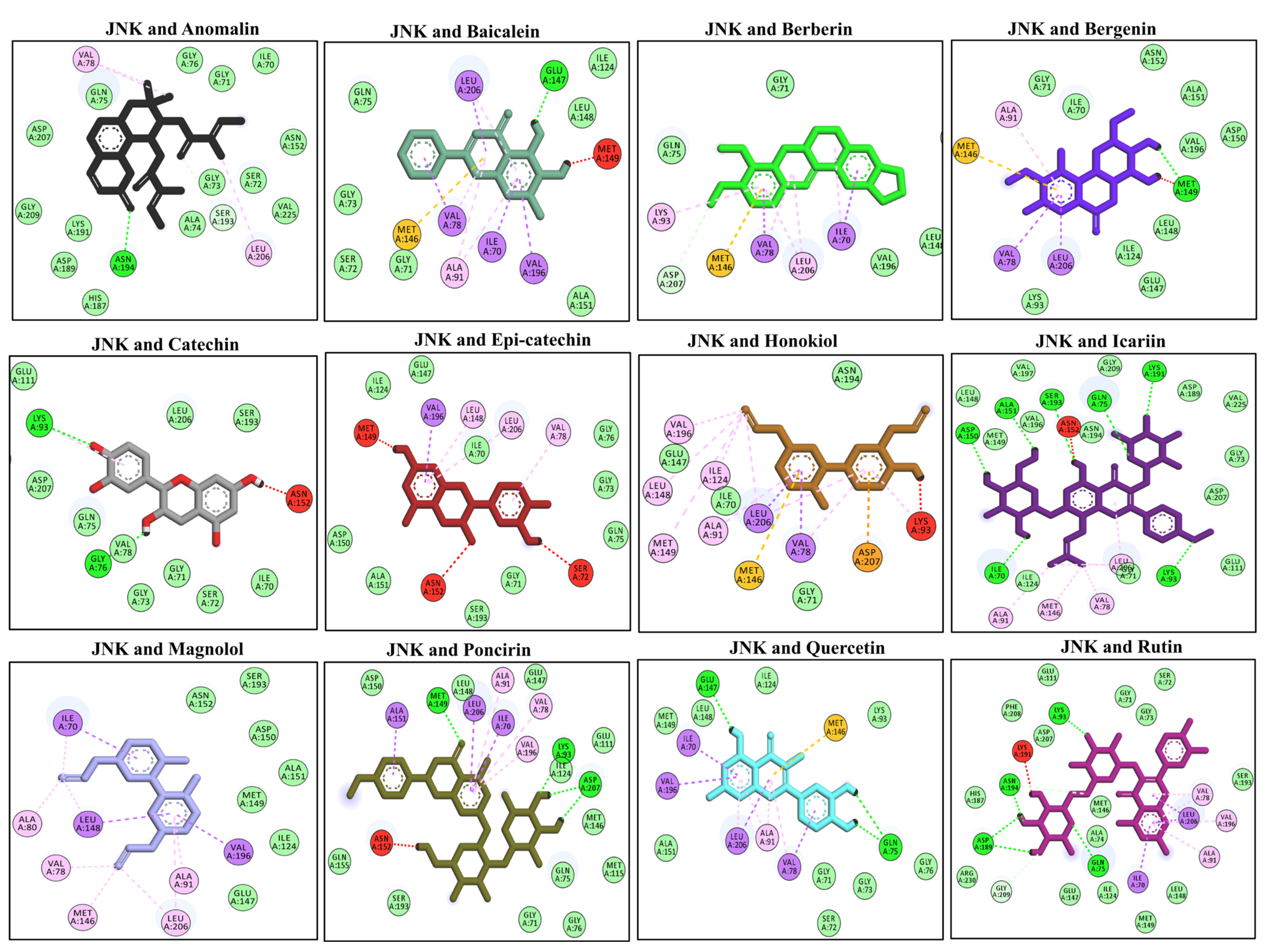
3.9. Docking Interaction with the MAPKs (JNK)
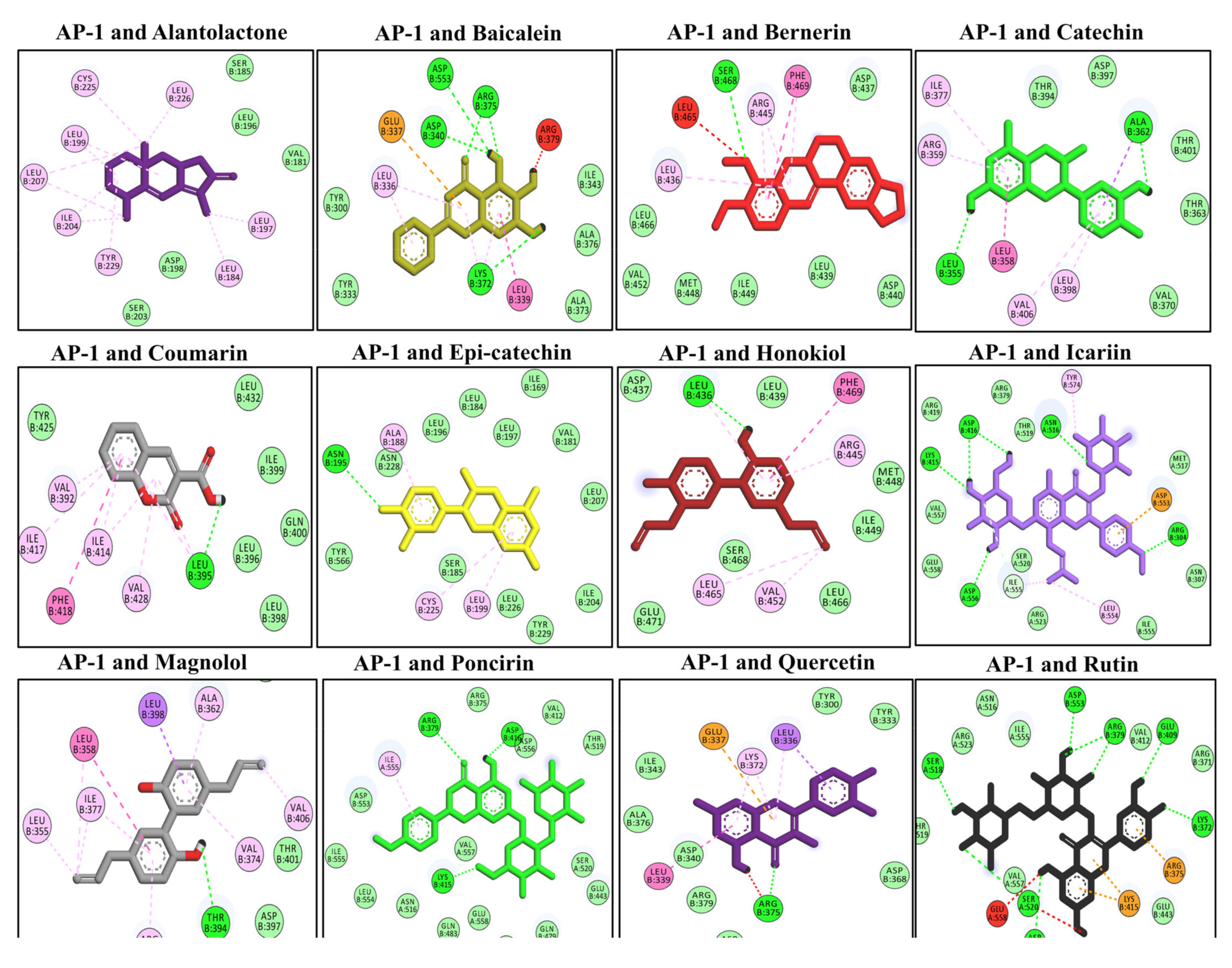
3.10. Docking Interaction with the AP-1
3.11. Analysis of MD Simulation and Calculation of Secondary Structure Content
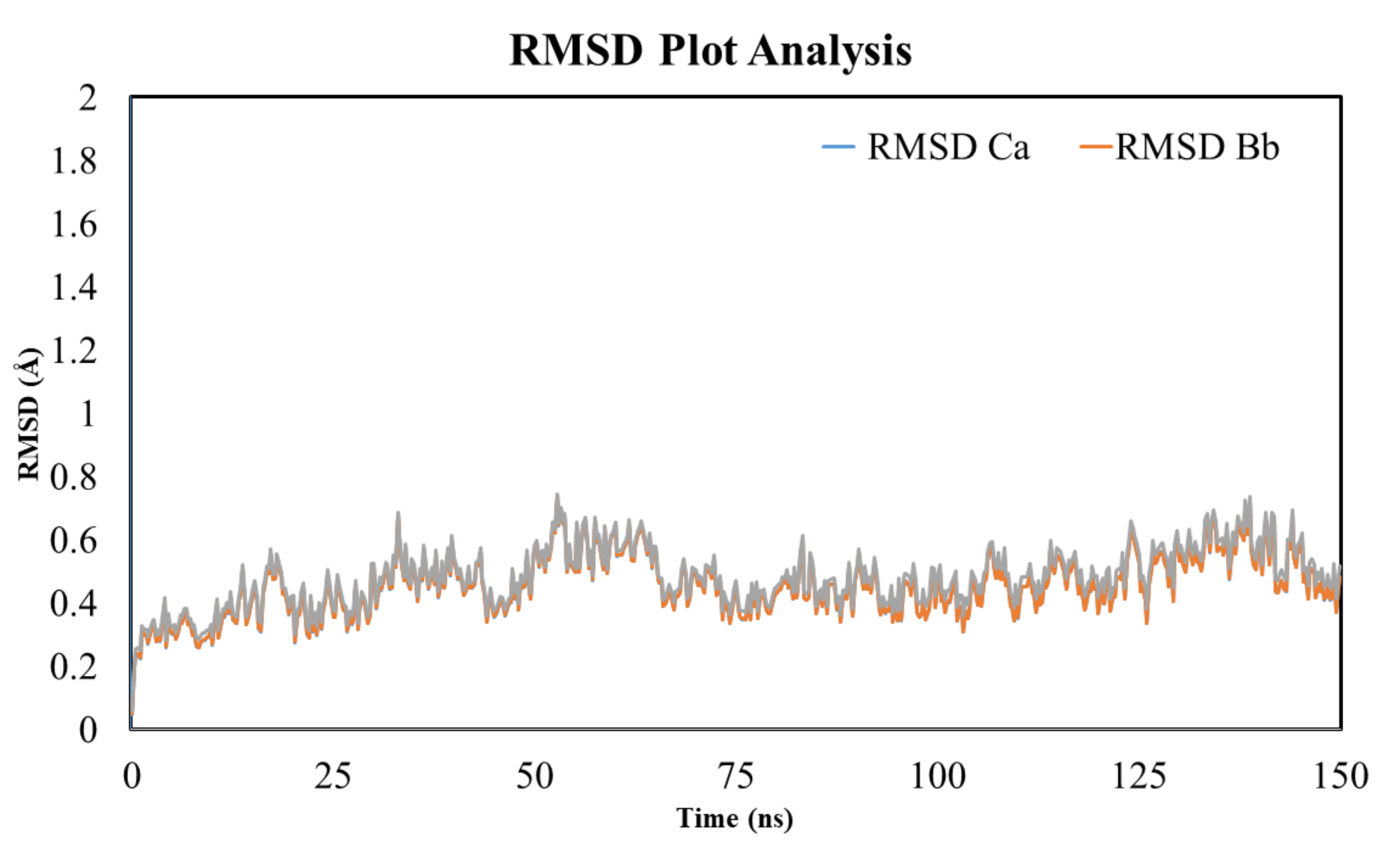
3.12. RMSD Analysis
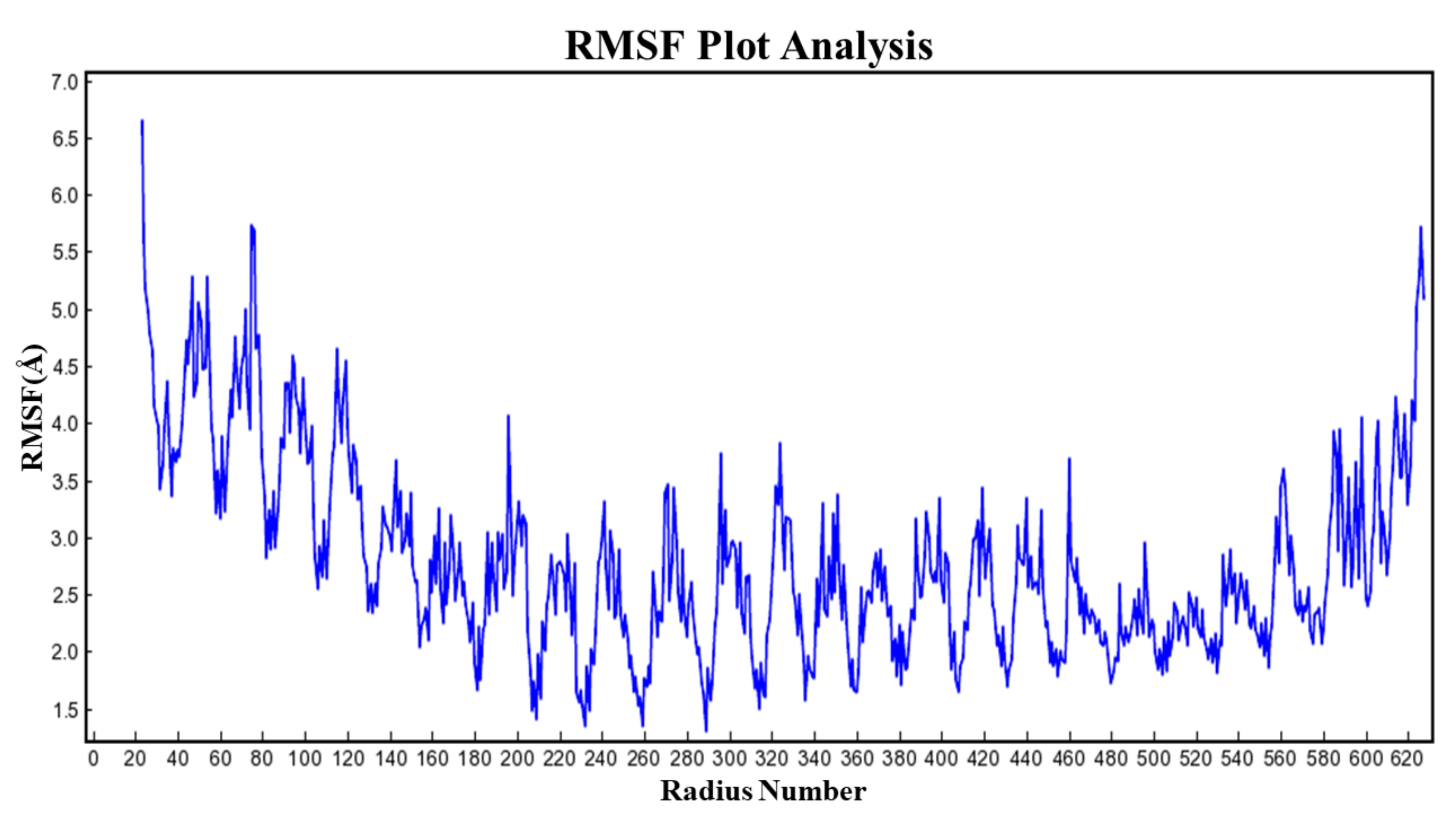
3.13. RMSF Analysis
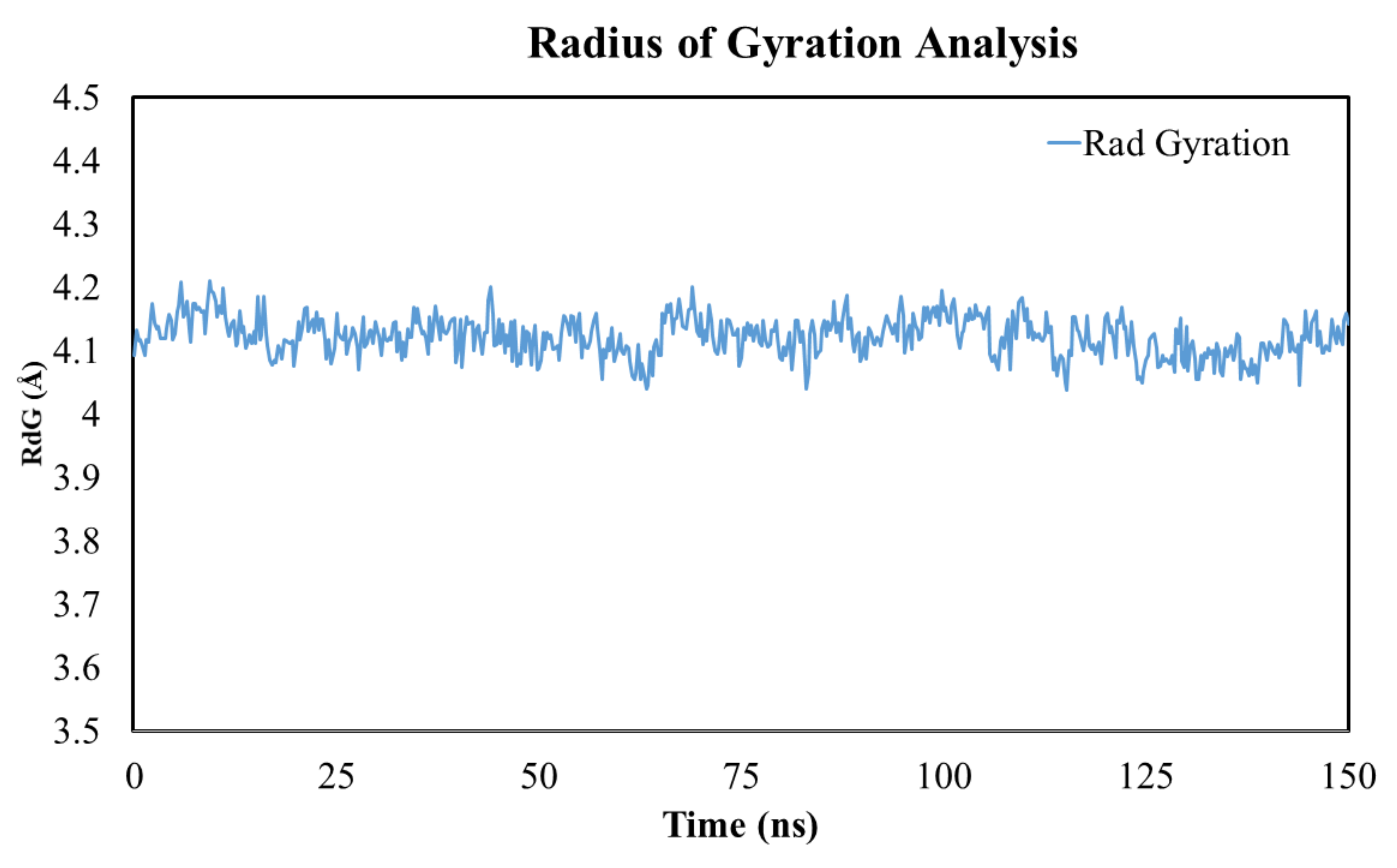
3.14. Radius of Gyration
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Khan, A.; Ullah, M.Z.; Afridi, R.; Rasheed, H.; Khalid, S.; Ullah, H.; Ali, H.; AlSharari, S.D.; Kim, Y.S.; Khan, S. Antinociceptive properties of 25-methoxy hispidol A, a triterpinoid isolated from Poncirus trifoliata (Rutaceae) through inhibition of nf-κb signalling in mice. Phytother. Res. 2019, 33, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Supandi, Y.; Dwita, L.P. Docking studies and molecular dynamics simulation of compounds contained in Kaempferia galanga L. to lipoxygenase (lox) for anti-inflammatory drugs. J. Math. Fundam. Sci. 2021, 53, 218–230. [Google Scholar] [CrossRef]
- Khalid, S.; Ullah, M.Z.; Khan, A.U.; Afridi, R.; Rasheed, H.; Khan, A.; Ali, H.; Kim, Y.S.; Khan, S. Antihyperalgesic properties of honokiol in inflammatory pain models by targeting of nf-κb and nrf2 signaling. Front. Pharmacol. 2018, 9, 140. [Google Scholar] [CrossRef]
- Ullah, H.; Khan, A.; Baig, M.W.; Ullah, N.; Ahmed, N.; Tipu, M.K.; Ali, H.; Khan, S. Poncirin attenuates CCL4-induced liver injury through inhibition of oxidative stress and inflammatory cytokines in mice. BMC Complement. Med. Ther. 2020, 20, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.; Khan, S.; Ali, H.; Shah, K.U.; Ali, H.; Shehzad, O.; Onder, A.; Kim, Y.S. Anomalin attenuates LPS-induced acute lungs injury through inhibition of AP-1 signaling. Int. Immunopharmacol. 2019, 73, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.M.; Khan, A.U.; Ali, H.; Islam, S.U.; Seo, E.K.; Khan, S. Continentalic acid exhibited nephroprotective activity against the LPS and E. coli-induced kidney injury through inhibition of the oxidative stress and inflammation. Int. Immunopharmacol. 2020, 80, 106209. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.U.; Khan, A.; Khan, A.; Shal, B.; Aziz, A.; Ahmed, M.N.; Islam, S.U.; Ali, H.; Shehzad, A.; Khan, S. Inhibition of nf-κb signaling and hsp70/hsp90 proteins by newly synthesized hydrazide derivatives in arthritis model. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2021, 394, 1497–1519. [Google Scholar] [CrossRef]
- Sana, E.; Zeeshan, M.; Ain, Q.U.; Khan, A.U.; Hussain, I.; Khan, S.; Lepeltier, E.; Ali, H. Topical delivery of curcumin-loaded transfersomes gel ameliorated rheumatoid arthritis by inhibiting NF-κβ pathway. Nanomedicine 2021, 16, 819–837. [Google Scholar] [CrossRef]
- Batool, M.; Choi, S. TLR4-targeting therapeutics: Structural basis and computer-aided drug discovery approaches. Molecules 2020, 25, 627. [Google Scholar] [CrossRef] [Green Version]
- Sinha, S.; Doble, M.; Manju, S.J.E. Design, synthesis and identification of novel substituted 2-amino thiazole analogues as potential anti-inflammatory agents targeting 5-lipoxygenase. Eur. J. Med. Chem. 2018, 158, 34–50. [Google Scholar] [CrossRef]
- Dahan, A.; Markovic, M.; Keinan, S.; Kurnikov, I.; Aponick, A.; Zimmermann, E.M.; Ben-Shabat, S. Computational modeling and in-vitro/in-silico correlation of phospholipid-based prodrugs for targeted drug delivery in inflammatory bowel disease. J. Comput. Mol. Des. 2017, 31, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Waltenberger, B.; Atanasov, A.G.; Heiss, E.H.; Bernhard, D.; Rollinger, J.; Breuss, J.M.; Schuster, D.; Bauer, R.; Kopp, B.; Franz, C.; et al. Drugs from nature targeting inflammation (DNTI): A successful Austrian interdisciplinary network project. Monatsh Chem. 2016, 147, 479–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Zhou, Z.; Ding, K.; Yuan, Y.; Loftin, C.; Zheng, F.; Zhan, C.-G. DREAM-in-CDM approach and identification of a new generation of anti-inflammatory drugs targeting mPGES-1. Sci. Rep. 2020, 10, 10187. [Google Scholar] [CrossRef]
- Hassan, S.S.U.; Zhang, W.-D.; Jin, H.-Z.; Basha, S.H.; Priya, S. In-silico anti-inflammatory potential of guaiane dimers from Xylopia vielana targeting COX-2. J. Biomol. Struct. Dyn. 2020, 40, 484–498. [Google Scholar] [CrossRef]
- Quasdorf, K.W.; Antoft-Finch, A.; Liu, P.; Silberstein, A.L.; Komaromi, A.; Blackburn, T.; Ramgren, S.D.; Houk, K.N.; Snieckus, V.; Garg, N.K. Suzuki−Miyaura cross-coupling of aryl carbamates and sulfamates: Experimental and computational studies. J. Am. Chem. Soc. 2011, 133, 6352–6363. [Google Scholar] [CrossRef] [Green Version]
- Fouda, A.M.; Hassan, A.H.; Eliwa, E.M.; Ahmed, H.E.; Al-Dies, A.-A.M.; Omar, A.M.; Nassar, H.S.; Halawa, A.H.; Aljuhani, N.; El-Agrody, A.M.J.B.C. Targeted potent antimicrobial benzochromene-based analogues: Synthesis, computational studies, and inhibitory effect against 14α-demethylase and DNA gyrase. Bioorg. Chem. 2020, 105, 104387. [Google Scholar] [CrossRef] [PubMed]
- Naveja, J.J.; Madariaga-Mazón, A.; Flores-Murrieta, F.; Granados-Montiel, J.; Maradiaga-Ceceña, M.; Alaniz, V.D.; Maldonado-Rodriguez, M.; García-Morales, J.; Senosiain-Peláez, J.P.; Martinez-Mayorga, K.J.D.d.t. Union is strength: Antiviral and anti-inflammatory drugs for COVID-19. Drug Discov. Today 2021, 26, 229. [Google Scholar] [CrossRef]
- Hussain, M.S.; Azam, F.; Eldarrat, H.A.; Alkskas, I.; Mayoof, J.A.; Dammona, J.M.; Ismail, H.; Ali, M.; Arif, M.; Haque, A.J.A.J.o.C. Anti-inflammatory, analgesic and molecular docking studies of lanostanoic acid 3-o-α-d-glycopyranoside isolated from helichrysum stoechas. Arabian J. Chem. 2020, 13, 9196–9206. [Google Scholar] [CrossRef]
- Herowati, R.; Widodo, G.P. Molecular Docking Analysis: Interaction Studies of Natural Compounds to Anti-Inflammatory Targets Quantitative Structure-Activity Relationship; IntechOpen: Rijeka, Croatia, 2017. [Google Scholar] [CrossRef] [Green Version]
- Fu, W.; Chen, L.; Wang, Z.; Zhao, C.; Chen, G.; Liu, X.; Cai, Y.; Zhou, J.; Dai, Y.; Cai, Y.; et al. Determination of the binding mode for anti-inflammatory natural product xanthohumol with myeloid differentiation protein 2. Drug Des. Dev. Ther. 2016, 10, 455–463. [Google Scholar] [CrossRef] [Green Version]
- Ramezanpour, M.; Leung, S.S.; Delgado-Magnero, K.H.; Bashe, B.Y.; Thewalt, J.; Tieleman, D.P. Computational and experimental approaches for investigating nanoparticle-based drug delivery systems. Biochim. Biophys. Acta BBA Biomembr. 2016, 1858, 1688–1709. [Google Scholar] [CrossRef]
- Namas, R.; Mi, Q.; Namas, R.; Almahmoud, K.; Zaaqoq, A.M.; Abdul-Malak, O.; Azhar, N.; Day, J.; Abboud, A.; Zamora, R.; et al. Insights into the role of chemokines, damage-associated molecular patterns, and lymphocyte-derived mediators from computational models of trauma-induced inflammation. Antioxid. Redox Signal. 2015, 23, 1370–1387. [Google Scholar] [CrossRef] [PubMed]
- Asiedu, S.O.; Kwofie, S.K.; Broni, E.; Wilson, M.D. Computational identification of potential anti-inflammatory natural compounds targeting the p38 mitogen-activated protein kinase (MAPK): Implications for COVID-19-induced cytokine storm. Biomolecules 2021, 11, 653. [Google Scholar] [CrossRef] [PubMed]
- Grienke, M. Targeting inflammation and influenza: Integration of computational methods into lead finding from natural sources. Planta Med. 2012, 78, AL3. [Google Scholar] [CrossRef]
- Zanfirescu, A.; Ungurianu, A.; Mihai, D.P.; Radulescu, D.; Nitulescu, G.M. Targeting monoacylglycerol lipase in pursuit of therapies for neurological and neurodegenerative diseases. Molecules 2021, 26, 5668. [Google Scholar] [CrossRef] [PubMed]
- Norouzi, R.; Hojati, Z.; Dehbashi, M. In vitro and computational studies on the effects of ARE deletion and targeted mutations on the expression of interferon beta-1a in HEK293T cells. Appl. Microbiol. Biotechnol. 2018, 102, 7047–7059. [Google Scholar] [CrossRef]
- Yang, J.; Tian, B.; Brasier, A.R. Targeting chromatin remodeling in inflammation and fibrosis. Adv. Protein Chem. Struct. Biol. 2017, 107, 1–36. [Google Scholar] [CrossRef]
- Baron, G.; Altomare, A.; Regazzoni, L.; Redaelli, V.; Grandi, S.; Riva, A.; Morazzoni, P.; Mazzolari, A.; Carini, M.; Vistoli, G.; et al. Pharmacokinetic profile of bilberry anthocyanins in rats and the role of glucose transporters: LC–MS/MS and computational studies. J. Pharm. Biomed. Anal. 2017, 144, 112–121. [Google Scholar] [CrossRef]
- Birari, R.B.; Gupta, S.; Mohan, C.G.; Bhutani, K.K. Antiobesity and lipid lowering effects of Glycyrrhiza chalcones: Experimental and computational studies. Phytomedicine 2011, 18, 795–801. [Google Scholar] [CrossRef]
- Rao, B.G. Recent developments in the design of specific matrix metalloproteinase inhibitors aided by structural and computational studies. Curr. Pharm. Des. 2005, 11, 295–322. [Google Scholar] [CrossRef]
- Alam, M.M.; Nazreen, S.; Almalki, A.S.; Elhenawy, A.A.; Alsenani, N.I.; Elbehairi, S.E.I.; Malebari, A.M.; Alfaifi, M.Y.; Alsharif, M.A.; Alfaifi, S.Y. Naproxen based 1,3,4-oxadiazole derivatives as EGFR inhibitors: Design, synthesis, anticancer, and computational studies. Pharmaceuticals 2021, 14, 870. [Google Scholar] [CrossRef]
- Tripathi, A.C.; Upadhyay, S.; Paliwal, S.; Saraf, S.K. N1-benzenesulfonyl-2-pyrazoline hybrids in neurological disorders: Syntheses, biological screening and computational studies. EXCLI J. 2018, 17, 126–148. [Google Scholar] [CrossRef] [PubMed]
- Grzegorzewski, J.; Brandhorst, J.; Green, K.; Eleftheriadou, D.; Duport, Y.; Barthorscht, F.; Köller, A.; Ke, D.Y.J.; De Angelis, S.; König, M. PK-DB: Pharmacokinetics database for individualized and stratified computational modeling. Nucleic Acids Res. 2020, 49, D1358–D1364. [Google Scholar] [CrossRef] [PubMed]
- Rajulapati, V.; Sharma, K.; Dhillon, A.; Goyal, A. SAXS and homology modelling based structure characterization of pectin methylesterase a family 8 carbohydrate esterase from Clostridium thermocellum ATCC 27405. Arch. Biochem. Biophys. 2018, 641, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Yeni, Y.; Supandi, S.; Dwita, L.P.; Suswandari, S.; Shaharun, M.S.; Sambudi, N.S. Docking studies and molecular dynamics simulation of ipomoea batatas l. Leaves compounds as lipoxygenase (lox) inhibitor. Saudi J. Biol. Sci. 2020, 12, S836. [Google Scholar]
- Prasasty, V.D.; Cindana, S.; Ivan, F.X.; Zahroh, H.; Sinaga, E. Structure-based discovery of novel inhibitors of Mycobacterium tuberculosis CYP121 from Indonesian natural products. Comput. Biol. Chem. 2020, 85, 107205. [Google Scholar] [CrossRef]
- Raj, V.; Park, J.G.; Cho, K.-H.; Choi, P.; Kim, T.; Ham, J.; Lee, J. Assessment of antiviral potencies of cannabinoids against SARS-COV-2 using computational and in vitro approaches. Int. J. Biol. Macromol. 2021, 168, 474–485. [Google Scholar] [CrossRef]
- Prasasty, V.D.; Istyastono, E.P. Structure-based design and molecular dynamics simulations of pentapeptide AEYTR as a potential acetylcholinesterase inhibitor. Indones. J. Chem. 2020, 20, 953–959. [Google Scholar] [CrossRef]
- Skrt, M.; Benedik, E.; Podlipnik, Č.; Ulrih, N. Interactions of different polyphenols with bovine serum albumin using fluorescence quenching and molecular docking. Food Chem. 2012, 135, 2418–2424. [Google Scholar] [CrossRef]
- Hsieh, S.-R.; Reddy, P.M.; Chang, C.-J.; Kumar, A.; Wu, W.-C.; Lin, H.-Y. Exploring the behavior of bovine serum albumin in response to changes in the chemical composition of responsive polymers: Experimental and simulation studies. Polymers 2016, 8, 238. [Google Scholar] [CrossRef] [Green Version]
- Loza-Mejía, M.A.; Salazar, J.R.; Sánchez-Tejeda, J. In silico studies on compounds derived from calceolaria: Phenylethanoid glycosides as potential multitarget inhibitors for the development of pesticides. Biomolecules 2018, 8, 121. [Google Scholar] [CrossRef] [Green Version]
- Qu, H.; Ricklin, D.; Bai, H.; Chen, H.; Reis, E.S.; Maciejewski, M.; Tzekou, A.; DeAngelis, R.A.; Resuello, R.R.; Lupu, F.; et al. New analogs of the clinical complement inhibitor compstatin with subnanomolar affinity and enhanced pharmacokinetic properties. Immunobiology 2013, 218, 496–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, I.; Garikapati, K.R.; Setti, A.; Shaik, A.B.; Makani, V.K.K.; Shareef, M.A.; Rajpurohit, H.; Vangara, N.; Pal-Bhadra, M.; Kamal, A.; et al. Design, synthesis, in silico pharmacokinetics prediction and biological evaluation of 1,4-dihydroindeno [1,2-c]pyrazole chalcone as EGFR /Akt pathway inhibitors. Eur. J. Med. Chem. 2019, 163, 636–648. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Schwartz, J.B.; Verotta, D. A sample size computation method for non-linear mixed effects models with applications to pharmacokinetics models. Stat. Med. 2004, 23, 2551–2566. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.F.; Nahar, N.; Bin Rashid, R.; Chowdhury, A.; Rashid, M.A. Computational investigations of physicochemical, pharmacokinetic, toxicological properties and molecular docking of betulinic acid, a constituent of Corypha taliera (Roxb.) with Phospholipase A2 (PLA2). BMC Complement. Altern. Med. 2018, 18, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, T.R.; Renzelman, C.M.; Rand, A.C.; Rezai, T.; McEwen, C.M.; Gelev, V.M.; Turner, R.A.; Linington, R.; Leung, S.S.F.; Kalgutkar, A.S.; et al. On-resin N-methylation of cyclic peptides for discovery of orally bioavailable scaffolds. Nat. Chem. Biol. 2011, 7, 810–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akinpelu, O.I.; Lawal, M.M.; Kumalo, H.M.; Mhlongo, N.N. Computational studies of the properties and activities of selected trisubstituted benzimidazoles as potential antitubercular drugs inhibiting mtb-ftsz polymerization. J. Biomol. Struct. Dyn. 2020, 40, 1558–1570. [Google Scholar] [CrossRef]
- Jamil, W.; Shaikh, J.; Yousuf, M.; Taha, M.; Khan, K.M.; Shah, S.A.A. Synthesis, anti-diabetic and in silico QSAR analysis of flavone hydrazide Schiff base derivatives. J. Biomol. Struct. Dyn. 2021, 13, 1–16. [Google Scholar] [CrossRef]
- Abdulrahman, H.L.; Uzairu, A.; Uba, S. Computational pharmacokinetic analysis on some newly designed 2-anilinopyrimidine derivative compounds as anti-triple-negative breast cancer drug compounds. Bull. Natl. Res. Cent. 2020, 44, 63. [Google Scholar] [CrossRef]
- Tripathi, P.; Ghosh, S.; Talapatra, S.N. Bioavailability prediction of phytochemicals present in Calotropis procera (aiton) R. Br. by using Swiss-ADME tool. World Sci. News 2019, 131, 147–163. [Google Scholar]
- Siraj, M.A.; Rahman, M.S.; Tan, G.T.; Seidel, V. Molecular docking and molecular dynamics simulation studies of triterpenes from Vernonia patula with the cannabinoid type 1 receptor. Int. J. Mol. Sci. 2021, 22, 3595. [Google Scholar] [CrossRef]
- Khan, A.U.; Khan, A.M.; Khan, A.; Shal, B.; Aziz, A.; Ahmed, M.N.; Khan, S. The newly synthesized compounds (NCHDH and NTHDH) attenuates LPS-induced septicemia and multi-organ failure via Nrf2/HO1 and HSP/TRVP1 signaling in mice. Chem. Interact. 2020, 329, 109220. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.; Khan, A.; Ali, J.; Ullah, H.; Khan, A.; Ali, H.; Irshad, N.; Khan, S. Attenuation of lps-induced acute lung injury by continentalic acid in rodents through inhibition of inflammatory mediators correlates with increased nrf2 protein expression. BMC Pharamcol. Toxicol. 2020, 21, 81. [Google Scholar] [CrossRef]
- Halwes, M.E.; Tyo, K.M.; Steinbach-Rankins, J.M.; Frieboes, H.B. Computational modeling of antiviral drug diffusion from poly(lactic-co-glycolic-acid) fibers and multicompartment pharmacokinetics for application to the female reproductive tract. Mol. Pharm. 2018, 15, 1534–1547. [Google Scholar] [CrossRef]
- Shal, B.; Khan, A.; Khan, A.U.; Ullah, R.; Ali, G.; Islam, S.U.; Ali, H.; Seo, E.-K.; Khan, S. Alleviation of memory deficit by bergenin via the regulation of reelin and nrf-2/nf-κb pathway in transgenic mouse model. Int. J. Mol. Sci. 2021, 22, 6603. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Shal, B.; Khan, A.U.; Ullah, R.; Baig, M.W.; Haq, I.U.; Seo, E.K.; Khan, S. Suppression of TRPV1/TRPM8/P2Y nociceptors by withametelin via downregulating MAPK signaling in mouse model of vincristine-induced neuropathic pain. Int. J. Mol. Sci. 2021, 22, 6084. [Google Scholar] [CrossRef] [PubMed]
- Naveed, M.; Ullah, R.; Khan, A.; Shal, B.; Khan, A.U.; Khan, S.Z.; Rehman, Z.U.; Khan, S. Anti-neuropathic pain activity of a cationic palladium (II) dithiocarbamate by suppressing the inflammatory mediators in paclitaxel-induced neuropathic pain model. Mol. Biol. Rep. 2021, 48, 7647–7656. [Google Scholar] [CrossRef]
- Hou, L.-S.; Cui, Z.-Y.; Sun, P.; Piao, H.-Q.; Han, X.; Song, J.; Wang, G.; Zheng, S.; Dong, X.-X.; Gao, L.; et al. Rutin mitigates hepatic fibrogenesis and inflammation through targeting TLR4 and P2X7 receptor signaling pathway in vitro and in vivo. J. Funct. Foods 2020, 64, 103700. [Google Scholar] [CrossRef]
- Ullah, H.; Khan, A.; Bibi, T.; Ahmad, S.; Shehzad, O.; Ali, H.; Seo, E.K.; Khan, S. Comprehensive in vivo and in silico approaches to explore the hepatoprotective activity of poncirin against paracetamol toxicity. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2022, 395, 195–215. [Google Scholar] [CrossRef]
- Hosek, J.; Leláková, V.; Bobal, P.; Pizova, H.; Gazdová, M.; Malaník, M.; Jakubczyk, K.; Vesely, O.; Landa, P.; Temml, V. Prenylated stilbenoids affect inflammation by inhibiting the nf-κb/ap-1 signaling pathway and cyclooxygenases and lipoxygenase. J. Nat. Prod. 2019, 82, 1839–1848. [Google Scholar] [CrossRef]
- Hsieh, N.-H.; Reisfeld, B.; Bois, F.Y.; Chiu, W.A. Applying a global sensitivity analysis workflow to improve the computational efficiencies in physiologically-based pharmacokinetic modeling. Front. Pharmacol. 2018, 9, 588. [Google Scholar] [CrossRef]
- Ammar, S. In silico pharmacodynamics, toxicity profile and biological activities of the saharan medicinal plant Limoniastrum feei. Braz. J. Pharm. Sci. 2017, 53, e61. [Google Scholar] [CrossRef] [Green Version]
- Ntie-Kang, F.; Simoben, C.V.; Karaman, B.; Ngwa, V.F.; Judson, P.N.; Sippl, W.; Mbaze, L.M. Pharmacophore modeling and in silico toxicity assessment of potential anticancer agents from African medicinal plants. Drug Des. Dev. Ther. 2016, 10, 2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rim, K.-T. In silico prediction of toxicity and its applications for chemicals at work. Toxicol. Environ. Health Sci. 2020, 12, 191–202. [Google Scholar] [CrossRef]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided-Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef]
- Afridi, R.; Khan, A.U.; Khalid, S.; Shal, B.; Rasheed, H.; Ullah, M.Z.; Shehzad, O.; Kim, Y.S.; Khan, S. Anti-hyperalgesic properties of a flavanone derivative Poncirin in acute and chronic inflammatory pain models in mice. BMC Pharmacol. Toxicol. 2019, 20, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Zhu, W. Molecular docking for drug discovery and development: A widely used approach but far from perfect. Futur. Med. Chem. 2016, 8, 1707–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No | Molecular Weight | LogP | Rotatable Bonds | Acceptors | Donors | Surface Area |
|---|---|---|---|---|---|---|
| Anomalin | 426.465 | 4.3925 | 4 | 7 | 0 | 179.832 |
| Alantolactone | 232.323 | 3.2406 | 0 | 2 | 0 | 102.727 |
| Baicalein | 270.24 | 2.5768 | 1 | 5 | 3 | 112.519 |
| Berberin | 336.367 | 3.0963 | 2 | 4 | 0 | 144.867 |
| Bergenin | 328.273 | −1.2006 | 2 | 9 | 5 | 129.813 |
| Catechin | 290.271 | 1.5461 | 1 | 6 | 5 | 119.662 |
| Continentalic acid | 302.458 | 5.2062 | 2 | 1 | 1 | 134.232 |
| Coumarin | 146.145 | 1.793 | 0 | 2 | 0 | 63.079 |
| Epi-catechin | 290.271 | 1.5461 | 1 | 6 | 5 | 119.662 |
| Ferulic acid | 194.186 | 1.4986 | 3 | 3 | 2 | 81.065 |
| Gallic acid | 170.12 | 0.5016 | 1 | 4 | 4 | 67.135 |
| Honokiol | 266.34 | 4.2218 | 5 | 2 | 2 | 118.887 |
| Icariin | 676.668 | 0.0679 | 9 | 15 | 8 | 273.926 |
| Linoleic acid | 28.452 | 5.8845 | 14 | 1 | 1 | 124.520 |
| Magnolol | 266.34 | 4.2218 | 5 | 2 | 2 | 118.887 |
| Matrine | 248.37 | 1.8717 | 0 | 2 | 0 | 109.506 |
| P-coumaric acid | 164.16 | 1.49 | 2 | 2 | 2 | 69.587 |
| Pimelic acid | 160.169 | 1.1061 | 6 | 2 | 2 | 64.840 |
| Poncirin | 594.566 | −0.8622 | 7 | 14 | 7 | 239.713 |
| Quercetin | 302.238 | 1.988 | 1 | 7 | 5 | 122.108 |
| Rutin | 610.521 | −1.6871 | 6 | 16 | 10 | 240.901 |
| Vanillic acid | 168.148 | 1.099 | 2 | 3 | 2 | 69.025 |
| S. No | Ames Toxicity | herG I Inhibitor | herG II Inhibitor | Hepatotoxicity | Skin Sensitization | T. Pyriformis Toxicity | Minnow Toxicity | Max. Tolerated Dose (Human) | Oral Rat Acute Toxicity (LD50) | Oral Rat Chronic Toxicity (LOAEL) |
|---|---|---|---|---|---|---|---|---|---|---|
| Anomalin | NO | NO | NO | YES | NO | 0.365 | 0.325 | 0.135 | 3.266 | 2.137 |
| Alantolactone | NO | NO | NO | NO | YES | 1.237 | 0.936 | 0.042 | 1.597 | 1.856 |
| Baicalein | NO | NO | NO | NO | NO | 0.42 | 1.25 | 0.498 | 2.325 | 2.645 |
| Berberin | YES | NO | NO | YES | NO | 0.354 | −0.277 | 0.144 | 2.571 | 1.89 |
| Bergenin | NO | NO | NO | NO | NO | 0.285 | 5.688 | −0.013 | 1.879 | 3.614 |
| Catechin | NO | NO | NO | NO | NO | 0.347 | 3.585 | 0.438 | 2.428 | 2.5 |
| Continentalic acid | NO | NO | NO | YES | NO | 0.31 | −0.35 | 0.008 | 1.838 | 2.207 |
| Coumarin | NO | NO | NO | NO | NO | 0.365 | 1.555 | 0.435 | 2.112 | 1.903 |
| Epi-catechin | NO | NO | NO | NO | NO | 0.347 | 3.585 | 0.438 | 2.428 | 2.5 |
| Ferulic acid | NO | NO | NO | NO | NO | 0.271 | 1.825 | 1.082 | 2.282 | 2.065 |
| Gallic acid | NO | NO | NO | NO | NO | 0.285 | 3.188 | 0.7 | 2.218 | 3.06 |
| Honokiol | NO | NO | YES | NO | NO | 0.749 | 0.14 | 0.305 | 2.184 | 1.791 |
| Icariin | NO | NO | YES | NO | NO | 0.285 | 5.51 | 0.451 | 2.631 | 5.081 |
| Linoleic acid | NO | NO | NO | YES | YES | 0.701 | −1.31 | −0.827 | 1.429 | 3.187 |
| Magnolol | NO | NO | YES | NO | NO | 0.941 | −0.054 | 0.468 | 1.976 | 1.851 |
| Matrine | NO | NO | NO | NO | YES | 0.56 | 2.264 | 0.141 | 2.54 | 0.874 |
| P-cumaric acid | NO | NO | NO | NO | NO | 0.319 | 1.607 | 1.111 | 2.155 | 2.534 |
| Pimelic acid | NO | NO | NO | NO | NO | −0.103 | 2.006 | 0.106 | 1.338 | 3.11 |
| Poncirin | NO | NO | YES | NO | NO | 0.285 | 5.65 | 0.259 | 2.545 | 4.096 |
| Quercetin | NO | NO | NO | NO | NO | 0.288 | 3.721 | 0.499 | 2.471 | 2.612 |
| Rutin | NO | NO | YES | NO | NO | 0.452 | 2.491 | 3.673 | 0.285 | 7.677 |
| Vanillic acid | NO | NO | NO | NO | NO | 0.265 | 1.926 | 0719 | 2.454 | 2.032 |
| Compounds | GI Absorption | BBB Permeation | Caco-2 Permeability | P-Glycoprotein Substrate | P-Glycoprotein Inhibitor | CYP1A2 Inhibitor | CYP2C19 Inhibitor | CYP2C9 Inhibitor | CYP3A4 Inhibitor |
|---|---|---|---|---|---|---|---|---|---|
| Anomalin | High | NO | 0.938 | NO | YES | NO | YES | YES | YES |
| Alantolactone | High | YES | 1.603 | NO | YES | NO | YES | YES | NO |
| Baicalein | High | NO | 1.117 | NO | NO | YES | NO | NO | YES |
| Berberin | High | YES | 1.734 | YES | NO | YES | NO | NO | YES |
| Bergenin | Low | NO | 0.289 | YES | NO | NO | NO | NO | NO |
| Catechin | Low | NO | −0.283 | YES | NO | NO | NO | NO | NO |
| Continentalic acid | High | YES | 1.742 | NO | NO | NO | YES | YES | NO |
| Coumarin | High | YES | 1.649 | NO | NO | YES | NO | NO | NO |
| Epi-catechin | Low | NO | −0.283 | YES | NO | NO | NO | NO | NO |
| Ferulic acid | High | YES | 0.176 | NO | NO | NO | NO | NO | NO |
| Gallic acid | Low | NO | −0.081 | NO | NO | NO | NO | NO | YES |
| Honokiol | High | YES | 1.586 | YES | NO | YES | YES | YES | YES |
| Icariin | Low | NO | −0805 | YES | YES | NO | NO | NO | NO |
| Linoleic acid | High | YES | 1.57 | NO | NO | YES | NO | YES | NO |
| Magnolol | High | YES | 1.707 | YES | NO | YES | YES | YES | YES |
| Matrine | High | YES | 1.463 | YES | NO | NO | NO | NO | NO |
| P-cumaric acid | High | YES | 1.21 | NO | NO | NO | NO | NO | NO |
| Pimelic acid | High | NO | 0.598 | NO | NO | NO | NO | NO | NO |
| Poncirin | Low | NO | 0.62 | YES | YES | NO | NO | NO | NO |
| Quercetin | Low | NO | −0.229 | YES | NO | YES | NO | NO | YES |
| Rutin | Low | NO | −0.949 | YES | NO | NO | NO | NO | NO |
| Vanillic acid | High | NO | 0.33 | NO | NO | NO | NO | NO | NO |
| Compounds | Lipinski | Ghose | Veber | Egan | Muegge |
|---|---|---|---|---|---|
| Anomalin | YES | YES | YES | YES | YES |
| Alantolactone | YES | YES | YES | YES | YES |
| Baicalein | YES | YES | YES | YES | YES |
| Berberin | YES | YES | YES | YES | YES |
| Bergenin | YES | NO | NO | NO | YES |
| Catechin | YES | YES | YES | YES | YES |
| Continentalic acid | YES | YES | YES | YES | NO |
| Coumarin | YES | NO | YES | YES | NO |
| Epi-catechin | YES | YES | YES | YES | YES |
| Ferulic acid | YES | YES | YES | YES | NO |
| Gallic acid | YES | NO | YES | YES | NO |
| Honokiol | YES | YES | YES | YES | YES |
| Icariin | NO | NO | NO | NO | NO |
| Linoleic acid | YES | NO | NO | NO | NO |
| Magnolol | YES | YES | YES | YES | YES |
| Matrine | YES | YES | YES | YES | YES |
| P-cumaric acid | YES | YES | YES | YES | NO |
| Pimelic acid | YES | NO | YES | YES | NO |
| Poncirin | NO | NO | NO | NO | NO |
| Quercetin | YES | YES | YES | YES | YES |
| Rutin | NO | NO | NO | NO | NO |
| Vanillic acid | YES | YES | YES | YES | NO |
| Sample | TLR-4 (4g8e) Energy (kcal/mol) | JNK (30xi) Energy (kcal/mol) | P65 (1le5) Energy (kcal/mol) | AP-1 (4hmy) Energy (kcal/mol) |
|---|---|---|---|---|
| Anomalin | −7.0 | −7.0 | −6.6 | −7.0 |
| Alantalactone | −6.2 | −7.1 | −7.1 | −8.7 |
| Baicalein | −7.7 | −8.1 | −8.0 | −8.6 |
| Berberin | −7.6 | −8.2 | −6.9 | −8.1 |
| Bergenin | −7.5 | −7.5 | −6.7 | −6.9 |
| Catechin | −7.5 | −7.3 | −7.3 | −7.9 |
| Continentalic acid | −7.3 | −6.8 | −6.9 | −7.5 |
| Coumarin | −6.2 | −6.9 | −6.3 | −7.9 |
| Epi-catechin | −7.9 | −7.6 | −7.6 | −8.6 |
| Ferulic acid | −5.9 | −6.2 | −5.7 | −6.9 |
| Gallic acid | −6.0 | −5.6 | −6.0 | −6.1 |
| Honokiol | −6.7 | −7.2 | −7.1 | −7.8 |
| Icariin | −9.1 | −9.4 | −7.0 | −9.1 |
| Linoleic acid | −4.7 | −5.9 | −4.9 | −5.9 |
| Magnolol | −6.9 | −7.5 | −6.9 | −8.9 |
| Matrine | −6.1 | −6.8 | −7.1 | −6.6 |
| P-coumaric acid | −5.8 | −6.1 | −5.2 | −6.6 |
| Pimelic acid | −4.5 | −4.6 | −5.3 | −5.4 |
| Poncirin | −10.1 | −9.5 | −9.4 | −8.0 |
| Quercetin | −8.0 | −7.8 | −7.3 | −8.0 |
| Rutin | −10.4 | −9.1 | −7.8 | −8.6 |
| Vanillic acid | −6.1 | −5.6 | −5.2 | −6.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, A.; Khan, S.U.; Khan, A.; Shal, B.; Rehman, S.U.; Rehman, S.U.; Htar, T.T.; Khan, S.; Anwar, S.; Alafnan, A.; et al. Anti-Inflammatory and Anti-Rheumatic Potential of Selective Plant Compounds by Targeting TLR-4/AP-1 Signaling: A Comprehensive Molecular Docking and Simulation Approaches. Molecules 2022, 27, 4319. https://doi.org/10.3390/molecules27134319
Khan A, Khan SU, Khan A, Shal B, Rehman SU, Rehman SU, Htar TT, Khan S, Anwar S, Alafnan A, et al. Anti-Inflammatory and Anti-Rheumatic Potential of Selective Plant Compounds by Targeting TLR-4/AP-1 Signaling: A Comprehensive Molecular Docking and Simulation Approaches. Molecules. 2022; 27(13):4319. https://doi.org/10.3390/molecules27134319
Chicago/Turabian StyleKhan, Ashrafullah, Shafi Ullah Khan, Adnan Khan, Bushra Shal, Sabih Ur Rehman, Shaheed Ur Rehman, Thet Thet Htar, Salman Khan, Sirajudheen Anwar, Ahmed Alafnan, and et al. 2022. "Anti-Inflammatory and Anti-Rheumatic Potential of Selective Plant Compounds by Targeting TLR-4/AP-1 Signaling: A Comprehensive Molecular Docking and Simulation Approaches" Molecules 27, no. 13: 4319. https://doi.org/10.3390/molecules27134319