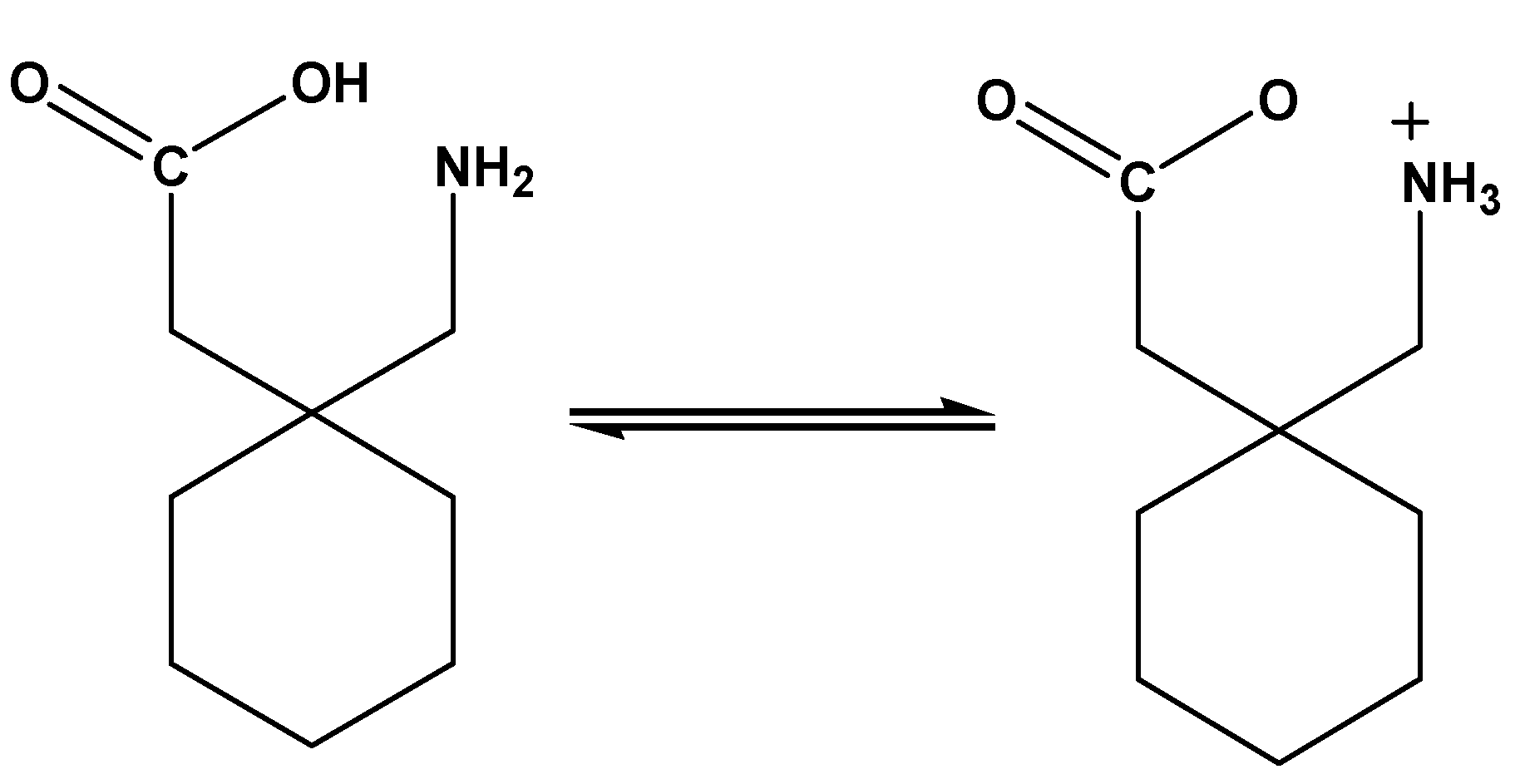
Figure 1.
The zwitterion structure of the gabapentin (Gpn) drug.
Figure 1.
The zwitterion structure of the gabapentin (Gpn) drug.
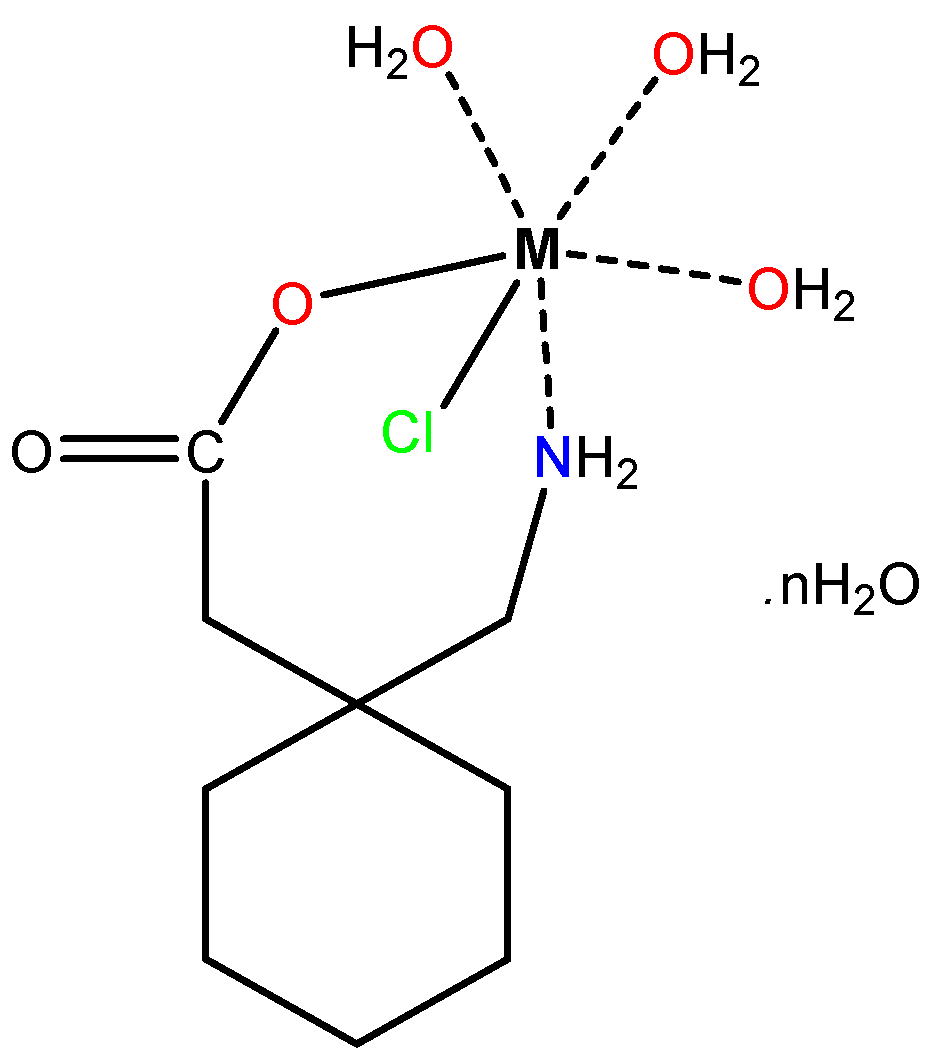
Figure 2.
The structures of the synthesized Mn(II), Co(II), Ni(II), and Cu(II)–Gpn complexes, where = 2 for Cu(II), 3 for Ni(II), 4 for Mn(II), and 6 for Co(II).
Figure 2.
The structures of the synthesized Mn(II), Co(II), Ni(II), and Cu(II)–Gpn complexes, where = 2 for Cu(II), 3 for Ni(II), 4 for Mn(II), and 6 for Co(II).
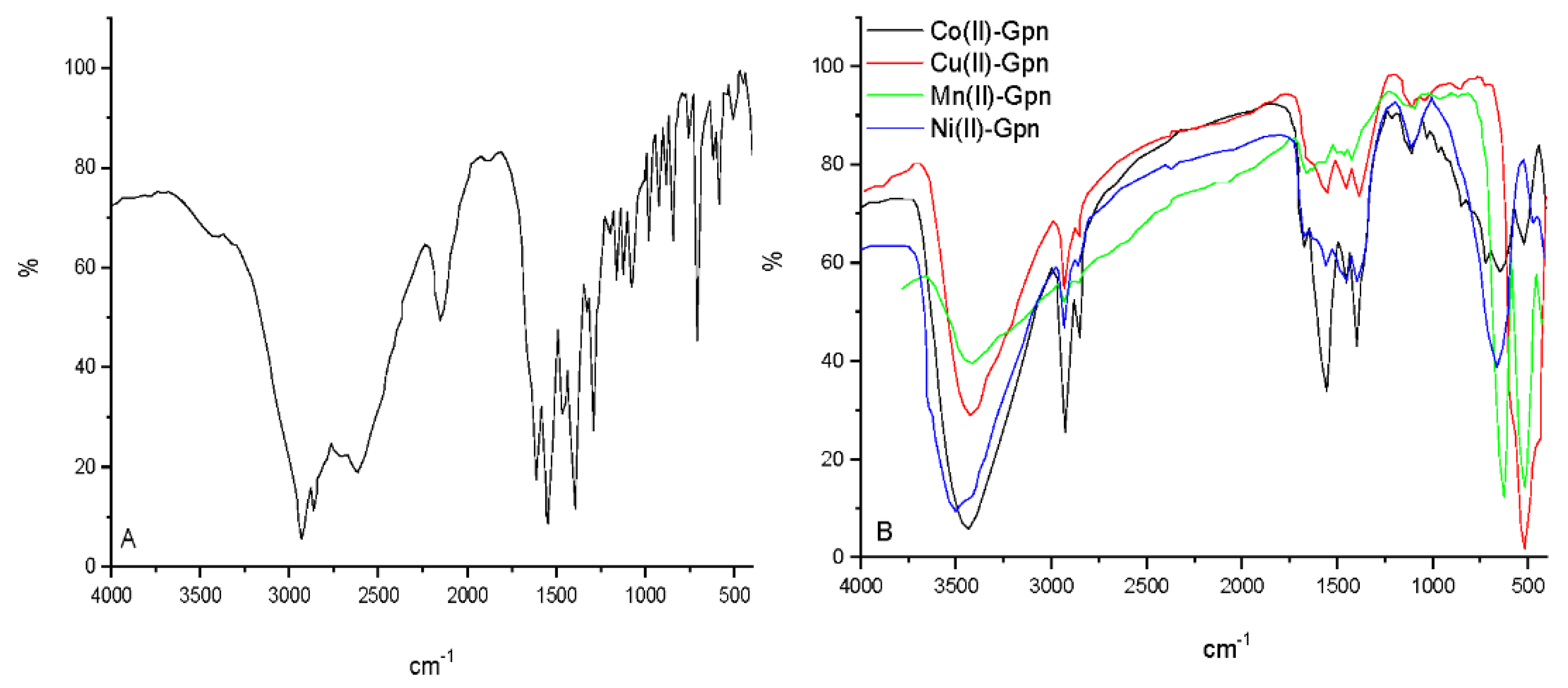
Figure 3.
The infrared spectra of (A) Gpn free drug and (B) Mn(II), Co(II), Ni(II), and Cu(II)–Gpn complexes.
Figure 3.
The infrared spectra of (A) Gpn free drug and (B) Mn(II), Co(II), Ni(II), and Cu(II)–Gpn complexes.
Figure 4.
The SEM images of (I) Mn(II), (II) Cu(II), (III) Co(II), and (IV) Ni(II)–Gpn.
Figure 4.
The SEM images of (I) Mn(II), (II) Cu(II), (III) Co(II), and (IV) Ni(II)–Gpn.
Figure 5.
The TEM images of the (I) Mn(II), (II) Cu(II), (III) Co(II), and (IV) Ni(II)–Gpn complexes.
Figure 5.
The TEM images of the (I) Mn(II), (II) Cu(II), (III) Co(II), and (IV) Ni(II)–Gpn complexes.
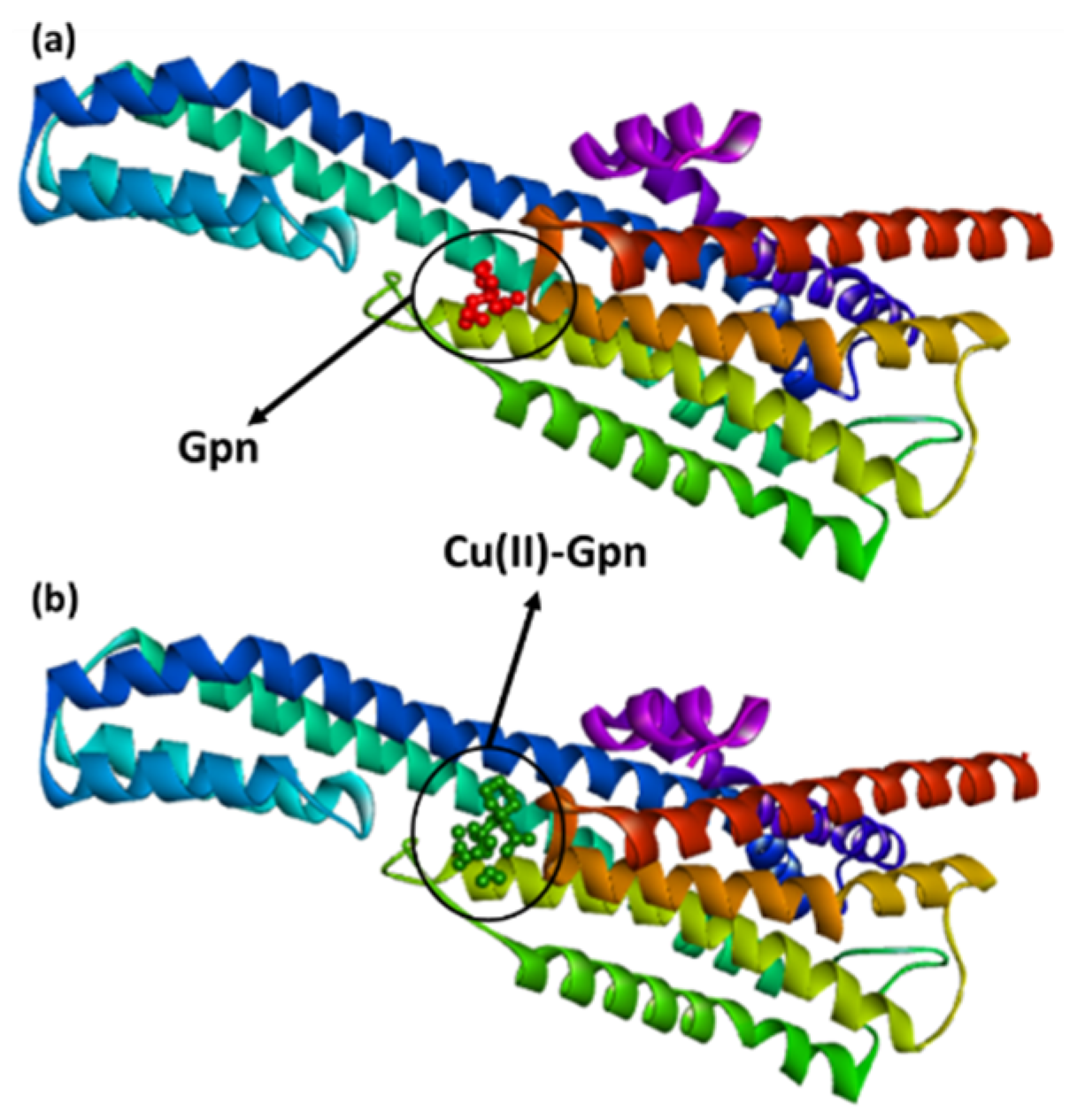
Figure 6.
The best docked pose showing a helical model of serotonin (PDB ID: 6BQH) docked with (a) the Gpn drug and (b) metal complex [Cu(II)–(Gpn)].
Figure 6.
The best docked pose showing a helical model of serotonin (PDB ID: 6BQH) docked with (a) the Gpn drug and (b) metal complex [Cu(II)–(Gpn)].
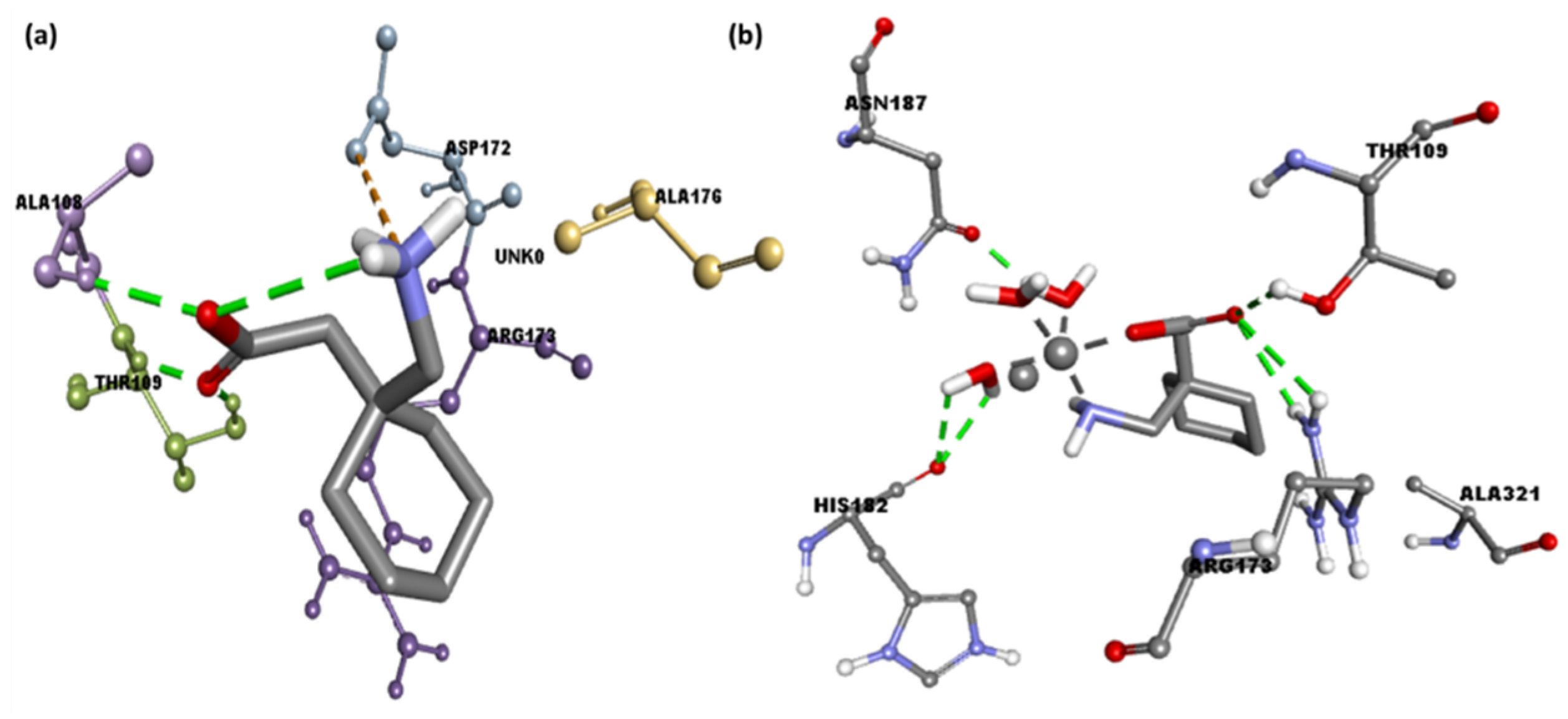
Figure 7.
The 3D representation of the interactions for serotonin (PDB ID: 6BQH) docked with (a) the Gpn drug and (b) metal complex [Cu(II)–(Gpn)].
Figure 7.
The 3D representation of the interactions for serotonin (PDB ID: 6BQH) docked with (a) the Gpn drug and (b) metal complex [Cu(II)–(Gpn)].
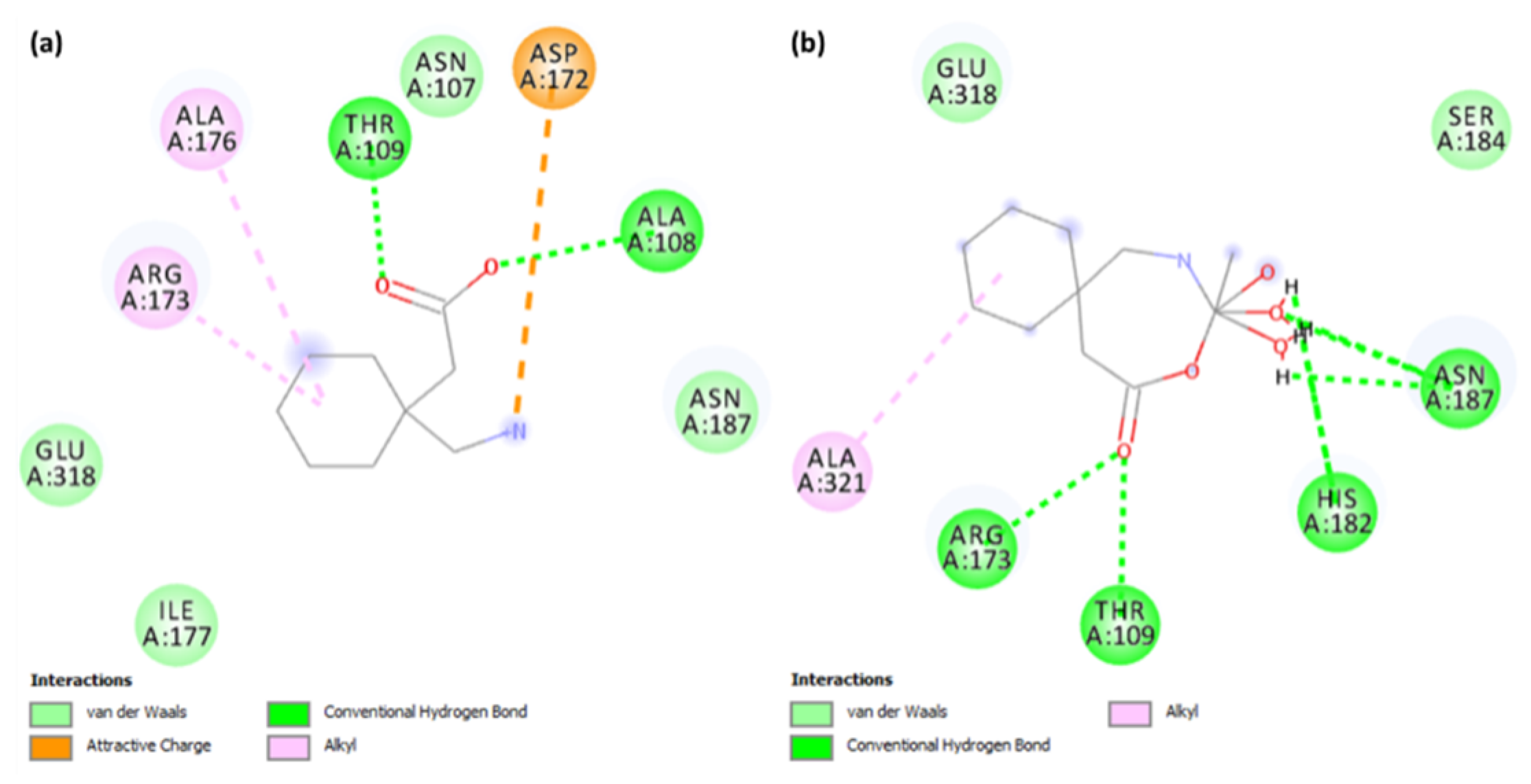
Figure 8.
The 2D representation of interactions for serotonin (PDB ID: 6BQH) docked with (a) the Gpn drug and (b) metal complex [Cu(II)–(Gpn)].
Figure 8.
The 2D representation of interactions for serotonin (PDB ID: 6BQH) docked with (a) the Gpn drug and (b) metal complex [Cu(II)–(Gpn)].
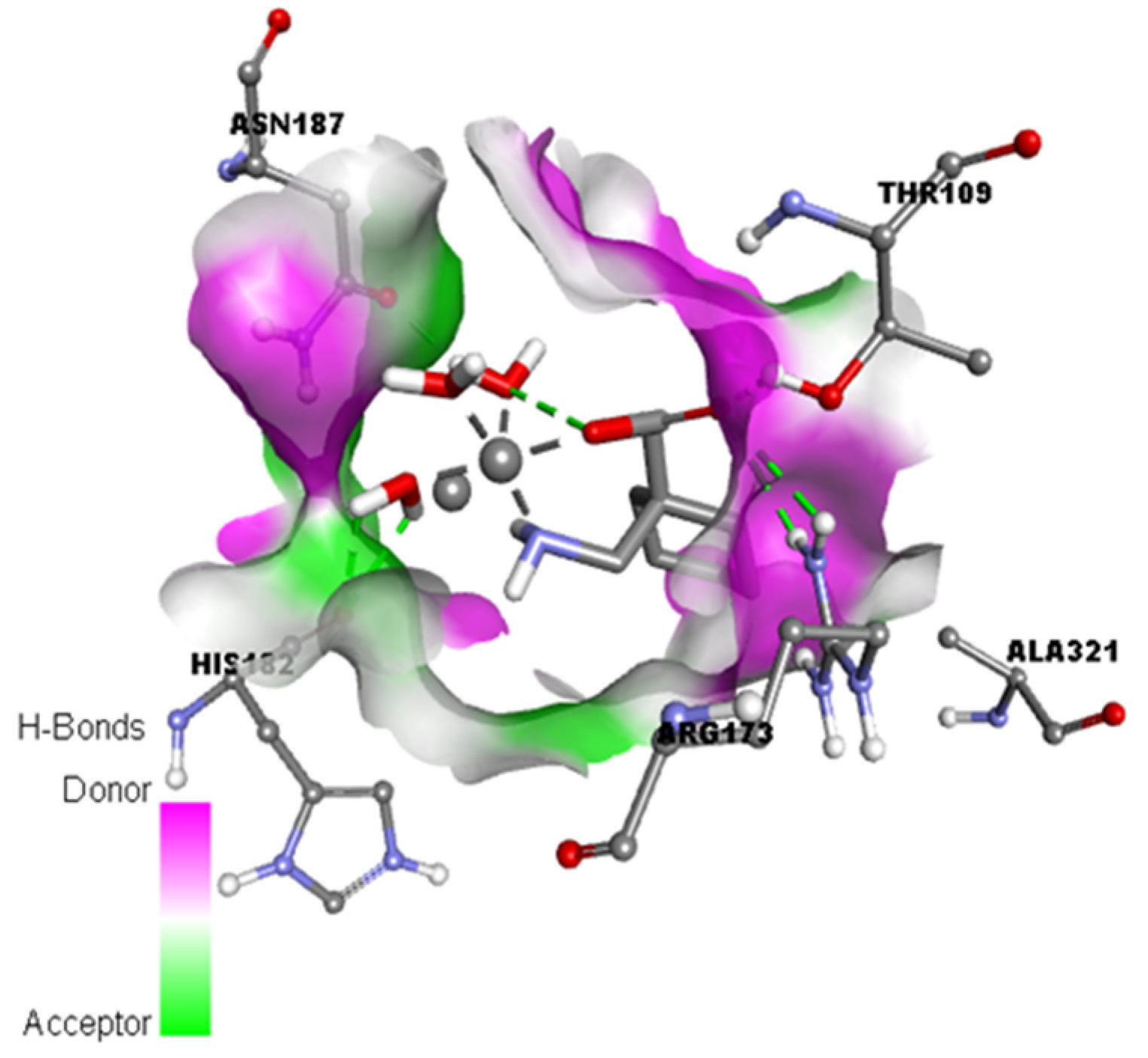
Figure 9.
Representation of the hydrogen binding surface between the serotonin and metal complex [Cu(II)–(Gpn)].
Figure 9.
Representation of the hydrogen binding surface between the serotonin and metal complex [Cu(II)–(Gpn)].
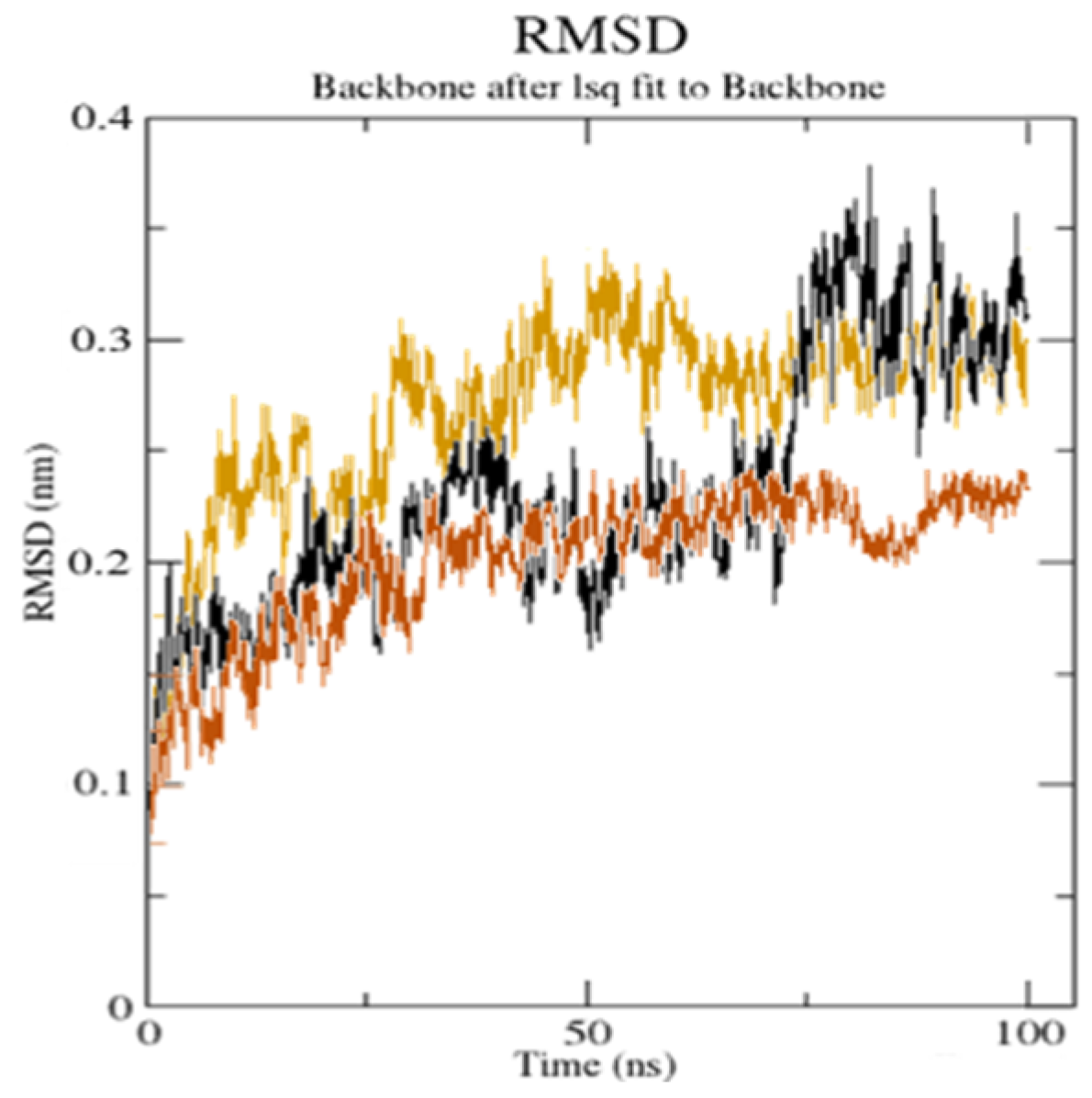
Figure 10.
The root mean square deviation (RMSD) of the solvated receptor backbone and ligand complex during the 100 ns MD simulation [unbound serotonin receptor (yellow),
GpnS complex (black), and
CuGS complex (brown)]. According to the literature, a RMSD value under <3.0 Å is the most acceptable [
44]. The drop in the RMSD value for
CuGS showed an alteration in the secondary structure conformation of the protein due to the [Cu(II)–(Gpn)] interaction. This finding shows that
CuGS developed a more stable combination. The results confirm that this interaction brings protein chains closer and reduces the gap between them, as shown in
Figure 11 [
45].
Figure 10.
The root mean square deviation (RMSD) of the solvated receptor backbone and ligand complex during the 100 ns MD simulation [unbound serotonin receptor (yellow),
GpnS complex (black), and
CuGS complex (brown)]. According to the literature, a RMSD value under <3.0 Å is the most acceptable [
44]. The drop in the RMSD value for
CuGS showed an alteration in the secondary structure conformation of the protein due to the [Cu(II)–(Gpn)] interaction. This finding shows that
CuGS developed a more stable combination. The results confirm that this interaction brings protein chains closer and reduces the gap between them, as shown in
Figure 11 [
45].
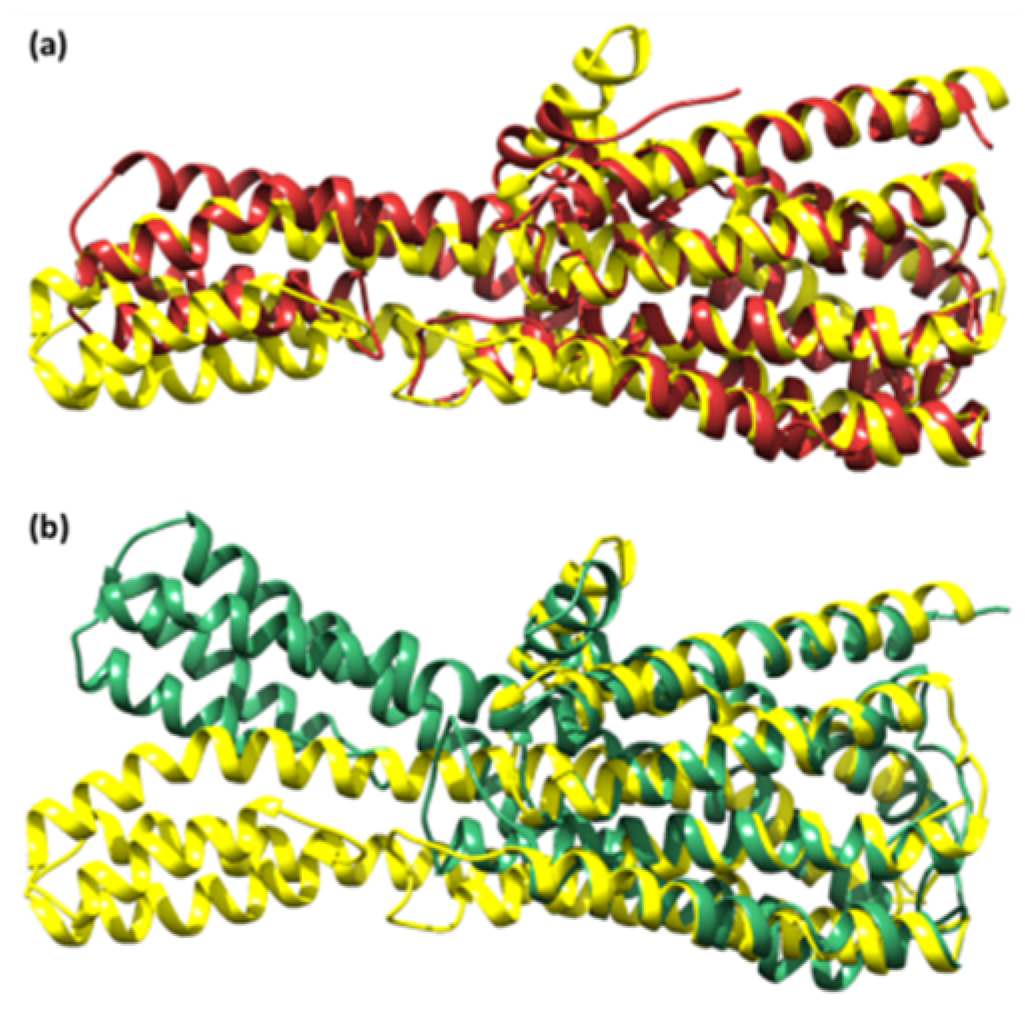
Figure 11.
The superimposed structure of (a) the unbound serotonin receptor (yellow) and serotonin receptor after simulation (brown) for GpnS and (b) the unbound serotonin receptor (yellow) and serotonin receptor after simulation (green) for CuGS.
Figure 11.
The superimposed structure of (a) the unbound serotonin receptor (yellow) and serotonin receptor after simulation (brown) for GpnS and (b) the unbound serotonin receptor (yellow) and serotonin receptor after simulation (green) for CuGS.
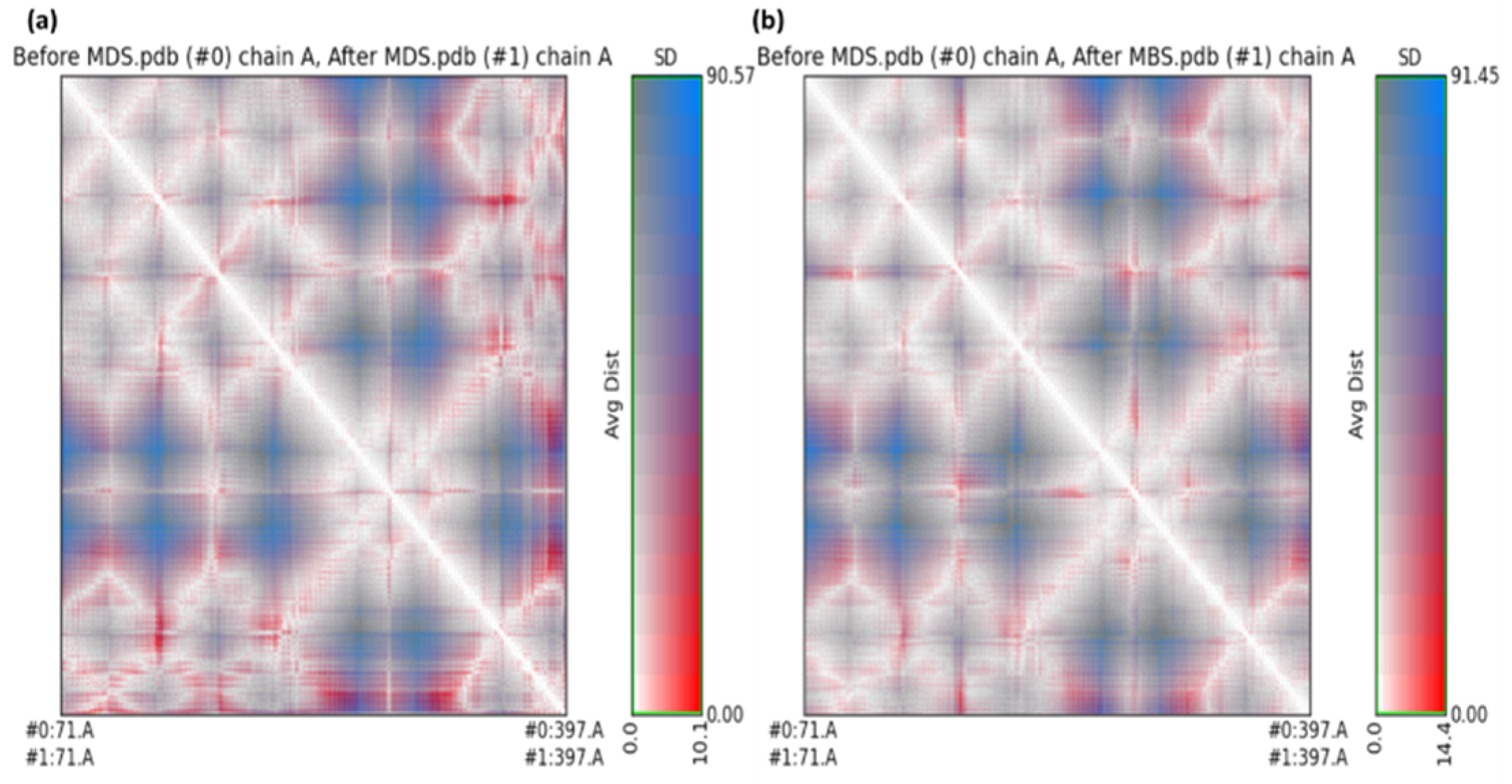
Figure 12.
The RR distance map displaying patterns of spatial interactions between (a) the unbound serotonin receptor and the serotonin receptor after the simulation for GpnS; and (b) the unbound serotonin receptor and serotonin receptor after simulation for CuGS, showing the average distance and standard deviation for all amino acid pairs. (MDS = Molecular Dynamics simulation).
Figure 12.
The RR distance map displaying patterns of spatial interactions between (a) the unbound serotonin receptor and the serotonin receptor after the simulation for GpnS; and (b) the unbound serotonin receptor and serotonin receptor after simulation for CuGS, showing the average distance and standard deviation for all amino acid pairs. (MDS = Molecular Dynamics simulation).
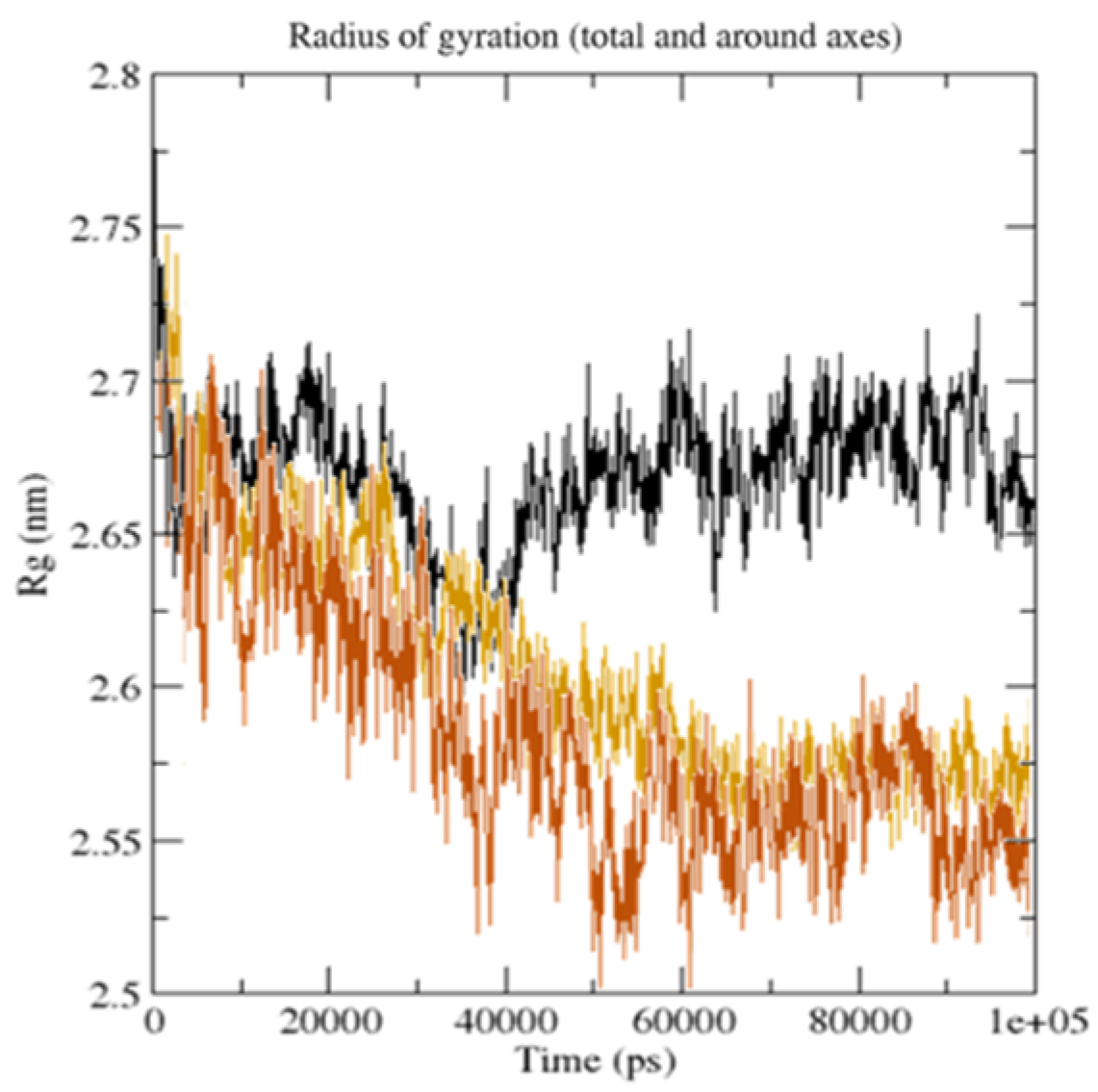
Figure 13.
The radius of gyration (Rg) for the unbound serotonin receptor (yellow), GpnS complex (black), and CuGS complex (brown) during the 100 ns simulation time.
Figure 13.
The radius of gyration (Rg) for the unbound serotonin receptor (yellow), GpnS complex (black), and CuGS complex (brown) during the 100 ns simulation time.
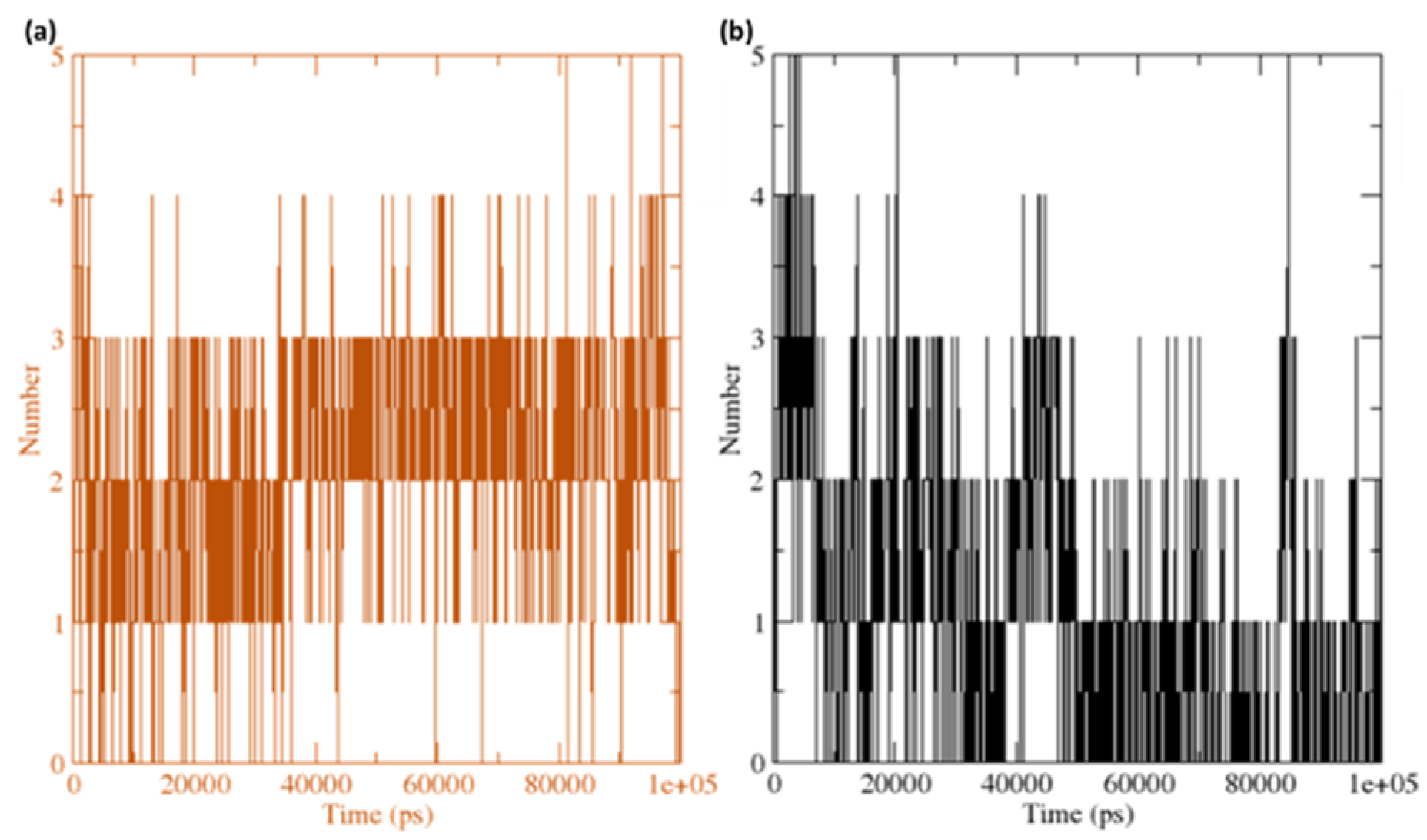
Figure 14.
The number of average hydrogen bonding interactions between (a) the CuGS complex and (b) GpnS complex during the 100 ns simulation time.
Figure 14.
The number of average hydrogen bonding interactions between (a) the CuGS complex and (b) GpnS complex during the 100 ns simulation time.
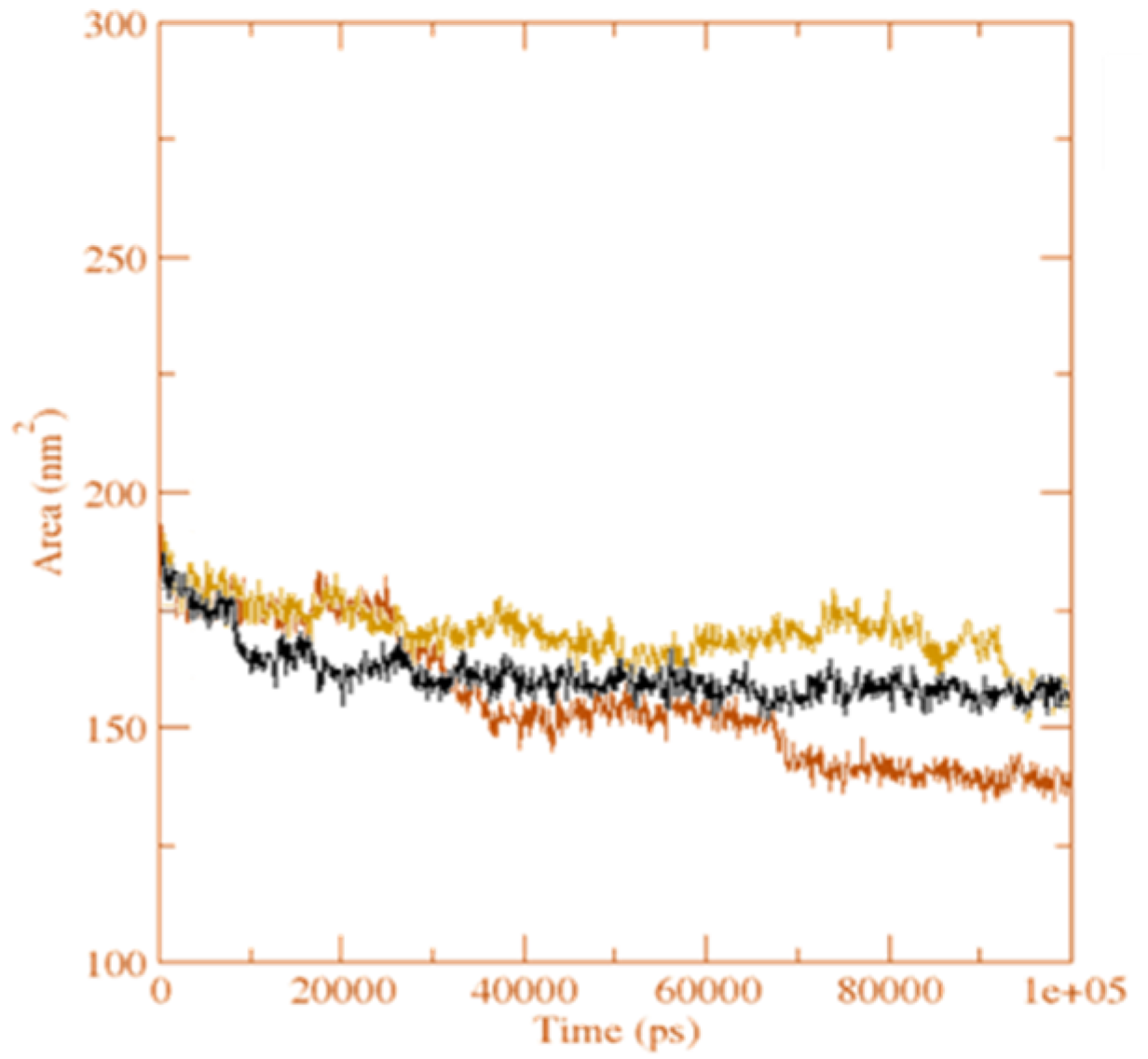
Figure 15.
The solvent accessible surface area analysis for the unbound serotonin receptor (yellow), GpnS complex (black), and CuGS complex (brown) during the 100 ns simulation time.
Figure 15.
The solvent accessible surface area analysis for the unbound serotonin receptor (yellow), GpnS complex (black), and CuGS complex (brown) during the 100 ns simulation time.
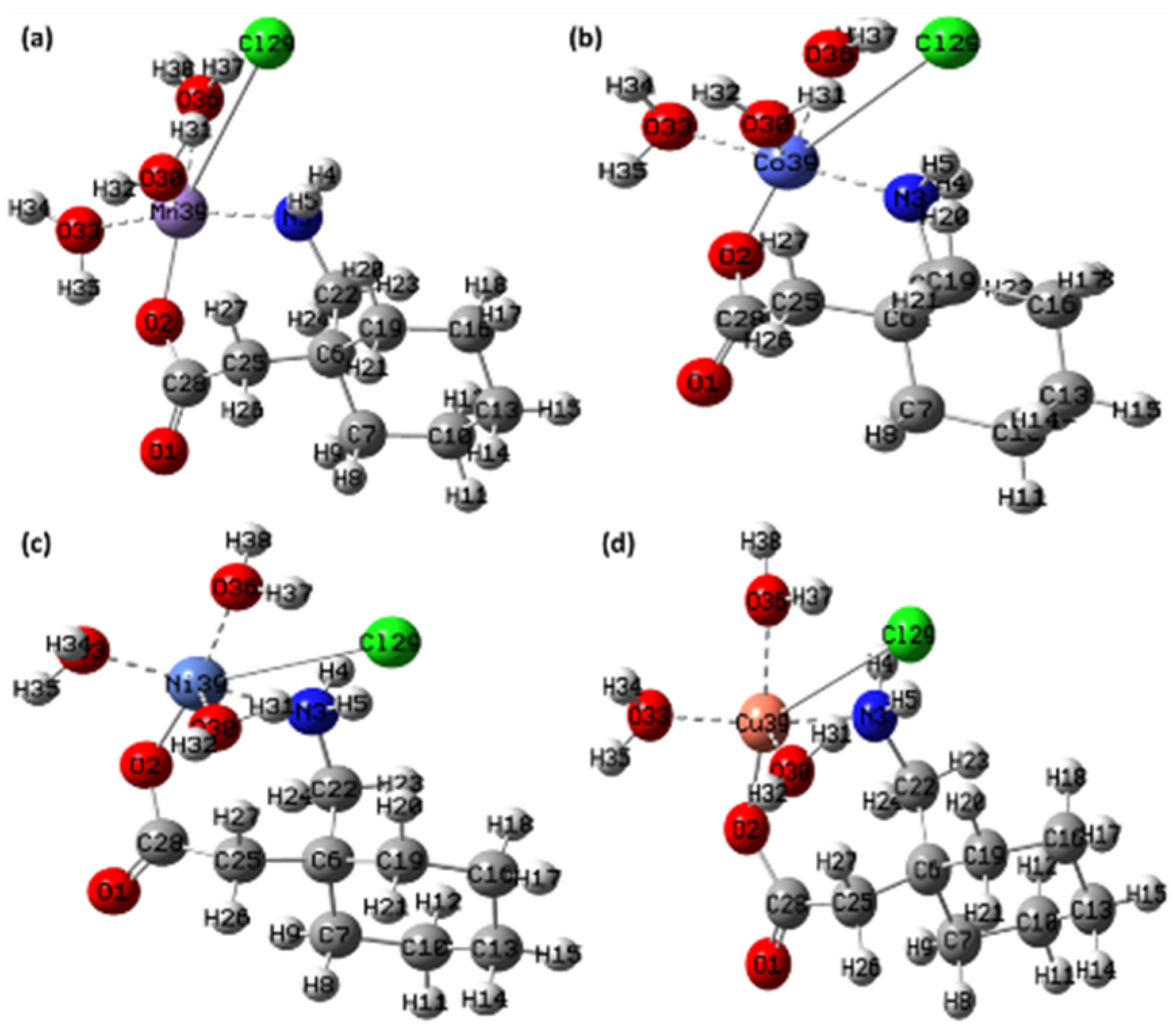
Figure 16.
The optimized structure of the synthesized metal complexes (a) [Mn(II)–(Gpn)], (b) [Co(II)–(Gpn)], (c) [Ni(II)–(Gpn)], and (d) [Cu(II)–(Gpn)] with the Mulliken atom numbering scheme.
Figure 16.
The optimized structure of the synthesized metal complexes (a) [Mn(II)–(Gpn)], (b) [Co(II)–(Gpn)], (c) [Ni(II)–(Gpn)], and (d) [Cu(II)–(Gpn)] with the Mulliken atom numbering scheme.
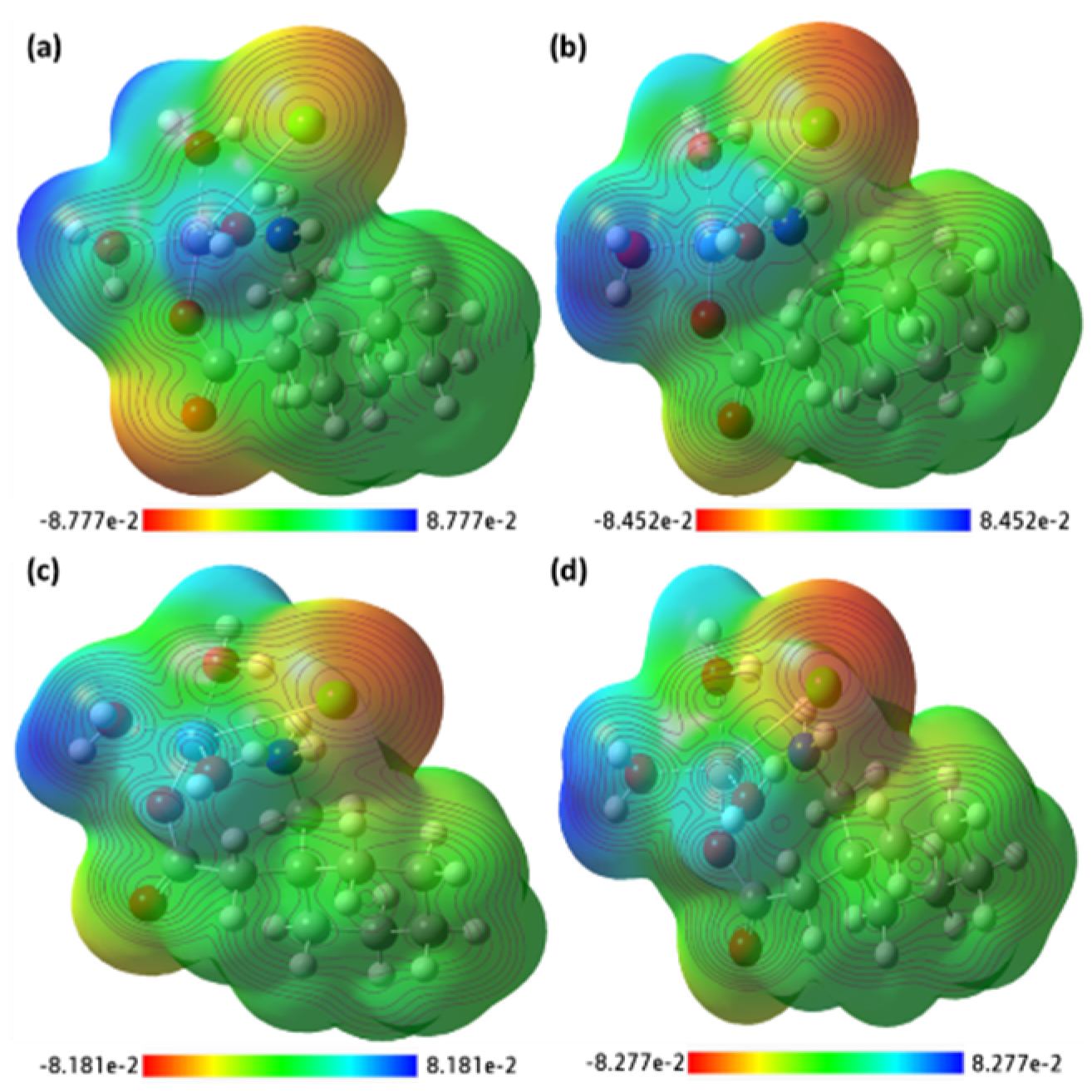
Figure 17.
The MEP surface map of the synthesized metal complexes (a) [Mn(II)–(Gpn)], (b) [Co(II)–(Gpn)], (c) [Ni(II)–(Gpn)], and (d) [Cu(II)–(Gpn)] with the respective color scales.
Figure 17.
The MEP surface map of the synthesized metal complexes (a) [Mn(II)–(Gpn)], (b) [Co(II)–(Gpn)], (c) [Ni(II)–(Gpn)], and (d) [Cu(II)–(Gpn)] with the respective color scales.
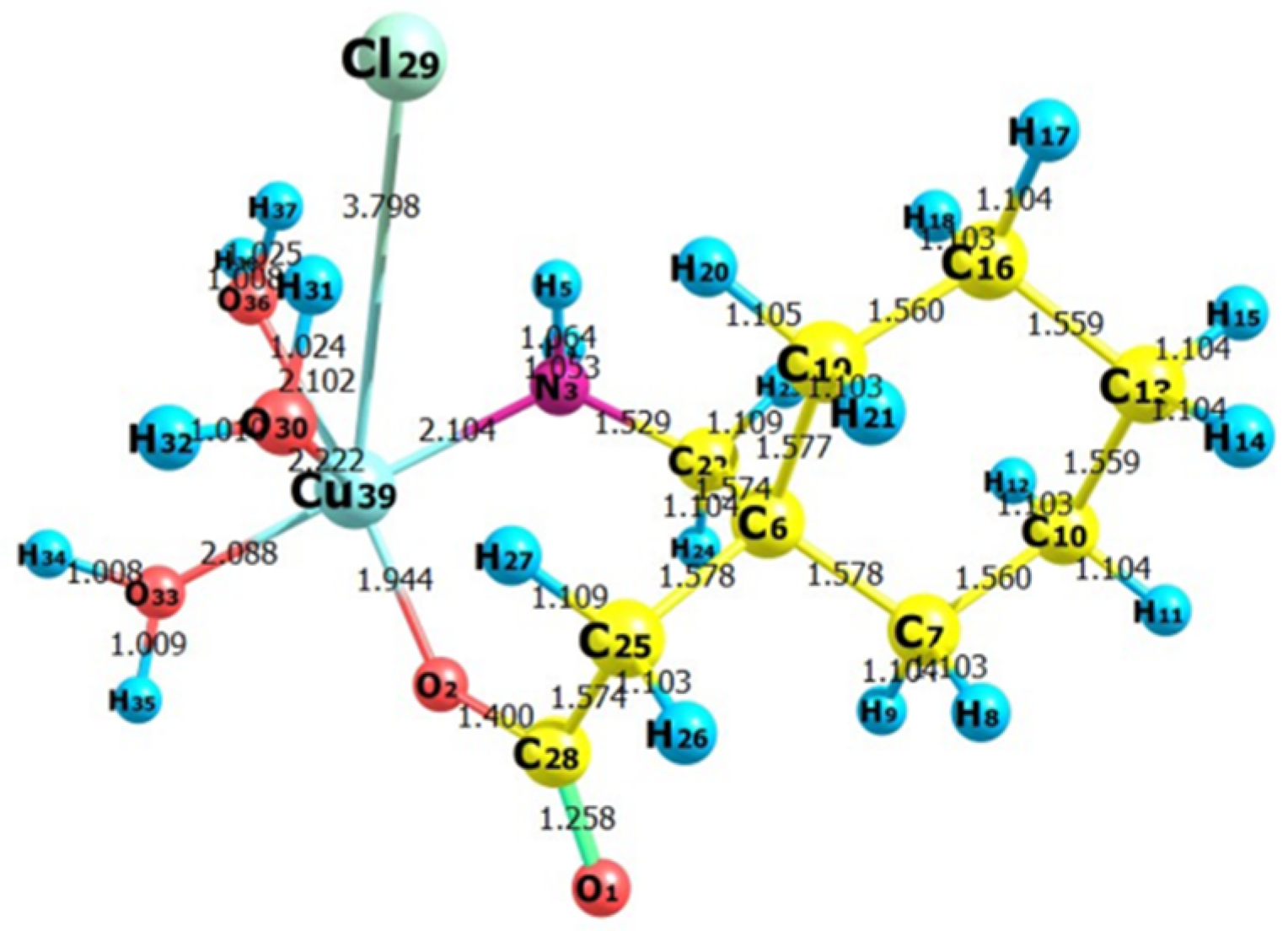
Figure 18.
The optimized structure of the synthesized metal complex [Cu(II)–(Gpn)] showing bond lengths.
Figure 18.
The optimized structure of the synthesized metal complex [Cu(II)–(Gpn)] showing bond lengths.
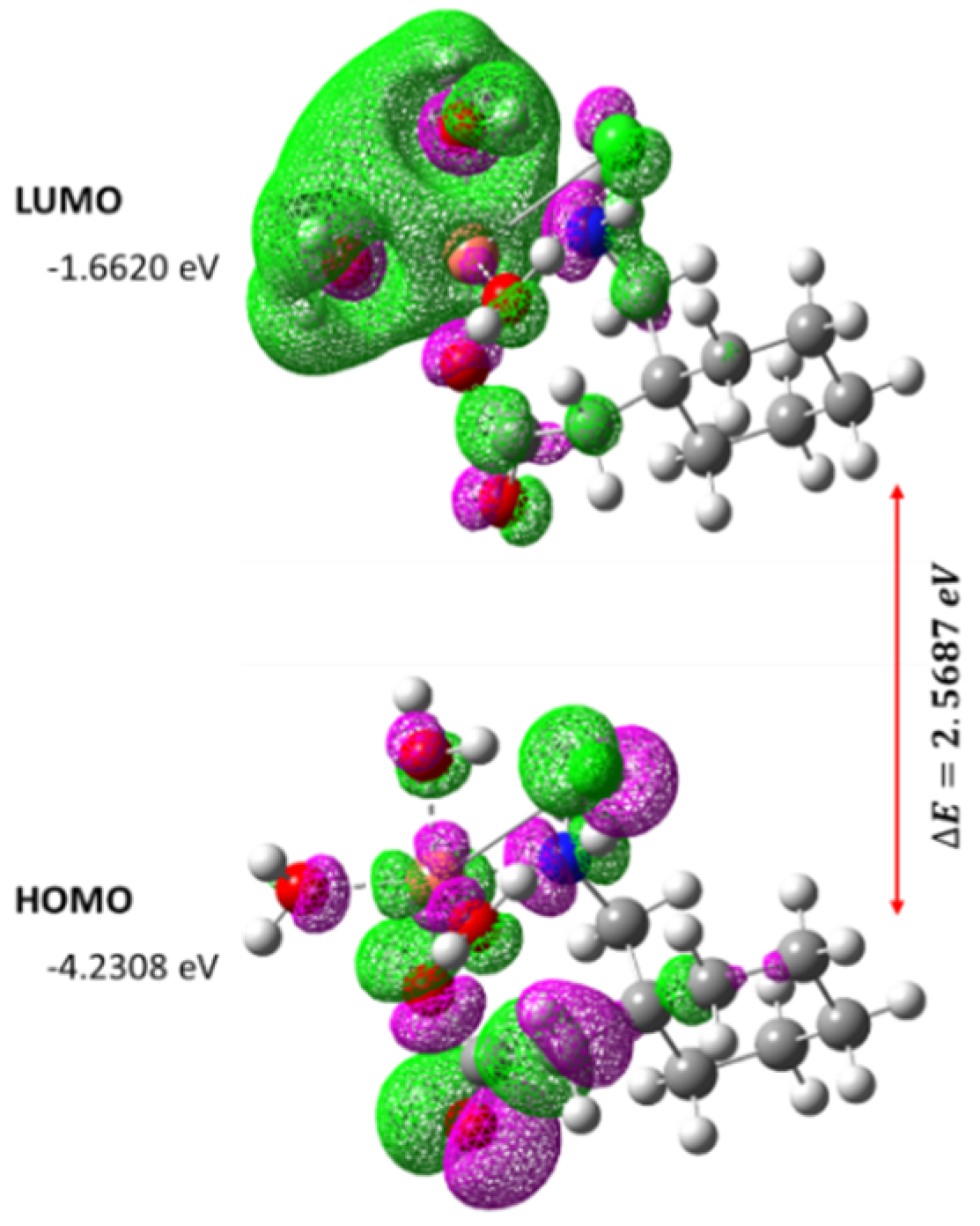
Figure 19.
The spatial plot of HOMO and LUMO with their energy gap for the synthesized metal complex [Cu(II)–(Gpn)].
Figure 19.
The spatial plot of HOMO and LUMO with their energy gap for the synthesized metal complex [Cu(II)–(Gpn)].
Table 1.
Infrared spectral data (cm−1) of Gpn and its complexes.
Table 1.
Infrared spectral data (cm−1) of Gpn and its complexes.
| Compounds | Frequencies, cm−1 |
|---|
| νas(COO) | νs(COO) | δ(NH2) | ν(M-O) | ν(M-N) |
|---|
| Mn(II) | 1650 | 1410 | 1568 | 617 | 502 |
| Cu(II) | 1660 | 1390 | 1558 | 612 | 516 |
| Co(II) | 1672 | 1398 | 1552 | 620 | 519 |
| Ni(II) | 1678 | 1395 | 1553 | 615 | 468 |
Table 2.
The docking score of the four metal complexes docked with two receptors [serotonin (6BQH) and dopamine (6CM4)].
Table 2.
The docking score of the four metal complexes docked with two receptors [serotonin (6BQH) and dopamine (6CM4)].
| S. No. | Receptor Complex | Binding Free Energy (kcal/mol) |
|---|
| | | PDB: 6BQH | PDB: 6CM4 |
|---|
| 1 | [Mn(II)–(Gpn)] | −6.9 | −6.6 |
| 2 | [Co(II)–(Gpn)] | −7.0 | −6.7 |
| 3 | [Ni(II)–(Gpn)] | −6.9 | −6.4 |
| 4 | [Cu(II)–(Gpn)] | −7.2 | −6.5 |
| 5 | Gpn | −5.1 | −4.8 |
Table 3.
The interactions of the [Cu(II)–(Gpn)] complex and Gpn with serotonin (6BQH).
Table 3.
The interactions of the [Cu(II)–(Gpn)] complex and Gpn with serotonin (6BQH).
| S. No. | Receptor | Binding Free Energy (kcal/mol) | Interactions |
|---|
| | | | H-Bond | Others |
| 1 | [Cu(II)–(Gpn)] | −7.2 | Arg173, Thr109, His182, and Asn187 | Asp172 (Attractive charge) and Ala176, Arg173 (Alkyl) |
| 2 | Gpn | −5.1 | Thr109 and Ala108 | Ala321 (Alkyl) |
Table 4.
The [Cu(II)–(Gpn)]–serotonin interaction results by Discovery Studio.
Table 4.
The [Cu(II)–(Gpn)]–serotonin interaction results by Discovery Studio.
| Name | Distance | Category |
|---|
| THR109:HG1–[Cu(II)–Gpn]:O | 2.50 | Hydrogen Bond |
| ARG173:HH11–[Cu(II)–Gpn]:O | 2.63 | Hydrogen Bond |
| ARG173:HH12–[Cu(II)–Gpn]:O | 2.78 | Hydrogen Bond |
| ASN187:HD22–[Cu(II)–Gpn]:O | 2.43 | Hydrogen Bond |
| [Cu(II)-Gpn]:H26–HIS182:O | 2.59 | Hydrogen Bond |
| [Cu(II)-Gpn]:H26–HIS182:O | 2.49 | Hydrogen Bond |
| [Cu(II)-Gpn]:H25–ASN187:OD1 | 2.35 | Hydrogen Bond |
| [Cu(II)-Gpn]:H25–ASN187:OD1 | 2.50 | Hydrogen Bond |
| ALA321–[Cu(II)–Gpn] | 5.31 | Hydrophobic |
Table 5.
The Gpn–serotonin interaction results by Discovery Studio.
Table 5.
The Gpn–serotonin interaction results by Discovery Studio.
| Name | Distance | Category |
|---|
| Gpn:N–ASP172:OD2 | 5.34 | Electrostatic |
| ALA108:HN–Gpn:O | 2.30 | Hydrogen Bond |
| THR109:HN–Gpn:O | 1.85 | Hydrogen Bond |
| THR109:HG1–Gpn:O | 1.97 | Hydrogen Bond |
| Cu(II)-Gpn:H–Gpn:O | 2.91 | Hydrogen Bond |
| ARG173–Gpn | 4.93 | Hydrophobic |
| ALA176–Gpn | 5.04 | Hydrophobic |
Table 6.
The bond lengths of the metal complex [Cu(II)–(Gpn)] obtained through DFT.
Table 6.
The bond lengths of the metal complex [Cu(II)–(Gpn)] obtained through DFT.
| S. No. | [Cu(II)–(Gpn)] (B3LYP/LanL2DZ) |
|---|
| | Atom No. | Bond Length (Å) | Atom No. | Bond Length (Å) |
|---|
| 1 | R(1–28) | 1.258 | R(16–17) | 1.104 |
| 2 | R(2–28) | 1.400 | R(16–18) | 1.103 |
| 3 | R(2–39) | 1.944 | R(16–19) | 1.56 |
| 4 | R(3–4) | 1.053 | R(19–20) | 1.105 |
| 5 | R(3–5) | 1.064 | R(19–21) | 1.103 |
| 6 | R(3–22) | 1.529 | R(22–23) | 1.109 |
| 7 | R(3–39) | 2.104 | R(22–24) | 1.104 |
| 8 | R(6–7) | 1.578 | R(25–26) | 1.103 |
| 9 | R(6–19) | 1.577 | R(25–27) | 1.109 |
| 10 | R(6–22) | 1.574 | R(25–28) | 1.574 |
| 11 | R(6–25) | 1.578 | R(30–31) | 1.024 |
| 12 | R(7–8) | 1.103 | R(30–32) | 1.01 |
| 13 | R(7–9) | 1.104 | R(30–39) | 2.222 |
| 14 | R(7–10) | 1.560 | R(33–34) | 1.008 |
| 15 | R(10–11) | 1.104 | R(33–35) | 1.009 |
| 16 | R(10–12) | 1.103 | R(33–39) | 2.088 |
| 17 | R(10–13) | 1.559 | R(36–37) | 1.025 |
| 18 | R(13–14) | 1.104 | R(36–38) | 1.008 |
| 19 | R(13–15) | 1.104 | R(36–39) | 2.102 |
| 20 | R(13–16) | 1.559 | R(29–39) | 3.798 |
Table 7.
The Mulliken atomic charges of the metal complex [Cu(II)–(Gpn)] atoms.
Table 7.
The Mulliken atomic charges of the metal complex [Cu(II)–(Gpn)] atoms.
| S. No. | Synthesized [Cu(II)-(Gpn)] Complex |
|---|
| Mulliken Atomic Numbers | Mulliken Atomic Charges | MullikenAtomic Numbers | Mulliken Atomic Charges |
|---|
| 1 | 1O | −0.19391 | 21H | 0.06922 |
| 2 | 2O | −0.11703 | 22C | −0.06068 |
| 3 | 3N | −0.34646 | 23H | 0.08103 |
| 4 | 4H | 0.20606 | 24H | 0.09072 |
| 5 | 5H | 0.23817 | 25C | −0.16982 |
| 6 | 6C | 0.02926 | 26H | 0.08199 |
| 7 | 7C | −0.13471 | 27H | 0.11175 |
| 8 | 8H | 0.07097 | 28C | 0.21118 |
| 9 | 9H | 0.07212 | 29Cl | −0.7276 |
| 10 | 10C | −0.13351 | 30O | −0.28504 |
| 11 | 11H | 0.07166 | 31H | 0.24647 |
| 12 | 12H | 0.06446 | 32H | 0.20389 |
| 13 | 13C | −0.13247 | 33O | −0.25834 |
| 14 | 14H | 0.06717 | 34H | 0.23457 |
| 15 | 15H | 0.06992 | 35H | 0.23367 |
| 16 | 16C | −0.1326 | 36O | −0.28583 |
| 17 | 17H | 0.07643 | 37H | 0.25414 |
| 18 | 18H | 0.06574 | 38H | 0.21341 |
| 19 | 19C | −0.13937 | C39u | −0.03369 |
| 20 | 20H | 0.08705 | | |
Table 8.
The various other theoretical molecular parameters of the metal complex [Cu(II)–(Gpn)].
Table 8.
The various other theoretical molecular parameters of the metal complex [Cu(II)–(Gpn)].
| Parameters | RB3LYP/LanL2DZ |
|---|
| Minimum SCF energy (a.u.) | −982.219370 |
| Polarizability (a) (a.u.) | 84.074494 |
| Dipole moment (Debye) | 7.662013 |
| Zero point vibrational energy (kcal/mol) | 241.62872 |
| Total thermal energy (kcal/mol) | 253.148 |
| Electronic spatial extent (a.u.) | 5378.5506 |
| Frontier MO energies (eV) | |
| LUMO | −1.6620 |
| HOMO | −4.2308 |
| Gap (HOMO–LUMO) | 2.5687 |


 ,
, 





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}