
K18- and CAG-hACE2 Transgenic Mouse Models and SARS-CoV-2: Implications for Neurodegeneration Research

 , , , and
, , , and
Abstract
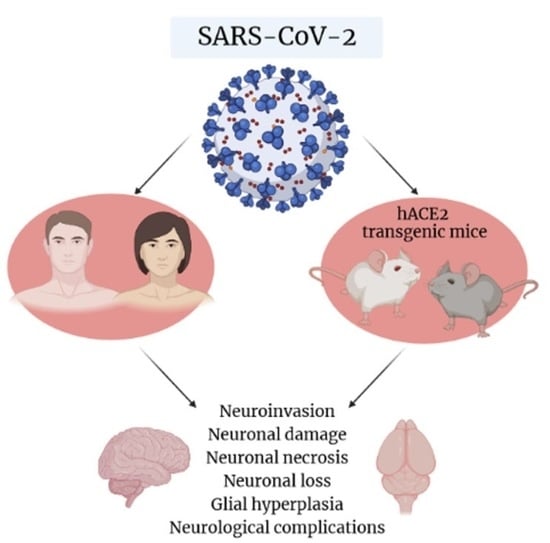
:
1. Introduction
2. SARS-CoV-2 and Possible Implications for Human Brain Neurodegeneration
3. SARS-CoV-2-Induced Increased Mortality Rate of K18-hACE2 Mice
4. K18-hACE2 Mice and SARS-CoV-2-Evoked Inflammation
5. SARS-CoV-2 Entry, Distribution, and Neuroinvasion in K18-hACE2 Mice
6. CAG-hACE2 Transgenic Mice
7. CAG-hACE2 Transgenic Mice: Research on Antiviral Drugs and Vaccines
8. Other hACE2-Expressing Mouse Models
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Achbani, A.; Sine, H.; Naciri, A.; Baba, M.A.; Kharbach, A.; Bouchriti, Y.; Nejmeddine, M. Can the 2019 Novel Coronavirus Cause Parkinson’s Disease? Mov. Disord. 2020, 35, 1102–1103. [Google Scholar] [CrossRef]
- Krey, L.; Huber, M.K.; Höglinger, G.U.; Wegner, F. Can SARS-CoV-2 Infection Lead to Neurodegeneration and Parkinson’s Disease? Brain Sci. 2021, 11, 1654. [Google Scholar] [CrossRef] [PubMed]
- Dziedzic, A.; Saluk-Bijak, J.; Miller, E.; Niemcewicz, M.; Bijak, M. The Impact of SARS-CoV-2 Infection on the Development of Neurodegeneration in Multiple Sclerosis. Int. J. Mol. Sci. 2021, 22, 1804. [Google Scholar] [CrossRef] [PubMed]
- LaRovere, K.L.; Riggs, B.J.; Poussaint, T.Y.; Young, C.C.; Newhams, M.M.; Maamari, M.; Walker, T.C.; Singh, A.R.; Dapul, H.; Hobbs, C.V.; et al. Overcoming COVID-19 Investigators. Neurologic Involvement in Children and Adolescents Hospitalized in the United States for COVID-19 or Multisystem Inflammatory Syndrome. JAMA Neurol. 2021, 78, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Solomon, I.H.; Normandin, E.; Bhattacharyya, S.; Mukerji, S.S.; Keller, K.; Ali, A.S.; Adams, G.; Hornick, J.L.; Padera, R.F., Jr.; Sabeti, P. Neuropathological Features of COVID-19. N. Engl. J. Med. 2020, 383, 989–992. [Google Scholar] [CrossRef]
- Desforges, M.; le Coupanec, A.; Brison, É.; Meessen-Pinard, M.; Talbot, P.J. Neuroinvasive and neurotropic human respiratory coronaviruses: Potential neurovirulent agents in humans. Adv. Exp. Med. Biol. 2014, 807, 75–96. [Google Scholar]
- Paniz-Mondolfi, A.; Bryce, C.; Grimes, Z.; Gordon, R.E.; Reidy, J.; Lednicky, J.; Sordillo, E.M.; Fowkes, M. Central nervous system involvement by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). J. Med. Virol. 2020, 92, 699–702. [Google Scholar] [CrossRef] [Green Version]
- Matschke, J.; Lutgehetmann, M.; Hagel, C.; Sperhake, J.P.; Schroder, A.S.; Edler, C.; Mushumba, H.; Fitzek, A.; Allweiss, L.; Dandri, M.; et al. Neuropathology of patients with COVID-19 in Germany: A post-mortem case series. Lancet Neurol. 2020, 19, 919–929. [Google Scholar] [CrossRef]
- Constant, O.; Barthelemy, J.; Bolloré, K.; Tuaillon, E.; Gosselet, F.; Chable-Bessia, C.; Merida, P.; Muriaux, D.; Van de Perre, P.; Salinas, S.; et al. SARS-CoV-2 Poorly Replicates in Cells of the Human Blood-Brain Barrier Without Associated Deleterious Effects. Front. Immunol. 2021, 12, 697329. [Google Scholar] [CrossRef]
- Buzhdygan, T.P.; DeOre, B.J.; Baldwin-Leclair, A.; Bullock, T.A.; McGary, H.M.; Khan, J.A.; Razmpour, R.; Hale, J.F.; Galie, P.A.; Potula, R.; et al. The SARS-CoV-2 spike protein alters barrier function in 2D static and 3D microfluidic in-vitro models of the human blood-brain barrier. Neurobiol. Dis. 2020, 146, 105131. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Y.; Li, L.; Zhang, Y.; Wang, X.S. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect. Dis. Poverty 2020, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Puelles, V.G.; Lütgehetmann, M.; Lindenmeyer, M.T.; Sperhake, J.P.; Wong, M.N.; Allweiss, L.; Chilla, S.; Heinemann, A.; Wanner, N.; Liu, S.; et al. Multiorgan and renal tropism of SARS-CoV-2. N. Engl. J. Med. 2020, 383, 590–592. [Google Scholar] [CrossRef]
- Moein, S.T.; Hashemian, S.M.; Mansourafshar, B.; Khorram-Tousi, A.; Tabarsi, P.; Doty, R.L. Smell dysfunction: A biomarker for COVID-19. Int. Forum Allergy Rhinol. 2020, 10, 944–950. [Google Scholar] [CrossRef]
- Giacomelli, A.; Pezzati, L.; Conti, F.; Bernacchia, D.; Siano, M.; Oreni, L.; Rusconi, S.; Gervasoni, C.; Ridolfo, A.L.; Rizzardini, G.; et al. Self-reported Olfactory and Taste Disorders in Patients with Severe Acute Respiratory Coronavirus 2 Infection: A Cross-sectional Study. Clin. Infect. Dis. 2020, 71, 889–890. [Google Scholar] [CrossRef] [Green Version]
- Chung, T.W.; Sridhar, S.; Zhang, A.J.; Chan, K.H.; Li, H.L.; Wong, F.K.; Ng, M.Y.; Tsang, R.K.; Lee, A.C.; Fan, Z.; et al. Olfactory Dysfunction in Coronavirus Disease 2019 Patients: Observational Cohort Study and Systematic Review. Open Forum Infect. Dis. 2020, 7, ofaa199. [Google Scholar] [CrossRef]
- Kumari, P.; Rothan, H.A.; Natekar, J.P.; Stone, S.; Pathak, H.; Strate, P.G.; Arora, K.; Brinton, M.A.; Kumar, M. Neuroinvasion and Encephalitis Following Intranasal Inoculation of SARS-CoV-2 in K18-hACE2 Mice. Viruses 2021, 13, 132. [Google Scholar] [CrossRef]
- Meinhardt, J.; Radke, J.; Dittmayer, C.; Franz, J.; Thomas, C.; Mothes, R.; Laue, M.; Schneider, J.; Brünink, S.; Greuel, S.; et al. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat. Neurosci. 2021, 2, 168–175. [Google Scholar] [CrossRef]
- Khan, M.; Yoo, S.J.; Clijsters, M.; Backaert, W.; Vanstapel, A.; Speleman, K.; Lietaer, C.; Choi, S.; Hether, T.D.; Marcelis, L.; et al. Visualizing in deceased COVID-19 patients how SARS-CoV-2 attacks the respiratory and olfactory mucosae but spares the olfactory bulb. Cell 2021, 184, 5932–5949.e15. [Google Scholar] [CrossRef]
- Pellegrini, L.; Albecka, A.; Mallery, D.L.; Kellner, M.J.; Paul, D.; Carter, A.P.; James, L.C.; Lancaster, M.A. SARS-CoV-2 Infects the Brain Choroid Plexus and Disrupts the Blood-CSF Barrier in Human Brain Organoids. Cell Stem Cell 2020, 27, 951–961.e5. [Google Scholar] [CrossRef]
- Pleasure, S.J.; Green, A.J.; Josephson, S.A. The spectrum of neurologic disease in the severe acute respiratory syndrome coronavirus 2 pandemic infection: Neurologists move to the frontlines. JAMA Neurol. 2020, 77, 679–680. [Google Scholar] [CrossRef] [PubMed]
- Nath, A. Neurologic complications of coronavirus infections. Neurology 2020, 94, 809–810. [Google Scholar] [CrossRef] [PubMed]
- Helms, J.; Kremer, S.; Merdji, H.; Clere-Jehl, R.; Schenck, M.; Kummerlen, C.; Collange, O.; Boulay, C.; Fafi-Kremer, S.; Ohana, M.; et al. Neurologic Features in Severe SARS-CoV-2 Infection. N. Engl. J. Med. 2020, 382, 2268–2270. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of Immune Response in Patients with Coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef]
- Park, M.D. Macrophages: A Trojan horse in COVID-19? Nat. Rev. Immunol. 2020, 13, 2178. [Google Scholar] [CrossRef]
- Khatoon, F.; Prasad, K.; Kumar, V. Neurological manifestations of COVID-19: Available evidences and a new paradigm. J. Neuro Virol. 2020, 26, 619–630. [Google Scholar] [CrossRef]
- Vojdani, A.; Vojdani, E.; Kharrazian, D. Reaction of Human Monoclonal Antibodies to SARS-CoV-2 Proteins with Tissue Antigens: Implications for Autoimmune Diseases. Front. Immunol. 2021, 11, 617089. [Google Scholar] [CrossRef]
- Prüss, H. Autoantibodies in neurological disease. Nat. Rev. Immunol. 2021, 21, 798–813. [Google Scholar] [CrossRef]
- Kreye, J.; Reincke, S.M.; Kornau, H.C.; Sánchez-Sendin, E.; Corman, V.M.; Liu, H.; Yuan, M.; Wu, N.C.; Zhu, X.; Lee, C.D.; et al. A Therapeutic Non-self-reactive SARS-CoV-2 Antibody Protects from Lung Pathology in a COVID-19 Hamster Model. Cell 2020, 183, 1058–1069.e19. [Google Scholar] [CrossRef]
- Yeh, E.A.; Collins, A.; Cohen, M.E.; Duffner, P.K.; Faden, H. Detection of coronavirus in the central nervous system of a child with acute disseminated encephalomyelitis. Pediatrics 2004, 113, e73–e76. [Google Scholar]
- Toscano, G.; Palmerini, F.; Ravaglia, S.; Ruiz, L.; Invernizzi, P.; Cuzzoni, M.G.; Franciotta, D.; Baldanti, F.; Daturi, R.; Postorino, P.; et al. Guillain-Barré Syndrome Associated with SARS-CoV-2. N. Engl. J. Med. 2020, 382, 2574–2576. [Google Scholar] [CrossRef] [PubMed]
- Poyiadji, N.; Shahin, G.; Noujaim, D.; Stone, M.; Patel, S.; Griffith, B. COVID-19-associated Acute Hemorrhagic Necrotizing Encephalopathy: Imaging Features. Radiology 2020, 296, e119–e120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, A.; Iliffe, S. Alzheimer’s disease. BMJ 2009, 338, b158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Douaud, G.; Lee, S.; Alfaro-Almagro, F.; Arthofer, C.; Wang, C.; McCarthy, P.; Lange, F.; Andersson, J.L.R.; Griffanti, L.; Duff, E.; et al. SARS-CoV-2 is associated with changes in brain structure in UK Biobank. Nature 2022, 604, 697–707. [Google Scholar] [CrossRef]
- Terry, R.D. Biologic differences between early- and late-onset Alzheimer disease. Alzheimer Dis. Assoc. Disord. 1995, 9, 26–27. [Google Scholar] [CrossRef]
- Harris, T.C.; de Rooij, R.; Kuhl, E. The Shrinking Brain: Cerebral Atrophy Following Traumatic Brain Injury. Ann. Biomed. Eng. 2019, 47, 1941–1959. [Google Scholar] [CrossRef] [Green Version]
- Ramani, A.; Müller, L.; Ostermann, P.N.; Gabriel, E.; Abida-Islam, P.; Müller-Schiffmann, A.; Mariappan, A.; Goureau, O.; Gruell, H.; Walker, A.; et al. SARS-CoV-2 targets neurons of 3D human brain organoids. EMBO J. 2020, 39, e106230. [Google Scholar] [CrossRef]
- Thakur, K.T.; Miller, E.H.; Glendinning, M.D.; Al-Dalahmah, O.; Banu, M.A.; Boehme, A.K.; Boubour, A.L.; Bruce, S.S.; Chong, A.M.; Claassen, J.; et al. COVID-19 neuropathology at Columbia University Irving Medical Center/New York Presbyterian Hospital. Brain 2021, 144, 2696–2708. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, J.; Hou, Y.; Leverenz, J.B.; Kallianpur, A.; Mehra, R.; Liu, Y.; Yu, H.; Pieper, A.A.; Jehi, L.; et al. Network medicine links SARS-CoV-2/COVID-19 infection to brain microvascular injury and neuroinflammation in dementia-like cognitive impairment. Alzheimers Res. Ther. 2021, 13, 110. [Google Scholar] [CrossRef]
- Cohen, M.E.; Eichel, R.; Steiner-Birmanns, B.; Janah, A.; Ioshpa, M.; Bar-Shalom, R.; Paul, J.J.; Gaber, H.; Skrahina, V.; Bornstein, N.M.; et al. A case of probable Parkinson’s disease after SARS-CoV-2 infection. Lancet Neurol. 2020, 19, 804–805. [Google Scholar] [CrossRef]
- Palao, M.; Fernández-Díaz, E.; Gracia-Gil, J.; Romero-Sánchez, C.M.; Díaz-Maroto, I.; Segura, T. Multiple sclerosis following SARS-CoV-2 infection. Mult. Scler. Relat. Disord. 2020, 45, 102377. [Google Scholar] [CrossRef] [PubMed]
- Arbour, N.; Day, R.; Newcombe, J.; Talbot, P.J. Neuroinvasion by human respiratory coronaviruses. J. Virol. 2000, 74, 8913–8921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, T.L.; Weitzer, D.J. Long COVID and Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS)-A Systemic Review and Comparison of Clinical Presentation and Symptomatology. Medicina 2021, 57, 418. [Google Scholar] [CrossRef]
- Leonardi, M.; Padovani, A.; McArthur, J.C. Neurological manifestations associated with COVID-19: A review and a call for action. J. Neurol. 2020, 267, 1573–1576. [Google Scholar] [CrossRef]
- Roberts, A.; Subbarao, K. Animal models for SARS. Adv. Exp. Med. Biol. 2006, 581, 463–471. [Google Scholar]
- Gembardt, F.; Sterner-Kock, A.; Imboden, H.; Spalteholz, M.; Reibitz, F.; Schultheiss, H.P.; Siems, W.E.; Walther, T. Organ-specific distribution of ACE2 mRNA and correlating peptidase activity in rodents. Peptides 2005, 26, 1270–1277. [Google Scholar] [CrossRef]
- Chen, R.; Wang, K.; Yu, J.; Howard, D.; French, L.; Chen, Z.; Wen, C.; Xu, Z. The Spatial and Cell-Type Distribution of SARS-CoV-2 Receptor ACE2 in the Human and Mouse Brains. Front. Neurol. 2021, 11, 573095. [Google Scholar] [CrossRef]
- Dong, W.; Mead, H.; Tian, L.; Park, J.G.; Garcia, J.I.; Jaramillo, S.; Barr, T.; Kollath, D.S.; Coyne, V.K.; Stone, N.E.; et al. The K18-Human ACE2 Transgenic Mouse Model Recapitulates Non-severe and Severe COVID-19 in Response to an Infectious Dose of the SARS-CoV-2 Virus. J. Virol. 2021, 12, e0096421. [Google Scholar] [CrossRef]
- McCray, P.B., Jr.; Pewe, L.; Wohlford-Lenane, C.; Hickey, M.; Manzel, L.; Shi, L.; Netland, J.; Jia, H.P.; Halabi, C.; Sigmund, C.D.; et al. Lethal infection of K18-hACE2 mice infected with severe acute respiratory syndrome coronavirus. J. Virol. 2007, 81, 813–821. [Google Scholar] [CrossRef] [Green Version]
- Coperchini, F.; Chiovato, L.; Croce, L.; Magri, F.; Rotondi, M. The cytokine storm in COVID-19: An overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev. 2020, 53, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Oladunni, F.S.; Park, J.G.; Pino, P.A.; Gonzalez, O.; Akhter, A.; Allué-Guardia, A.; Olmo-Fontánez, A.; Gautam, S.; Garcia-Vilanova, A.; Ye, C.; et al. Lethality of SARS-CoV-2 infection in K18 human angiotensin-converting enzyme 2 transgenic mice. Nat. Commun. 2020, 11, 6122. [Google Scholar] [CrossRef] [PubMed]
- Buszko, M.; Park, J.H.; Verthelyi, D.; Sen, R.; Young, H.A.; Rosenberg, A.S. The dynamic changes in cytokine responses in COVID-19: A snapshot of the current state of knowledge. Nat. Immunol. 2020, 21, 1146–1151. [Google Scholar] [CrossRef]
- Klingenstein, M.; Klingenstein, S.; Neckel, P.H.; Mack, A.F.; Wagner, A.P.; Kleger, A.; Liebau, S.; Milazzo, A. Evidence of SARS-CoV2 Entry Protein ACE2 in the Human Nose and Olfactory Bulb. Cells Tissues Organs 2020, 209, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Golden, J.W.; Cline, C.R.; Zeng, X.; Garrison, A.R.; Carey, B.D.; Mucker, E.M.; White, L.E.; Shamblin, J.D.; Brocato, R.L.; Liu, J.; et al. Human angiotensin-converting enzyme 2 transgenic mice infected with SARS-CoV-2 develop severe and fatal respiratory disease. JCI Insight 2020, 5, e142032. [Google Scholar] [CrossRef]
- Moreau, G.B.; Burgess, S.L.; Sturek, J.M.; Donlan, A.N.; Petri, W.A.; Mann, B.J. Evaluation of K18-hACE2 Mice as a Model of SARS-CoV-2 Infection. Am. J. Trop. Med. Hyg. 2020, 103, 1215–1219. [Google Scholar] [CrossRef]
- Perlman, S.; Evans, G.; Afifi, A. Effect of olfactory bulb ablation on spread of a neurotropic coronavirus into the mouse brain. J. Exp. Med. 1990, 172, 1127–1132. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhou, L.; Bao, L.; Liu, J.; Zhu, H.; Lv, Q.; Liu, R.; Chen, W.; Tong, W.; Wei, Q.; et al. SARS-CoV-2 crosses the blood-brain barrier accompanied with basement membrane disruption without tight junctions alteration. Sig. Transduct. Target Ther. 2021, 6, 337. [Google Scholar] [CrossRef]
- Reynolds, J.L.; Mahajan, S.D. SARS-CoV-2 alters blood brain barrier integrity contributing to neuro-inflammation. J. Neuroimmune Pharmacol. 2021, 16, 4–6. [Google Scholar] [CrossRef]
- Rhea, E.M.; Logsdon, A.F.; Hansen, K.M.; Williams, L.M.; Reed, M.J.; Baumann, K.K.; Holden, S.J.; Raber, J.; Banks, W.A.; Erickson, M.A. The S1 protein of SARS-CoV-2 crosses the blood-brain barrier in mice. Nat. Neurosci. 2021, 24, 368–378. [Google Scholar] [CrossRef]
- Crunfli, F.; Carregari, V.C.; Martins-de-Souza, D. SARS-CoV-2 infects brain astrocytes of COVID-19 patients and impairs neuronal viability. medRxiv 2021. preprint. [Google Scholar] [CrossRef]
- Rothan, H.A.; Kumari, P.; Stone, S.; Natekar, J.P.; Arora, K.; Auroni, T.T.; Kumar, M. SARS-CoV-2 Infects Primary Neurons from Human ACE2 Expressing Mice and Upregulates Genes Involved in the Inflammatory and Necroptotic Pathways. Pathogens 2022, 11, 257. [Google Scholar] [CrossRef]
- Carossino, M.; Kenney, D.; O’Connell, A.K.; Montanaro, P.; Tseng, A.E.; Gertje, H.P.; Grosz, K.A.; Ericsson, M.; Huber, B.R.; Kurnick, S.A.; et al. Fatal Neurodissemination and SARS-CoV-2 Tropism in K18-hACE2 Mice Is Only Partially Dependent on hACE2 Expression. Viruses 2022, 14, 535. [Google Scholar] [CrossRef] [PubMed]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef]
- Shapira, T.; Monreal, I.A.; Dion, S.P.; Jager, M.; Désilets, A.; Olmstead, A.D.; Vandal, T.; Buchholz, D.W.; Imbiakha, B.; Gao, G.; et al. A novel highly potent inhibitor of TMPRSS2-like proteases blocks SARS-CoV-2 variants of concern and is broadly protective against infection and mortality in mice. bioRxiv 2021. preprint. [Google Scholar] [CrossRef]
- Chen, R.E.; Winkler, E.S.; Case, J.B.; Aziati, I.D.; Bricker, T.L.; Joshi, A.; Darling, T.L.; Ying, B.; Errico, J.M.; Shrihari, S.; et al. In vivo monoclonal antibody efficacy against SARS-CoV-2 variant strains. Nature 2021, 596, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Wong, L.R.; Li, K.; Verma, A.K.; Ortiz, M.E.; Wohlford-Lenane, C.; Leidinger, M.R.; Knudson, C.M.; Meyerholz, D.K.; McCray, P.B., Jr.; et al. COVID-19 treatments and pathogenesis including anosmia in K18-hACE2 mice. Nature 2021, 589, 603–607. [Google Scholar] [CrossRef]
- Halfmann, P.J.; Iida, S.; Iwatsuki-Horimoto, K.; Maemura, T.; Kiso, M.; Scheaffer, S.M.; Darling, T.L.; Joshi, A.; Loeber, S.; Singh, G.; et al. SARS-CoV-2 Omicron virus causes attenuated disease in mice and hamsters. Nature 2022, 603, 687–692. [Google Scholar] [CrossRef]
- Shuai, H.; Chan, J.F.; Hu, B.; Chai, Y.; Yuen, T.T.; Yin, F.; Huang, X.; Yoon, C.; Hu, J.C.; Liu, H.; et al. Attenuated replication and pathogenicity of SARS-CoV-2 B.1.1.529 Omicron. Nature 2022, 603, 693–699. [Google Scholar] [CrossRef]
- Ku, M.W.; Authié, P.; Bourgine, M.; Anna, F.; Noirat, A.; Moncoq, F.; Vesin, B.; Nevo, F.; Lopez, J.; Souque, P.; et al. Brain cross-protection against SARS-CoV-2 variants by a lentiviral vaccine in new transgenic mice. EMBO Mol. Med. 2021, 13, e14459. [Google Scholar] [CrossRef]
- Fumagalli, V.; Ravà, M.; Marotta, D.; Di Lucia, P.; Laura, C.; Sala, E.; Grillo, M.; Bono, E.; Giustini, L.; Perucchini, C.; et al. Administration of aerosolized SARS-CoV-2 to K18-hACE2 mice uncouples respiratory infection from fatal neuroinvasion. Sci. Immunol. 2022, 7, eabl9929. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.T.; Huang, C.; Newman, P.; Wang, N.; Narayanan, K.; Watts, D.M.; Makino, S.; Packard, M.M.; Zaki, S.R.; Chan, T.S.; et al. Severe acute respiratory syndrome coronavirus infection of mice transgenic for the human Angiotensin-converting enzyme 2 virus receptor. J. Virol. 2007, 81, 1162–1173. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, N.; Yoshikawa, T.; Hill, T.; Huang, C.; Watts, D.M.; Makino, S.; Milligan, G.; Chan, T.; Peters, C.J.; Tseng, C.T. Differential virological and immunological outcome of severe acute respiratory syndrome coronavirus infection in susceptible and resistant transgenic mice expressing human angiotensin-converting enzyme 2. J. Virol. 2009, 83, 5451–5465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asaka, M.N.; Utsumi, D.; Kamada, H.; Nagata, S.; Nakachi, Y.; Yamaguchi, T.; Kawaoka, Y.; Kuba, K.; Yasutomi, Y. Highly susceptible SARS-CoV-2 model in CAG promoter-driven hACE2-transgenic mice. JCI Insight 2021, 6, e152529. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Yan, L.; Xu, W.; Agrawal, A.S.; Algaissi, A.; Tseng, C.K.; Wang, Q.; Du, L.; Tan, W.; Wilson, I.A.; et al. A pan-coronavirus fusion inhibitor targeting the HR1 domain of human coronavirus spike. Sci. Adv. 2019, 5, eaav4580. [Google Scholar] [CrossRef] [Green Version]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef] [Green Version]
- Xia, S.; Lan, Q.; Zhu, Y.; Wang, C.; Xu, W.; Li, Y.; Wang, L.; Jiao, F.; Zhou, J.; Hua, C.; et al. Structural and functional basis for pan-CoV fusion inhibitors against SARS-CoV-2 and its variants with preclinical evaluation. Signal Transduct. Target Ther. 2021, 6, 288. [Google Scholar] [CrossRef]
- Zhao, P.; Praissman, J.L.; Grant, O.C.; Cai, Y.; Xiao, T.; Rosenbalm, K.E.; Aoki, K.; Kellman, B.P.; Bridger, R.; Barouch, D.H.; et al. Virus-Receptor Interactions of Glycosylated SARS-CoV-2 Spike and Human ACE2 Receptor. Cell Host Microbe 2020, 28, 586–601.e6. [Google Scholar] [CrossRef]
- Watanabe, Y.; Berndsen, Z.T.; Raghwani, J.; Seabright, G.E.; Allen, J.D.; Pybus, O.G.; McLellan, J.S.; Wilson, I.A.; Bowden, T.A.; Ward, A.B.; et al. Vulnerabilities in coronavirus glycan shields despite extensive glycosylation. Nat. Commun. 2020, 11, 2688. [Google Scholar] [CrossRef]
- Huang, H.Y.; Liao, H.Y.; Chen, X.; Wang, S.W.; Cheng, C.W.; Shahed-Al-Mahmud, M.; Liu, Y.M.; Mohapatra, A.; Chen, T.H.; Lo, J.M.; et al. Vaccination with SARS-CoV-2 spike protein lacking glycan shields elicits enhanced protective responses in animal models. Sci. Transl. Med. 2022, 1, eabm0899. [Google Scholar] [CrossRef]
- McCallum, M.; De Marco, A.; Lempp, F.A.; Tortorici, M.A.; Pinto, D.; Walls, A.C.; Beltramello, M.; Chen, A.; Liu, Z.; Zatta, F.; et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. Cell 2021, 184, 2332–2347. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Yao, W.; Hu, Y.; Wu, S.; Li, J.; Zhou, H.; Hong, K.; Chen, J.; Liu, L.; Lan, K.; et al. A trimeric NTD and RBD SARS-CoV-2 subunit vaccine induced protective immunity in CAG-hACE2 transgenic mice and rhesus macaques. bioRxiv 2021, 11. [Google Scholar] [CrossRef]
- Menachery, V.D.; Yount, B.L., Jr.; Sims, A.C.; Debbink, K.; Agnihothram, S.S.; Gralinski, L.E.; Graham, R.L.; Scobey, T.; Plante, J.A.; Royal, S.R.; et al. SARS-like WIV1-CoV poised for human emergence. Proc. Natl. Acad. Sci. USA 2016, 113, 3048–3053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Xia, H.; Bindom, S.M.; Lazartigues, E. Neuron-targeted expression of ACE2 in the central nervous system prevents angiotensin-II-mediated hypertension. FASEB J. 2008, 22, 741.1. [Google Scholar] [CrossRef]
- Yang, X.H.; Deng, W.; Tong, Z.; Liu, Y.X.; Zhang, L.F.; Zhu, H.; Gao, H.; Huang, L.; Liu, Y.L.; Ma, C.M.; et al. Mice transgenic for human angiotensin-converting enzyme 2 provide a model for SARS coronavirus infection. Comp. Med. 2007, 57, 450–459. [Google Scholar]
- Bruter, A.V.; Korshunova, D.S.; Kubekina, M.V.; Sergiev, P.V.; Kalinina, A.A.; Ilchuk, L.A.; Silaeva, Y.Y.; Korshunov, E.N.; Soldatov, V.O.; Deykin, A.V. Novel transgenic mice with Cre-dependent co-expression of GFP and human ACE2: A safe tool for study of COVID-19 pathogenesis. Transgenic Res. 2021, 30, 289–301. [Google Scholar] [CrossRef]
- Rockx, B.; Kuiken, T. Comparative pathogenesis of COVID-19, MERS, and SARS in a nonhuman primate model. Science 2020, 368, 1012–1015. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.I.; Kim, S.G.; Kim, S.M.; Kim, E.H.; Park, S.J.; Yu, K.M.; Chang, J.H.; Kim, E.J.; Lee, S.; Casel, M.A.B.; et al. Infection and rapid transmission of SARS-CoV-2 in ferrets. Cell Host Microbe 2020, 27, 704–709.e2. [Google Scholar] [CrossRef]
- Sia, S.F.; Yan, L.M.; Chin, A.W.H.; Fung, K.; Choy, K.T.; Wong, A.Y.L.; Kaewpreedee, P.; Perera, R.A.P.M.; Poon, L.L.M.; Nicholls, J.M.; et al. Pathogenesis and transmission of SARS-CoV-2 in golden hamsters. Nature 2020, 583, 834–838. [Google Scholar] [CrossRef]
- Wong, T.Y.; Horspool, A.M.; Russ, B.P.; Ye, C.; Lee, K.S.; Winters, M.T.; Bevere, J.R.; Miller, O.A.; Rader, N.A.; Cooper, M.; et al. Evaluating Antibody Mediated Protection against Alpha, Beta, and Delta SARS-CoV-2 Variants of Concern in K18-hACE2 Transgenic Mice. J. Virol. 2022, 23, e0218421. [Google Scholar] [CrossRef]
- Rosenfeld, R.; Noy-Porat, T.; Mechaly, A.; Makdasi, E.; Levy, Y.; Alcalay, R.; Falach, R.; Aftalion, M.; Epstein, E.; Gur, D.; et al. Post-exposure protection of SARS-CoV-2 lethal infected K18-hACE2 transgenic mice by neutralizing human monoclonal antibody. Nat. Commun. 2021, 11, 944. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Model | Promoter |
|---|---|
| K18-hACE | Cytokeratine |
| CAG-hACE | Cytomegalovirus, β-actin, beta-rabbit globine (CAG) |
| HFH4-hACE | HFH4 /FOXJ |
| syn-hACE2 | Synapse |
| CMV-hACE2 | Chicken β-actin |
| mAce2-hACE | Mouse Ace2 |
| hACE2 KI | CRISPR/Cas9 technology |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dedoni, S.; Avdoshina, V.; Camoglio, C.; Siddi, C.; Fratta, W.; Scherma, M.; Fadda, P. K18- and CAG-hACE2 Transgenic Mouse Models and SARS-CoV-2: Implications for Neurodegeneration Research. Molecules 2022, 27, 4142. https://doi.org/10.3390/molecules27134142
Dedoni S, Avdoshina V, Camoglio C, Siddi C, Fratta W, Scherma M, Fadda P. K18- and CAG-hACE2 Transgenic Mouse Models and SARS-CoV-2: Implications for Neurodegeneration Research. Molecules. 2022; 27(13):4142. https://doi.org/10.3390/molecules27134142
Chicago/Turabian StyleDedoni, Simona, Valeria Avdoshina, Chiara Camoglio, Carlotta Siddi, Walter Fratta, Maria Scherma, and Paola Fadda. 2022. "K18- and CAG-hACE2 Transgenic Mouse Models and SARS-CoV-2: Implications for Neurodegeneration Research" Molecules 27, no. 13: 4142. https://doi.org/10.3390/molecules27134142