
Development of a Novel Class of Pyridazinone Derivatives as Selective MAO-B Inhibitors

 ,
,  , ,
, ,  , , ,
, , ,  , and
, and
Abstract
:1. Introduction
2. Results and Discussion
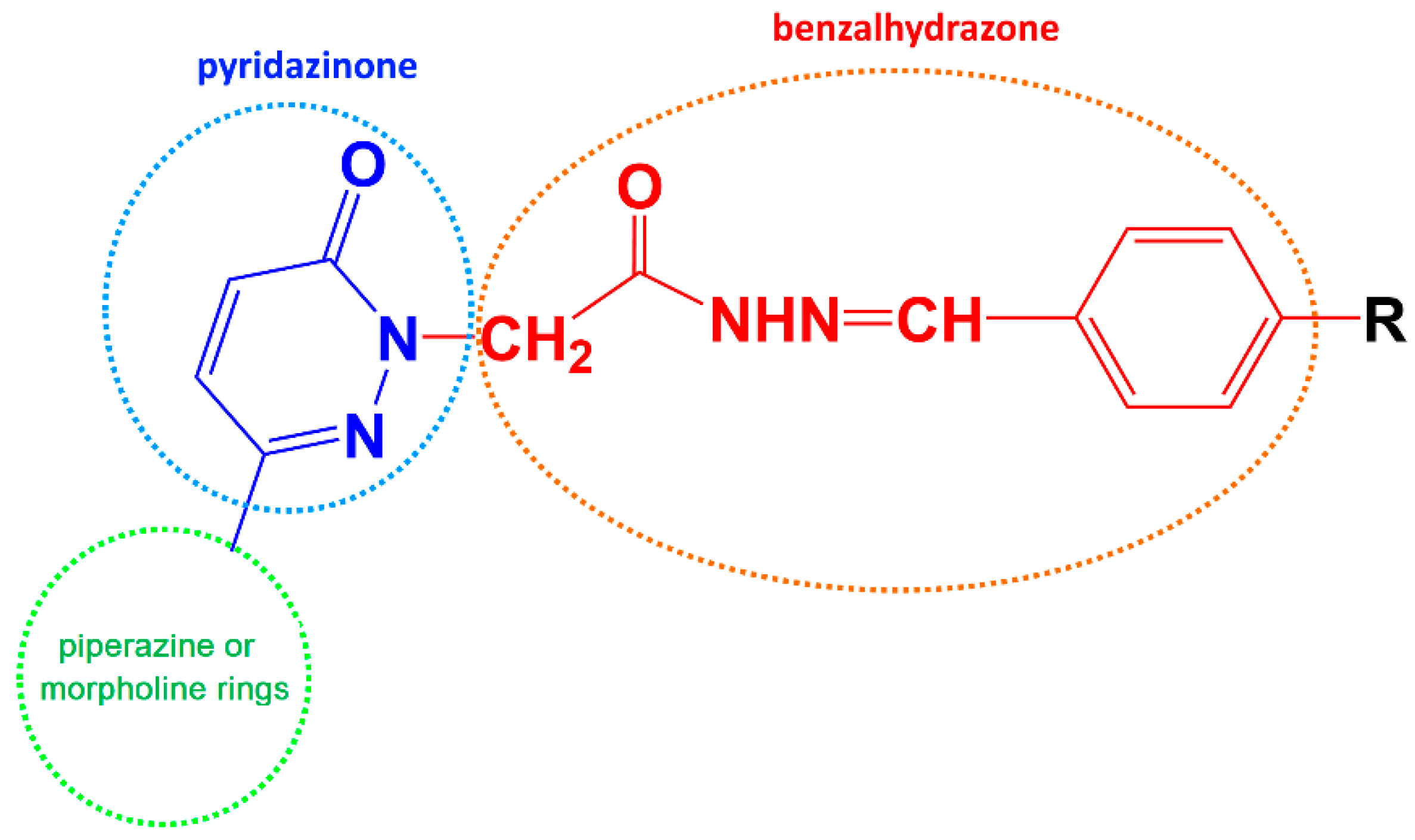
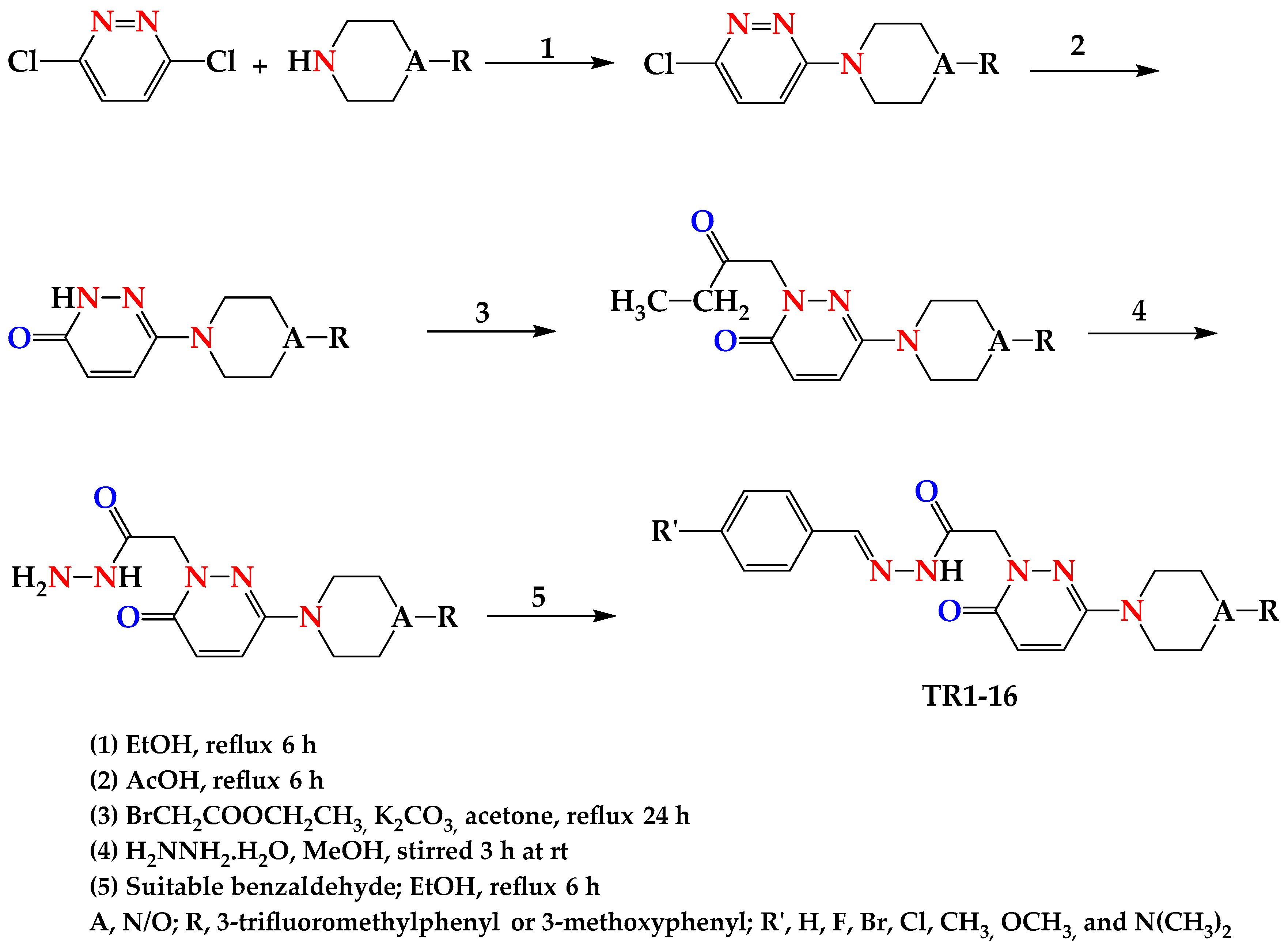
2.1. General Chemistry of Synthetic Scheme
2.2. Biochemistry
2.2.1. MAO Inhibition Studies
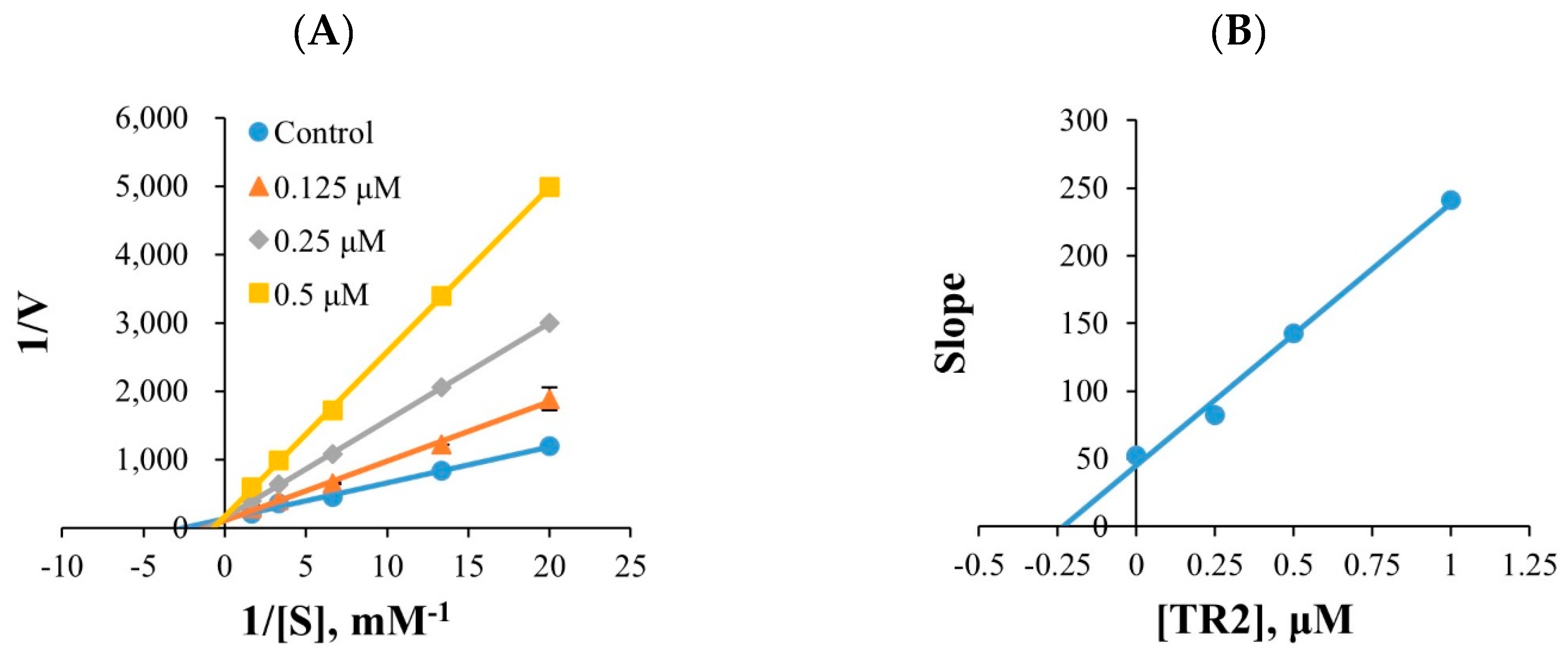
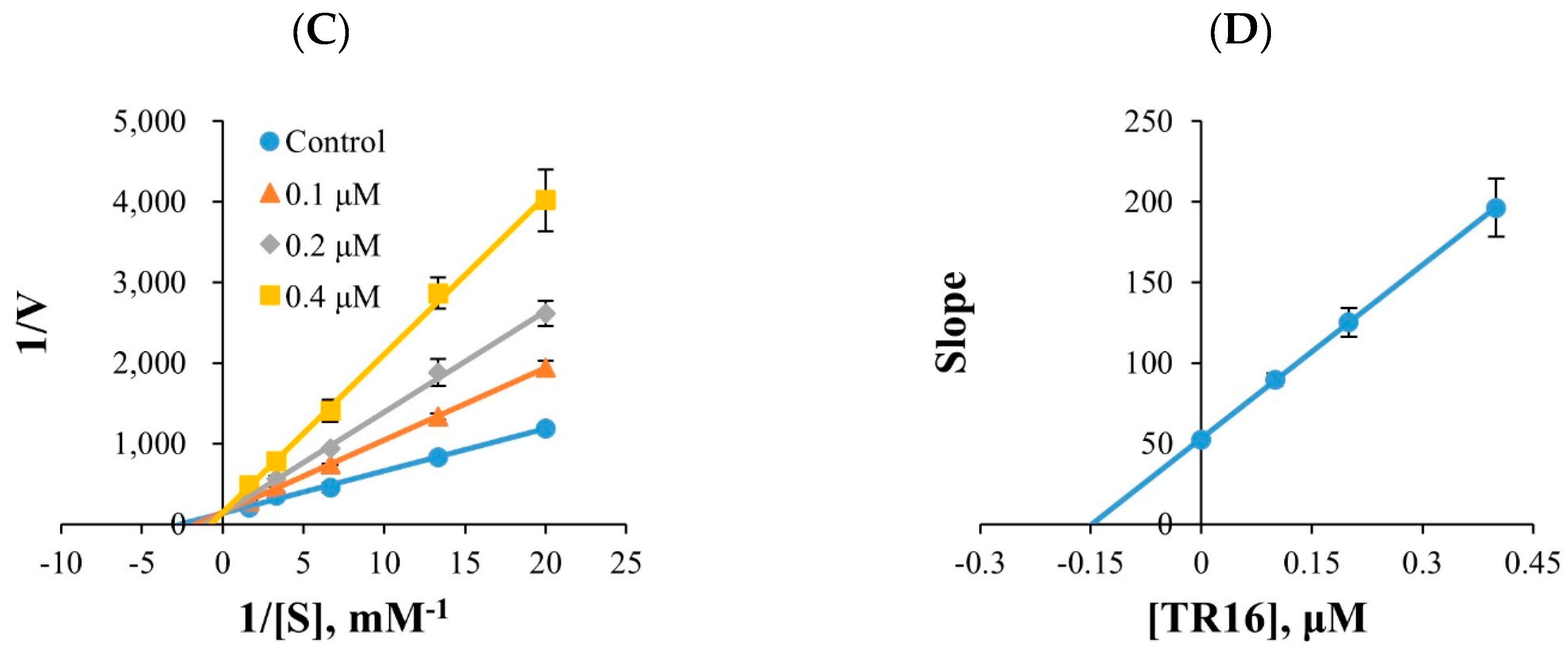
2.2.2. Kinetic Study
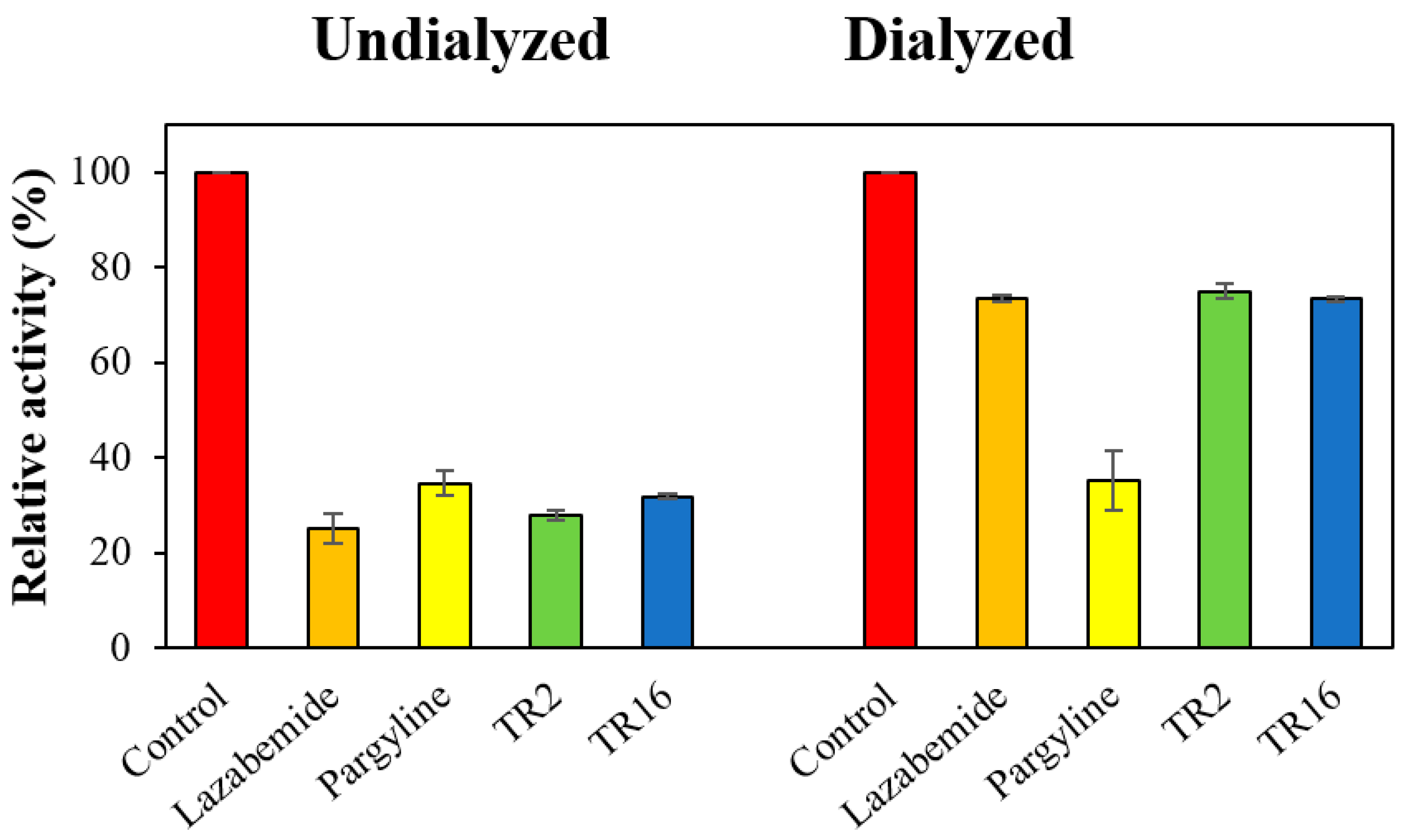
2.2.3. Reversibility Studies
2.3. Parallel Artificial Membrane Permeability Assay (PAMPA)
2.4. Computational Studies
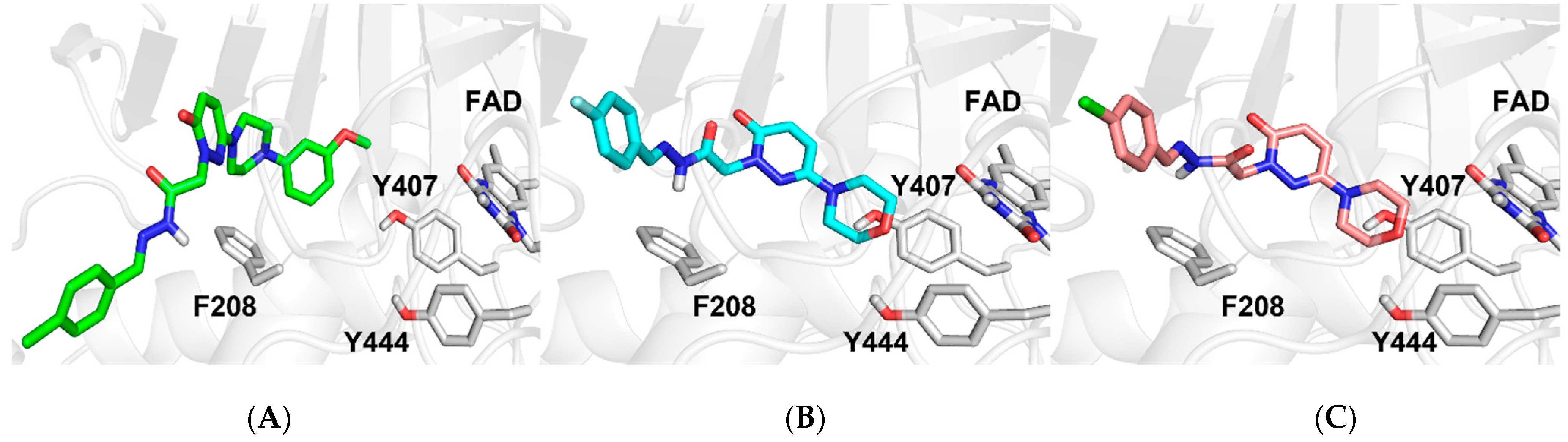
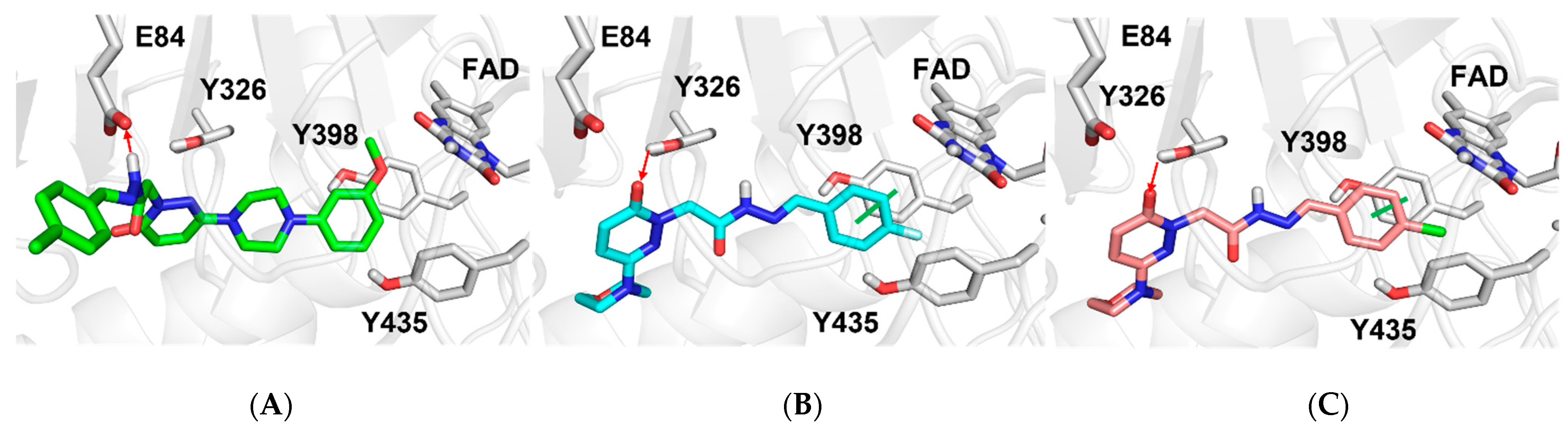
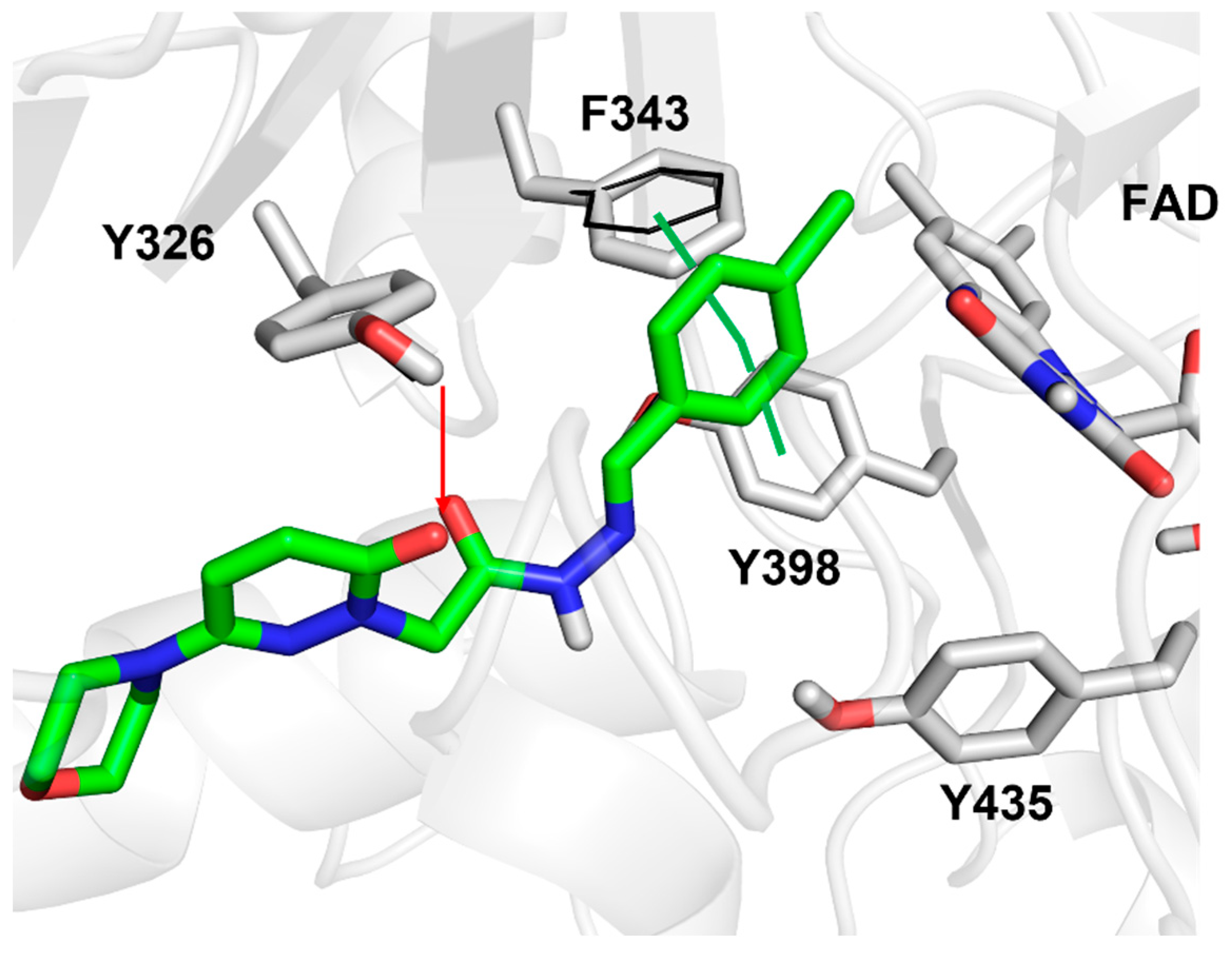
2.4.1. Docking Studies
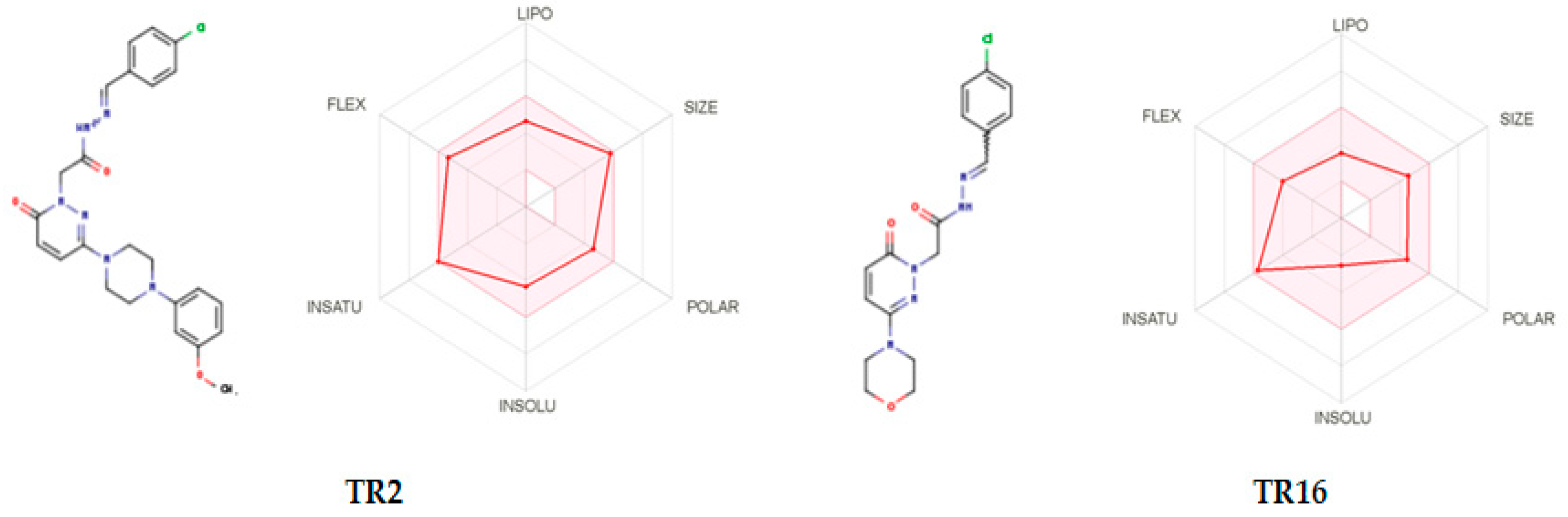
2.4.2. Bioavailability Prediction
3. Materials and Methods
3.1. Chemistry
3.2. Biological Analysis
3.2.1. Enzyme Assays
3.2.2. Inhibition Profile of MAOs and Kinetic Studies
3.2.3. Inhibition Reversibility of TR2 and TR16
3.3. BBB Study by PAMPA Method
3.4. Docking Studies
3.5. Bioavailiability Predicition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Jankovic, J.; Sherer, T. The future of research in Parkinson disease. JAMA Neurol. 2014, 71, 1351–1352. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson disease epidemiology, pathology, genetics, and pathophysiology. Clin. Geriatr. Med. 2020, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Tanji, K.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease: Molecules implicated in the formation and degradation of α-synuclein aggregates. Neuropathology 2007, 27, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Entzeroth, M.; Ratty, A.K. Monoamine oxidase inhibitors-revisiting a therapeutic principle. Open J. Depress. 2017, 6, 31–68. [Google Scholar] [CrossRef] [Green Version]
- Kumar, B.; Sheetal Mantha, A.K.; Kumar, V. Recent developments on the structure-activity relationship studies of MAO inhibitors and their role in different neurological disorders. RSC Adv. 2016, 6, 42660–42683. [Google Scholar] [CrossRef]
- Rehuman, N.A.; Mathew, B.; Jat, R.K.; Nicolotti, O.; Kim, H. A Comprehensive review of monoamine oxidase-A inhibitors in their syntheses and potencies. Comb. Chem. High Throughput Screen. 2020, 23, 898–914. [Google Scholar] [CrossRef]
- Özdemir, Z.; Alagöz, M.A.; Bahçecioğlu, Ö.F.; Gök, S. Monoamine oxidase-B (MAO-B) inhibitors in the treatment of Alzheimer’s and Parkinson’s disease. Curr. Med. Chem. 2021, 28, 6045–6065. [Google Scholar] [CrossRef]
- Guglielmi, P.; Carradori, S.; Ammazzalorso, A.; Secci, D. Novel approaches to the discovery of selective human monoamine oxidase-B inhibitors: Is there room for improvement? Expert Opin. Drug Discov. 2019, 14, 995–1035. [Google Scholar] [CrossRef]
- Tripathi, A.C.; Upadhyay, S.; Paliwal, S.; Saraf, S.K. Privileged scaffolds as MAO inhibitors: Retrospect and prospects. Eur. J. Med. Chem. 2018, 145, 445–497. [Google Scholar] [CrossRef]
- Rangarajan, T.M.; Mathew, B. Recent updates on pyrazoline derivatives as promising candidates for neuropsychiatric and neurodegenerative disorders. Curr. Top. Med. Chem. 2021, 21, 2695–2714. [Google Scholar] [CrossRef]
- Guglielmi, P.; Mathew, B.; Secci, D.; Carradori, S. Chalcones: Unearthing their therapeutic possibility as monoamine oxidase B inhibitors. Eur. J. Med. Chem. 2020, 205, 112650. [Google Scholar] [CrossRef] [PubMed]
- Koyiparambath, V.P.; Rajappan, K.P.; Rangarajan, T.M.; Al-Sehemi, A.G.; Pannipara, M.; Bhaskar, V.; Nair, A.S.; Sudevan, S.T.; Kumar, S.; Mathew, B. Deciphering the detailed structure-activity relationship of coumarins as monoamine oxidase enzyme inhibitors—An updated review. Chem. Biol. Drug Des. 2021, 98, 655–673. [Google Scholar] [CrossRef] [PubMed]
- Mellado, M.; González, C.; Mella, J.; Aguilar, L.F.; Viña, D.; Uriarte, E.; Cuellar, M.; Matos, M.J. Combined 3D-QSAR and docking analysis for the design and synthesis of chalcones as potent and selective monoamine oxidase B inhibitors. Bioorg. Chem. 2021, 108, 104689. [Google Scholar] [CrossRef] [PubMed]
- Mellado, M.; Mella, J.; González, C.; Viña, D.; Uriarte, E.; Matos, M.J. 3-Arylcoumarins as highly potent and selective monoamine oxidase B inhibitors: Which chemical features matter? Bioorg. Chem. 2020, 101, 103964. [Google Scholar] [CrossRef]
- Özdemir, Z.; Yılmaz, H.; Sarı, S.; Karakurt, A.; Şenol, F.S.; Uysal, M. Design, synthesis, and molecular modeling of new 3(2H)-pyridazinone derivatives as acetylcholinesterase/butyrylcholinesterase inhibitors. Med. Chem. Res. 2017, 26, 2293–2308. [Google Scholar] [CrossRef]
- Özdemir, Z.; Alagöz, M.A.; Uslu, H.; Karakurt, A.; Erikci, A.; Ucar, G.; Uysal, M. Synthesis, molecular modelling and biological activity of some pyridazinone derivatives as selective human monoamine oxidase-B inhibitors. Pharmacol. Rep. 2020, 72, 692–704. [Google Scholar] [CrossRef]
- Çeçen, M.; Oh, J.M.; Özdemir, Z.; Büyüktuncel, S.E.; Uysal, M.; Abdelgawad, M.A.; Musa, A.; Gambacorta, N.; Nicolotti, O.; Mathew, B.; et al. Design, synthesis, and biological evaluation of pyridazinones containing the (2-fluorophenyl) piperazine moiety as selective MAO-B inhibitors. Molecules 2020, 25, 5371. [Google Scholar] [CrossRef]
- Özçelik, A.B.; Özdemir, Z.; Sari, S.; Utku, S.; Uysal, M. A new series of pyridazinone derivatives as cholinesterases inhibitors: Synthesis, in vitro activity and molecular modeling studies. Pharmacol. Rep. 2019, 71, 1253–1263. [Google Scholar] [CrossRef]
- Bozbey, İ.; Özdemir, Z.; Uslu, H.; Özçelik, A.B.; Şenol, F.S.; Erdoğan-Orhan, İ.; Uysal, M. A series of new hydrazone derivatives: Synthesis, molecular docking and anticholinesterase activity studies. Mini-Rev. Med. Chem. 2020, 20, 1042–1060. [Google Scholar] [CrossRef]
- Alagöz, M.A.; Özdemir, Z.; Uysal, M.; Carradori, S.; Gallorini, M.; Ricci, A.; Zara, S.; Mathew, B. Synthesis, cytotoxicity and anti-proliferative activity against AGS cells of new 3(2H)-pyridazinone derivatives endowed with a piperazinyl linker. Pharmaceuticals 2021, 14, 183. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H.; Fan, K.; McConnell, O.J.; Carter, G.T. High throughput artificial membrane permeability assay for blood-brain barrier. Eur. J. Med. Chem. 2003, 38, 223–232. [Google Scholar] [CrossRef]
- Al-Sehemi, A.G.; Olotu, F.A.; Dev, S.; Pannipara, M.; Soliman, M.E.S.; Carradori, S.; Mathew, B. Natural products database screening for the discovery of naturally occurring SARS-Cov-2 spike glycoprotein blockers. ChemistrySelect 2020, 5, 13309–13317. [Google Scholar] [CrossRef] [PubMed]
- Venkidath, A.; Oh, J.M.; Dev, S.; Amin, E.; Rasheed, S.P.; Vengamthodi, A.; Gambacorta, N.; Khames, A.; Abdelgawad, M.A.; George, G.; et al. Selected class of enamides bearing nitro functionality as dual-acting with highly selective monoamine oxidase-B and BACE1 inhibitors. Molecules 2021, 26, 6004. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.P.; Kang, M.G.; Lee, J.Y.; Oh, J.M.; Baek, S.C.; Leem, H.H.; Park, D.; Cho, M.L.; Kim, H. Potent inhibition of acetylcholinesterase by sargachromanol I from Sargassum siliquastrum and by selected natural compounds. Bioorg. Chem. 2019, 89, 103043. [Google Scholar] [CrossRef] [PubMed]
- Mathew, B.; Oh, J.M.; Khames, A.; Abdelgawad, M.A.; Rangarajan, T.M.; Nath, L.R.; Agoni, C.; Soliman, M.E.S.; Mathew, G.E.; Kim, H. Replacement of chalcone-ethers with chalcone-thioethers as potent and highly selective monoamine oxidase-B inhibitors and their protein-ligand interactions. Pharmaceuticals 2021, 14, 1148. [Google Scholar] [CrossRef]
- Heo, J.H.; Eom, B.H.; Ryu, H.W.; Kang, M.G.; Park, J.E.; Kim, D.Y.; Kim, J.H.; Park, D.; Oh, S.R.; Kim, H. Acetylcholinesterase and butyrylcholinesterase inhibitory activities of khellactone coumarin derivatives isolated from Peucedanum japonicum Thurnberg. Sci. Rep. 2020, 10, 21695. [Google Scholar] [CrossRef]
- Jeong, G.; Kang, M.-G.; Lee, J.; Lee, S.; Park, D.; Cho, M.; Kim, H. Inhibition of butyrylcholinesterase and human monoamine oxidase-B by the coumarin glycyrol and liquiritigenin isolated from Glycyrrhiza uralensis. Molecules 2020, 25, 3896. [Google Scholar] [CrossRef]
- Jeong, G.S.; Kang, M.G.; Han, S.A.; Noh, J.I.; Park, J.E.; Nam, S.J.; Park, D.; Yee, S.T.; Kim, H. Selective inhibition of human monoamine oxidase B by 5-hydroxy-2-methyl-chroman-4-one isolated from an endogenous lichen fungus Daldinia fissa. J. Fungi 2021, 7, 84. [Google Scholar] [CrossRef]
- Lee, H.W.; Ryu, H.W.; Kang, M.G.; Park, D.; Oh, S.R.; Kim, H. Potent selective monoamine oxidase B inhibition by maackiain, a pterocarpan from the roots of Sophora flavescens. Bioorg. Med. Chem. Lett. 2016, 26, 4714–4719. [Google Scholar] [CrossRef]
- Baek, S.C.; Lee, H.W.; Ryu, H.W.; Kang, M.G.; Park, D.; Kim, S.H.; Cho, M.L.; Oh, S.R.; Kim, H. Selective inhibition of monoamine oxidase A by hispidol. Bioorg. Med. Chem. Lett. 2018, 15, 584–588. [Google Scholar] [CrossRef]
- Son, S.Y.; Ma, J.; Kondou, Y.; Yoshimura, M.; Yamashita, E.; Tsukihara, T. Structure of human monoamine oxidase A at 2.2-A resolution: The control of opening the entry for substrates/inhibitors. Proc. Natl. Acad. Sci. USA 2008, 105, 5739–5744. [Google Scholar] [CrossRef] [Green Version]
- Binda, C.; Wang, J.; Pisani, L.; Caccia, C.; Carotti, A.; Salvati, P.; Edmondson, D.E.; Mattevi, A. Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: Safinamide and coumarin analogs. J. Med. Chem. 2007, 50, 5848–5852. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger. Protein Preparation Wizard; Schrödinger, L.L.C.: New York, NY, USA, 2020. [Google Scholar]
- Schrödinger. LigPrep; Schrödinger, L.L.C.: New York, NY, USA, 2020. [Google Scholar]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Compound | Activity |
|---|---|---|
 | Zardaverine | Cardiotonic |
 | Levosimendan | Vasodilator |
 | Pimobendan | Vasodilator |
 | Imazodan | Cardiotonic |
 | Emorfazone | Analgesic |
 | Medazomide | Antitussif |
 | |||||
| Entry | R1 | R2 | Yield (%) | M.P. (°C) | Molecular Formula |
| TR1 | OCH3 | H | 79.23 | 232–235 | C24H26N6O3 |
| TR2 | OCH3 | 4-Cl | 82.49 | 205 | C24H25ClN6O3 |
| TR3 | OCH3 | 4-F | 57.10 | 172 | C24H25FN6O3 |
| TR4 | OCH3 | 4-OCH3 | 67.08 | 154 | C25H28N6O4 |
| TR5 | OCH3 | 4-CH3 | 76.73 | 201–202 | C25H28N6O3 |
| TR6 | OCH3 | 4-Br | 82.54 | 214 | C24H25BrN6O3 |
| TR7 | CF3 | H | 76.90 | 126 | C24H23F3N6O2 |
| TR8 | CF3 | 4-Cl | 91.67 | 242 | C24H22ClF3N6O2 |
| TR9 | CF3 | 4-F | 78.09 | 224 | C24H22F4N6O2 |
| TR10 | CF3 | 4-OCH3 | 86.36 | 195 | C25H25F3N6O3 |
| TR11 | CF3 | 4-CH3 | 83.43 | 142 | C25H25F3N6O2 |
| TR12 | CF3 | 4-N(CH3)2 | 80.21 | 227 | C26H28F3N7O2 |
| TR13 | CF3 | 4-Br | 82.64 | 241 | C24H22BrF3N6O2 |
 | |||||
| Entry | R2 | Yield (%) | M.P. (°C) | Molecular Formula | |
| TR14 | H | 71.22 | 206 | C17H19N5O3 | |
| TR15 | 4-F | 61.17 | 119 | C17H18FN5O3 | |
| TR16 | 4-Cl | 80.73 | 152 | C17H18ClN5O3 | |
| Compounds | Residual Activity at 10 µM (%) | IC50 (µM) | SI b | ||
|---|---|---|---|---|---|
| MAO-A | MAO-B | MAO-A | MAO-B | ||
| TR1 | 82.04 ± 1.20 | 80.54 ± 1.01 | 17.32 ± 1.20 | >40 | <0.43 |
| TR2 | 65.85 ± 0.66 | −4.37 ± 4.13 | 22.94 ± 0.24 | 0.27 ± 0.02 | 84.96 |
| TR3 | 71.66 ± 0.71 | 20.11 ± 1.15 | 19.65 ± 0.30 | 2.11 ± 0.51 | 9.31 |
| TR4 | 61.00 ± 3.54 | 88.64 ± 8.61 | 16.53 ± 1.66 | >40 | <0.41 |
| TR5 | 72.83 ± 1.02 | 45.56 ± 2.56 | 18.19 ± 0.09 | 4.42 ± 0.23 | 4.12 |
| TR6 | 51.73 ± 4.55 | 70.23 ± 3.80 | 12.33 ± 0.11 | 15.12 ± 0.18 | 0.81 |
| TR7 | 55.40 ± 0.84 | 90.66 ± 6.64 | 14.43 ± 3.55 | >40 | <0.36 |
| TR8 | 52.87 ± 0.82 | 69.09 ± 0.95 | 12.02 ± 1.75 | 20.00 ± 0.00 | 0.60 |
| TR9 | 72.28 ± 0.40 | 46.51 ± 4.93 | 17.08 ± 0.21 | 1.00 ± 0.15 | 17.08 |
| TR10 | 56.00 ± 2.26 | 91.86 ± 7.09 | 13.28 ± 1.81 | >40 | <0.33 |
| TR11 | 55.65 ± 2.28 | 83.87 ± 0.56 | 18.54 ± 5.83 | 32.00 ± 1.21 | 0.57 |
| TR12 | 52.55 ± 1.77 | 85.33 ± 1.59 | 14.72 ± 2.96 | >40 | <0.37 |
| TR13 | 74.24 ± 6.43 | 100.32 ± 5.43 | 31.00 ± 4.33 | >40 | <0.78 |
| TR14 | 97.47 ± 0.71 | 106.69 ± 2.01 | >40 | >40 | 1.00 |
| TR15 | 85.59 ± 1.04 | 12.50 ± 3.54 | >40 | 0.43 ± 0.05 | >93.02 |
| TR16 | 85.00 ± 3.08 | 13.06 ± 0.64 | >40 | 0.17 ± 0.04 | >235.29 |
| Toloxatone | 1.08 ± 0.03 | - | |||
| Lazabemide | - | 0.11 ± 0.02 | |||
| Clorgyline | 0.007 ± 0.001 | - | |||
| Pargyline | - | 0.14 ± 0.01 | |||
| Compounds | Bibliography Pe (×10−6 cm/s) a | Experimental Pe (×10−6 cm/s) | Prediction |
|---|---|---|---|
| Dopamine | 0.2 | 0.21 ± 0.01 | CNS- |
| Lomefloxacin | 1.1 | 1.13 ± 0.01 | CNS- |
| Verapamil | 16.0 | 15.35 ± 0.33 | CNS+ |
| Progesterone | 9.3 | 9.02 ± 0.17 | CNS+ |
| TR2 | 9.33 ± 0.33 | CNS+ | |
| TR16 | 10.62 ± 0.26 | CNS+ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alagöz, M.A.; Oh, J.M.; Zenni, Y.N.; Özdemir, Z.; Abdelgawad, M.A.; Naguib, I.A.; Ghoneim, M.M.; Gambacorta, N.; Nicolotti, O.; Kim, H.; et al. Development of a Novel Class of Pyridazinone Derivatives as Selective MAO-B Inhibitors. Molecules 2022, 27, 3801. https://doi.org/10.3390/molecules27123801
Alagöz MA, Oh JM, Zenni YN, Özdemir Z, Abdelgawad MA, Naguib IA, Ghoneim MM, Gambacorta N, Nicolotti O, Kim H, et al. Development of a Novel Class of Pyridazinone Derivatives as Selective MAO-B Inhibitors. Molecules. 2022; 27(12):3801. https://doi.org/10.3390/molecules27123801
Chicago/Turabian StyleAlagöz, Mehmet Abdullah, Jong Min Oh, Yaren Nur Zenni, Zeynep Özdemir, Mohamed A. Abdelgawad, Ibrahim A. Naguib, Mohammed M. Ghoneim, Nicola Gambacorta, Orazio Nicolotti, Hoon Kim, and et al. 2022. "Development of a Novel Class of Pyridazinone Derivatives as Selective MAO-B Inhibitors" Molecules 27, no. 12: 3801. https://doi.org/10.3390/molecules27123801