
Asymmetric Michael Addition in Synthesis of β-Substituted GABA Derivatives

 ,
,  , and
, and
Abstract
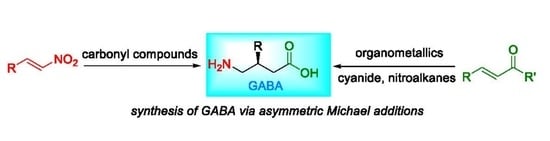
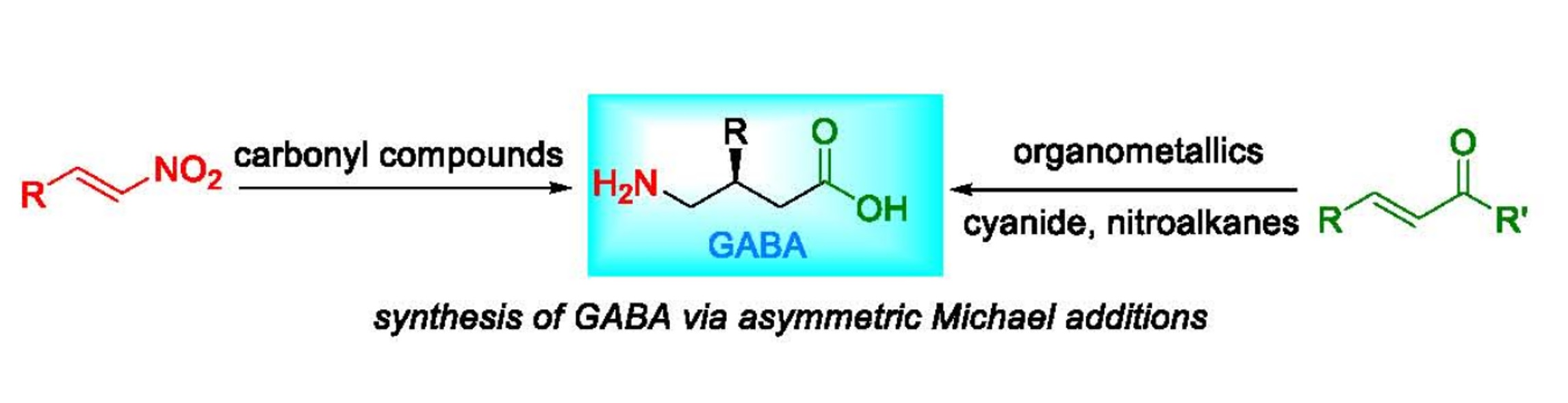
:
{kind=link}
{kind=link}
{kind=link}
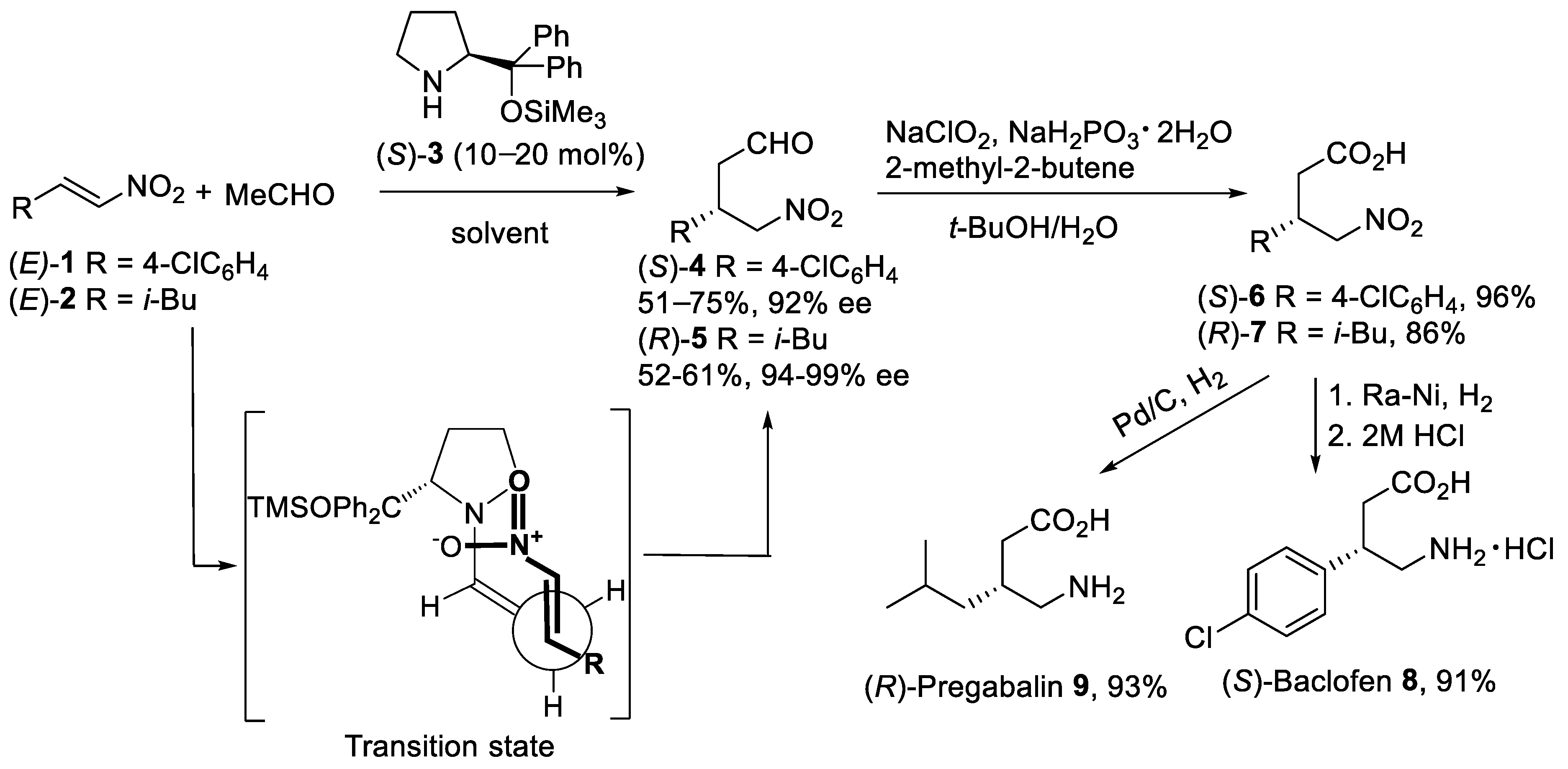{kind=link}
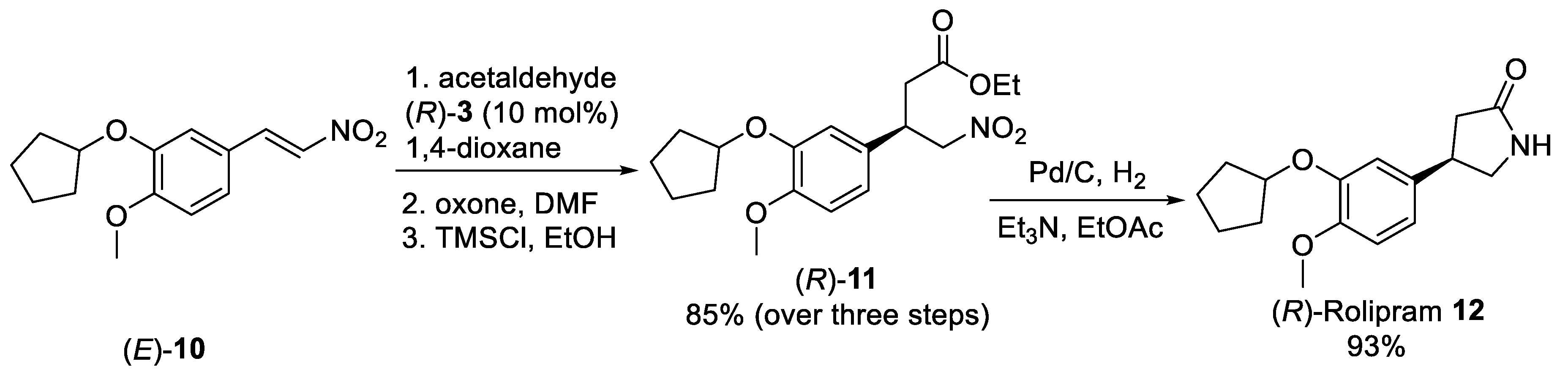{kind=link}
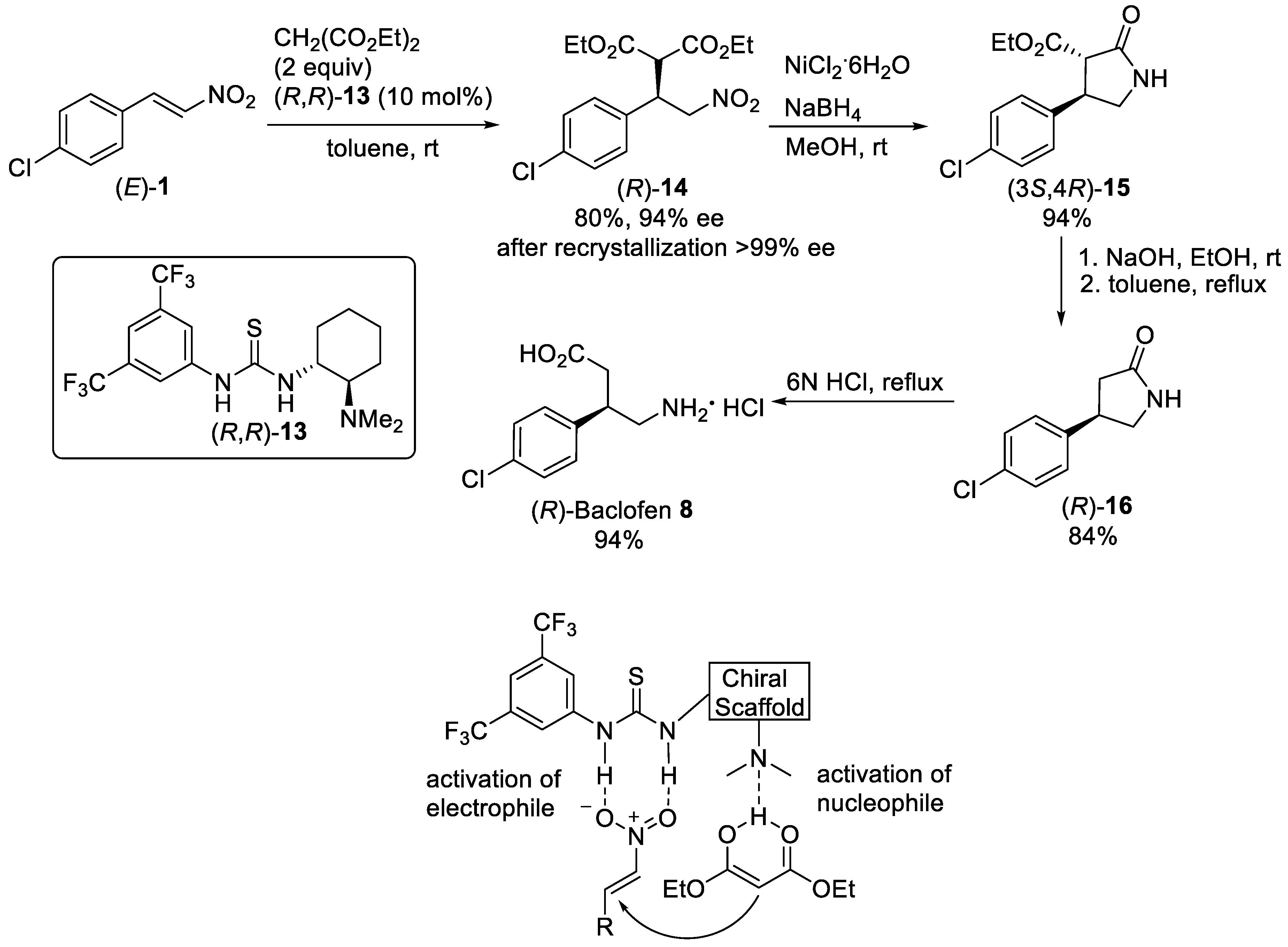{kind=link}
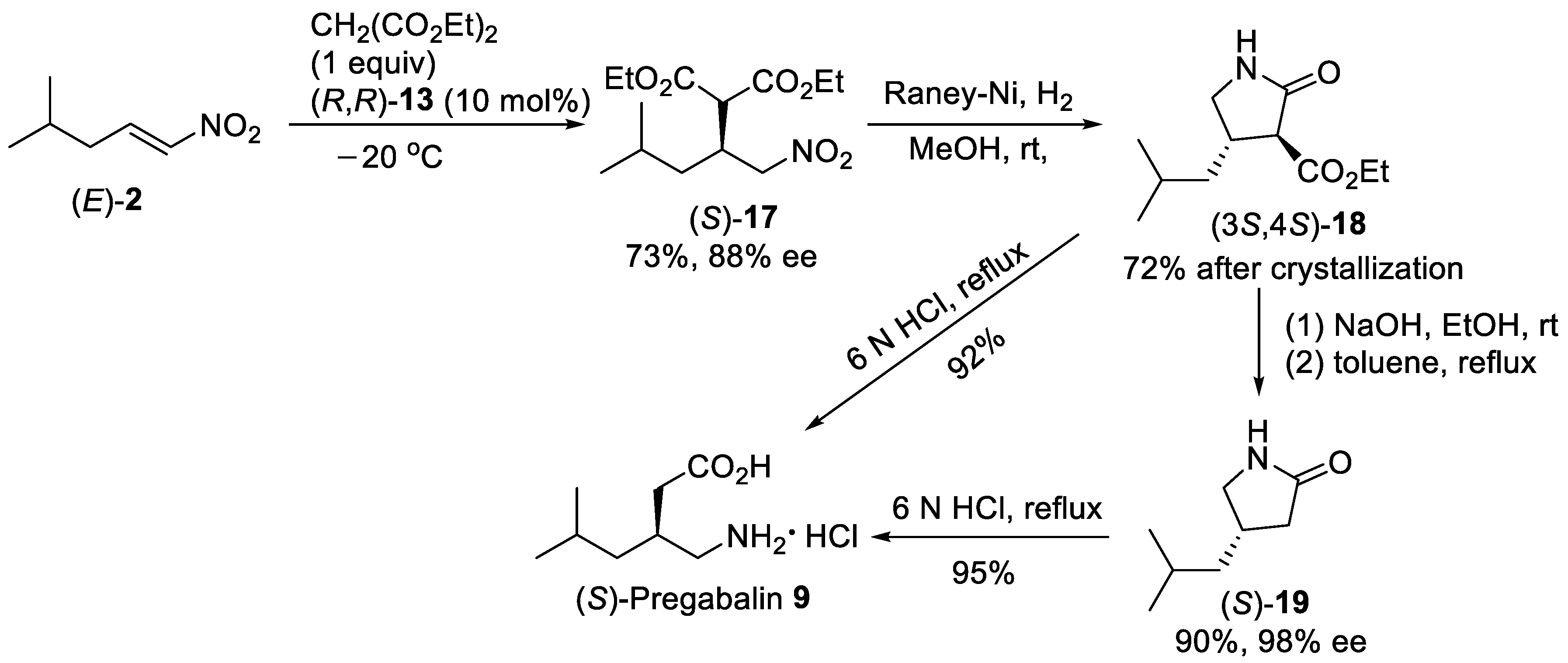{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
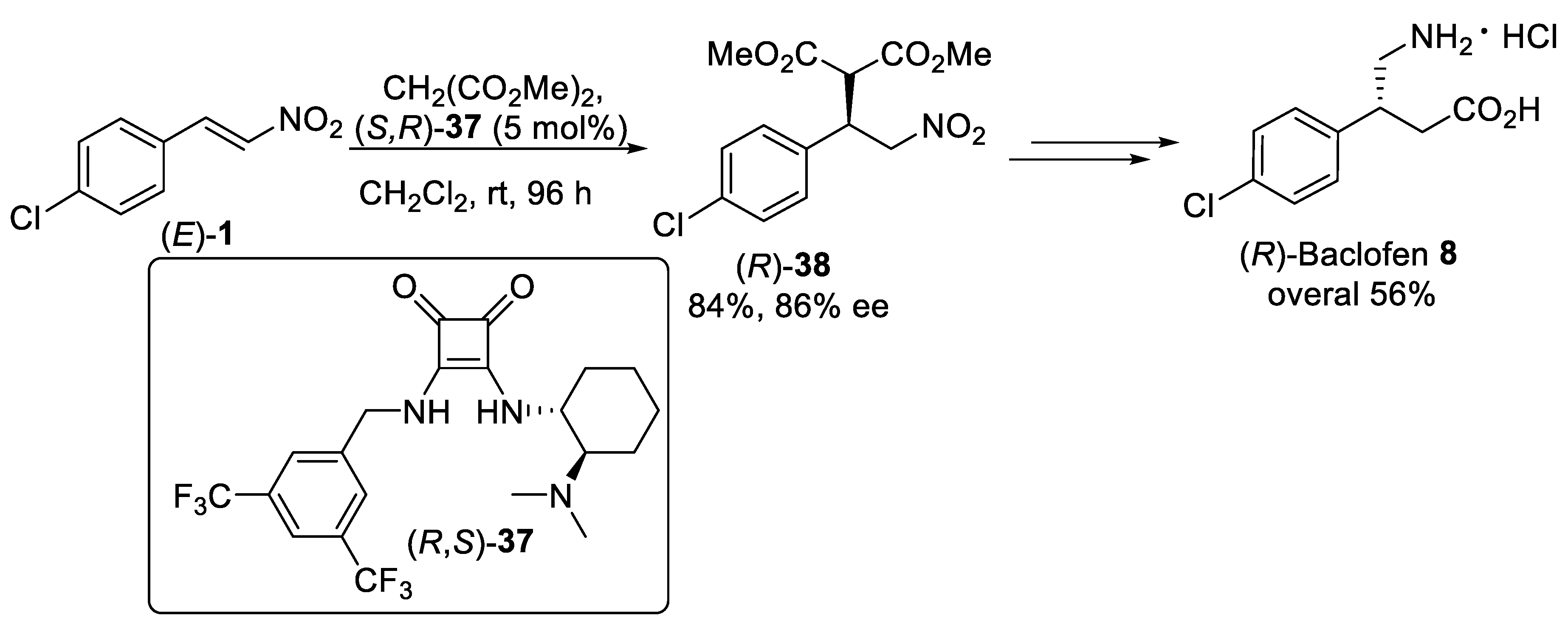{kind=link}
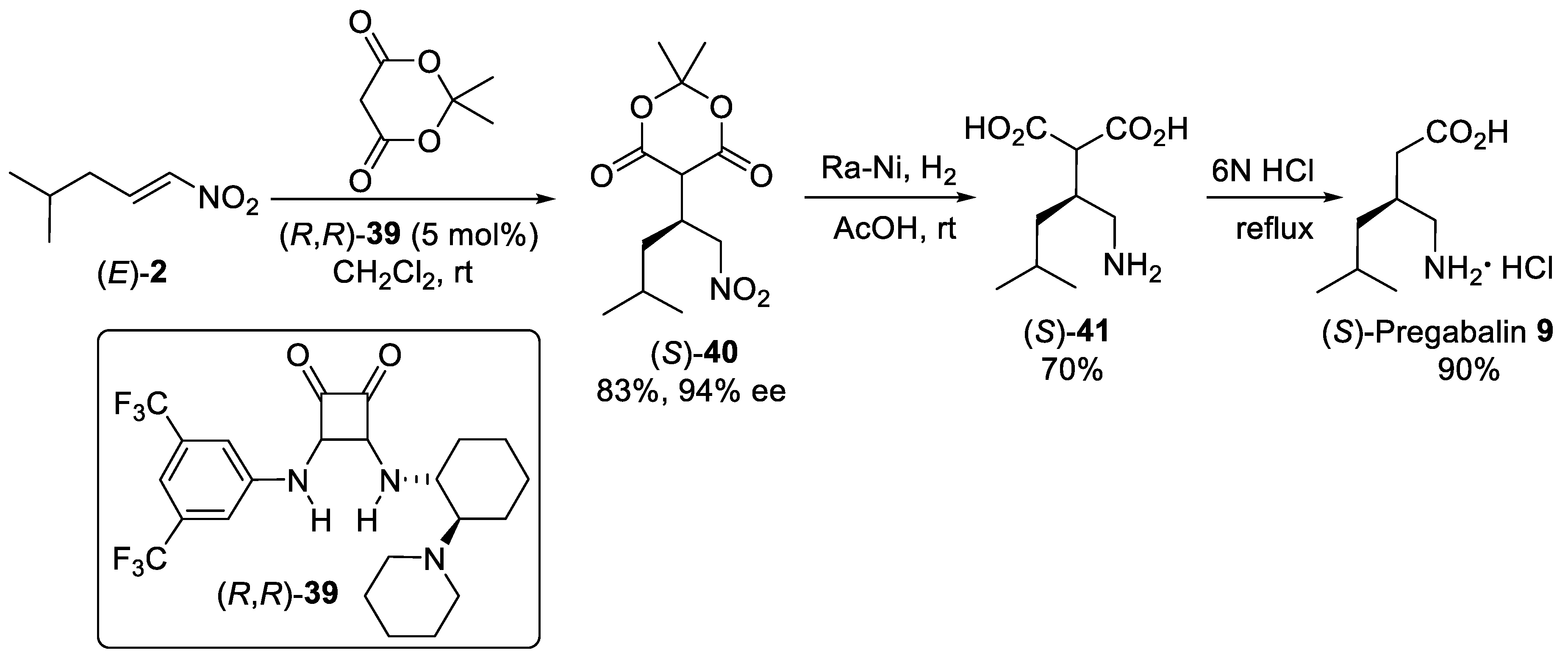{kind=link}
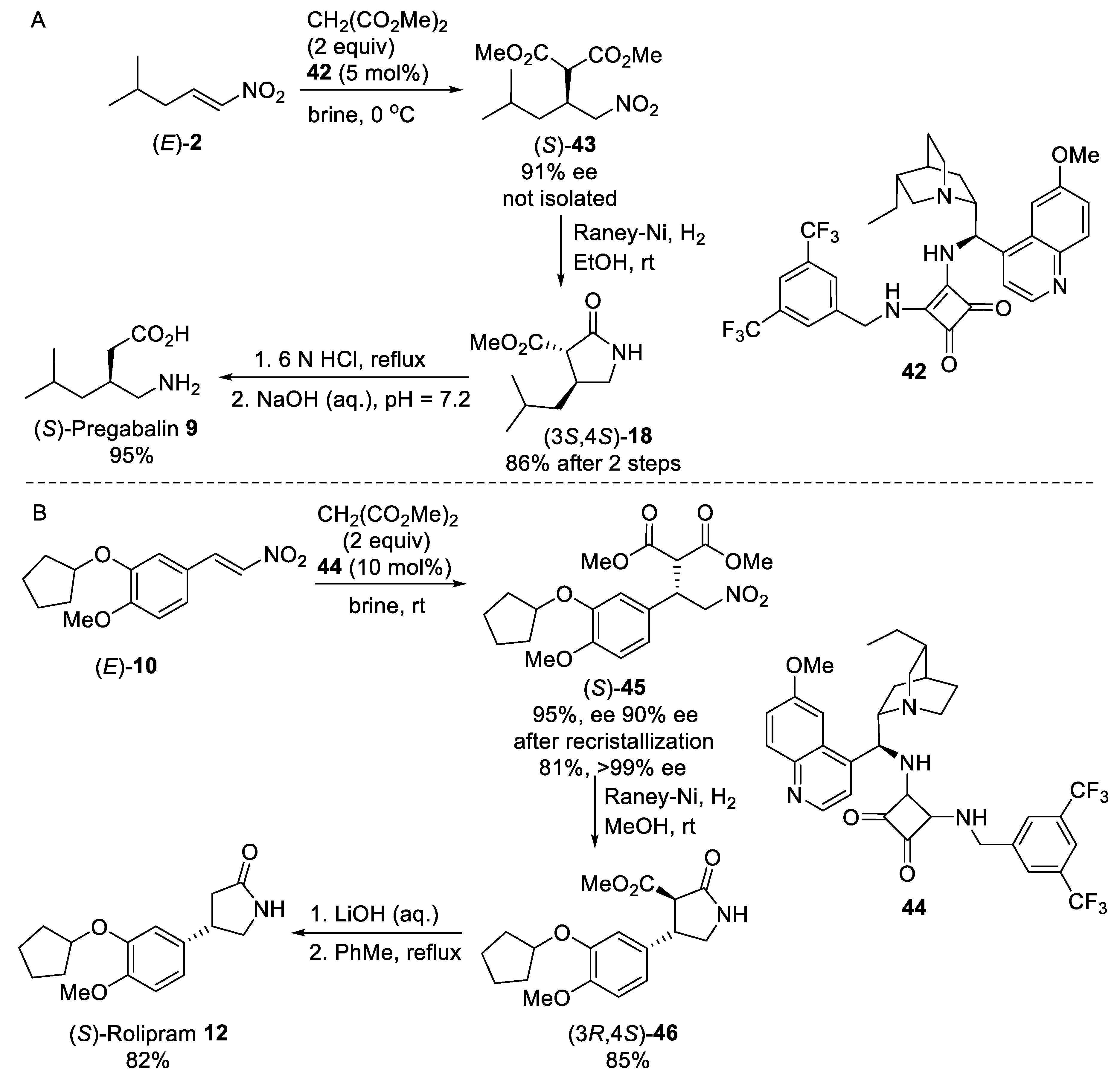{kind=link}
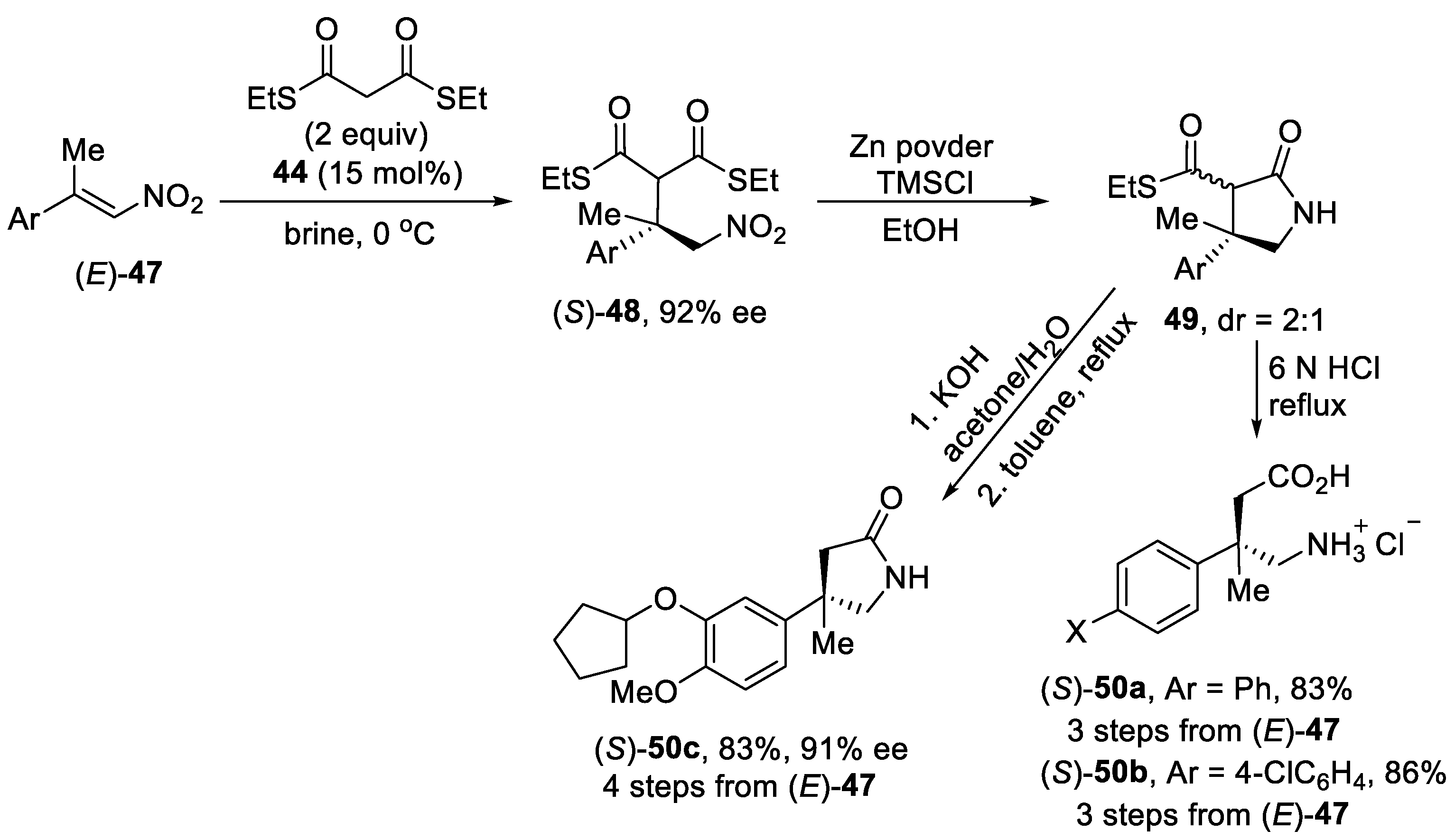{kind=link}
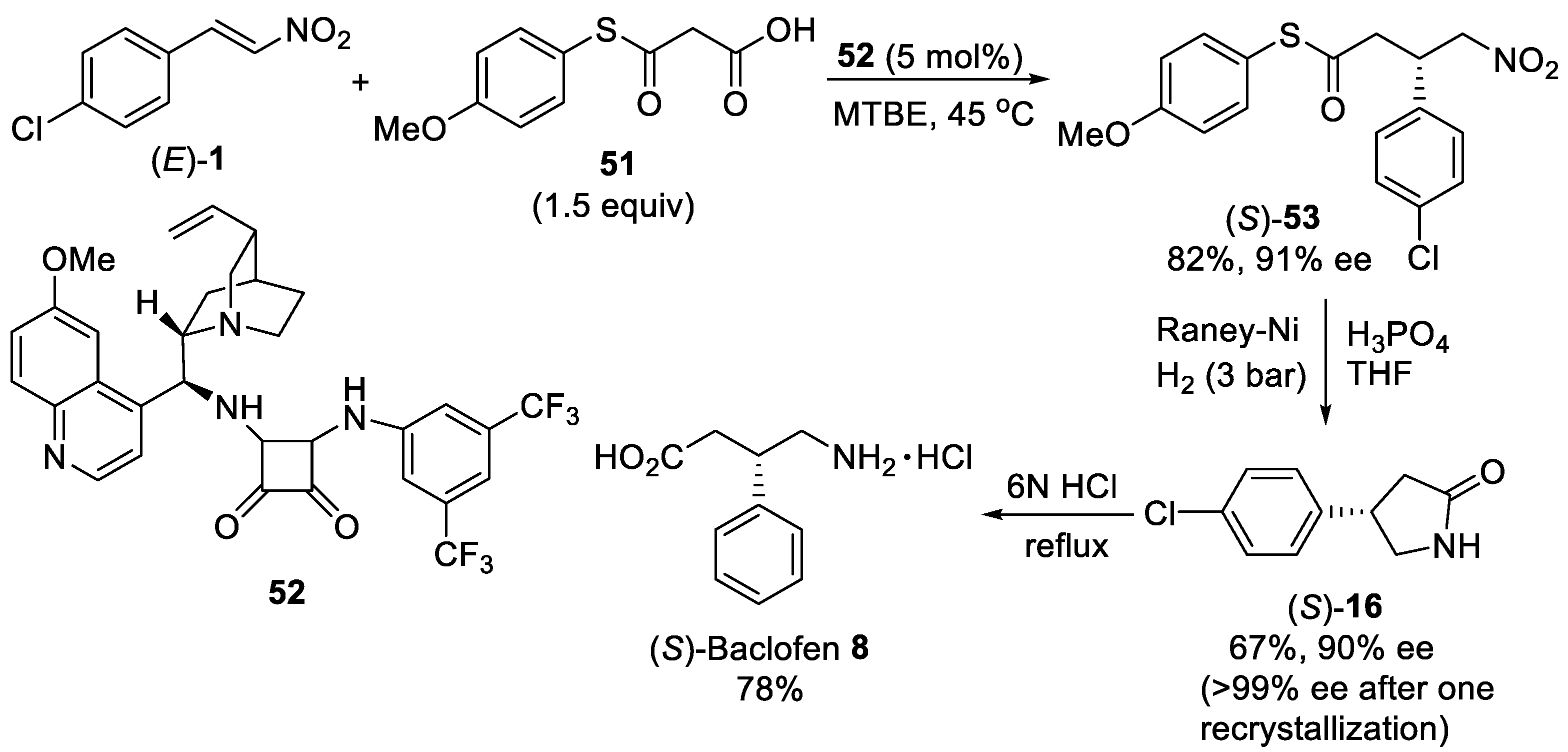{kind=link}
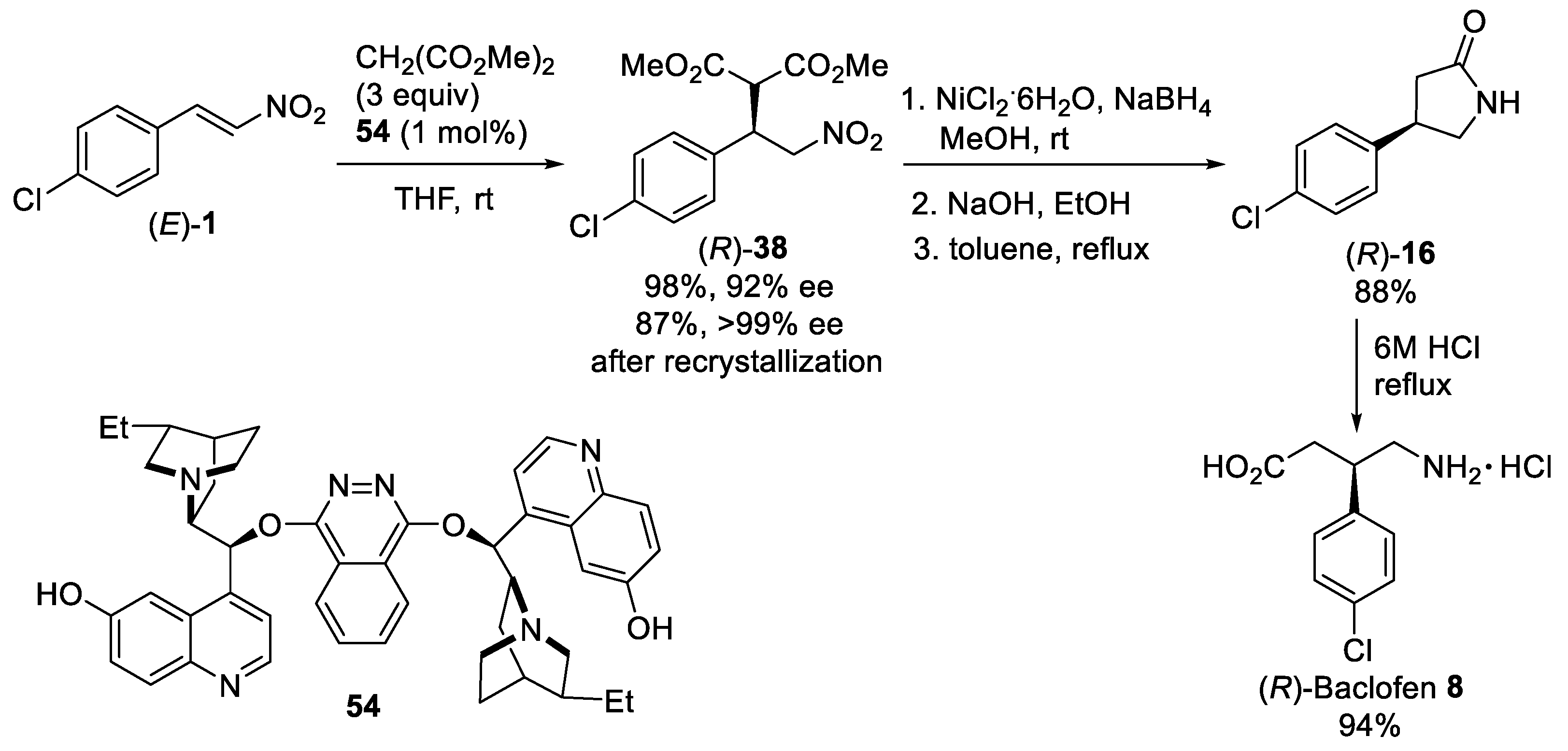{kind=link}
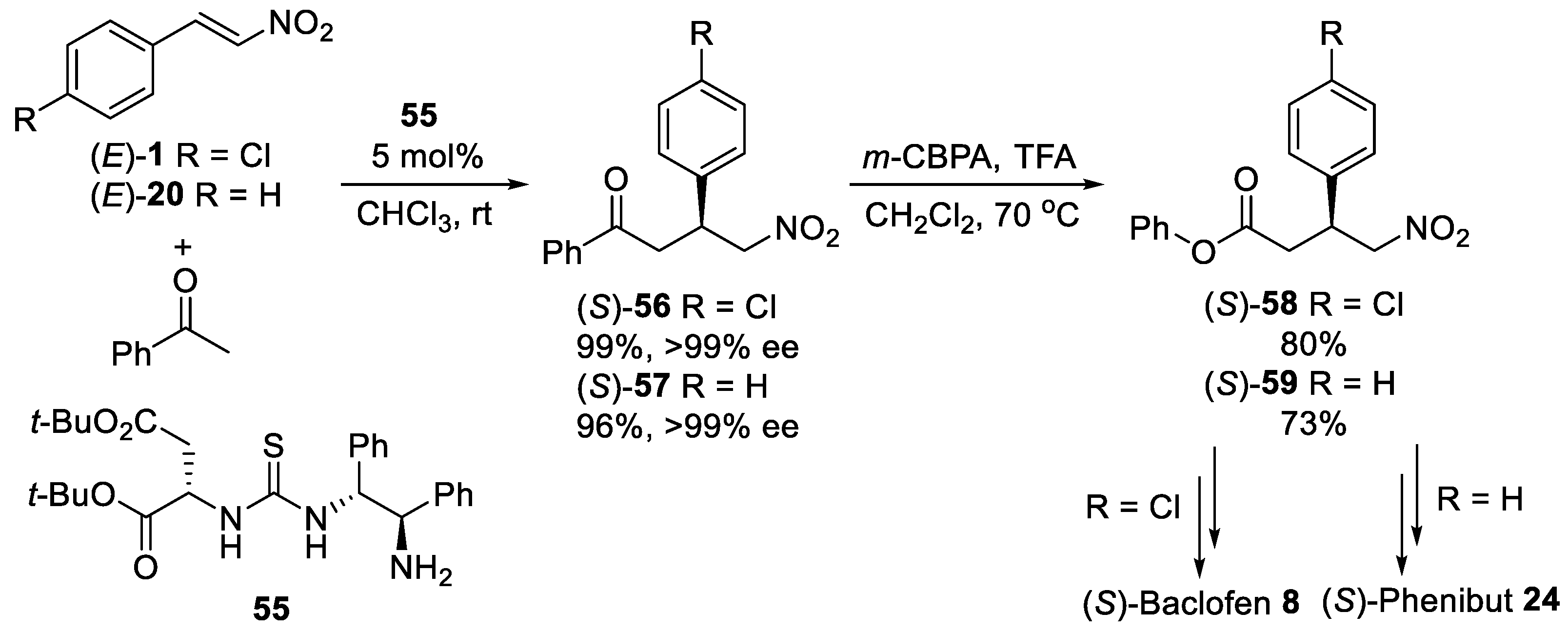{kind=link}
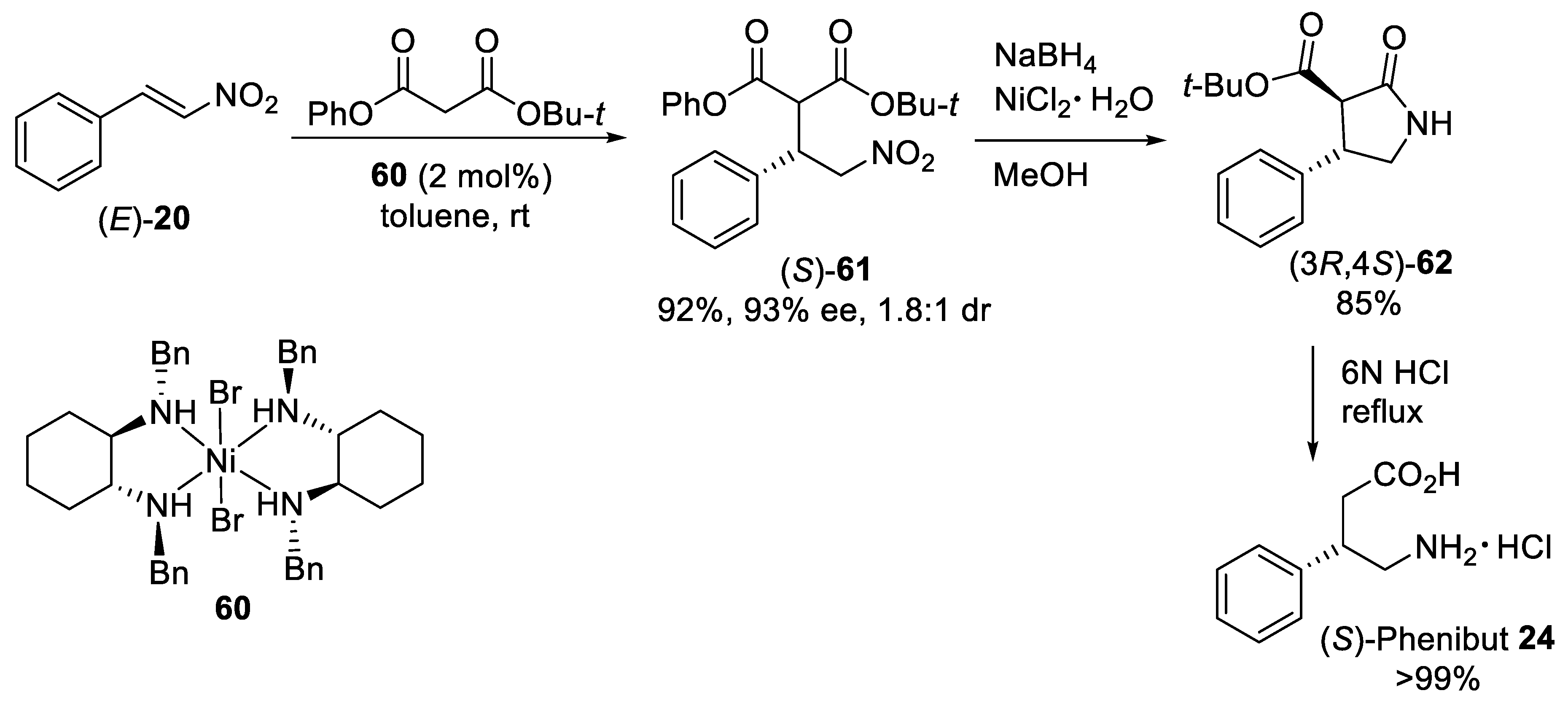{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
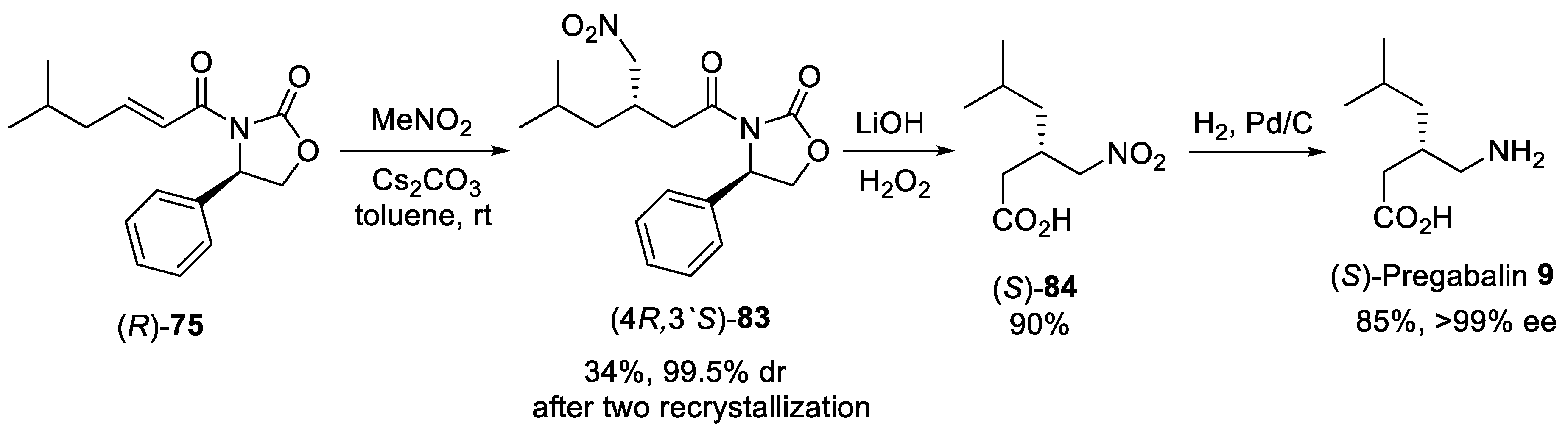{kind=link}
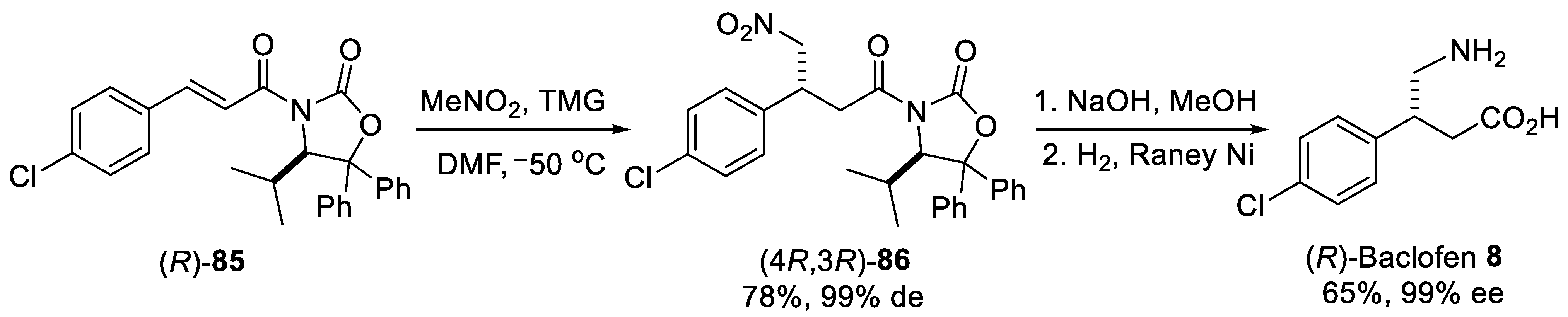{kind=link}
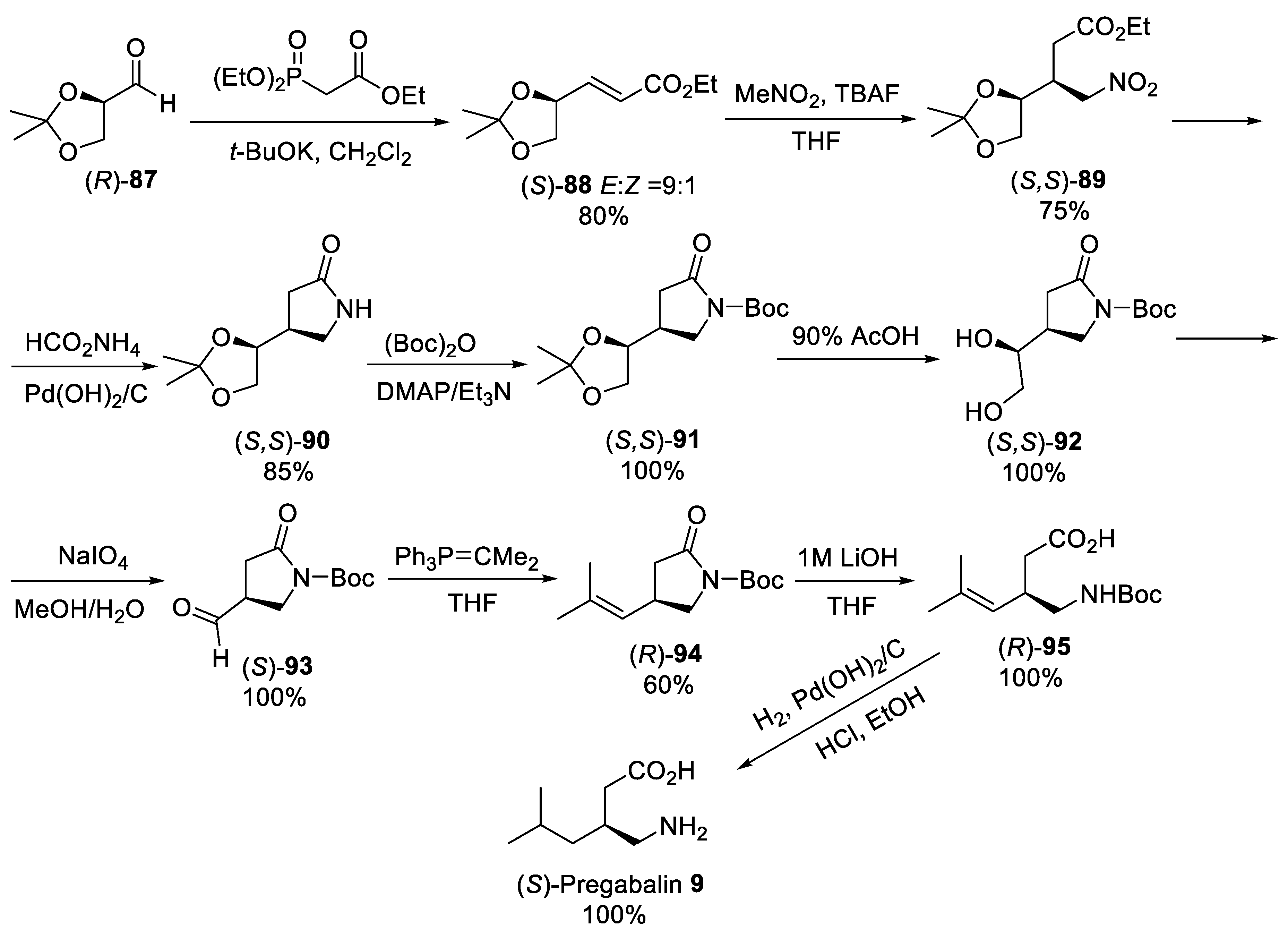{kind=link}
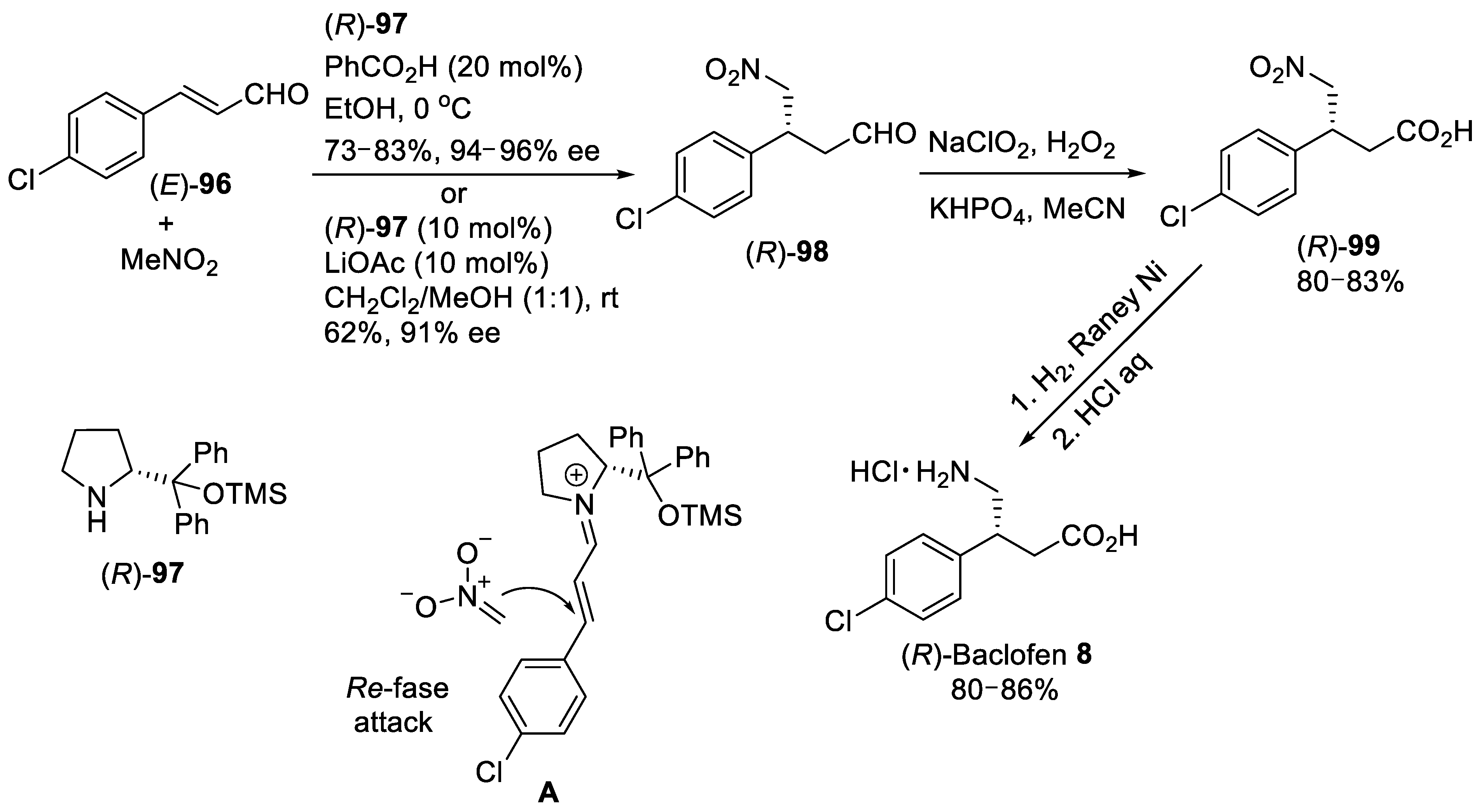{kind=link}
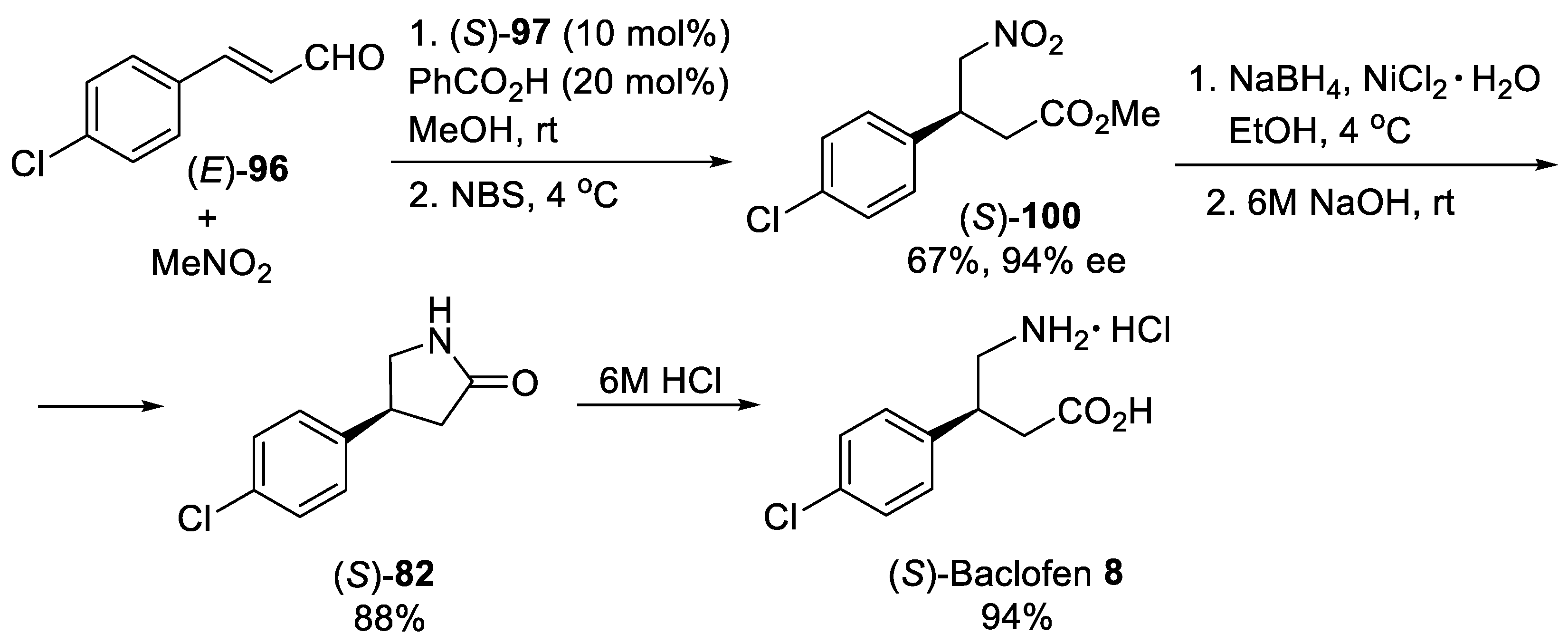{kind=link}
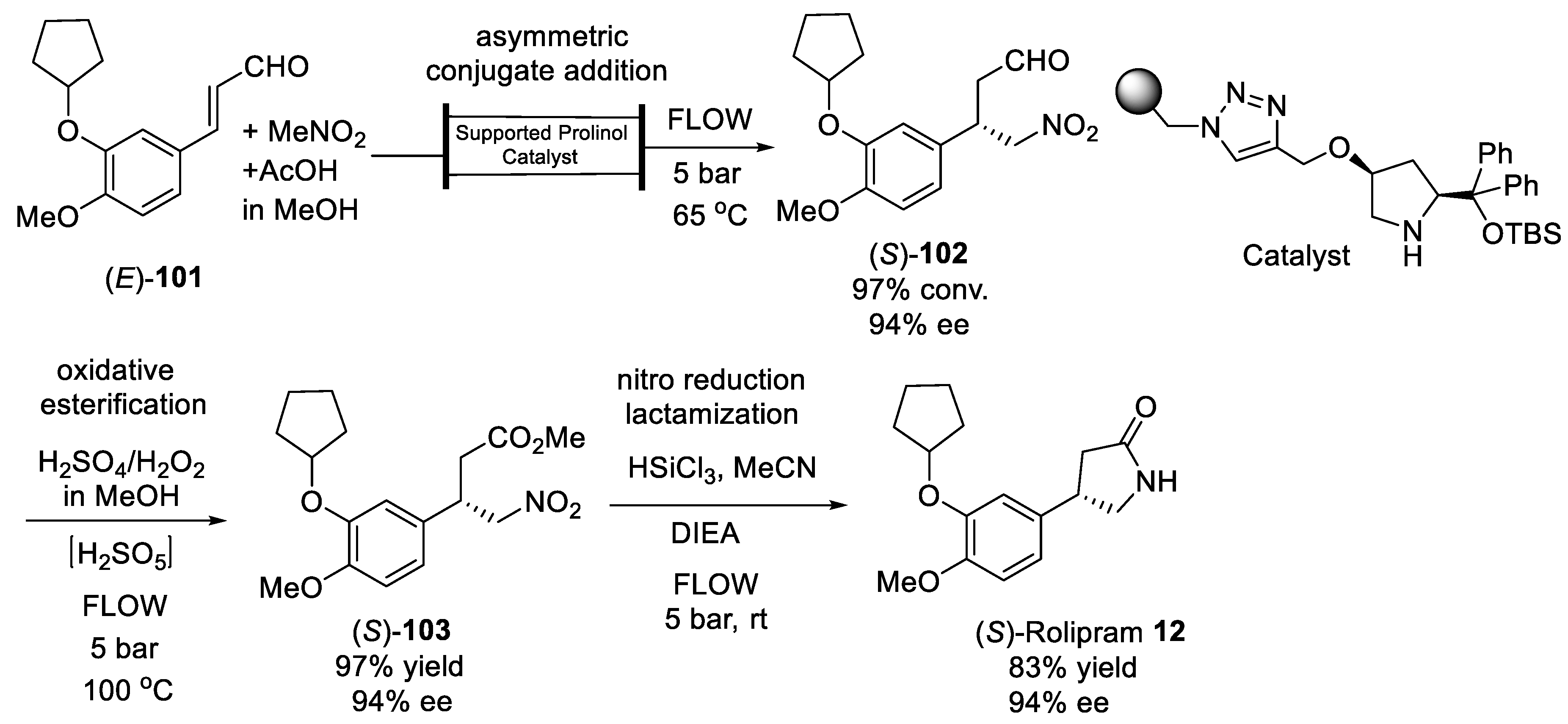{kind=link}
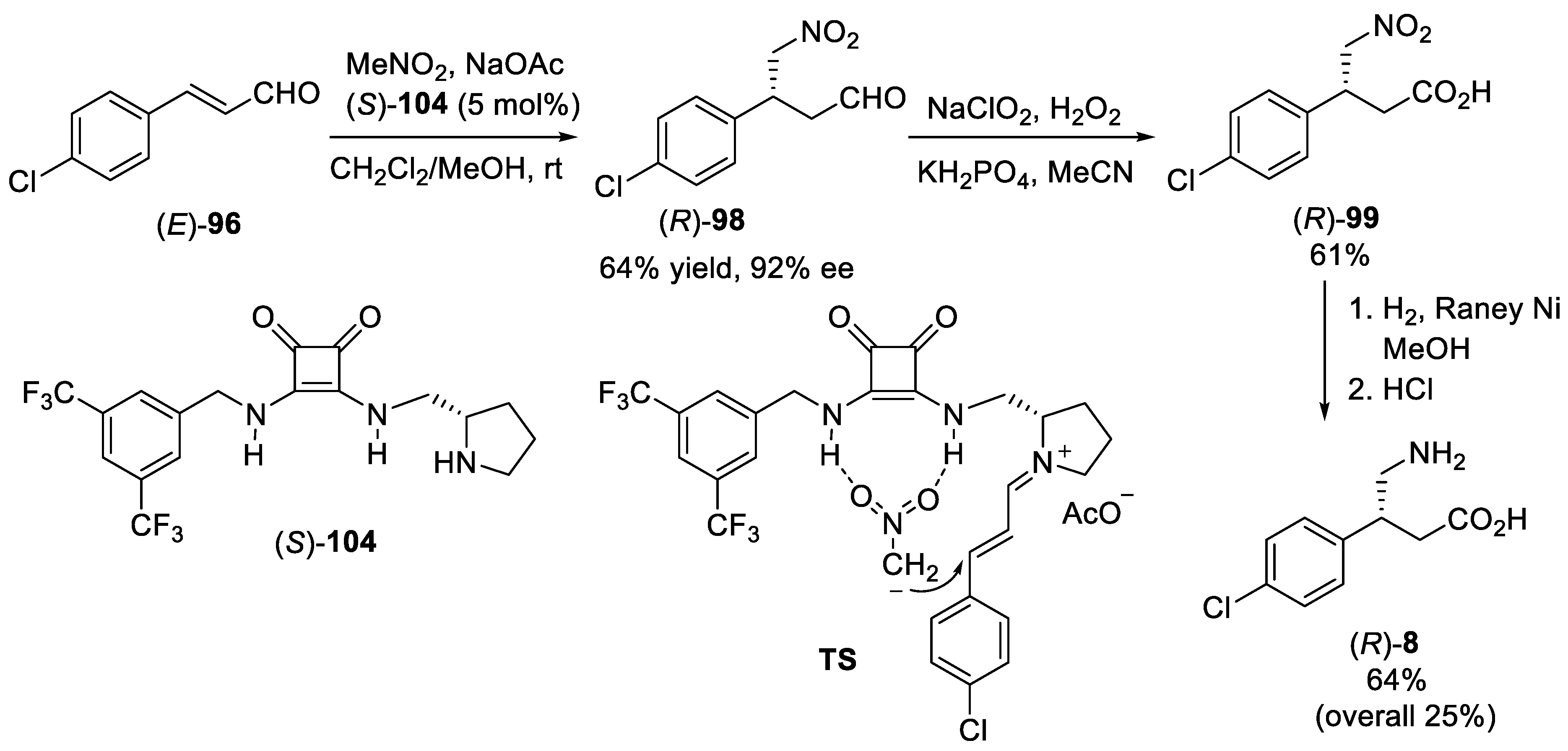{kind=link}
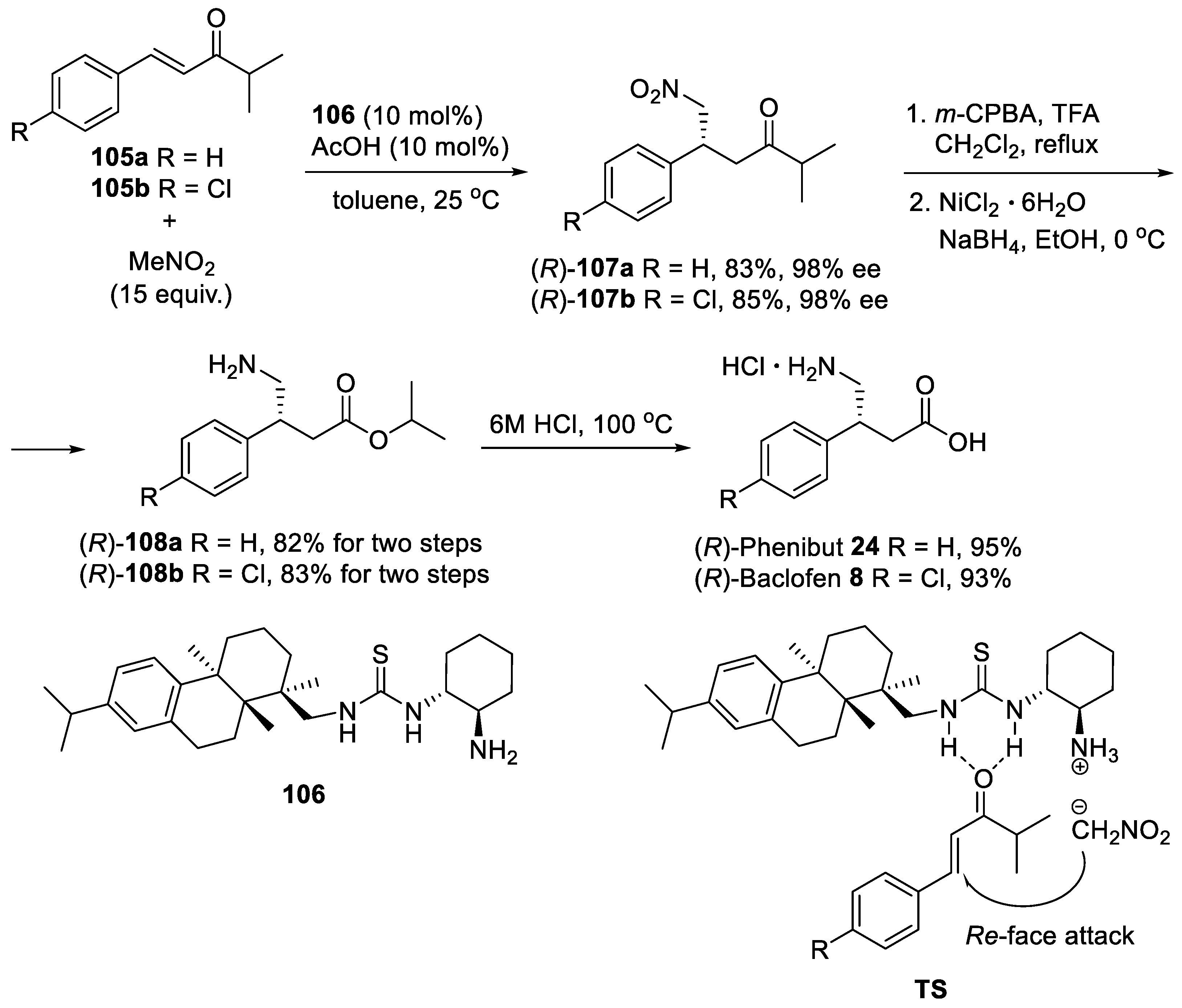{kind=link}
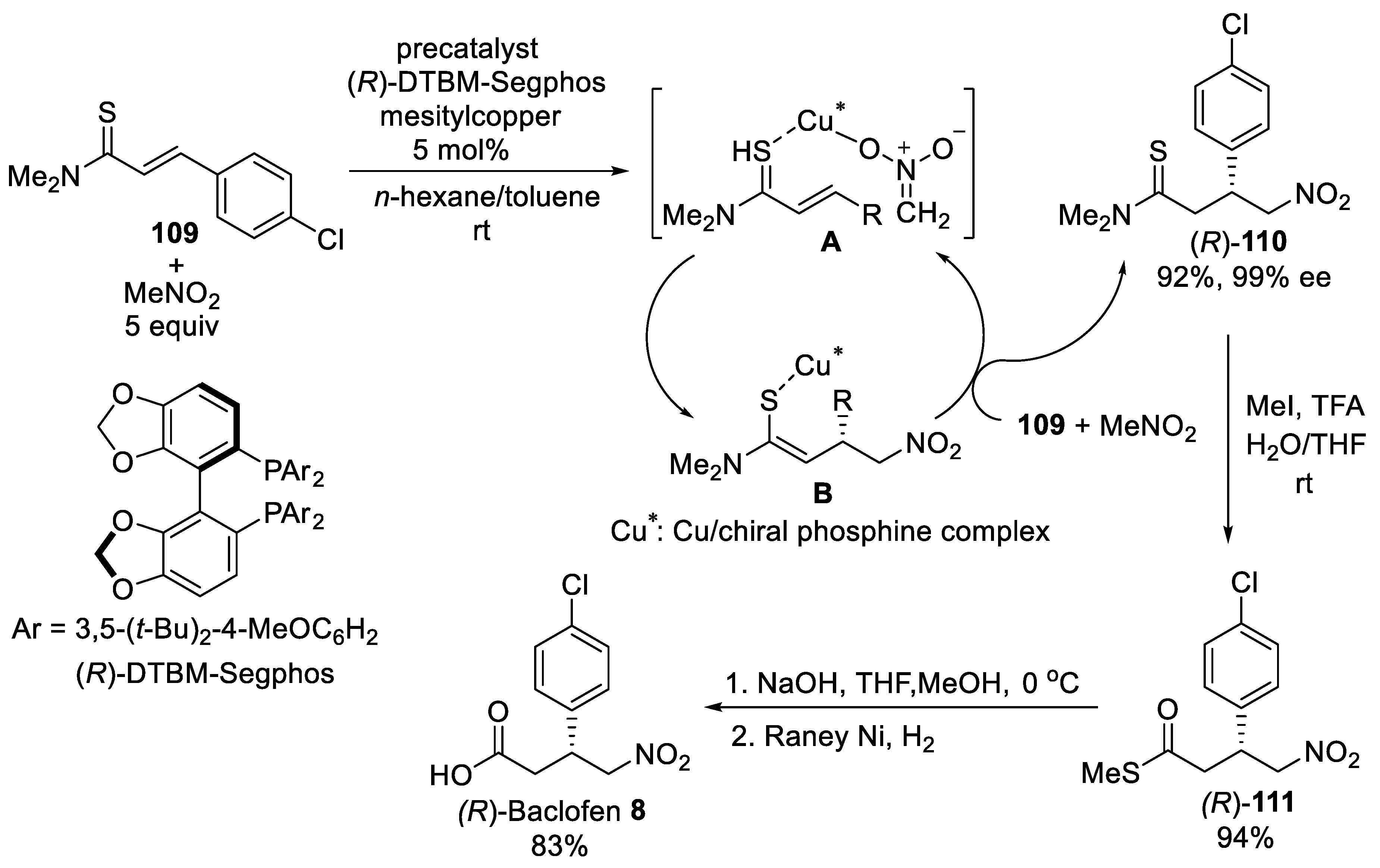{kind=link}
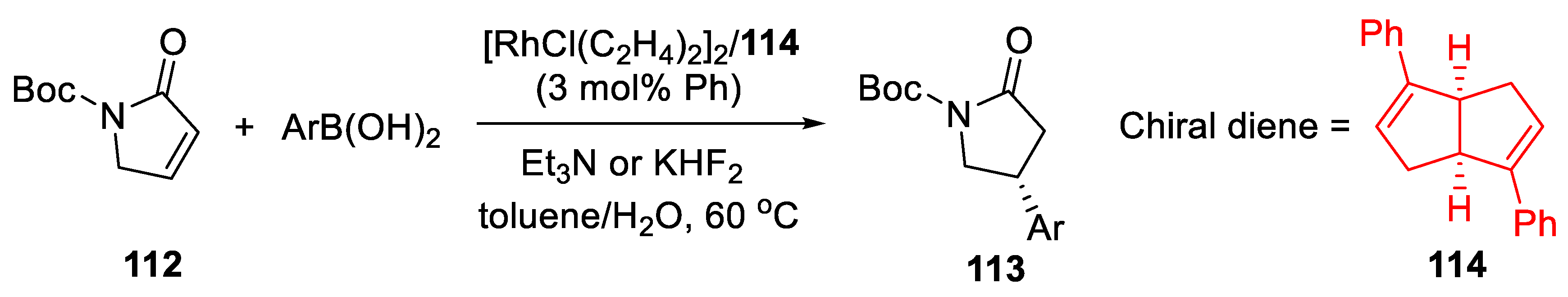{kind=link}
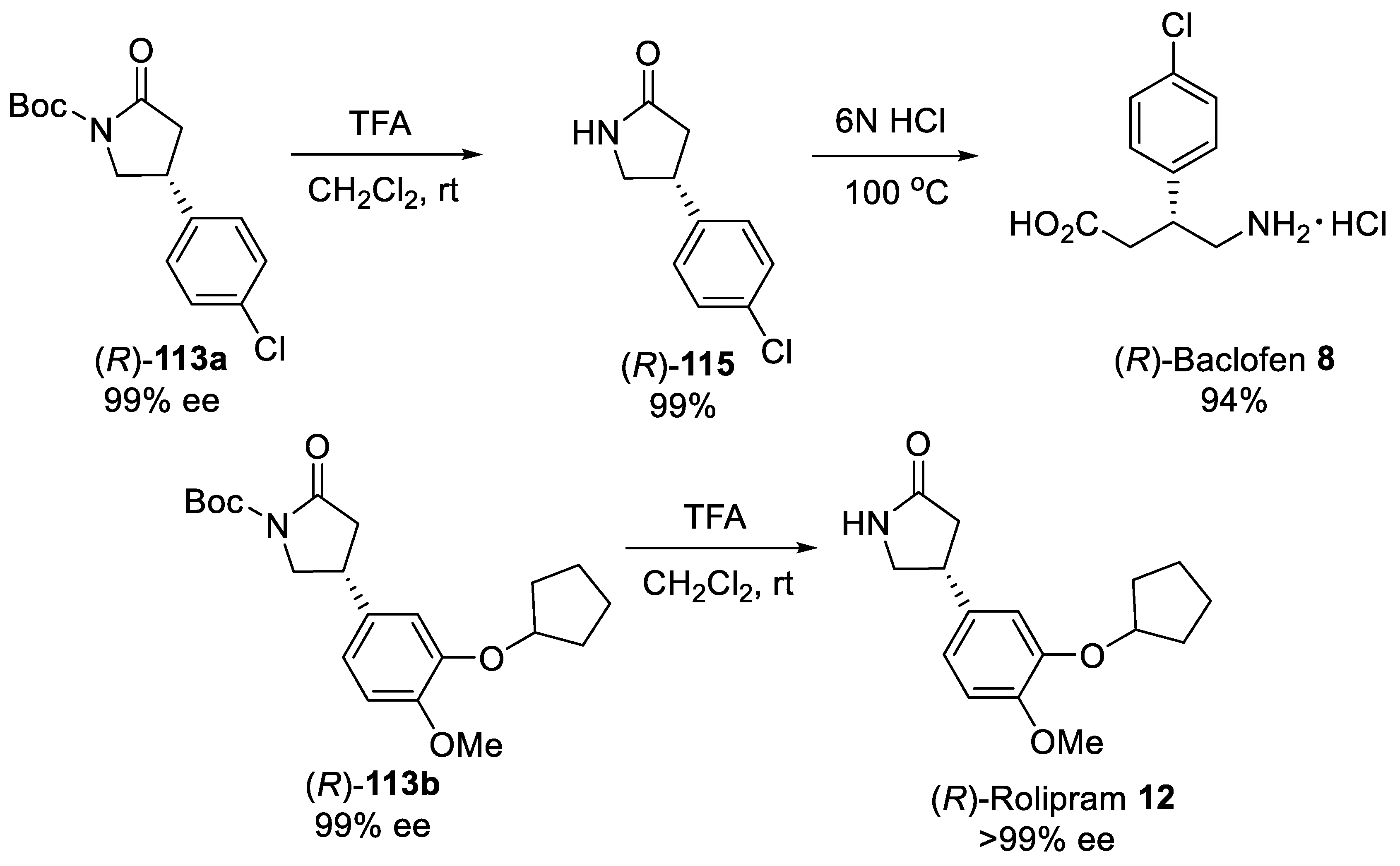{kind=link}
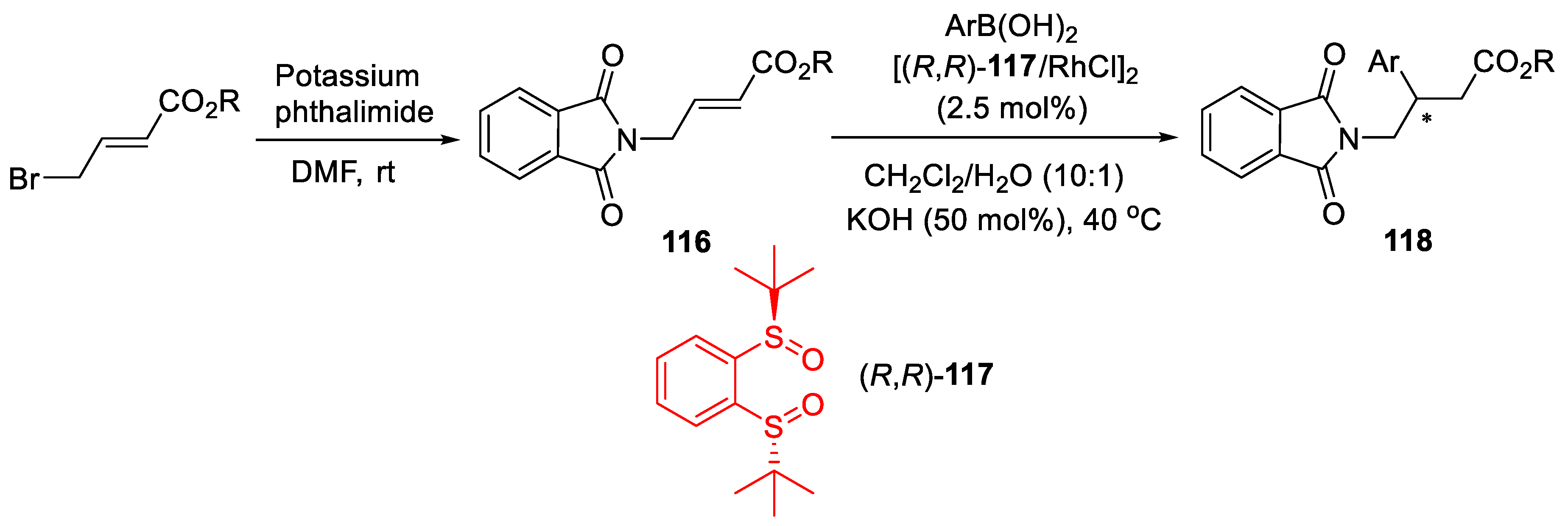{kind=link}
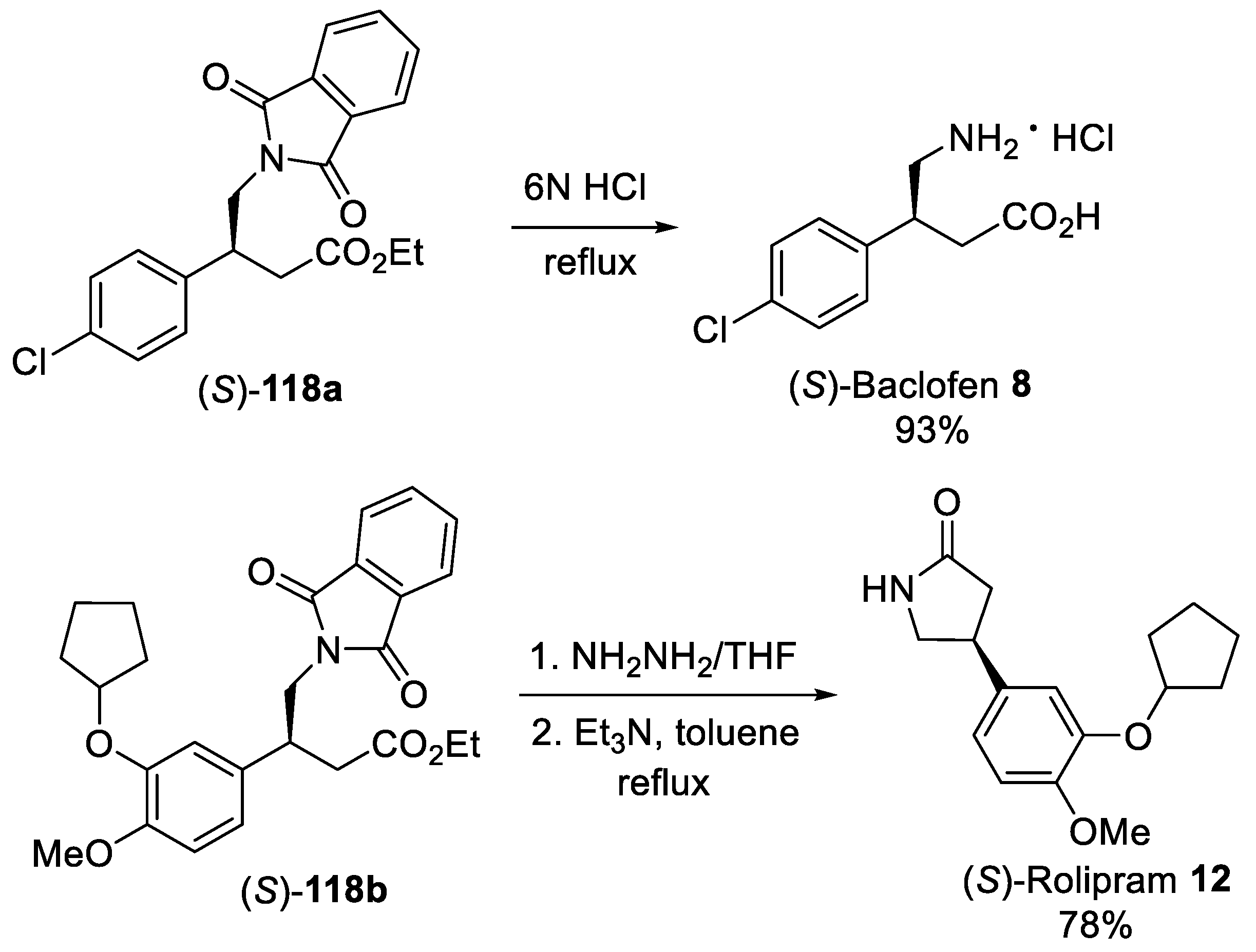{kind=link}
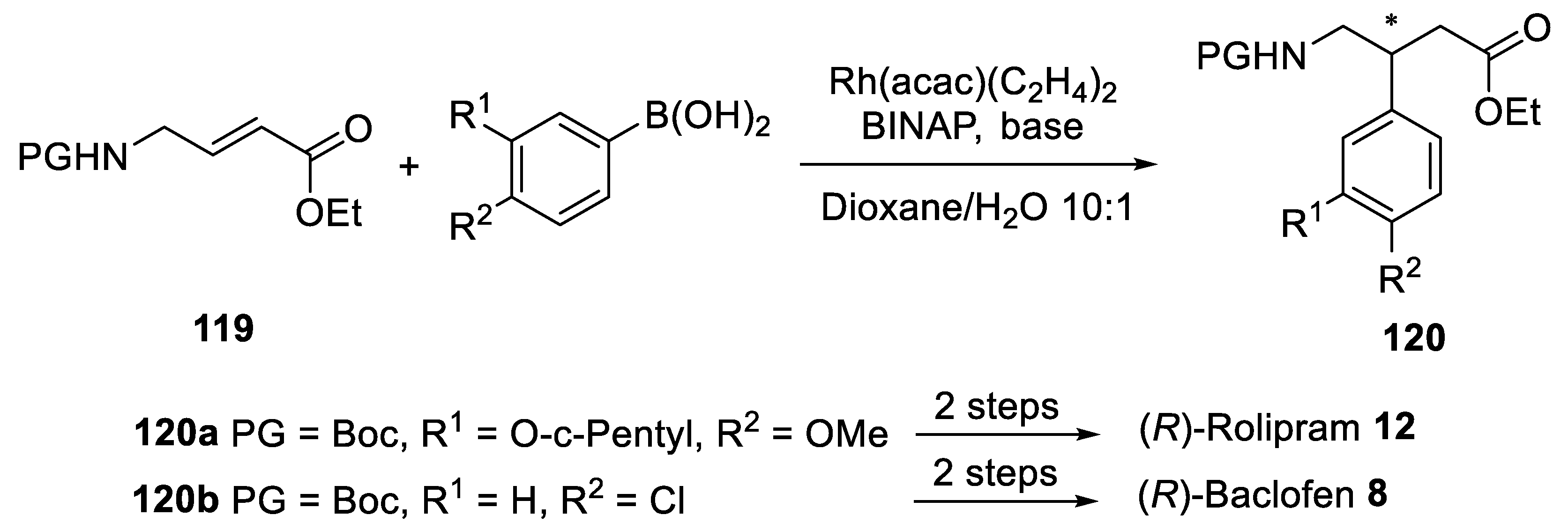{kind=link}
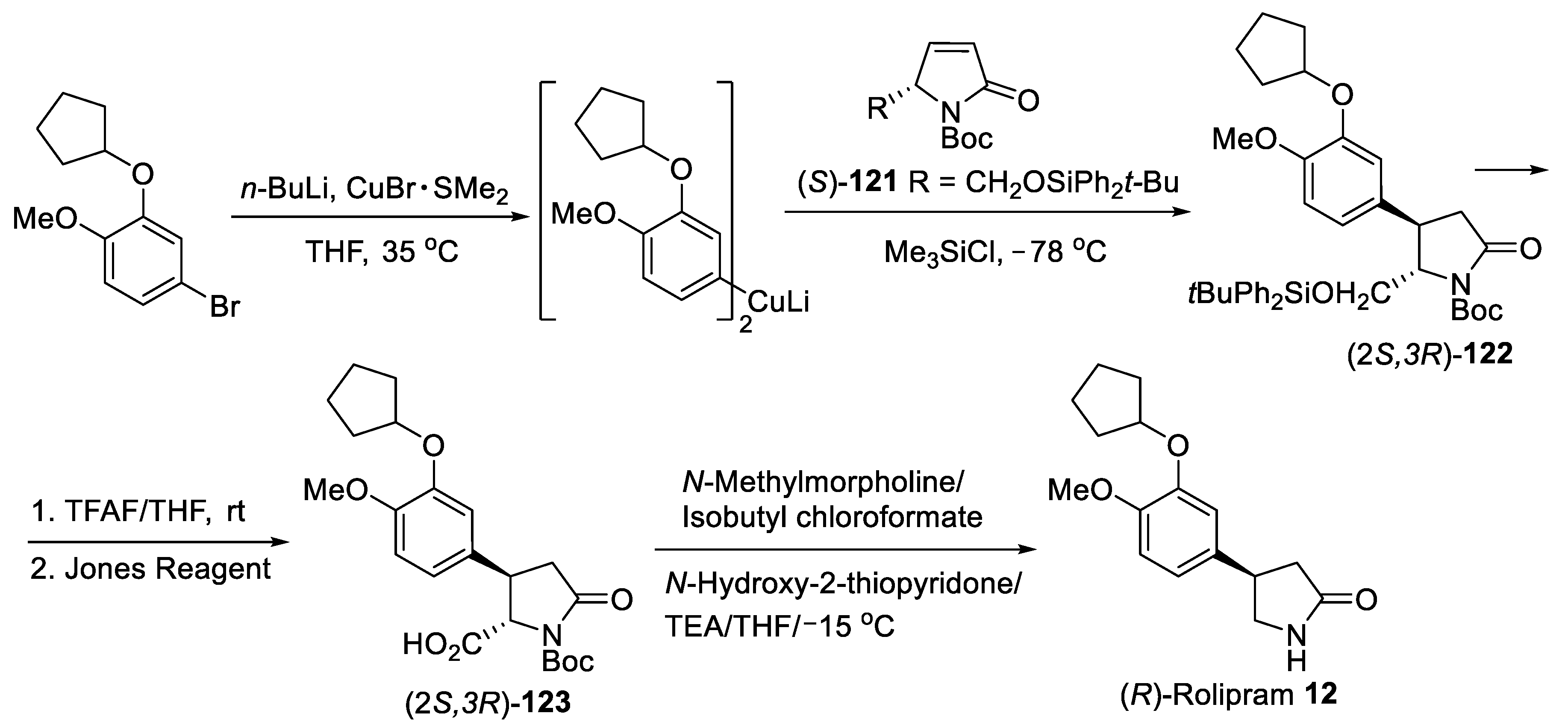{kind=link}
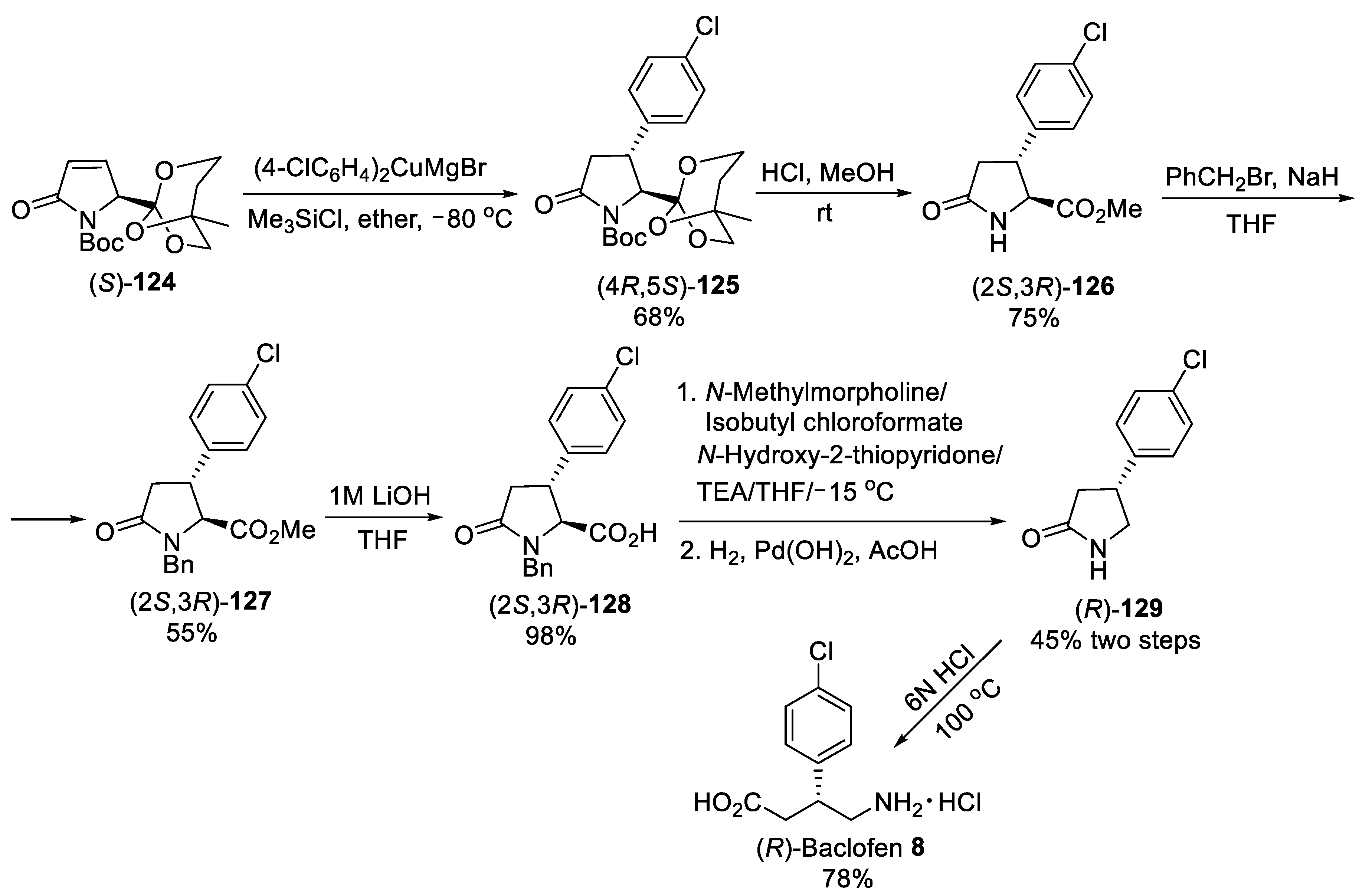{kind=link}
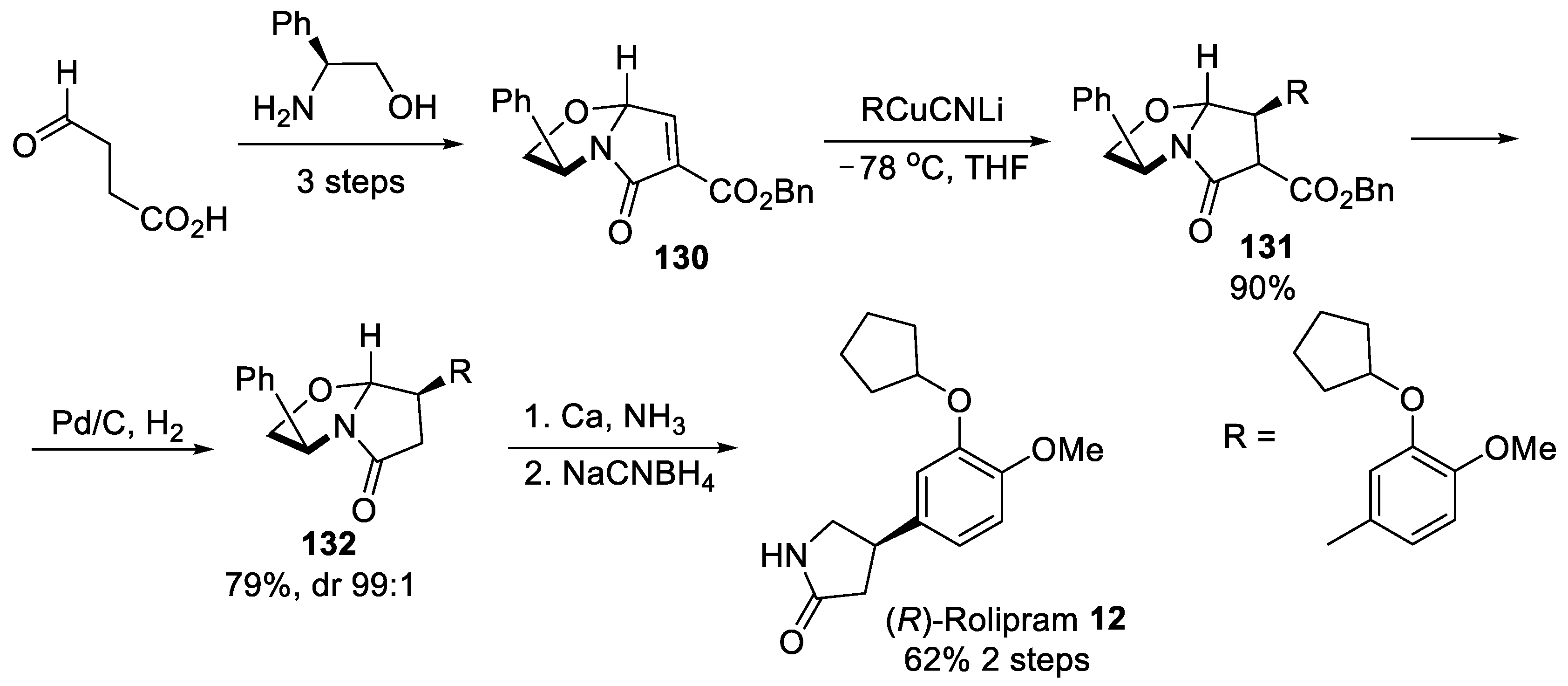{kind=link}
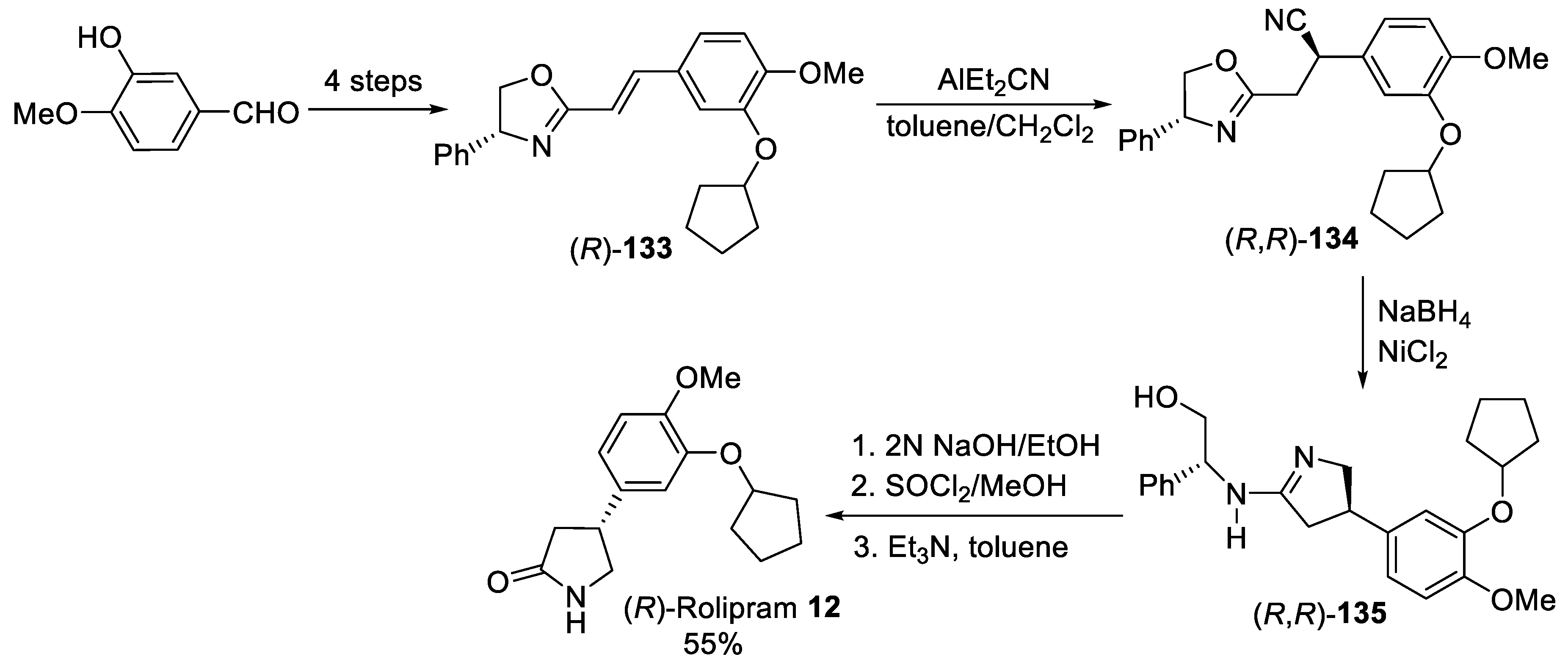{kind=link}
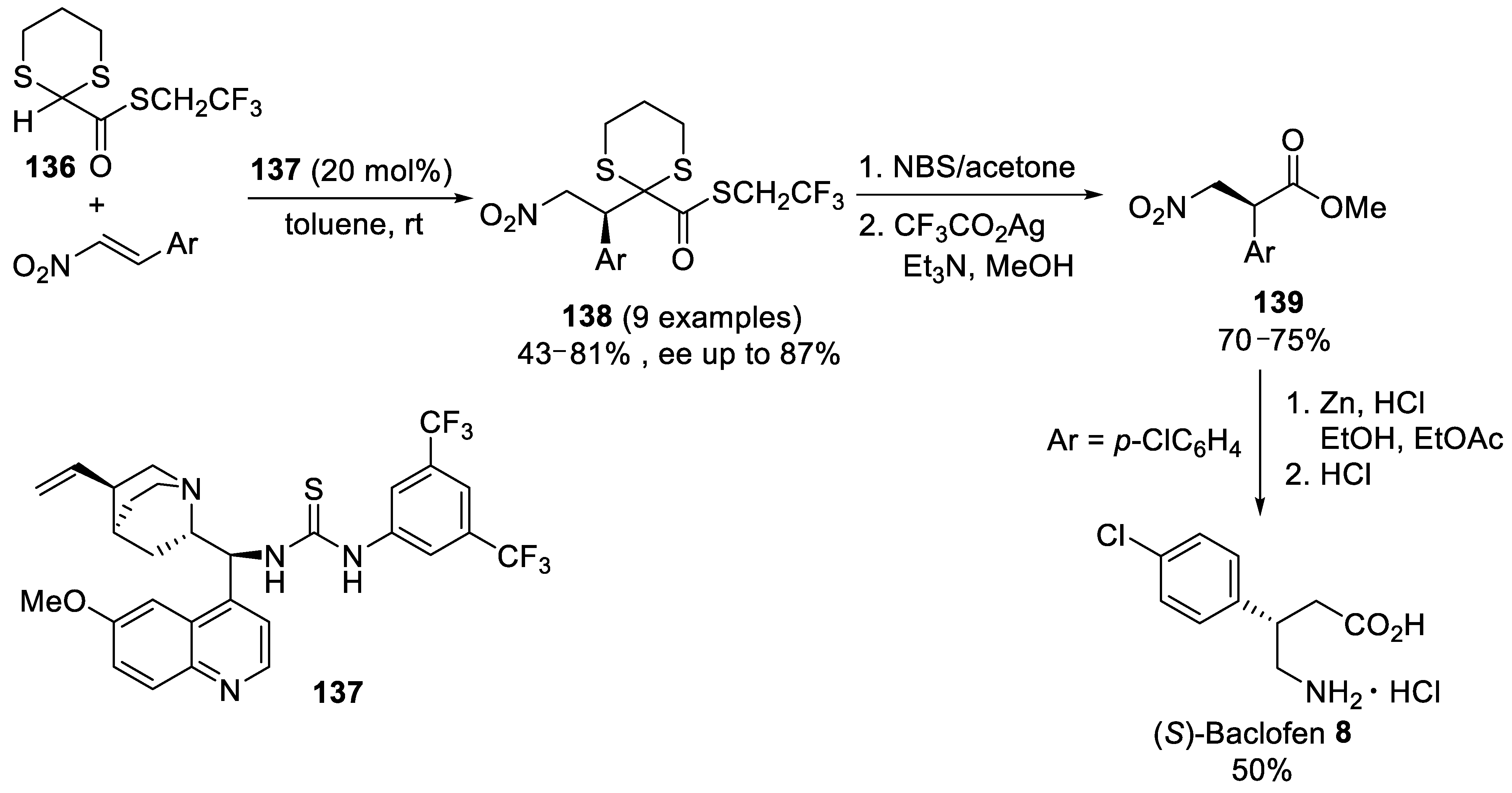{kind=link}
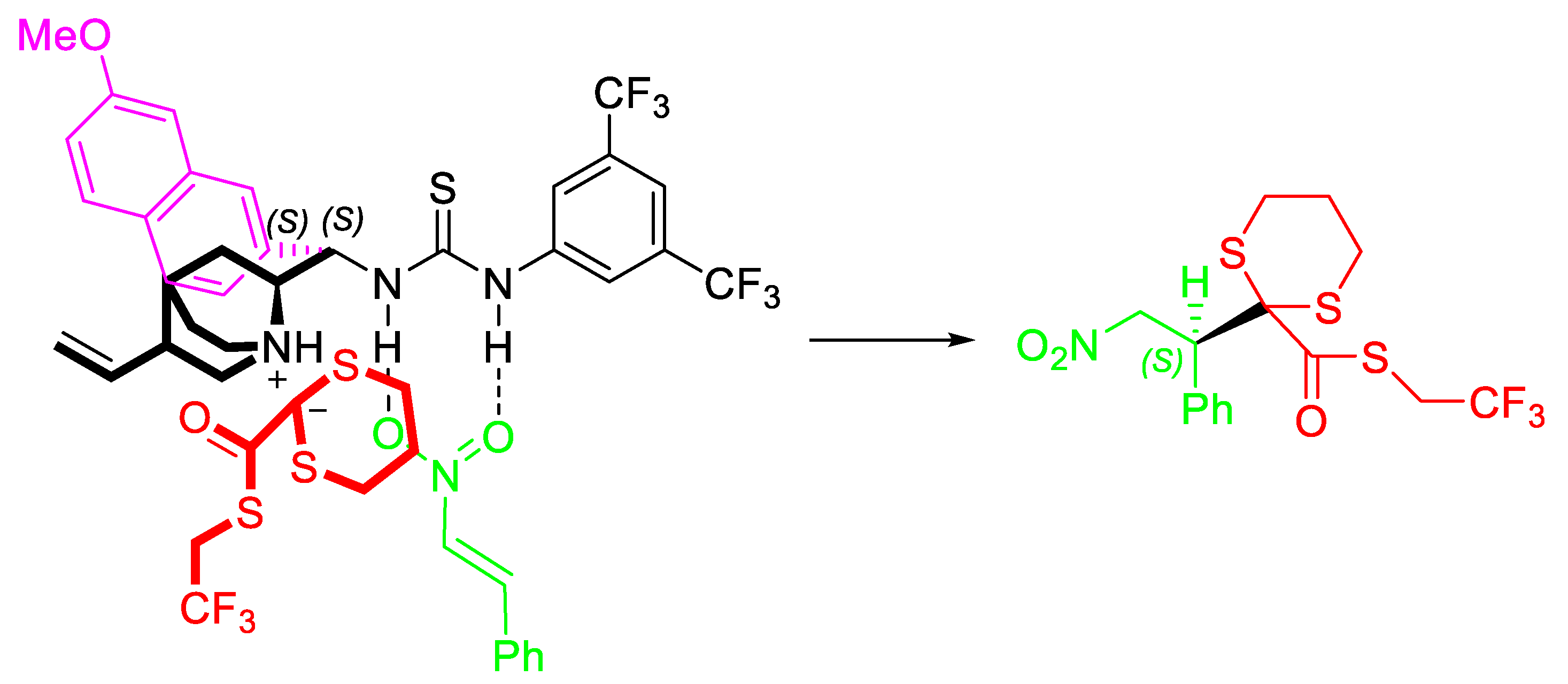{kind=link}
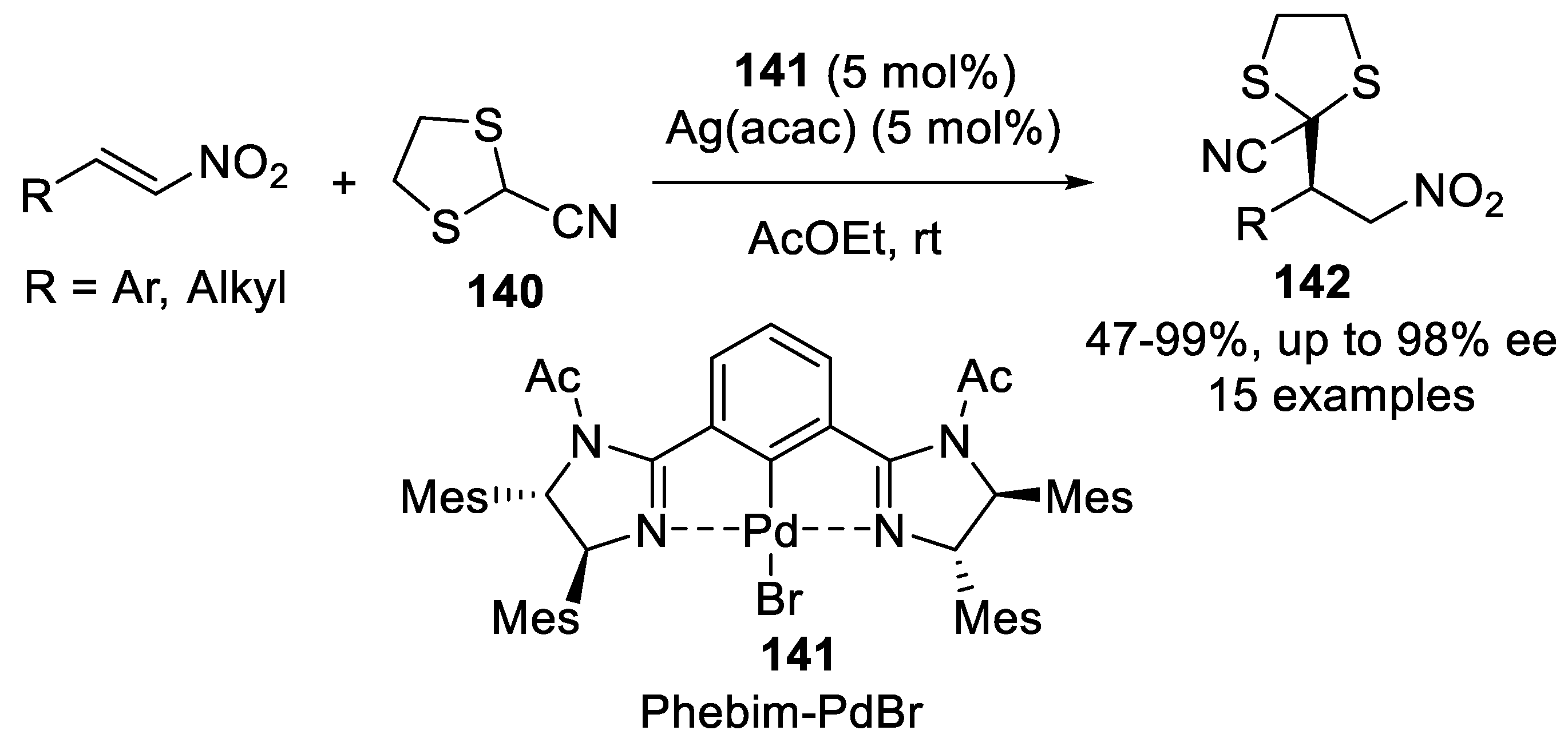{kind=link}
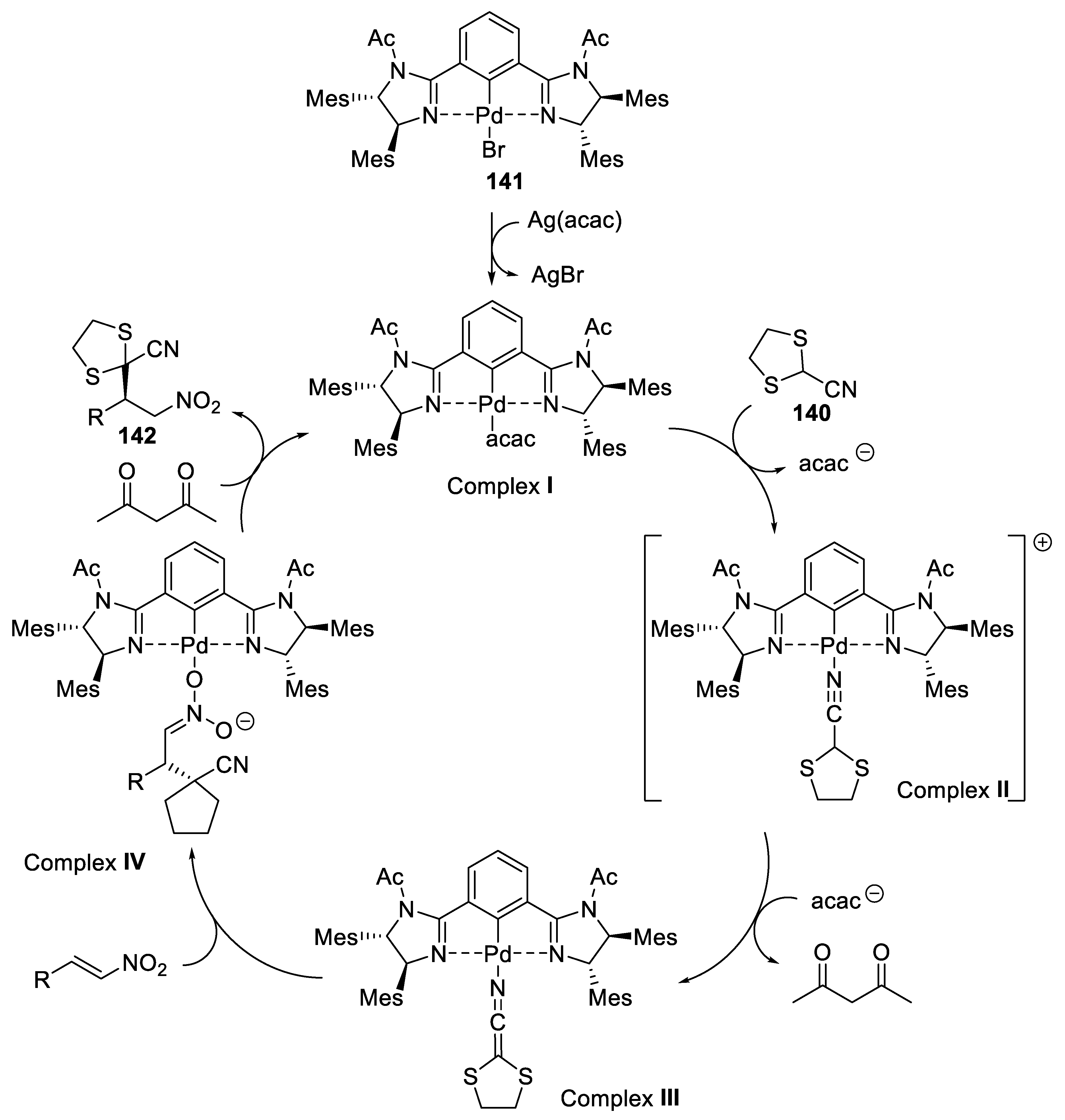{kind=link}
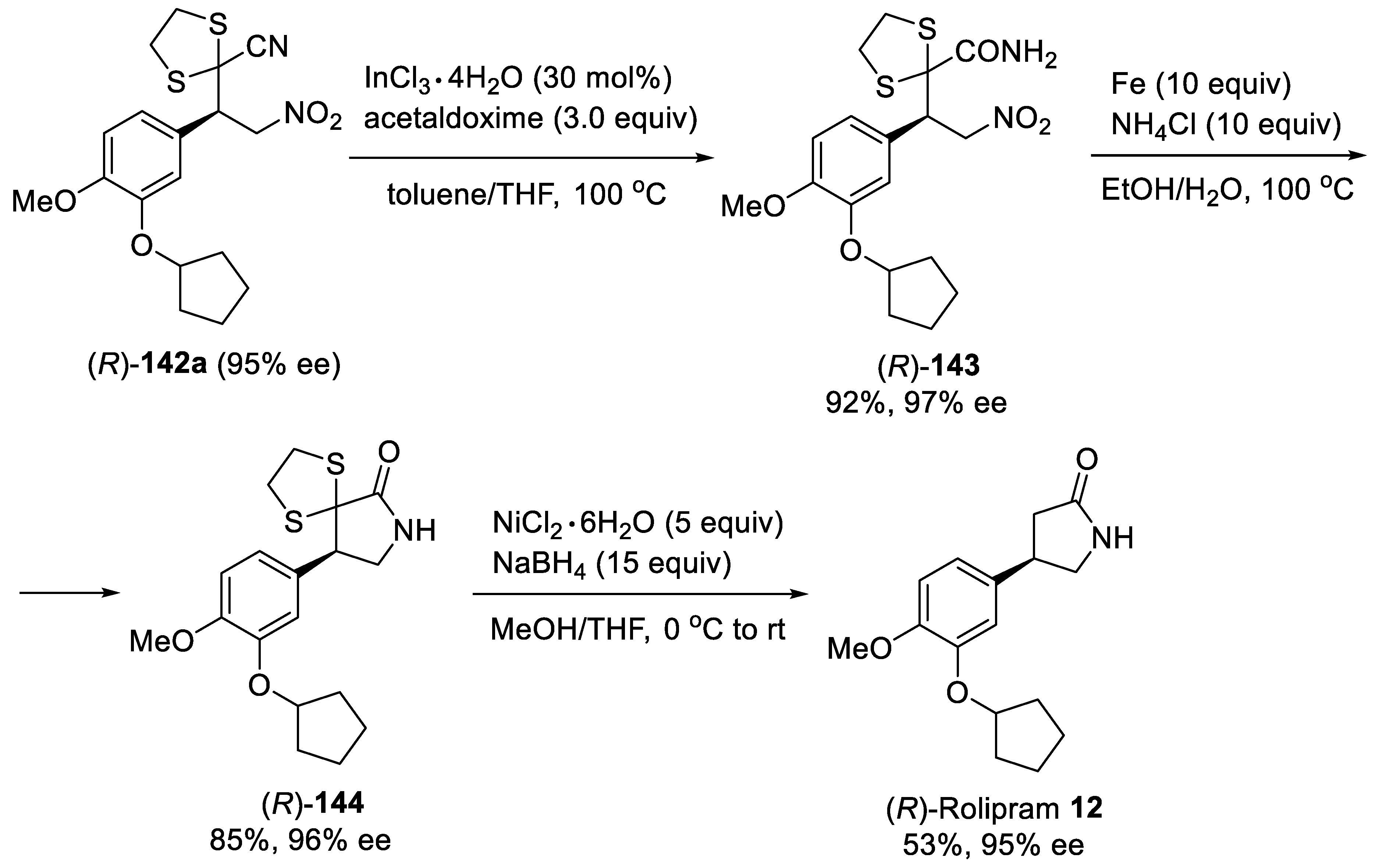{kind=link}
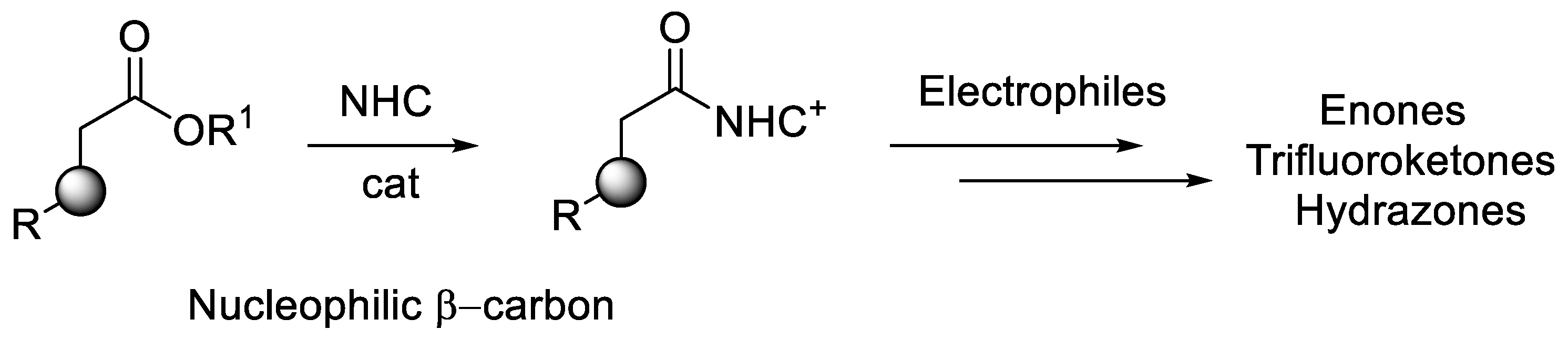{kind=link}
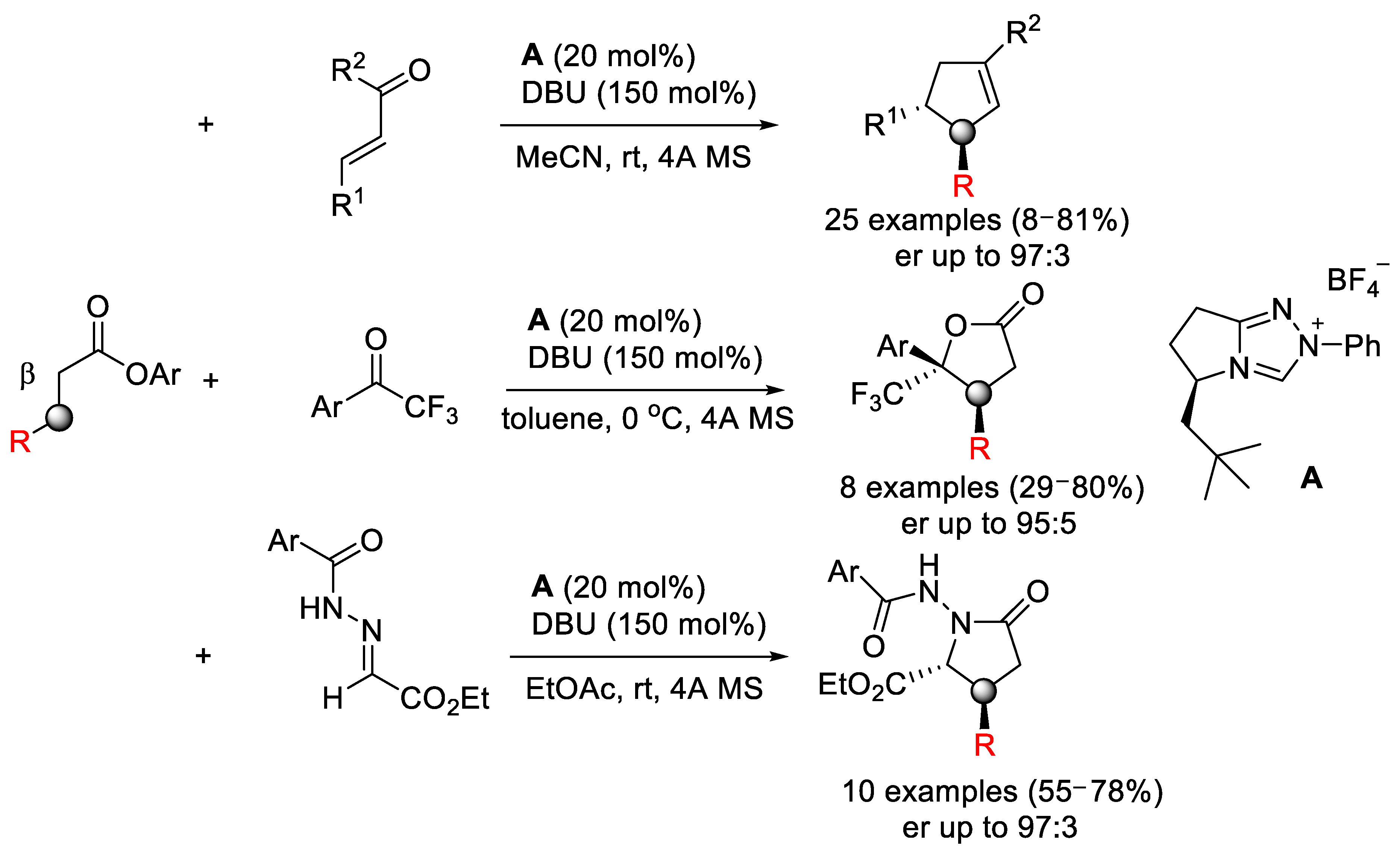{kind=link}
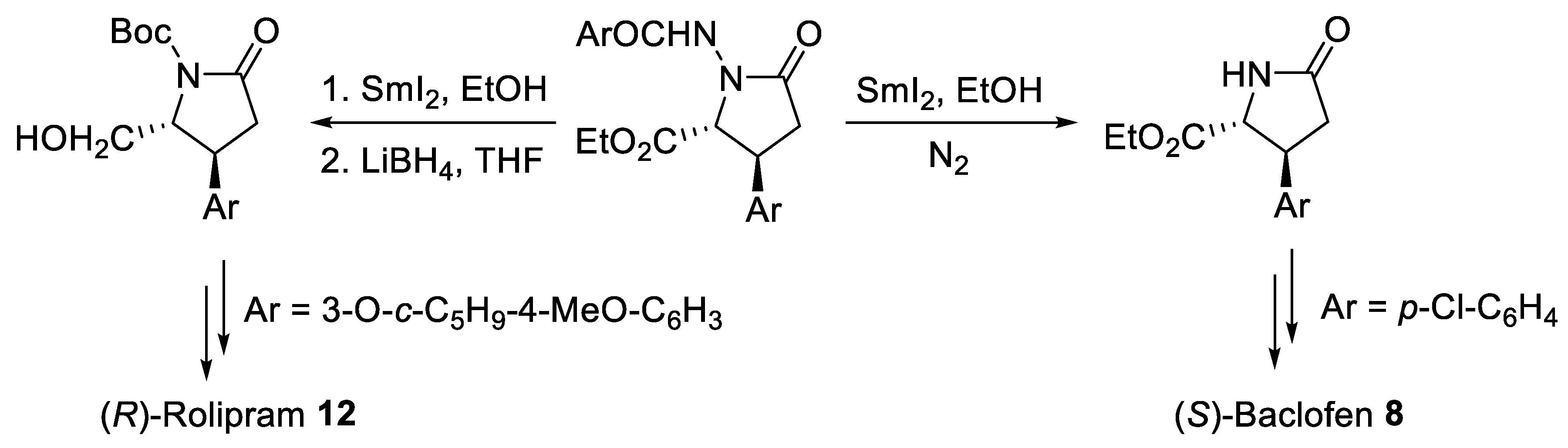{kind=link}
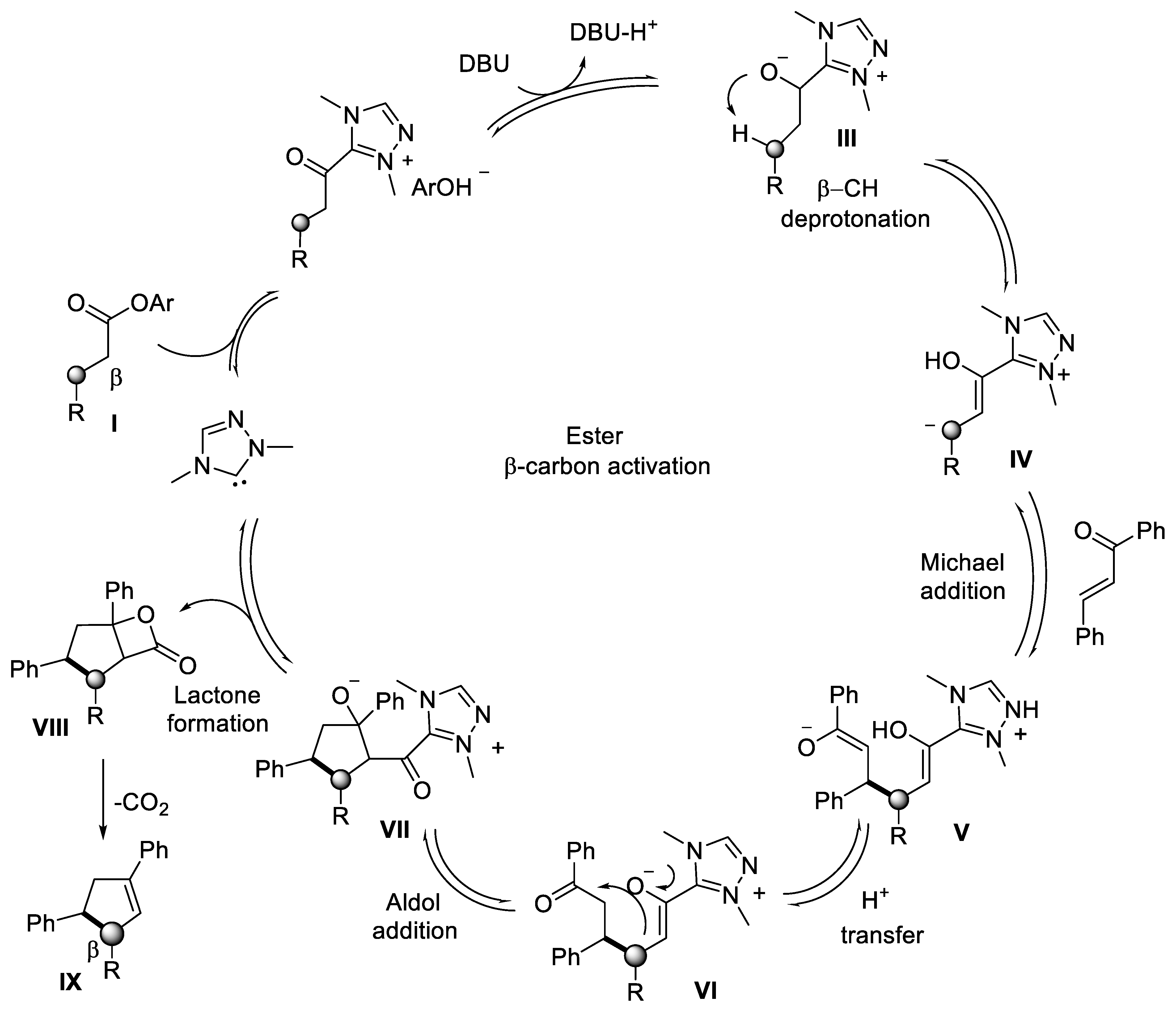{kind=link}
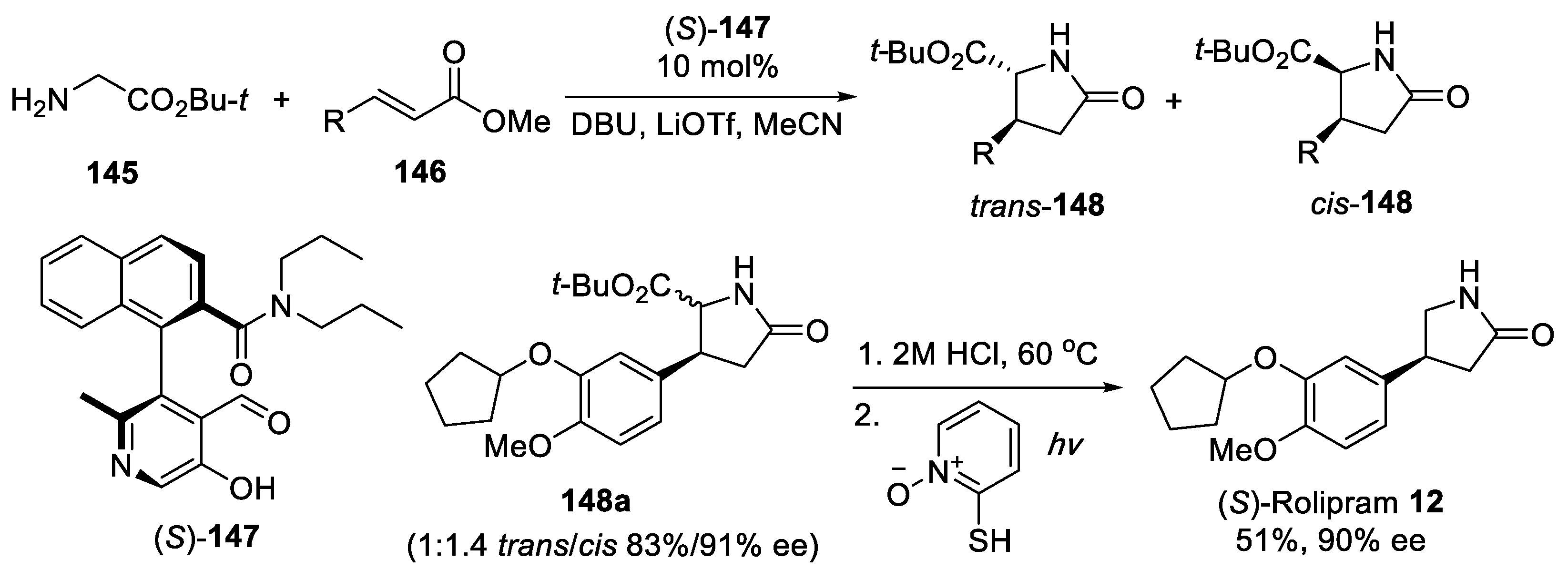{kind=link}
{kind=link}
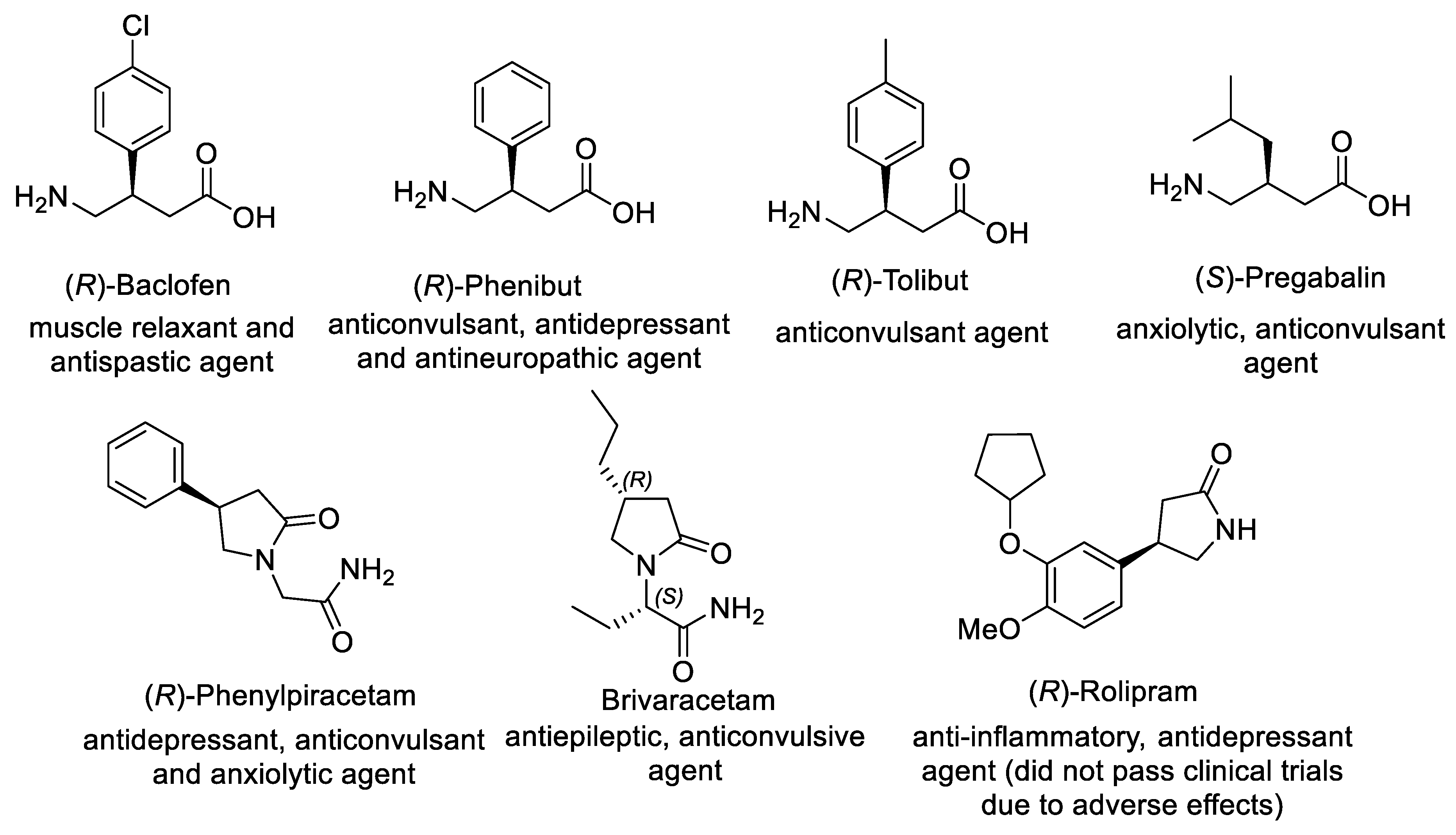
1. Introduction
2. Michael Addition of Carbonyl Compounds to α,β-Unsaturated Nitroalkenes
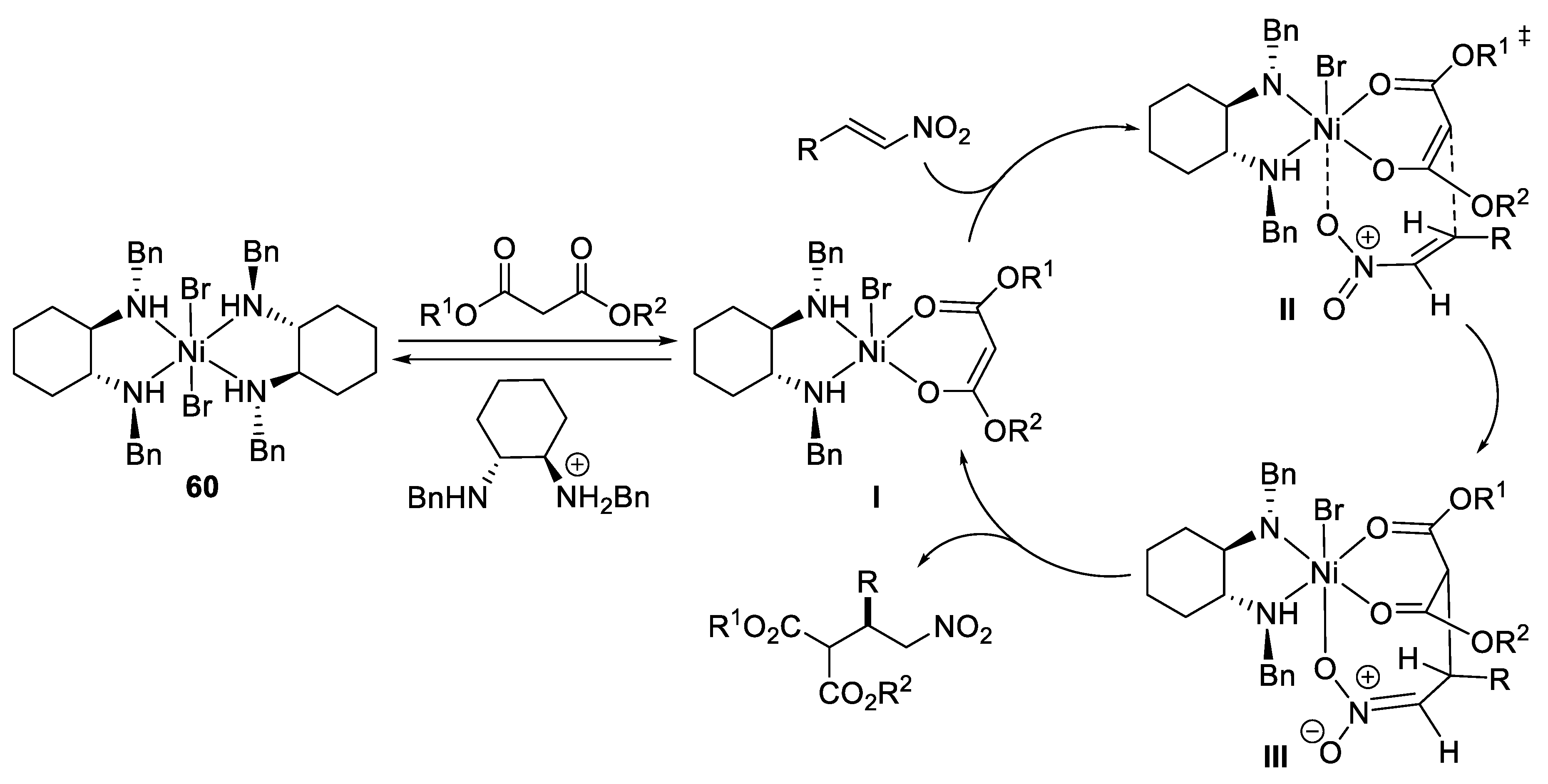
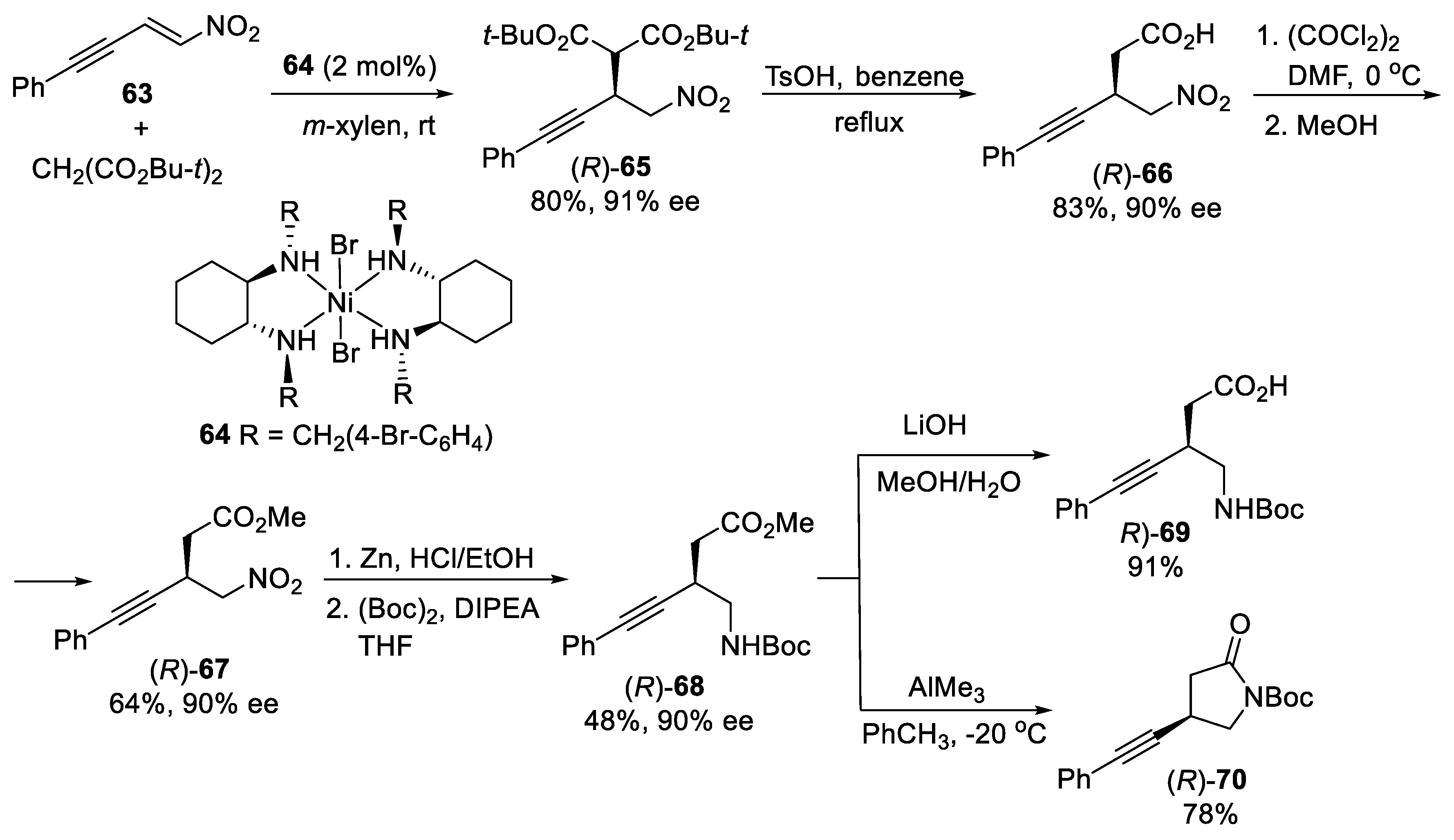
3. Michael Additions of Cyanide or Nitroalkanes to α,β-Unsaturated Carbonyl Compounds
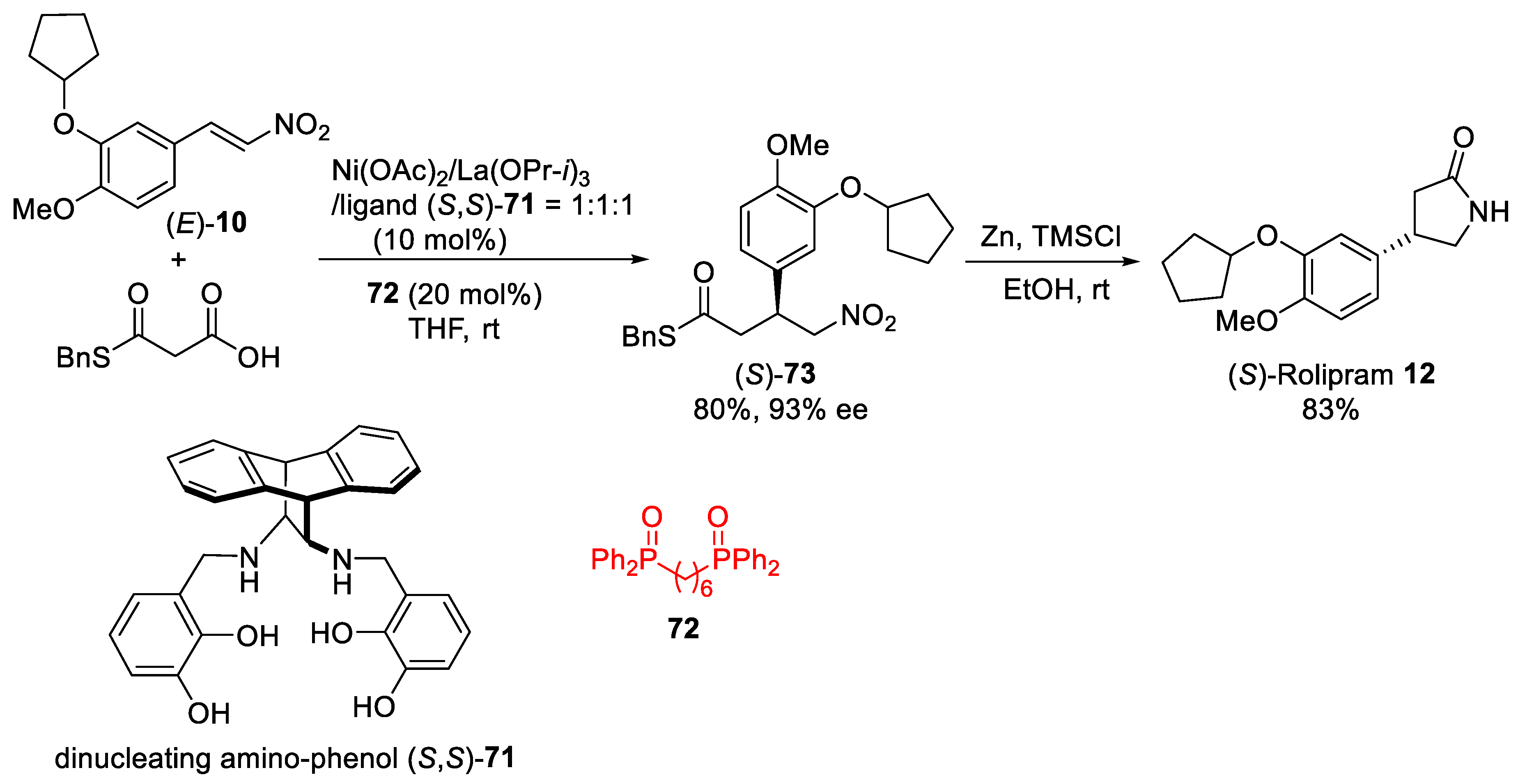
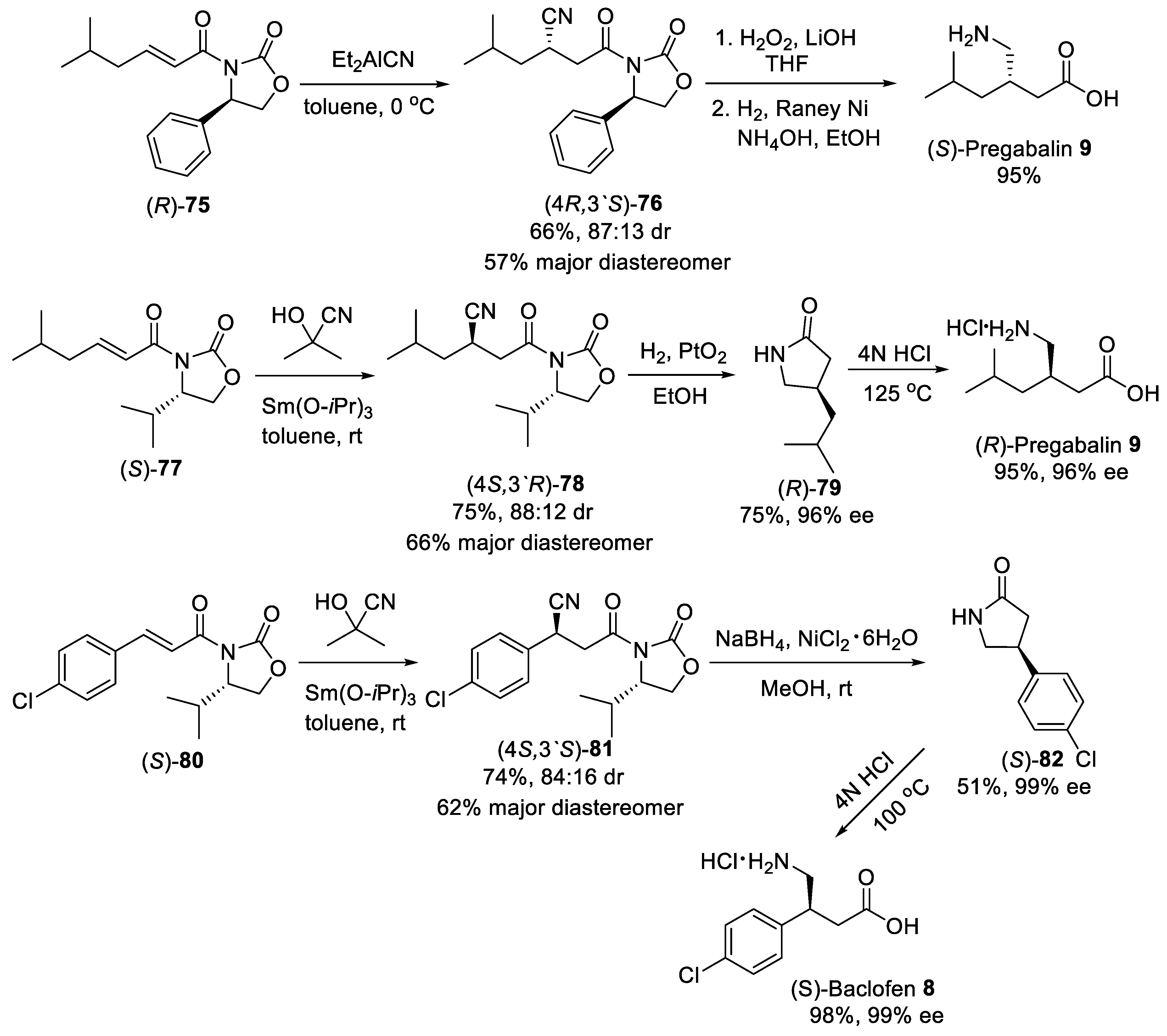
4. Michael Addition of Organometallic Reagents to α,β-Unsaturated Carbonyl Compounds
5. Miscellaneous Types of Michael Addition
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Soloshonok, V.A.; Cai, C.; Hruby, V.J.; Van Meervelt, L. Asymmetric synthesis of novel highly sterically constrained (2S, 3S)-3-methyl-3-trifluoromethyl-and (2S, 3S, 4R)-3-trifluoromethyl-4-methylpyroglutamic acids. Tetrahedron 1999, 55, 12045–12058. [Google Scholar] [CrossRef]
- Han, J.; Lyutenko, N.V.; Sorochinsky, A.E.; Okawara, A.; Konno, H.; White, S.; Soloshonok, V.A. Tailor-Made Amino Acids in Pharmaceutical Industry: Synthetic Approaches to Aza-Tryptophan Derivatives. Chem. Eur. J. 2021, 27, 17510–17528. [Google Scholar] [CrossRef]
- Han, J.; Konno, H.; Sato, T.; Soloshonok, V.A.; Izawa, K. Tailor-made amino acids in the design of small-molecule blockbuster drugs. Eur. J. Med. Chem. 2021, 220, 113448. [Google Scholar] [CrossRef]
- Han, J.; Konno, H.; Sato, T.; Izawa, K.; Soloshonok, V.A. Peptidomimetics and Peptide-Based Blockbuster Drugs. Curr. Org. Chem. 2021, 25, 1627–1658. [Google Scholar] [CrossRef]
- Liu, J.; Han, J.; Izawa, K.; Sato, T.; White, S.; Meanwell, N.A.; Soloshonok, V.A. Cyclic tailor-made amino acids in the design of modern pharmaceuticals. Eur. J. Med. Chem. 2020, 208, 112736. [Google Scholar] [CrossRef]
- Mei, H.; Han, J.; White, S.; Graham, D.J.; Izawa, K.; Sato, T.; Fustero, S.; Meanwell, N.A.; Soloshonok, V.A. Tailor-made amino acids and fluorinated motifs as prominent traits in modern pharmaceuticals. Chem. Eur. J. 2020, 26, 11349–11390. [Google Scholar] [CrossRef]
- Liu, A.; Han, J.; Nakano, A.; Konno, H.; Moriwaki, H.; Abe, H.; Izawa, K.; Soloshonok, V.A. New pharmaceuticals approved by FDA in 2020: Small-molecule drugs derived from amino acids and related compounds. Chirality 2022, 34, 86–103. [Google Scholar] [CrossRef]
- Yin, Z.; Hu, W.; Zhang, W.; Konno, H.; Moriwaki, H.; Izawa, K.; Han, J.; Soloshonok, V.A. Tailor-made amino acid-derived pharmaceuticals approved by the FDA in 2019. Amino Acids 2020, 52, 1227–1261. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Izawa, K. (Eds.) Asymmetric Synthesis and Application of α-Amino Acids; ACS Symposium Series #1009; Oxford University Press: Washington, DC, USA, 2009. [Google Scholar]
- Kent, C.N.; Park, C.; Lindsley, C.W. Classics in chemical neuroscience: Baclofen. ACS Chem. Neurosci. 2020, 11, 1740–1755. [Google Scholar] [CrossRef]
- Lapin, I. Phenibut (β-phenyl-GABA): A tranquilizer and nootropic drug. CNS Drug Rev. 2001, 7, 471–481. [Google Scholar] [CrossRef]
- Zvejniece, L.; Vavers, E.; Svalbe, B.; Veinberg, G.; Rizhanova, K.; Liepins, V.; Kalvinsh, I.; Dambrova, M. R-phenibut binds to the α2–δ subunit of voltage-dependent calcium channels and exerts gabapentin-like anti-nociceptive effects. Pharmacol. Biochem. Behav. 2015, 137, 23–29. [Google Scholar] [CrossRef]
- Dambrova, M.; Zvejniece, L.; Liepinsh, E.; Cirule, H.; Zharkova, O.; Veinberg, G.; Kalvinsh, I. Comparative pharmacological activity of optical isomers of phenibut. Eur. J. Pharmacol. 2008, 583, 128–134. [Google Scholar] [CrossRef]
- Tyurenkov, I.N.; Borodkina, L.E.; Bagmetova, V.V.; Berestovitskaya, V.M.; Vasil’eva, O.S. Comparison of nootropic and neuroprotective features of aryl-substituted analogs of gamma-aminobutyric acid. Bull. Exp. Biol. Med. 2016, 160, 465–469. [Google Scholar] [CrossRef]
- Silverman, R.B. From basic science to blockbuster drug: The discovery of Lyrica. Angew. Chem. Int. Ed. 2008, 47, 3500–3504. [Google Scholar] [CrossRef]
- Reinares, M.; Rosa, A.R.; Franco, C.; Goikolea, J.M.; Fountoulakis, K.; Siamouli, M.; Gonda, X.; Frangou, S.; Vieta, E. A systematic review on the role of anticonvulsants in the treatment of acute bipolar depression. Int. J. Neuropsychop. 2013, 16, 485. [Google Scholar] [CrossRef]
- Calandre, E.P.; Rico-Villademoros, F.; Slim, M. Alpha2delta ligands, gabapentin, pregabalin and mirogabalin: A review of their clinical pharmacology and therapeutic use. Expert Rev. Neurother. 2016, 16, 1263–1277. [Google Scholar] [CrossRef]
- Zvejniece, L.; Zvejniece, B.; Videja, M.; Stelfa, G.; Vavers, E.; Grinberga, S.; Svalbe, B.; Dambrova, M. Neuroprotective and anti-inflammatory activity of DAT inhibitor R-phenylpiracetam in experimental models of inflammation in male mice. Infammopharmacology 2020, 28, 1283–1292. [Google Scholar] [CrossRef]
- Malykh, A.G.; Sadaie, M.R. Piracetam and piracetam-like drugs. Drugs 2010, 70, 287–312. [Google Scholar] [CrossRef]
- Kenda, B.M.; Matagne, A.C.; Talaga, P.E.; Pasau, P.M.; Differding, E.; Lallemand, B.I.; Frycia, A.M.; Moureau, F.G.; Klitgaard, H.V.; Gillard, M.R.; et al. Discovery of 4-substituted pyrrolidone butanamides as new agents with significant antiepileptic activity. J. Med. Chem. 2004, 47, 530–549. [Google Scholar] [CrossRef]
- von Rosenstiel, P. Brivaracetam (ucb 34714). Neurotherapeutics 2007, 4, 84–87. [Google Scholar] [CrossRef]
- Zhu, J.; Mix, E.; Winblad, B. The antidepressant and antiinflammatory effects of rolipram in the central nervous system. CNS Drug Rev. 2001, 7, 387–398. [Google Scholar] [CrossRef]
- Gajcy, K.; Lochyński, S.; Librowski, T. A role of GABA analogues in the treatment of neurological diseases. Curr. Med. Chem. 2010, 17, 2338–2347. [Google Scholar] [CrossRef]
- Ordóñez, M.; Cativiela, C.; Romero-Estudillo, I. An update on the stereoselective synthesis of γ-amino acids. Tetrahedron Asymmetry 2016, 27, 999–1055. [Google Scholar] [CrossRef]
- Ramesh, P.; Suman, D.; Reddy, K.S.N. Asymmetric Synthetic Strategies of (R)-(–)-Baclofen: An Antispastic Drug. Synthesis 2018, 50, 211–226. [Google Scholar] [CrossRef]
- Martínez, C.A.; Hu, S.; Dumond, Y.; Tao, J.; Kelleher, P.; Tully, L. Development of a chemoenzymatic manufacturing process for pregabalin. Org. Process Res. Dev. 2008, 12, 392–398. [Google Scholar] [CrossRef]
- Shagufta, K.; Ameer, F.Z.; Sajjad, A.; Rabia, A.; Iqra, K.; Wajiha, Q.; Attia, M. Recent trends in the development of novel catalysts for asymmetric Michael reaction. Curr. Org. Chem. 2020, 24, 1397–1458. [Google Scholar]
- García-García, P.; Ladépêche, A.; Halder, R.; List, B. Catalytic asymmetric Michael reactions of acetaldehyde. Angew. Chem. Int. Ed. 2008, 47, 4719–4721. [Google Scholar] [CrossRef]
- Hayashi, Y.; Itoh, T.; Ohkubo, M.; Ishikawa, H. Asymmetric Michael reaction of acetaldehyde catalyzed by diphenylprolinol silyl ether. Angew. Chem. Int. Ed. 2008, 47, 4722–4724. [Google Scholar] [CrossRef]
- Gotoh, H.; Ishikawa, H.; Hayashi, Y. Diphenylprolinol silyl ether as catalyst of an asymmetric, catalytic, and direct Michael reaction of nitroalkanes with α, β-unsaturated aldehydes. Org. Lett. 2007, 9, 5307–5309. [Google Scholar] [CrossRef]
- Seebach, D.; Golinski, J. Synthesis of Open-Chain 2,3-Disubstituted 4-nitroketones by Diastereoselective Michael-addition of (E)-Enamines to (E)-Nitroolefins. A topological rule for C, C-bond forming processes between prochiral centres. Preliminary communication. Helv. Chim. Acta 1981, 64, 1413–1423. [Google Scholar] [CrossRef]
- Kaur, R.; Pandey, S.K. Efficient synthesis of (−)-(R)-and (+)-(S)-rolipram. Tetrahedron Lett. 2017, 58, 4333–4335. [Google Scholar] [CrossRef]
- Qiao, Y.; He, J.; Ni, B.; Headley, A.D. Asymmetric Michael reaction of acetaldehyde with nitroolefins catalyzed by highly water-compatible organocatalysts in aqueous media. Adv. Synth. Catal. 2012, 354, 2849–2853. [Google Scholar] [CrossRef]
- Nori, V.; Sinibaldi, A.; Giorgianni, G.; Pesciaioli, F.; Di Donato, F.; Cocco, E.; Biancolillo, A.; Landa, A.; Carlone, A. DoE-Driven Development of an Organocatalytic Enantioselective Addition of Acetaldehyde to Nitrostyrenes in Water. Chem. Eur. J. 2022, 28, e202104524. [Google Scholar] [CrossRef]
- Palomo, C.; Landa, A.; Mielgo, A.; Oiarbide, M.; Puente, A.; Vera, S. Water-compatible iminium activation: Organocatalytic Michael reactions of carbon-centered nucleophiles with enals. Angew. Chem. Int. Ed. 2007, 46, 8431–8435. [Google Scholar] [CrossRef]
- Alonso, D.A.; Baeza, A.; Chinchilla, R.; Gómez, C.; Guillena, G.; Pastor, I.M.; Ramón, D.R. Recent advances in asymmetric organocatalyzed conjugate additions to nitroalkenes. Molecules 2017, 22, 895. [Google Scholar] [CrossRef] [Green Version]
- Odagi, M.; Nagasawa, K. Recent Advances in Natural Products Synthesis Using Bifunctional Organocatalysts bearing a Hydrogen-Bonding Donor Moiety. Asian J. Org. Chem. 2019, 8, 1766–1774. [Google Scholar] [CrossRef]
- Okino, T.; Hoashi, Y.; Furukawa, T.; Xu, X.; Takemoto, Y. Enantio-and diastereoselective Michael reaction of 1, 3-dicarbonyl compounds to nitroolefins catalyzed by a bifunctional thiourea. J. Am. Chem. Soc. 2005, 127, 119–125. [Google Scholar] [CrossRef]
- Perlikowska, R.; Piekielna, J.; Mazur, M.; Koralewski, R.; Olczak, J.; do Rego, J.C.; Fichna, J.; Modranka, J.; Janecki, T.; Janecka, A. Antinociceptive and antidepressant-like action of endomorphin-2 analogs with proline surrogates in position 2. Bioorg. Med. Chem. 2014, 22, 4803–4809. [Google Scholar] [CrossRef]
- Sukhanova, A.A.; Nelyubina, Y.V.; Zlotin, S.G. Asymmetric synthesis of 3-prenyl-substituted pyrrolidin-2-ones. Mendeleev Commun. 2016, 26, 471–473. [Google Scholar] [CrossRef]
- Steppeler, F.; Dominika Iwan, D.; Elżbieta Wojaczyńska, E.; Wojaczyński, J. Chiral thioureas—Preparation and significance in asymmetric synthesis and medicinal chemistry. Molecules 2020, 25, 401. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, X.; Ge, Z.; Sun, Q.; Cheng, T.; Li, R. Solvent-free organocatalytic Michael addition of diethyl malonate to nitroalkenes: The practical synthesis of Pregabalin and γ-nitrobutyric acid derivatives. Tetrahedron 2011, 67, 636–640. [Google Scholar] [CrossRef]
- Bae, H.Y.; Song, C.E. Unprecedented Hydrophobic Amplification in Noncovalent Organocatalysis “on Water”: Hydrophobic Chiral Squaramide Catalyzed Michael Addition of Malonates to Nitroalkenes. ACS Catal. 2015, 5, 3613–3619. [Google Scholar] [CrossRef]
- Jin, H.; Kim, S.T.; Hwang, G.S.; Ryu, D.H. l-Proline Derived Bifunctional Organocatalysts: Enantioselective Michael Addition of Dithiomalonates to trans-β-Nitroolefins. J. Org. Chem. 2016, 81, 3263–3274. [Google Scholar] [CrossRef]
- Kimmel, K.L.; Weaver, J.D.; Ellman, J.A. Enantio-and diastereoselective addition of cyclohexyl Meldrum’s acid to β-and α, β-disubstituted nitroalkenes via N-sulfinyl urea catalysis. Chem. Sci. 2012, 3, 121–125. [Google Scholar] [CrossRef]
- Mondal, A.; Bhowmick, S.; Ghosh, A.; Chanda, T.; Bhowmick, K.C. Advances on asymmetric organocatalytic 1,4-conjugate addition reactions in aqueous and semi-aqueous media. Tetrahedron Asymmetry 2017, 28, 849–875. [Google Scholar] [CrossRef]
- Sim, J.H.; Park, J.H.; Maity, P.; Song, C.E. Access to Chiral GABA Analogues Bearing a Trifluoromethylated All-Carbon Quaternary Stereogenic Center through Water-Promoted Organocatalytic Michael Reactions. Org. Lett. 2019, 21, 6715–6719. [Google Scholar] [CrossRef]
- Leyva-Pérez, A.; García-García, P.; Corma, A. Multisite organic–inorganic hybrid catalysts for the direct sustainable synthesis of GABAergic drugs. Angew. Chem. Int. Ed. 2014, 53, 8687–8690. [Google Scholar] [CrossRef]
- García-García, P.; Zagdoun, A.; Copèret, C.; Lesage, A.; Díaz, U.; Corma, A. In situ preparation of a multifunctional chiral hybrid organic–inorganic catalyst for asymmetric multicomponent reactions. Chem. Sci. 2013, 4, 2006–2012. [Google Scholar] [CrossRef]
- Veverková, E.; Bilka, S.; Baran, R.; Šebesta, R. Squaramide-Catalyzed Michael Addition as a Key Step for the Direct Synthesis of GABAergic Drugs. Synthesis 2016, 48, 1474–1482. [Google Scholar]
- Marchetti, L.A.; Kumawat, L.K.; Mao, N.; Stephens, J.C.; Elmes, R.B.P. The versatility of squaramides: From supramolecular chemistry to chemical biology. Chem 2019, 5, 1398–1485. [Google Scholar] [CrossRef]
- Baran, R.; Veverková, E.; Škvorcová, A.; Šebesta, R. Enantioselective Michael addition of 1, 3-dicarbonyl compounds to a nitroalkene catalyzed by chiral squaramides—A key step in the synthesis of pregabalin. Org. Biomol. Chem. 2013, 11, 7705–7711. [Google Scholar] [CrossRef] [PubMed]
- Bassas, O.; Huuskonen, J.; Rissanen, K.; Koskinen, A.M.P. A simple organocatalytic enantioselective synthesis of pregabalin. Eur. J. Org. Chem. 2009, 2009, 1340–1351. [Google Scholar] [CrossRef] [Green Version]
- Kitanosono, T.; Kobayashi, S. Synthetic Organic “Aquachemistry” that Relies on Neither Cosolvents nor Surfactants. ACS Cent. Sci. 2021, 7, 739–747. [Google Scholar] [CrossRef]
- Bae, H.Y.; Some, S.; Oh, J.S.; Lee, Y.S.; Song, C.E. Hydrogen bonding mediated enantioselective organocatalysis in brine: Significant rate acceleration and enhanced stereoselectivity in enantioselective Michael addition reactions of 1,3-dicarbonyls to β-nitroolefins. Chem. Commun. 2011, 47, 9621–9623. [Google Scholar] [CrossRef] [PubMed]
- Sim, J.H.; Song, C.E. Water-Enabled Catalytic Asymmetric Michael Reactions of Unreactive Nitroalkenes: One-Pot Synthesis of Chiral GABA-Analogs with All-Carbon Quaternary Stereogenic Centers. Angew. Chem. Int. Ed. 2017, 56, 1835–1839. [Google Scholar] [CrossRef]
- Wang, Z.L. Recent advances in catalytic asymmetric decarboxylative addition reactions. Adv. Synth. Catal. 2013, 355, 2745–2755. [Google Scholar] [CrossRef]
- Bae, H.Y.; Some, S.; Lee, J.H.; Kim, J.Y.; Song, M.J.; Lee, S.; Zhang, Y.J.; Song, C.E. Organocatalytic Enantioselective Michael-Addition of Malonic Acid Half-Thioesters to β-Nitroolefins: From Mimicry of Polyketide Synthases to Scalable Synthesis of γ-Amino Acids. Adv. Synth. Catal. 2011, 353, 3196–3202. [Google Scholar] [CrossRef]
- Lubkoll, J.; Wennemers, H. Mimicry of Polyketide Synthases—Enantioselective 1, 4-Addition Reactions of Malonic Acid Half-Thioesters to Nitroolefins. Angew. Chem. Int. Ed. 2007, 46, 6841–6844. [Google Scholar] [CrossRef]
- Li, F.; Li, Y.Z.; Jia, Z.S.; Xu, M.H.; Tian, P.; Lin, G.Q. Biscinchona alkaloids as highly efficient bifunctional organocatalysts for the asymmetric conjugate addition of malonates to nitroalkenes at ambient temperature. Tetrahedron 2011, 67, 10186–10194. [Google Scholar] [CrossRef]
- Serdyuk, O.V.; Heckel, C.M.; Tsogoeva, S.B. Bifunctional primary amine-thioureas in asymmetric organocatalysis. Org. Biomol. Chem. 2013, 11, 7051–7071. [Google Scholar] [CrossRef] [Green Version]
- Tsakos, M.; Kokotos, C.G.; Kokotos, G. Primary Amine-Thioureas with Improved Catalytic Properties for “Difficult” Michael Reactions: Efficient Organocatalytic Syntheses of (S)-Baclofen, (R)-Baclofen and (S)-Phenibut. Adv. Synth. Catal. 2012, 354, 740–746. [Google Scholar] [CrossRef]
- Zheng, K.; Liu, X.; Feng, X. Recent advances in metal-catalyzed asymmetric 1,4-conjugate addition (ACA) of nonorganometallic nucleophiles. Chem. Rev. 2018, 118, 7586–7656. [Google Scholar] [CrossRef] [PubMed]
- Hui, C.; Pu, F.; Xu, J. Metal-Catalyzed Asymmetric Michael Addition in Natural Product Synthesis. Chem. Eur. J. 2017, 23, 4023–4036. [Google Scholar] [CrossRef]
- Evans, D.A.; Mito, S.; Seidel, D. Scope and mechanism of enantioselective Michael additions of 1,3-dicarbonyl compounds to nitroalkenes catalyzed by nickel (II)–diamine complexes. J. Am. Chem. Soc. 2007, 129, 11583–11592. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Jin, R.; Ceng, T.; Xu, X.; Gao, F.; Liu, G.; Li, H. Functionalized Periodic Mesoporous Organosilica: A Highly Enantioselective Catalyst for the Michael Addition of 1,3-Dicarbonyl Compounds to Nitroalkenes. Chem. Eur. J. 2012, 18, 15546–15553. [Google Scholar] [CrossRef]
- Bissessar, D.; Achard, T.; Bellemin-Laponnaz, S. Robust and Recyclable Self-Supported Chiral Nickel Catalyst for the Enantioselective Michael Addition. Adv. Synth. Catal. 2016, 358, 1982–1988. [Google Scholar] [CrossRef]
- Poe, S.L.; Kobašlija, M.; McQuade, D.T. Mechanism and application of a microcapsule enabled multicatalyst reaction. J. Am. Chem. Soc. 2007, 129, 9216–9221. [Google Scholar] [CrossRef]
- Ishitani, H.; Kanai, K.; Yoo, W.J.; Yoshida, T.; Kobayashi, S. A Nickel-Diamine/Mesoporous Silica Composite as a Heterogeneous Chiral Catalyst for Asymmetric 1, 4-Addition Reactions. Angew. Chem. Int. Ed. 2019, 58, 13313–13317. [Google Scholar] [CrossRef]
- Buendia, M.B.; Kegnæs, S.; Kramera, S. A Nickel-Bisdiamine Porous Organic Polymer as Heterogeneous Chiral Catalyst for Asymmetric Michael Addition to Aliphatic Nitroalkenes. Adv. Synth. Catal. 2020, 362, 5506–5512. [Google Scholar] [CrossRef]
- Li, X.; Li, X.; Peng, F.; Shao, Z. Mutually Complementary Metal- and Organocatalysis with Collective Synthesis: Asymmetric Conjugate Addition of 1,3-Carbonyl Compounds to Nitroenynes and Further Reactions of the Products. Adv. Synth. Catal. 2012, 354, 2873–2885. [Google Scholar] [CrossRef]
- Matsunaga, S.; Shibasaki, M. Recent advances in cooperative bimetallic asymmetric catalysis: Dinuclear Schiff base complexes. Chem. Commun. 2014, 50, 1044–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furutachi, M.; Mouri, S.; Matsunaga, S.; Shibasaki, M. A Heterobimetallic Ni/La-salan Complex for Catalytic Asymmetric Decarboxylative 1,4-Addition of Malonic Acid Half-Thioester. Chem. Asian J. 2010, 5, 2351–2354. [Google Scholar] [CrossRef] [PubMed]
- Tsubogo, T.; Yamashita, Y.; Kobayashi, S. Toward efficient asymmetric carbon–carbon bond formation: Continuous flow with chiral heterogeneous catalysts. Chem. Eur. J. 2012, 18, 13624–13628. [Google Scholar] [CrossRef] [PubMed]
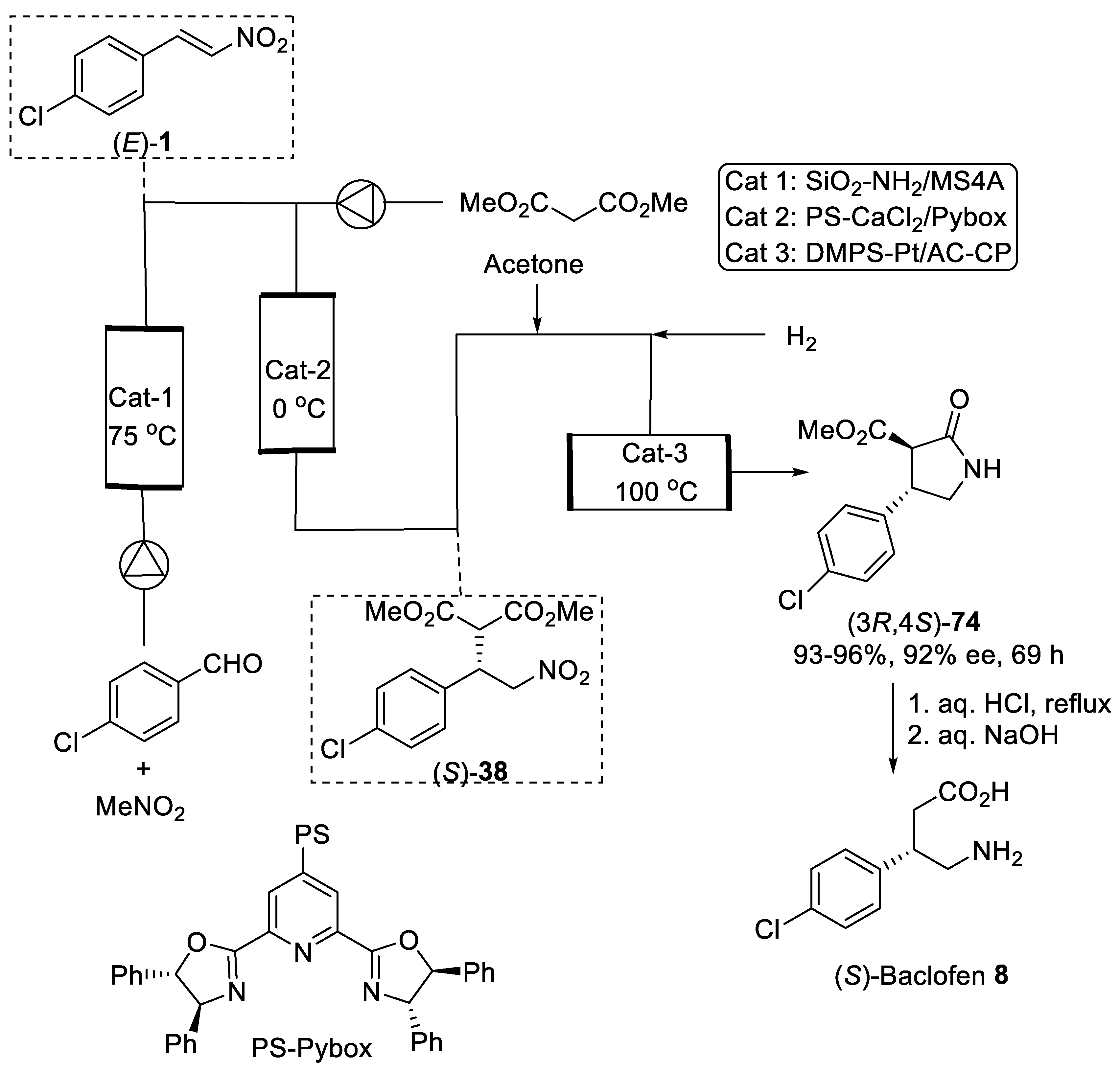
- Ishitani, H.; Furiya, Y.; Kobayashi, S. Enantioselective Sequential-Flow Synthesis of Baclofen Precursor via Asymmetric 1,4-Addition and Chemoselective Hydrogenation on Platinum/Carbon/Calcium Phosphate Composites. Chem. Asian. J. 2020, 15, 1688–1691. [Google Scholar] [CrossRef]
- Ishitani, H.; Saito, Y.; Tsubogo, T.; Kobayashi, S. Synthesis of Nitro-Containing Compounds through Multistep Continuous Flow with Heterogeneous Catalysts. Org. Lett. 2016, 18, 1346–1349. [Google Scholar] [CrossRef]
- Tsubogo, T.; Oyamada, H.; Kobayashi, S. Multistep continuous-flow synthesis of (R)-and (S)-rolipram using heterogeneous catalysts. Nature 2015, 520, 329–332. [Google Scholar] [CrossRef]
- Zadsirjan, V.; Heravi, M.M. Oxazolidinones as Chiral Auxiliaries in the Asymmetric 1,4-Conjugate Addition Reaction Applied to the Total Synthesis of Natural Products: A Supplemental Mini-Review. Curr. Org. Synth. 2018, 15, 3–20. [Google Scholar] [CrossRef]
- Tovar-Gudiño, E.; Morales-Nava, R.; Fernández-Zertuche, M. Diasteroselective conjugate addition of diethylaluminum cyanide to a conjugated N-enoyl system: An alternative synthesis of (S)-pregabalin. Can. J. Chem. 2014, 92, 45–48. [Google Scholar] [CrossRef]
- Armstrong, A.; Convine, N.J.; Popkin, M.E. Diastereoselective conjugate addition of cyanide to α, β-unsaturated oxazolidinones: Enantioselective synthesis of ent-pregabalin and baclofen. Synlett 2006, 2006, 1589–1591. [Google Scholar] [CrossRef]
- He, C.; Zhai, Z.; Zhou, Y.; Li, J.; Wang, G. A new synthetic route for the preparation of pregabalin. Synth. Commun. 2021, 51, 2034–2040. [Google Scholar] [CrossRef]
- Kuo, J.H.; Wong, W.C. Method for Preparing Chiral Baclofen. U.S. Patent 20,120,029,230, 25 September 2012. [Google Scholar]
- Izquierdo, S.; Aguilera, J.; Buschmann, H.H.; García, M.; Torrens, A.; Ortuño, R.M. Stereoselective and efficient synthesis of (S)-pregabalin from d-mannitol. Tetrahedron Asymmetry 2008, 19, 651–653. [Google Scholar] [CrossRef]
- Hayashi, Y. Domino and one-pot syntheses of biologically active compounds using diphenylprolinol silyl ether. Phys. Sci. Rev. 2020, 5, 20180088. [Google Scholar] [CrossRef]
- Donslund, B.S.; Johansen, T.K.; Poulsen, P.H.; Halskov, K.S.; Jørgensen, K.A. The diarylprolinol silyl ethers: Ten years after. Angew. Chem. Int. Ed. 2015, 54, 13860–13874. [Google Scholar] [CrossRef]
- Zu, L.; Xie, H.; Li, H.; Wang, J.; Wang, W. Highly enantioselective organocatalytic conjugate addition of nitromethane to α, β-unsaturated aldehydes: Three-step synthesis of optically active baclofen. Adv. Synth. Catal. 2007, 349, 2660–2664. [Google Scholar] [CrossRef]
- Wang, Y.; Li, P.; Liang, X.; Zhang, T.Y.; Ye, J. An efficient enantioselective method for asymmetric Michael addition of nitroalkanes to α, β-unsaturated aldehydes. Chem. Commun. 2008, 1232–1234. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Sakamoto, D.; Okamura, D. One-Pot Synthesis of (S)-Baclofen via Aldol Condensation of Acetaldehyde with Diphenylprolinol Silyl Ether Mediated Asymmetric Michael Reaction as a Key Step. Org. Lett. 2016, 18, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.L.; Poulsen, P.H.; Donslund, B.S.; Morana, F.; Jorgensen, K.A. Asymmetric synthesis of γ-nitroesters by an organocatalytic one-pot strategy. Org. Lett. 2012, 14, 1516–1519. [Google Scholar] [CrossRef]
- Ötvös, S.B.; Kappe, C.O. Continuous flow asymmetric synthesis of chiral active pharmaceutical ingredients and their advanced intermediates. Green Chem. 2021, 23, 6117–6138. [Google Scholar] [CrossRef]
- Jiao, J.; Nie, W.; Yu, T.; Yang, F.; Zhang, Q.; Aihemaiti, F.; Yang, T.; Liu, X.; Wang, J.; Li, P. Multi-Step Continuous-Flow Organic Synthesis: Opportunities and Challenges. Chem. Eur. J. 2021, 27, 4817–4838. [Google Scholar] [CrossRef]
- Bloemendal, V.R.L.J.; Janssen, M.A.C.H.; van Hest, J.C.M.; Rutjes, F.P.J.T. Continuous one-flow multi-step synthesis of active pharmaceutical ingredients. React. Chem. Eng. 2020, 5, 1186–1197. [Google Scholar] [CrossRef]
- Nagy, B.S.; Llanes, P.; Pericas, M.A.; Kappe, O.; Ötvös, S.B. Enantioselective Flow Synthesis of Rolipram Enabled by a Telescoped Asymmetric Conjugate Addition–Oxidative Aldehyde Esterification Sequence Using in Situ-Generated Persulfuric Acid as Oxidant. Org. Lett. 2022, 24, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Ötvös, S.B.; Llanes, P.; Pericás, M.A.; Kappe, O. Telescoped continuous flow synthesis of optically active γ-nitrobutyric acids as key intermediates of baclofen, phenibut, and fluorophenibut. Org. Lett. 2020, 22, 8122–8126. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.T.; Shen, J.; Sha, F.; Wu, X.Y. Highly enantioselective Michael addition of nitroalkanes to enones and its application in syntheses of (R)-Baclofen and (R)-Phenibut. Synthesis 2015, 47, 2063–2072. [Google Scholar] [CrossRef]
- Ogawa, T.; Mouri, S.; Yazaki, R.; Kumagai, N.; Shibasaki, M. Intermediate as catalyst: Catalytic asymmetric conjugate addition of nitroalkanes to α, β-unsaturated thioamides. Org. Lett. 2012, 14, 110–113. [Google Scholar] [CrossRef]
- Shao, C.; Yu, H.J.; Wu, N.Y.; Tian, P.; Wang, R.; Feng, C.G.; Lin, G.Q. Asymmetric Synthesis of β-Substituted γ-Lactams via Rhodium/Diene-Catalyzed 1,4-Additions: Application to the Synthesis of (R)-Baclofen and (R)-Rolipram. Org. Lett. 2011, 13, 788–791. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Feng, C.G.; Zhang, S.S.; Xu, M.H.; Lin, G.Q. Rhodium-Catalyzed Asymmetric Conjugate Addition of Organoboronic Acids to Nitroalkenes Using Chiral Bicyclo [3.3.0] Diene Ligands. Angew. Chem. Int. Ed. 2010, 49, 5780–5783. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Feng, C.G.; Xu, M.H.; Lin, G.Q. Design of C2-Symmetric Tetrahydropentalenes as New Chiral Diene Ligands for Highly Enantioselective Rh-Catalyzed Arylation of N-Tosylarylimines with Arylboronic Acids. J. Am. Chem. Soc. 2007, 129, 5336–5337. [Google Scholar] [CrossRef]
- Han, F.; Chen, J.; Zhang, X.; Liu, J.; Cun, L.; Zhu, J.; Deng, J.; Liao, J. Rhodium-catalyzed asymmetric addition of arylboronic acids to γ-phthalimido-substituted-α, β-unsaturated carboxylic acid esters: An approach to γ-amino acids. Tetrahedron Lett. 2011, 52, 830–833. [Google Scholar] [CrossRef]
- Chen, J.; Chen, J.; Lang, F.; Zhang, X.; Cun, L.; Zhu, J.; Deng, J.; Liao, J. A C2-Symmetric Chiral Bis-Sulfoxide Ligand in a Rhodium-Catalyzed Reaction: Asymmetric 1,4-Addition of Sodium Tetraarylborates to Chromenones. J. Am. Chem. Soc. 2010, 132, 4552–4553. [Google Scholar] [CrossRef]
- Becht, J.M.; Meyer, O.; Helmchen, G. Enantioselective syntheses of (-)-(R)-rolipram,(-)-(R)-baclofen and other GABA analogues via rhodium-catalyzed conjugate addition of arylboronic acids. Synthesis 2003, 18, 2805–2810. [Google Scholar] [CrossRef]
- Diaz, A.; Siro, J.G.; García-Navío, J.L.; Vaquero, J.J.; Alvarez-Builla, J. A stereoselective synthesis of (R)-(-)-rolipram from L-glutamic acid. Synthesis 1997, 1997, 559–562. [Google Scholar] [CrossRef] [Green Version]
- Oba, M.; Saegusa, T.; Nishiyama, N.; Nishiyama, K. Synthesis of non-proteinogenic amino acids using Michael addition to unsaturated orthopyroglutamate derivative. Tetrahedron 2009, 65, 128–133. [Google Scholar] [CrossRef]
- Chang, M.Y.; Sun, P.P.; Chena, S.T.; Chang, N.C. A facile synthesis of 3-aryl pyroglutamic acid. Facile synthesis of baclofen and chlorpheg. Tetrahedron Lett. 2003, 44, 5271–5273. [Google Scholar] [CrossRef]
- Meyers, A.I.; Snyder, L. The synthesis of aracemic 4-substituted pyrrolidinones and 3-substituted pyrrolidines. An asymmetric synthesis of (-)-rolipram. J. Org. Chem. 1993, 58, 36–42. [Google Scholar] [CrossRef]
- Meyers, A.I.; Snyder, L. Asymmetric conjugate additions to chiral bicyclic lactams. Synthesis of aracemic trans-2,3-disubstituted pyrrolidines. J. Org. Chem. 1992, 57, 3814–3819. [Google Scholar] [CrossRef]
- Langlois, N.; Wang, H.S. Synthesis of the Novel Antidepressant (R)-(-)-Rolipram. Synth. Commun. 1997, 27, 3133–3144. [Google Scholar] [CrossRef]
- Momioka, K.; Sudani, M.; Shinmi, Y.; Koga, K. Enantioselective conjugate addition reaction mediated by chiral ligands. Chem. Lett. 1985, 14, 329–332. [Google Scholar]
- Massolo, E.; Benaglia, M.; Genoni, A.; Annunziata, R.; Celentano, G.; Gaggero, N. Stereoselective reaction of 2-carboxythioesters-1,3-dithiane with nitroalkenes: An organocatalytic strategy for the asymmetric addition of a glyoxylate anion equivalent. Org. Biomol. Chem. 2015, 13, 5591–5596. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, S.; Tokunaga, A.; Saito, H.; Kondo, M. Enantioselective conjugate addition of an α, α-dithioacetonitrile with nitroalkenes using chiral bis (imidazoline)–Pd complexes. Chem. Commun. 2019, 55, 5391–5394. [Google Scholar] [CrossRef]
- Kondo, M.; Saito, H.; Nakamura, S. Direct catalytic enantioselective Mannich-type reaction of α, α-dithioacetonitriles with imines using chiral bis (imidazoline)–Pd complexes. Chem. Commun. 2017, 53, 6776–6779. [Google Scholar] [CrossRef]
- Baures, P.W.; Eggleston, D.S.; Erhard, K.F.; Cieslinski, L.B.; Torphy, T.J.; Christensen, S.B. Crystal structure, absolute configuration, and phosphodiesterase inhibitory activity of (+)-1-(4-bromobenzyl)-4-[(3-cyclopentyloxy)-4-methoxyphenyl]-2-pyrrolidinone. J. Med. Chem. 1993, 36, 3274–3277. [Google Scholar] [CrossRef] [PubMed]
- Pirnot, M.T.; Rankic, D.A.; Martin, D.B.C.; MacMillan, D.W.C. Photoredox activation for the direct β-arylation of ketones and aldehydes. Science 2013, 339, 1593–1596. [Google Scholar] [CrossRef]
- Fu, Z.; Xu, J.; Zhu, T.; Leong, W.W.Y.; Chi, Y.R. β-Carbon activation of saturated carboxylic esters through N-heterocyclic carbene organocatalysis. Nat. Chem. 2013, 5, 835–839. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhou, Q.; Song, G.; Song, Y.; Zhao, G.; Ding, K.; Zhao, B. Enantioselective synthesis of pyroglutamic acid esters from glycinate via carbonyl catalysis. Angew. Chem. Int. Ed. 2021, 60, 10588–10592. [Google Scholar] [CrossRef]
- Sorochinsky, A.E.; Aceña, J.L.; Soloshonok, V.A. Self-Disproportionation of Enantiomers of Chiral, Non-Racemic Fluoroorganic Compounds: Role of Fluorine as Enabling Element. Synthesis 2013, 45, 141–152. [Google Scholar] [CrossRef]
- Sorochinsky, A.E.; Katagiri, T.; Ono, T.; Wzorek, A.; Aceña, J.L.; Soloshonok, V.A. Optical purifications via Self-Disproportionation of Enantiomers by achiral chromatography; Case study of a series of α-CF3-containing secondary alcohols. Chirality 2013, 25, 365–368. [Google Scholar] [CrossRef]
- Nakamura, T.; Tateishi, K.; Tsukagoshi, S.; Hashimoto, S.; Watanabe, S.; Soloshonok, V.A.; Aceña, J.L.; Kitagawa, O. Self-Disproportionation of Enantiomers of Non-racemic Chiral Amine Derivatives Through Achiral Chromatography. Tetrahedron 2012, 68, 4013–4017. [Google Scholar] [CrossRef]
- Suzuki, Y.; Han, J.; Kitagawa, O.; Aceña, J.L.; Klika, K.D.; Soloshonok, V.A. A comprehensive examination of the self-disproportionation of enantiomers (SDE) of chiral amides via achiral, laboratory-routine, gravity-driven column chromatography. RSC Adv. 2015, 5, 2988–2993. [Google Scholar] [CrossRef]
- Han, J.; Wzorek, A.; Kwiatkowska, M.; Soloshonok, V.A.; Klika, K.D. The self-disproportionation of enantiomers (SDE) of amino acids and their derivatives. Amino Acids 2019, 51, 865–889. [Google Scholar] [CrossRef]
- Hosaka, T.; Imai, T.; Wzorek, A.; Marcinkowska, M.; Kolbus, A.; Kitagawa, O.; Soloshonok, V.A.; Klika, K.D. The self-disproportionation of enantiomers (SDE) of α-amino acid derivatives; facets of steric and electronic properties. Amino Acids 2019, 51, 283–294. [Google Scholar] [CrossRef]
- Wzorek, A.; Sato, A.; Drabowicz, J.; Soloshonok, V.A.; Klika, K.D. Remarkable magnitude of the self-disproportionation of enantiomers (SDE) via achiral chromatography; application to the practical-scale enantiopurification of β-amino acid esters. Amino Acids 2016, 48, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Kitagawa, O.; Wzorek, A.; Klika, K.D.; Soloshonok, V.A. The self-disproportionation of enantiomers (SDE): A menace or an opportunity? Chem. Sci. 2018, 9, 1718–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soloshonok, V.A.; Wzorek, A.; Klika, K.D. A question of policy: Should tests for the self-disproportionation of enantiomers (SDE) be mandatory for reports involving scalemates? Tetrahedron Asymmetry 2017, 28, 1430–1434. [Google Scholar] [CrossRef]
- Han, J.; Nelson, D.J.; Sorochinsky, A.E.; Soloshonok, V.A. Self-Disproportionation of Enantiomers via Sublimation; New and Truly Green Dimension in Optical Purification. Curr. Org. Synth. 2011, 8, 310–317. [Google Scholar] [CrossRef]
- Han, J.; Wzorek, A.; Klika, K.D.; Soloshonok, V.A. Recommended Tests for the Self-Disproportionation of Enantiomers (SDE) to Ensure Accurate Reporting of the Stereochemical Outcome of Enantioselective Reactions. Molecules 2021, 26, 2757. [Google Scholar] [CrossRef]
- Han, J.; Dembinski, R.; Soloshonok, V.A.; Klika, K.D. A Call for a Change in Policy Regarding the Necessity for SDE Tests to Validate the Veracity of the Outcome of Enantioselective Syntheses, the Inherent Chiral State of Natural Products, and Other Cases Involving Enantioenriched Samples. Molecules 2021, 26, 3994. [Google Scholar] [CrossRef]
- Chi, Y.; Guo, L.; Kopf, N.A.; Gellman, S.H. Enantioselective Organocatalytic Michael Addition of Aldehydes to Nitroethylene: Efficient Access to γ2-Amino Acids. J. Am. Chem. Soc. 2008, 130, 5608–5609. [Google Scholar] [CrossRef] [Green Version]
- Ye, W.; Jiang, Z.; Zhao, Y.; Goh, S.L.M.; Leow, D.; Soh, Y.T.; Tan, C.H. Chiral Bicyclic Guanidine as a Versatile Brønsted Base Catalystfor the Enantioselective Michael Reactions of Dithiomalonatesandb-Ket oThi oesters. Adv. Synth. Catal. 2007, 349, 2454–2458. [Google Scholar] [CrossRef]



















































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, J.; Escorihuela, J.; Fustero, S.; Landa, A.; Soloshonok, V.A.; Sorochinsky, A. Asymmetric Michael Addition in Synthesis of β-Substituted GABA Derivatives. Molecules 2022, 27, 3797. https://doi.org/10.3390/molecules27123797
Han J, Escorihuela J, Fustero S, Landa A, Soloshonok VA, Sorochinsky A. Asymmetric Michael Addition in Synthesis of β-Substituted GABA Derivatives. Molecules. 2022; 27(12):3797. https://doi.org/10.3390/molecules27123797
Chicago/Turabian StyleHan, Jianlin, Jorge Escorihuela, Santos Fustero, Aitor Landa, Vadim A. Soloshonok, and Alexander Sorochinsky. 2022. "Asymmetric Michael Addition in Synthesis of β-Substituted GABA Derivatives" Molecules 27, no. 12: 3797. https://doi.org/10.3390/molecules27123797