
Current Status of the X + C2H6 [X ≡ H, F(2P), Cl(2P), O(3P), OH] Hydrogen Abstraction Reactions: A Theoretical Review


Abstract
:1. Introduction
2. Theory/Experiment Comparison
2.1. An Update of the H + CH4 Reaction
2.2. The F(2P) + NH3 Reaction: Does a Huge Number of Points Guarantee the Accuracy of the Final Kinetics Results?
3. Analysis of Medium-Size Systems
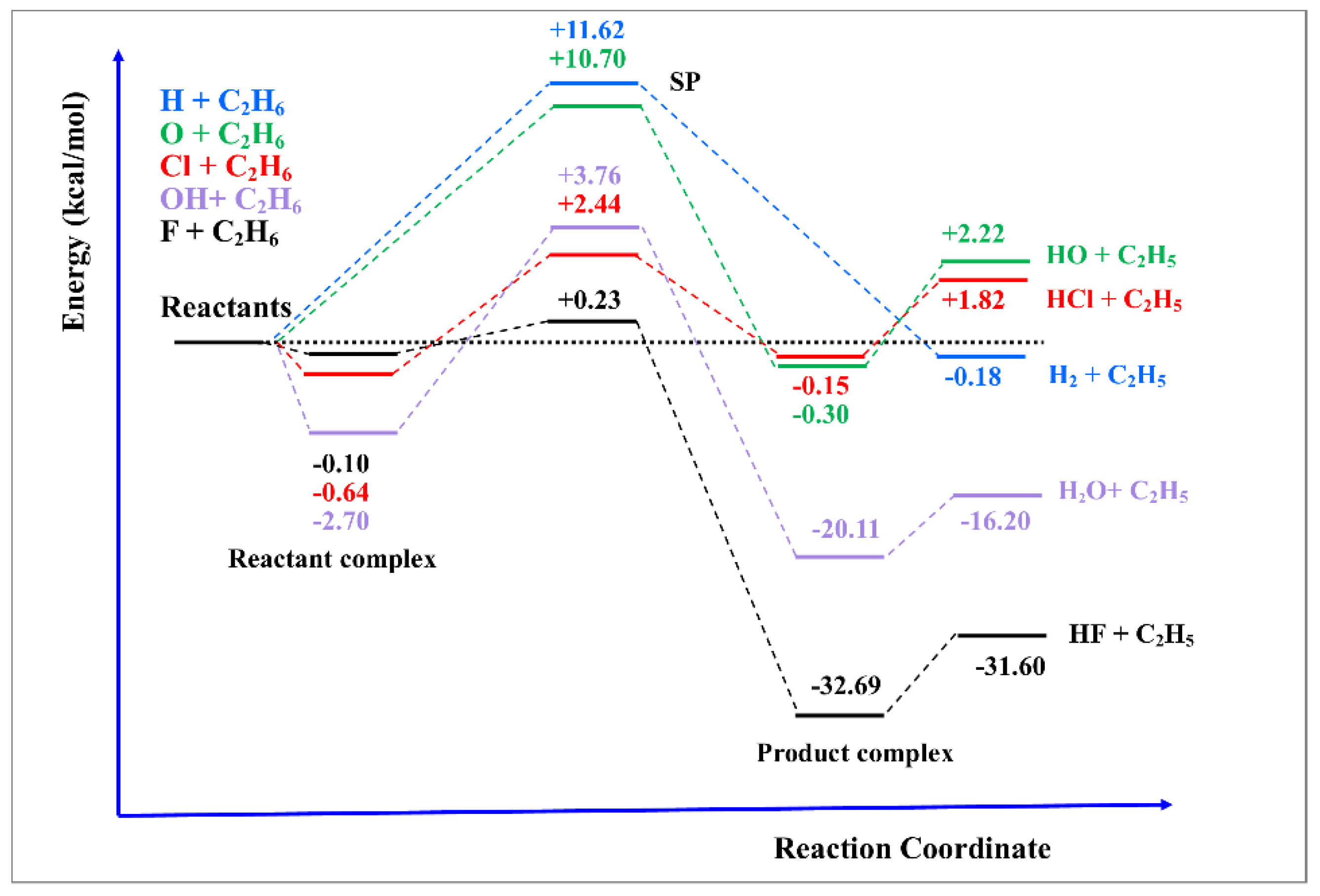
3.1. The Development of Potential Energy Surfaces
3.2. The Theoretical Tools: A Compromise between Accuracy and Capacity
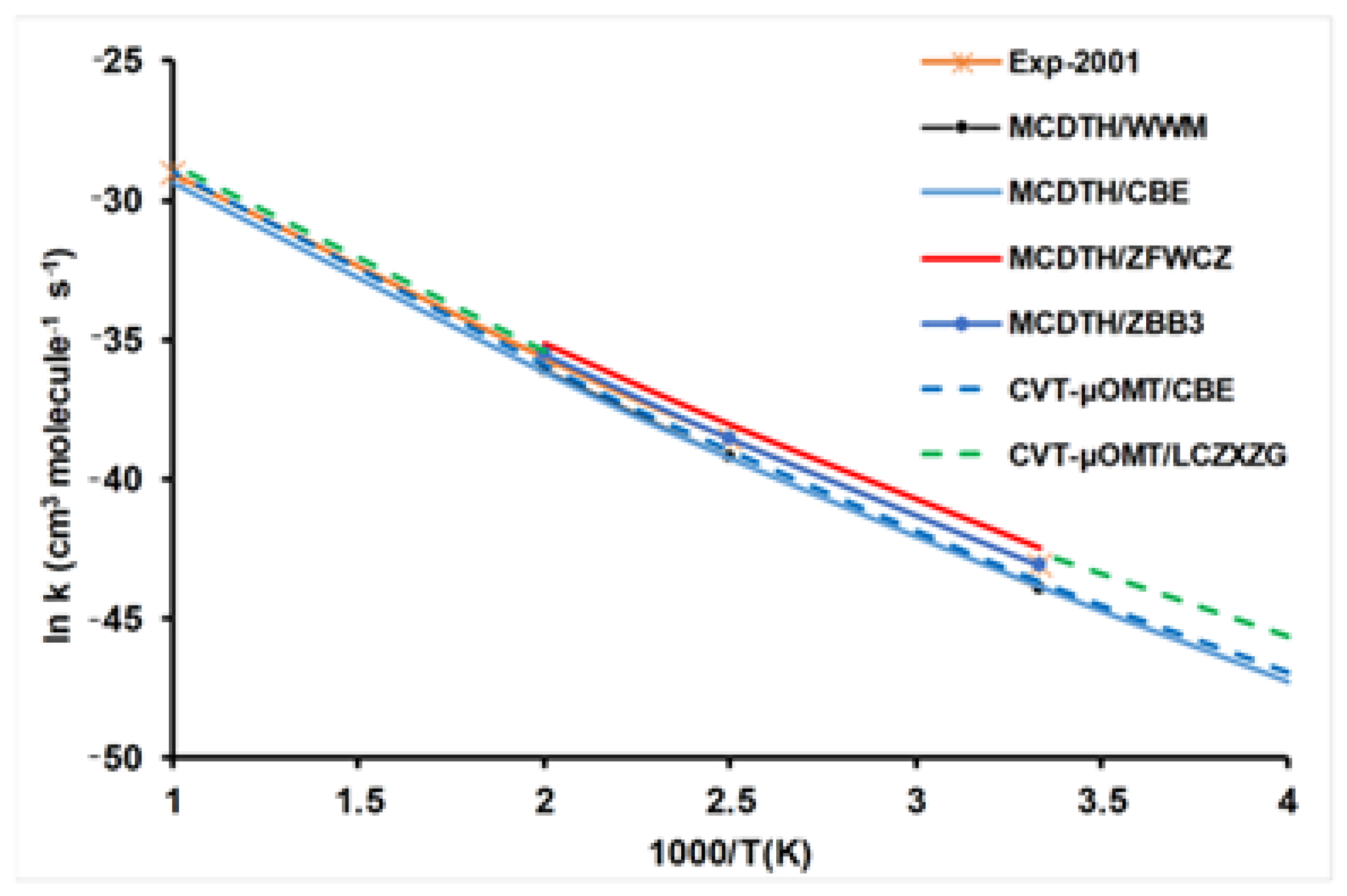
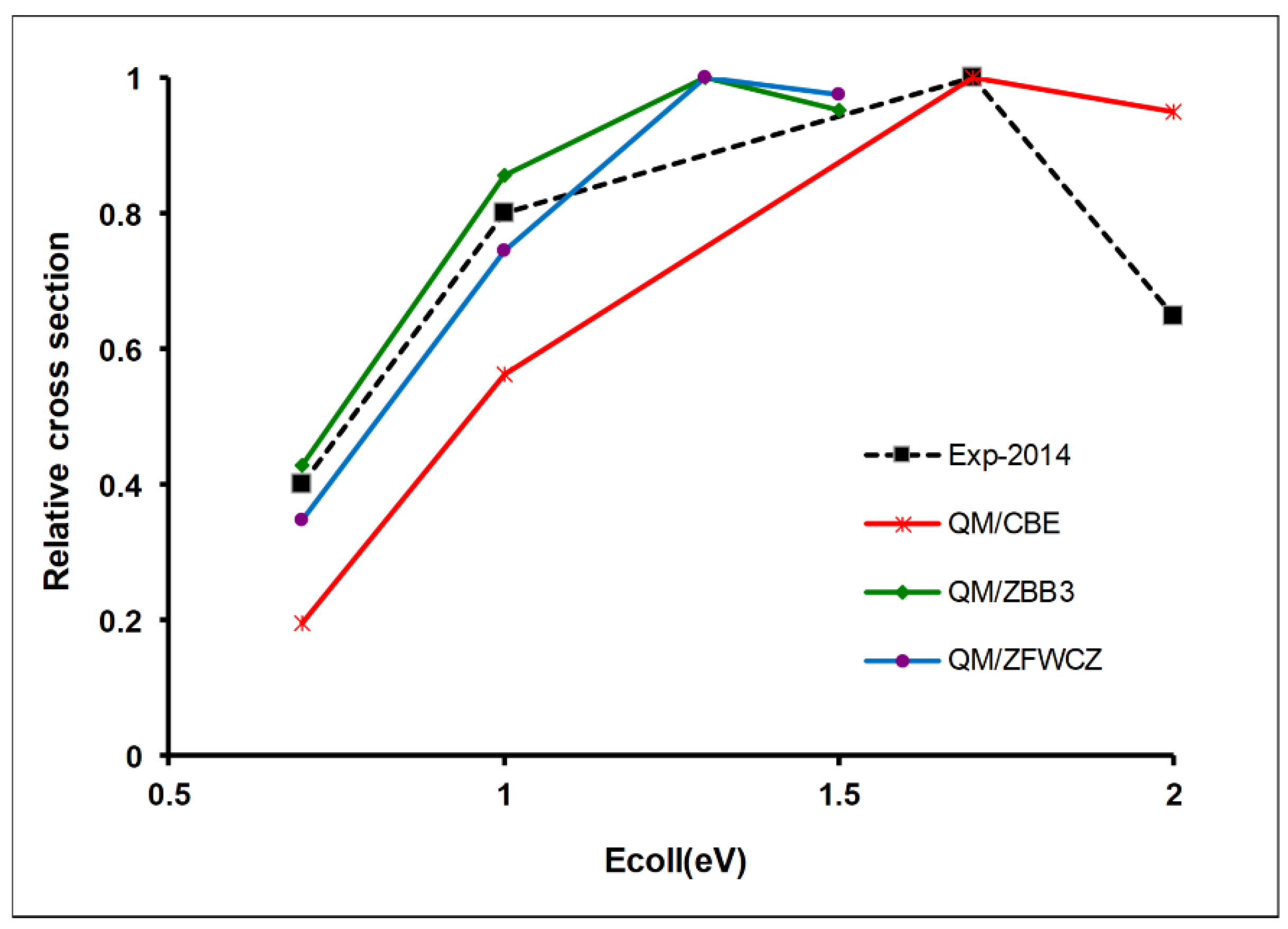
3.3. Theoretical Calculations Simulate Kinetics Experiments: The H + C2H6 Reaction
3.4. The Theoretical Dynamics Description Worsens the Agreement with Experiments
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Eyring, H.; Polanyi, M. On Simple Gas Reactions. Z. Physik. Chem. 1931, B12, 279–311. [Google Scholar]
- London, F.Z. Quantenmechanische deutung des vorgangs der aktivierung. Elektrochemie 1929, 35, 552. [Google Scholar]
- Sato, S. A New Method of Drawing the Potential Energy Surface. Bull. Chem. Soc. Jpn. 1955, 28, 450–453. [Google Scholar] [CrossRef]
- Wang, S.; Yuan, J.; Li, H.; Chen, M. A Neural Network Potential Energy Surface for the NaH2 System and Dynamics Studies on the H(2S) + NaH(X1Σ+) → Na(2S) + H2(X1Σg+) Reaction. Phys. Chem. Chem. Phys. 2017, 19, 19873–19880. [Google Scholar] [CrossRef]
- Yuan, J.; Duan, Z.; Wang, S.; Liu, J.; Han, K. Significant Effects of Vibrational Excitation of Reactant in K + H2 → H + KH Reaction based on a New Neural Network Potential Energy Surface. Phys. Chem. Chem. Phys. 2018, 20, 20641–20649. [Google Scholar] [CrossRef]
- Liu, X.; Xie, C.; Guo, H. A New Potential Energy Surface and State-to-State Quantum Dynamics of the Li + HF → H + LiF Reaction. Chem. Phys. 2018, 509, 66–71. [Google Scholar] [CrossRef]
- Cao, J.; Wu, Y.; Ma, H.; Shen, Z.; Bian, W. Dynamics and Kinetics of the Si(1D) + H2/D2 Reactions on a New Global Ab initio Potential Energy Surface. Phys. Chem. Chem. Phys. 2021, 23, 6141–6153. [Google Scholar] [CrossRef] [PubMed]
- Jankuna, J.; Sneha, M.; Zare, R.N.; Bonakline, F.; Althorpe, S.C.; Herraez-Aguilar, D.; Aoiz, F.J. Is the Simplest Chemical Reaction Really so Simple? Proc. Natl. Acad. Sci. USA 2014, 111, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Polanyi, J.C. Concepts in Reaction Dynamics. Acc. Chem. Res. 1972, 5, 161–168. [Google Scholar] [CrossRef]
- Elkowitz, A.B.; Wyat, R.E. Quantum Mechanical Reaction Cross Sections for the Three-dimensional Hydrogen Exchange Reaction. J. Chem. Phys. 1975, 62, 2504–2510. [Google Scholar] [CrossRef]
- Schatz, G.C.; Kupperman, A. Quantum Mechanical Reactive Scattering for Three-dimensional Atom Plus Diatom Systems. I. Theory. J. Chem. Phys. 1976, 65, 4642–4649. [Google Scholar] [CrossRef]
- Walker, R.B.; Stechel, E.B.; Light, J.C. Accurate H3 Dynamics on an Accurate H3 Potential Surface. J. Chem. Phys. 1978, 69, 2922. [Google Scholar] [CrossRef]
- Albu, T.; Espinosa-Garcia, J.; Truhlar, D.G. Computational Chemistry of Polyatomic Reaction Kinetics and Dynamics: The Quest for an Accurate CH5 Potential Energy Surface. Chem. Rev. 2007, 107, 5101–5132. [Google Scholar] [CrossRef] [PubMed]
- Bechtel, H.A.; Camden, J.P.; Brown, D.J.A.; Zare, R.N. Comparing the Dynamical Effects of Symmetric and Antisymmetric Stretch Excitation of Methane in the Cl + CH4 Reaction. J. Chem. Phys. 2004, 120, 5096, And References Therein. [Google Scholar] [CrossRef] [PubMed]
- Kim, Z.H.; Bechtel, H.A.; Camden, J.P.; Zare, R.N. Effect of Bending and Torsional Mode Excitation on the Reaction Cl + CH4 → HCl + CH3. J. Chem. Phys. 2005, 122, 084303, And References Therein. [Google Scholar] [CrossRef]
- Camden, J.P.; Bechtel, H.A.; Brown, D.J.A.; Zare, R.N. Effects of C-H Stretch Excitation on the H + CH4 Reaction. J. Chem. Phys. 2005, 123, 134301, And References Therein. [Google Scholar] [CrossRef] [PubMed]
- Camden, J.P.; Bechtel, H.A.; Brown, D.J.A.; Zare, R.N. Comparing Reactions of H and Cl with C–H Stretch-excited CHD3. J. Chem. Phys. 2006, 124, 034311. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Henton, S.; Zirkovic, A.N.; Crim, F.F. The Relative Reactivity of the Stretch–Bend Combination Vibrations of CH4 in the Cl (2P3/2) + CH4 Reaction. J. Chem. Phys. 2002, 116, 10744–10752. [Google Scholar] [CrossRef]
- Yoon, S.; Holiday, R.J.; Crim, F.F. Control of Bimolecular Reactions: Bond-selected Reaction of Vibrationally Excited CH3D with Cl(2P3/2). J. Chem. Phys. 2003, 119, 4755–4761. [Google Scholar] [CrossRef]
- Yoon, S.; Holiday, R.J.; Silbert III, E.L.; Crim, F.F. The Relative Reactivity of CH3D Molecules with Excited Symmetric and Antisymmetric Stretching Vibrations. J. Chem. Phys. 2003, 119, 9568–9576. [Google Scholar] [CrossRef]
- Fu, B.; Shan, X.; Zhang, D.H.; Clary, D.C. Recent Advances in Quantum Scattering Calculations on Polyatomic Bimolecular Reactions. Chem. Soc. Rev. 2017, 46, 7625–7649. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Liu, K. Control of Chemical Reactivity by Transition State and Beyond. Chem. Sci. 2016, 7, 3992–4003. [Google Scholar] [CrossRef]
- Nyman, G.; Yu, H.-G. Quantum Theory of Bimolecular Chemical Reactions. Rep. Prog. Phys. 2000, 63, 1001–1059. [Google Scholar] [CrossRef]
- Nyman, G.; Yu, H.-G. Quantum Approaches to Polyatomic Reaction Dynamics. Int. Rev. Phys. Chem. 2013, 32, 39–95. [Google Scholar] [CrossRef]
- Gorin, E.; Kauzmann, W.; Walter, J.; Eyring, H. Reactions Involving Hydrogen and the Hydrocarbons. J. Chem. Phys. 1939, 7, 633–640. [Google Scholar] [CrossRef]
- Bowman, J.M.; Czako, G.; Fu, B. High-dimensional Ab initio Potential Energy Surfaces for Reaction Dynamics Calculations. Phys. Chem. Chem. Phys. 2011, 13, 8094–8111. [Google Scholar] [CrossRef]
- Chakraborty, A.; Zhao, Y.; Lin, H.; Truhlar, D.G. Combined Valence Bond-Molecular Mechanics Potential-Energy Surface and Direct Dynamics Study of Rate Constants and Kinetic Isotope Effects for the H + C2H6 Reaction. J. Chem. Phys. 2006, 124, 044315. [Google Scholar] [CrossRef]
- Wu, T.; Werner, H.-J.; Manthe, U. First-Principles Theory for the H + CH4 → H2 + CH3 Reaction. Science 2004, 306, 2227–2229. [Google Scholar] [CrossRef]
- Zhang, X.; Braams, B.J.; Bowman, J.M. An Ab initio Potential Surface Describing Abstraction and Exchange for H + CH4. J. Chem. Phys. 2006, 124, 021104. [Google Scholar] [CrossRef]
- Corchado, J.C.; Bravo, J.L.; Espinosa-Garcia, J. The Hydrogen Abstraction Reaction H + CH4. I. New Analytical Potential Energy Surface based on Fitting to Ab initio Calculations. J. Chem. Phys. 2009, 130, 184314. [Google Scholar] [CrossRef]
- Zhou, Y.; Fu, B.; Wang, C.; Collins, M.A.; Zhang, D.H. Ab initio Potential Energy Surface and Quantum Dynamics for the H + CH4 → H2 + CH3 Reaction. J. Chem. Phys. 2011, 134, 064323. [Google Scholar] [CrossRef]
- Xu, X.; Chen, J.; Zhang, D.H. Global Potential Energy Surface for the H + CH4 → H2 + CH3 Reaction using Neural Networks. Chin. J. Chem. Phys. 2014, 27, 373–379. [Google Scholar] [CrossRef]
- Li, J.; Chen, J.; Zhao, Z.; Xie, D.; Zhang, D.H.; Guo, H. A Permutationally Invariant Full-dimensional Ab initio Potential Energy Surface for the Abstraction and Exchange Channels of the H + CH4 System. J. Chem. Phys. 2015, 142, 204302. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Werner, H.-J.; Manthe, U. Accurate Potential Energy Surface and Quantum Reaction Rate Calculations for the H+CH4→H2+CH3 Reaction. J. Chem. Phys. 2006, 124, 164307. [Google Scholar] [CrossRef]
- Sutherland, J.W.; Su, M.C.; Michael, J.V. Rate Constants for H + CH4, CH3 + H2, and CH4 Dissociation at High Temperature. Int. J. Chem. Kinet. 2001, 33, 669–684. [Google Scholar] [CrossRef]
- Pan, H.; Yang, J.; Shuai, Q.; Zhang, D.; Zhang, W.; Wu, G.; Dai, D.; Jiang, B.; Zhang, D.H.; Yang, X. Velocity Map Imaging Study of the Reaction Dynamics of the H + CH4 → H2 + CH3 Reaction: The Isotope Effects. J. Phys. Chem. A 2014, 118, 2426–2430. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Garcia, J.; Corchado, J.C. On the Energy-dependence of the Excitation Functions of the H + CH4 and H + CD4 Reactions. Comput. Theor. Chem. 2015, 1069, 1–3. [Google Scholar] [CrossRef]
- Walther, C.D.; Wagner, H.G. Uber die Reaktionen von F Atomen mit H2O, H2O2 und NH3. Ber. Bunsenges. Phys. Chem. 1983, 87, 403–409. [Google Scholar] [CrossRef]
- Persky, A. The Rate Constant of the F + NH3 Reaction: Inverse Temperature Dependence. Chem. Phys. Lett. 2007, 439, 3–7. [Google Scholar] [CrossRef]
- Goddard, J.D.; Donaldson, D.J.; Sloan, J.J. Hydrogen Bonded Complexes and the HF Vibrational Energy Distributions from the Reaction of F-Atoms with NH2 and NH3. Chem. Phys. 1987, 114, 321–329. [Google Scholar] [CrossRef]
- Leroy, G.; Sana, M.; Tinant, A. Etude Theorique des Reactions d’Abstractions d’Hydrogene RH + X = R + HX avec R = H, CH3, NH2, OH et F. Can. J. Chem. 1985, 63, 1447–1456. [Google Scholar] [CrossRef]
- Espinosa-Garcia, J.; Corchado, J.C. Analytical Surface for the Reaction with No Saddle-Point NH3 + F →NH2 + FH. Application of Variational Transition State Theory. J. Phys. Chem. A 1997, 101, 7336–7344. [Google Scholar] [CrossRef]
- Xiao, G.; Shen, G.; Wang, X.; Fan, H.; Yang, X. Crossed Beams Study on the Dynamics of the F-Atom Reaction with Ammonia. J. Phys. Chem. A 2010, 114, 4520–4523. [Google Scholar] [CrossRef]
- Feng, H.; Sun, W.; Xie, Y.; Schaefer, H.F. Is There an Entrance Complex for the F + NH3 Reaction? Chem. Asian J. 2011, 6, 3152–3156. [Google Scholar] [CrossRef]
- Espinosa-Garcia, J.; Monge-Palacios, M. Theoretical Study of the F + NH3 and F + ND3 Reactions: Mechanism and Comparison with Experiment. J. Phys. Chem. A 2011, 115, 13759–13763. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Garcia, J.; Fernandez-Ramos, A.; Suleimanov, Y.V.; Corchado, J.C. Theoretical Kinetics Study of the F(2P) + NH3 Hydrogen Abstraction Reaction. J. Phys. Chem. A 2014, 118, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Zhu, Y.; Song, H.; Yang, M. Theoretical Study of the F(2P) + NH3 → HF + NH2 Reaction on an Accurate Potential Energy Surface: Dynamics and Kinetics. Phys. Chem. Chem. Phys. 2019, 21, 11385–11394. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Mavri, J.; Warshel, A. Transition State Theory can be used in Studies of Enzyme Catalysis: Lessons from Simulations of Tunnelling and Dynamical Effects in Lipoxygenase and other Systems. Philos. Trans. R. Soc. B 2006, 361, 1417–1432. [Google Scholar] [CrossRef]
- Braams, B.J.; Bowman, J.M. Permutationally Invariant Potential Energy Surfaces in High Dimensionality. Int. Rev. Phys. Chem. 2009, 28, 577–606. [Google Scholar] [CrossRef]
- Czako, G.; Bowman, J.M. Reaction Dynamics of Methane with F, O, Cl, and Br on ab initio Potential Energy Surfaces. J. Phys. Chem. A 2014, 118, 2839–2864. [Google Scholar] [CrossRef] [PubMed]
- Qu, C.; Yu, Q.; Bowman, J.M. Permutationally Invariant Potential Energy Surfaces. Annu. Rev. Phys. Chem. 2018, 69, 151–175. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Li, J.; Guo, H. Potential Energy Surfaces from High Fidelity Fitting of ab initio Points: The Permutation Invariant Polynomial—Neural Network Approach. Int. Rev. Phys. Chem. 2016, 35, 479–506. [Google Scholar] [CrossRef]
- Shao, K.; Chen, J.; Zhou, Z.; Zhang, D.H. Communication: Fitting Potential Energy Surfaces with Fundamental Invariant Neural Network. J. Chem. Phys. 2016, 145, 071101. [Google Scholar] [CrossRef] [PubMed]
- Fu, B.; Zhang, D.H. Ab initio Potential Energy Surfaces and Quantum Dynamics for Polyatomic Bimolecular Reactions. J. Chem. Theory Comput. 2018, 14, 2289–2303. [Google Scholar] [CrossRef] [PubMed]
- Behler, J. First Principles Neural Network Potentials for Reactive Simulations of Large Molecular and Condensed Systems. Angew. Chem. Int. Ed. 2017, 56, 12828–12840. [Google Scholar] [CrossRef]
- Bartók, A.P.; Kondor, R.; Csányi, G. On Representing Chemical Environments. Phys. Rev. B 2013, 87, 184115. [Google Scholar] [CrossRef]
- Papp, D.; Gruber, B.; Czakó, G. Detailed Benchmark ab initio Mapping of the Potential Energy Surfaces of the X + C2H6 [X = F, Cl, Br, I] Reactions. Phys. Chem. Chem. Phys. 2019, 21, 396. [Google Scholar] [CrossRef]
- Czakó, G.; Győri, T.; Olasz, B.; Papp, D.; Szabó, I.; Tajti, V.; Tasi, D.A. Benchmark ab initio and Dynamical Characterization of the Stationary Points of Reactive Atom + Alkane and SN2 Potential Energy Surfaces. Phys. Chem. Chem. Phys. 2020, 22, 4298. [Google Scholar] [CrossRef]
- Papp, D.; Tajti, V.; Győri, T.; Czakó, G. Theory Finally Agrees with Experiment for the Dynamics of the Cl + C2H6 Reaction. J. Phys. Chem. Lett. 2020, 11, 4762. [Google Scholar] [CrossRef]
- Gruber, B.; Czakó, G. Benchmark ab initio Characterization of the Abstraction and Substitution Pathways of the OH + CH4/C2H6 Reactions. Phys. Chem. Chem. Phys. 2020, 22, 14560. [Google Scholar] [CrossRef]
- Papp, D.; Czakó, G. Full-dimensional MRCI-F12 Potential Energy Surface and Dynamics of the F(2P3/2) + C2H6 → HF + C2H5 Reaction. J. Chem. Phys. 2020, 153, 064305. [Google Scholar] [CrossRef] [PubMed]
- Czakó, G.; Győri, T.; Papp, D.; Tajti, V.; Tasi, D.A. First-principles Reaction Dynamics Beyond Six-atom Systems. J. Phys. Chem. A 2021, 125, 2385. [Google Scholar] [CrossRef] [PubMed]
- Papp, D.; Li, J.; Guo, H.; Czakó, G. Vibrational Mode-specificity in the Dynamics of the Cl + C2H6 → HCl + C2H5 Reaction. J. Chem. Phys. 2021, 155, 114303. [Google Scholar] [CrossRef] [PubMed]
- Papp, D.; Czakó, G. Vibrational Mode-specific Dynamics of the F(2P3/2) + C2H6 → HF + C2H5 Reaction. J. Chem. Phys. 2021, 155, 154302. [Google Scholar] [CrossRef]
- Rangel, C.; Espinosa-Garcia, J. Full-dimensional Analytical Potential Energy Surface Describing the Gas-phase Cl + C2H6 Reaction and Kinetics Study of Rate Constants and Kinetic Isotope Effects. Phys. Chem. Chem. Phys. 2018, 20, 3925–3938. [Google Scholar] [CrossRef]
- Corchado, J.C.; Bravo, J.L.; Espinosa-Garcia, J. The Hydrogen Abstraction Reaction H + CH4. II. Theoretical investigation of the kinetics and dynamics. J. Chem. Phys. 2009, 130, 184315. [Google Scholar]
- Gonzalez-Lavado, E.; Corchado, J.C.; Espinosa-Garcia, J. The Hydrogen Abstraction Reaction O(3P) + CH4: A New Analytical Potential Energy Surface Based on Fit to ab initio Calculations. J. Chem. Phys. 2014, 140, 064310. [Google Scholar] [CrossRef]
- Espinosa-Garcia, J.; Corchado, J.C. QCT Dynamics Study of the Reaction of Hydroxyl Radical and Methane using a New ab initio Fitted Full-dimensional Analytical Potential Energy Surface. Theor. Chem. Acc. 2015, 134, 6. [Google Scholar] [CrossRef]
- Monge, M.; Rangel, C.; Espinosa-Garcia, J. Ab initio Based Potential Energy Surface and Kinetics Study of the OH + NH3 Hydrogen Abstraction Reaction. J. Chem. Phys. 2013, 138, 084305. [Google Scholar] [CrossRef]
- Espinosa-Garcia, J.; Corchado, J.C.; Garcia-Chamorro, M.; Rangel, C. F(2P) + C2H6 → HF + C2H5 Kinetics Study Based on a New Analytical Potential Energy Surface. Phys. Chem. Chem. Phys. 2018, 20, 19860–19870. [Google Scholar] [CrossRef]
- Espinosa-Garcia, J.; Garcia-Chamorro, M.; Corchado, J.C. The Hydrogen Abstraction Reaction H + C2H6 → H2(v,j) + C2H5. Part I. A Full-dimensional Analytical Potential Energy Surface based on ab initio Calculations. Phys. Chem. Chem. Phys. 2019, 21, 13165–13173. [Google Scholar] [CrossRef]
- Espinosa-Garcia, J.; Rangel, C.; Corchado, J.C.; Garcia-Chamorro, M. Theoretical Study of the O(3P) + C2H6 Reaction based on a New ab initio-based Global Potential Energy Surface. Phys. Chem. Chem. Phys. 2019, 21, 22591–22601. [Google Scholar] [CrossRef] [PubMed]
- Rangel, C.; Garcia-Chamorro, M.; Corchado, J.C.; Espinosa-Garcia, J. Kinetics and Dynamics Study of the OH + C2H6 → H2O + C2H5 Reaction based on an Analytical Global Potential Energy Surface. Phys. Chem. Chem. Phys. 2020, 22, 14796–14810. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Garcia, J.; Corchado, J.C. Recalibration of Two Earlier Potential Energy Surfaces for the CH4 + H → CH3 + H2 Reaction. Application of Variational Transition-State Theory and Analysis of the Kinetic Isotope Effects Using Rectilinear and Curvilinear Coordinates. J. Phys. Chem. 1996, 100, 16561–16567. [Google Scholar] [CrossRef]
- Corchado, J.C.; Espinosa-Garcia, J. Theoretical Study of the CH4 + F → CH3 + FH Reaction. II. Semiempirical Surfaces. J. Chem. Phys. 1996, 105, 3160–3167. [Google Scholar] [CrossRef]
- JANAF. Thermochemical Tables, 3rd ed.; Chase, M.W., Jr., Davies, C.A., Downey, J.R., Frurip, D.J., McDonald, R.A., Syverud, A.N., Eds.; National Bureau of Standards: Washington, DC, USA, 1985; Volume 14. [Google Scholar]
- Duchovic, R.J.; Volobuev, Y.L.; Lynch, G.C.; Jasper, A.W.; Truhlar, D.G.; Allison, T.C.; Wagner, A.F.; Garrett, B.C.; Espinosa-Garcia, J.; Corchado, J.C. POTLIB Online Potential Energy Surface Library. 2022. Available online: http://comp.chem.umn.edu/potlib (accessed on 19 May 2022).
- Hu, X.; Hase, W.L.; Pirraglia, T. Vectorization of the general Monte Carlo Classical Trajectory Program VENUS. J. Comp. Chem. 1991, 12, 1014–1020. [Google Scholar] [CrossRef]
- Hase, W.L.; Duchovic, R.J.; Hu, X.; Komornicki, A.; Lim, K.F.; Lu, D.; Peslherbe, G.H.; Swamy, K.N.; Vande Linde, S.R.; Varandas, A.J.C.; et al. VENUS96: A General Chemical Dynamics Computer Program. QCPE Bull. 1996, 16, 43. [Google Scholar]
- Truhlar, D.G.; Isaacson, A.D.; Garrett, B.C. Generalized Transition State Theory. In The Theory of Chemical Reactions; Baer, M., Ed.; CRC: Boca Raton, FL, USA, 1985; Volume 4. [Google Scholar]
- Zheng, J.; Bao, J.L.; Meana-Paneda, R.; Zhang, S.; Lynch, B.J.; Corchado, J.C.; Chuang, Y.Y.; Fast, P.L.; Hu, W.P.; Liu, Y.P.; et al. POLYRATE-2016-2A; University of Minnesota: Minneapolis, MN, USA, 2016. [Google Scholar]
- Suleimanov, Y.V.; Collepardo-Guevara, R.; Manolopoulos, D.E. Bimolecular Reaction Rates from Ring Polymer Molecular Dynamics: Application to H + CH4 → H2 + CH3. J. Chem. Phys. 2011, 134, 044131. [Google Scholar] [CrossRef]
- Suleimanov, Y.V.; Allen, J.W.; Green, W.H. RPMDRATE: Bimolecular Chemical Reaction Rates from Ring Polymer Molecular Dynamics. Comput. Phys. Commun. 2013, 184, 833–840. [Google Scholar] [CrossRef]
- Espinosa-Garcia, J.; Garcia-Chamorro, M.; Corchado, J.C.; Bhowmick, S.; Suleimanov, Y. VTST and RPMD Kinetics Study of the Nine-body X + C2H6 (X = H, Cl, F) Reactions based on Analytical Potential Energy Surfaces. Phys. Chem. Chem. Phys. 2020, 22, 13790–13801. [Google Scholar] [CrossRef]
- Baulch, D.L.; Cobos, C.; Cox, R.A.; Esser, C.; Frank, P.; Just, T.; Kerr, J.A.; Pilling, M.J.; Troe, J.; Walker, R.W.; et al. Evaluated kinetic data for combustion modelling. J. Phys. Chem. Ref. Data 1992, 21, 411–737. [Google Scholar] [CrossRef]
- Meana-Pañeda, R.; Truhlar, D.G.; Fernandez-Ramos, A. Least-action Tunneling Transmission Coefficient for Polyatomic Reactions. J. Chem. Theory Comput. 2010, 6, 6–17. [Google Scholar] [CrossRef]
- Bryukov, M.G.; Slagle, I.R.; Knyazev, V.D. Kinetics of Reactions of Cl Atoms with Ethane, Chloroethane, and 1,1-Dichloroethane. J. Phys. Chem. A 2003, 107, 6565–6573, Which Is a Review of the Literature until 2003. [Google Scholar] [CrossRef]
- Fettis, G.C.; Knox, J.H.; Trotman-Dickenson, A.F. The Reactions of Fluorine Atoms with Alkanes. J. Chem. Soc. 1960, 1064–1070. [Google Scholar] [CrossRef]
- Maricq, M.M.; Szente, J.J. A Kinetic Study of the Reaction between Ethylperoxy Radicals and HO2. J. Phys. Chem. 1994, 98, 2078–2082. [Google Scholar] [CrossRef]
- Persky, A. The Temperature Dependence of the Rate Constant for the Reaction F+ C2H6. Chem. Phys. Lett. 2003, 380, 286–291. [Google Scholar] [CrossRef]
- Garcia-Chamorro, M.; Corchado, J.C.; Espinosa-Garcia, J. Kinetics Theoretical Study of the O(3P) + C2H6 Reaction on an ab initio-based Global Potential Energy Surface. Theor. Chem. Acc. 2020, 139, 182. [Google Scholar] [CrossRef]
- Tsang, W.; Hampson, R.F. Chemical Kinetic Data Base for Combustion Chemistry. Part I. Methane and Related Compounds. J. Phys. Chem. Ref. Data 1986, 15, 1087. [Google Scholar] [CrossRef]
- Cohen, N.; Westberg, K.R. Chemical Kinetic Data Sheets for High-Temperature Reactions. Part II. J. Phys. Chem. Ref. Data 1991, 20, 1211. [Google Scholar] [CrossRef]
- Krasnoperov, L.N.; Michael, J.V. Shock Tube Studies using a Novel Multipass Absorption Cell: Rate Constant Results for OH + H2 and OH + C2H6. J. Phys. Chem. A 2004, 108, 5643–5648. [Google Scholar] [CrossRef]
- Espinosa-Garcia, J.; Garcia-Chamorro, M.; Corchado, J.C. Rethinking the Description of Water Product in Polyatomic OH/OD + XH (X ≡ D, Br, NH2 and GeH3) Reactions: Theory/Experimental Comparison. Theor. Chem. Acc. 2020, 139, 63. [Google Scholar] [CrossRef]
- Germann, G.J.; Huh, Y.D.; Valentini, J.J. State-to-state Dynamics of Atom+Polyatom Abstraction Reactions. II. The H + C2H6/C3H8 → H2 (v′, J′) + C2H5/C3H7 Reactions. J. Chem. Phys. 1992, 96, 5746–5757. [Google Scholar] [CrossRef]
- Whitney, E.S.; Zolot, A.M.; McCoy, A.B.; Francisco, J.S.; Nesbitt, D.J. Reactive scattering dynamics in atom + polyatomicsystems: F + C2H6 → HF (v, J) + C2H5. J. Chem. Phys. 2005, 122, 124310. [Google Scholar] [CrossRef]
- Huang, C.; Li, W.; Suits, A.G. Rotationally Resolved Reactive Scattering: Imaging Detailed Cl + C2H6 Reaction Dynamics. J. Chem. Phys. 2006, 125, 133107. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, G.M.; McKendrick, K.G. Rotational and Spin-orbit Effects in the Dynamics of O(3Pj) + Hydrocarbon Reactions. II. Models for Spin-orbit Propensities. J. Chem. Phys. 1997, 106, 9182–9190. [Google Scholar] [CrossRef]
- Ausfelder, F.; McKendric, K.G. Progress in Reaction Kinetics and Mechanism. Sci. Technol. Lett. 2000, 25, 299–370. [Google Scholar]
- Sweeney, G.M.; Watson, A.; McKendrick, K.G. Rotational and Spin-orbit Effects in the Dynamics of O(3Pj) + Hydrocarbon Reactions. I. Experimental Results. J. Chem. Phys. 1997, 106, 9172–9181. [Google Scholar] [CrossRef]
- Butkovskaya, N.I.; Setser, D.W. Infrared Chemiluminescence from Water-forming Reactions: Characterization of Dynamics and Mechanisms. Int. Rev. Phys. Chem. 2003, 22, 1–72. [Google Scholar] [CrossRef]
- Espinosa-Garcia, J.; Garcia-Chamorro, M. Role of an Ethyl Radical and the Problem of HF(v) Bimodal Vibrational Distribution in the F(2P) + C2H6 → HF(v) + C2H5 Reaction. Phys. Chem. Chem. Phys. 2018, 20, 26634–26642. [Google Scholar] [CrossRef]
- Adler, T.B.; Knizia, G.; Werner, H.-J. A Simple and Efficient CCSD(T)-F12 Approximation. J. Chem. Phys. 2007, 127, 221106. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method PES a | Ab Initio Level | Points | Barrier b | Reference |
|---|---|---|---|---|
| Shepard interpolation | CCSD(T)/VTZ | 14.93 | WWM c | |
| PIP | CCSD(T)/AVTZ | ~20,000 | 14.78 | ZBB3 d |
| VB/MM | CCSD(T)/VTZ | ~20,000 | 15.01 | CBE e |
| Shepard interpolation | CCSD(T)/AVTZ | ~30,000 | 15.03 | ZFWCZ f |
| NN | CCSD(T)-F12a/AVTZ | ~48,000 | 14.69 | XCZ g |
| PIP-NN | CCSD(T)-F12a/AVTZ | ~63,000 | 14.69 | LCZXZG h |
| Level | |||||
|---|---|---|---|---|---|
| MP2 a | CC/TZ b | CC/augTZ c | CC/5Z d | CC-F12/TZ e | |
| H + C2H6 | |||||
| Saddle point | 19.43 | 12.35 | 11.89 | 11.97 | 11.83 |
| Products | 5.08 | −0.36 | −0.51 | −0.61 | −0.60 |
| ΔHr(298K) | 2.67 | −3.36 | −3.51 | −3.61 | −3.60 |
| Cl(2P) + C2H6 | |||||
| Saddle point | 9.82 | 4.59 | 2.68 | 1.87 | 1.77 |
| Products | 9.11 | 4.11 | 3.11 | 1.73 | 1.81 |
| ΔHr(298K) | 4.56 | −0.44 | −1.44 | −2.82 | −2.74 |
| F(2P) + C2H6 | |||||
| Saddle point | 4.24 | 0.19 | −1.00 | - | −0.93 |
| Products | −25.01 | −28.89 | −31.21 | - | −32.54 |
| ΔHr(298K) | −28.03 | −31.91 | −34.23 | - | −35.56 |
| H a | F(2P) b | Cl(2P) c | O(3P) d | OH e | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Theo | Exp | Theo | Exp | Theo | Exp | Theo | Exp | Theo | Exp | |
| fint(HX) | 26 | 18 | 70 | 70 | 6 | 2 | 12 | - | 70 | 51 |
| fint(C2H5) | 12 | - | 13 | - | 27 | 30 | 27 | - | 20 | - |
| ftrans | 62 | - | 17 | - | 67 | 68 | 61 | - | 10 | - |
| H a | F(2P) b | O(3P) c | |||||
|---|---|---|---|---|---|---|---|
| Theo | Exp | Theo | Exp | MO d | Theo | Exp | |
| v = 0 | 81 | - | 0 | 16 | 2 | 90 | - |
| v = 1 | 19 | - | 3 | 12 | 8 | 10 | - |
| v = 2 | 58 | 43 | 52 | ||||
| v = 3 | 39 | 28 | 38 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Espinosa-Garcia, J.; Rangel, C.; Corchado, J.C. Current Status of the X + C2H6 [X ≡ H, F(2P), Cl(2P), O(3P), OH] Hydrogen Abstraction Reactions: A Theoretical Review. Molecules 2022, 27, 3773. https://doi.org/10.3390/molecules27123773
Espinosa-Garcia J, Rangel C, Corchado JC. Current Status of the X + C2H6 [X ≡ H, F(2P), Cl(2P), O(3P), OH] Hydrogen Abstraction Reactions: A Theoretical Review. Molecules. 2022; 27(12):3773. https://doi.org/10.3390/molecules27123773
Chicago/Turabian StyleEspinosa-Garcia, Joaquin, Cipriano Rangel, and Jose C. Corchado. 2022. "Current Status of the X + C2H6 [X ≡ H, F(2P), Cl(2P), O(3P), OH] Hydrogen Abstraction Reactions: A Theoretical Review" Molecules 27, no. 12: 3773. https://doi.org/10.3390/molecules27123773