
UV-Vis Spectroscopy, Electrochemical and DFT Study of Tris(β-diketonato)iron(III) Complexes with Application in DSSC: Role of Aromatic Thienyl Groups


Abstract
:
1. Introduction
2. Results and Discussion
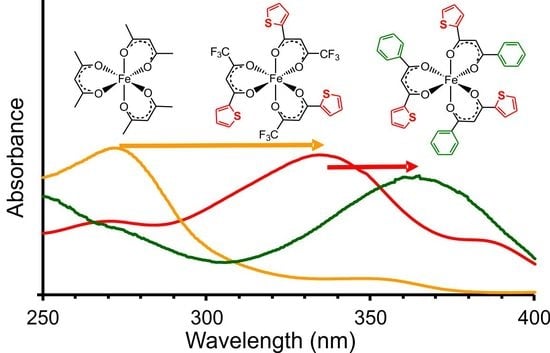
2.1. UV-Vis Spectroscopy
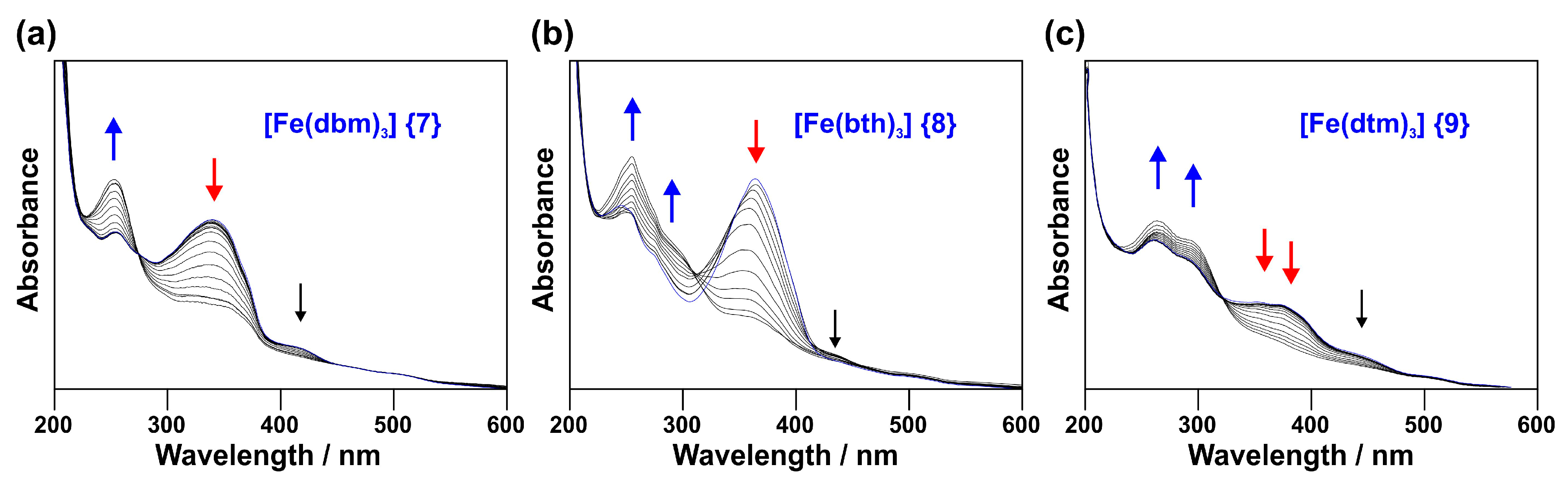
2.1.1. Experimental Spectra
- [Fe(acac)3] (1) (270 nm) → [Fe(ba)3] (3) (298 nm) → [Fe(dbm)3] (7) (336 nm)
- [Fe(tfaa)3] (2) (271 nm) → [Fe(tfba)3] (4) (304 nm) → [Fe(dbm)3] (7) (336 nm)
- [Fe(hfaa)3] (10) (278 nm, calc) → [Fe(tta)3] (6) (333 nm) → [Fe(dtm)3] (9) (376 nm)
2.1.2. TDDFT
2.1.3. Application as Dye to DSSC
2.2. Electrochemistry
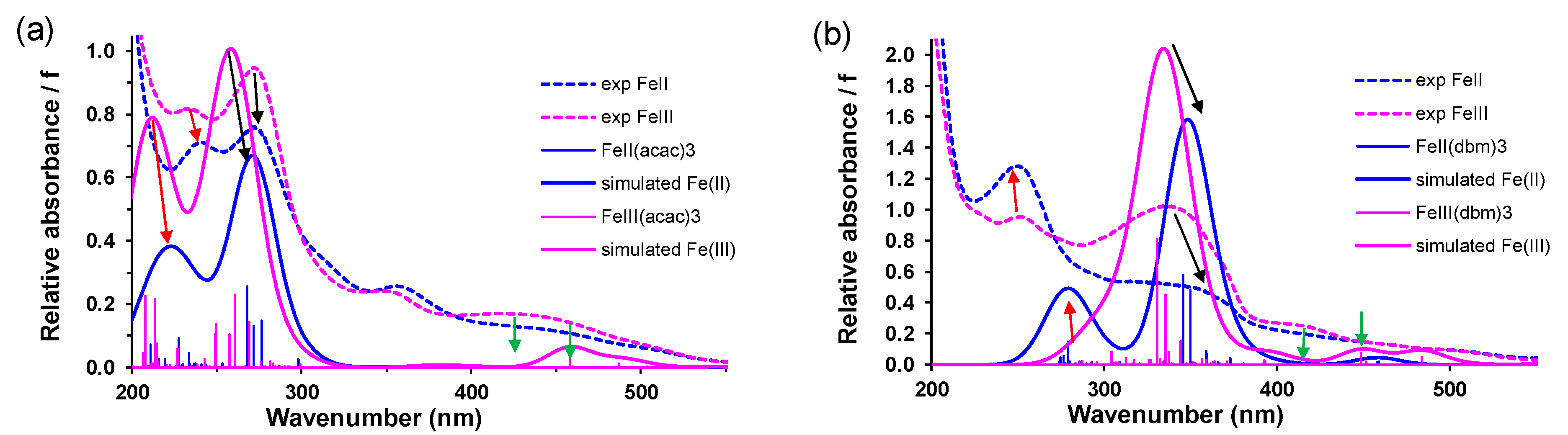
2.2.1. SEC
- (i)
- (ii)
2.2.2. Application as Redox Mediator in DSSC

3. Materials and Methods
3.1. General
3.2. DFT Methods
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Hosseini, M.S.; Abbasi, A.; Masteri-Farahani, M. Improving the photocatalytic activity of NH2-UiO-66 by facile modification with Fe(acac)3 complex for photocatalytic water remediation under visible light illumination. J. Hazard. Mater. 2022, 425, 127975. [Google Scholar] [CrossRef] [PubMed]
- Perera, I.R.; Daeneke, T.; Makuta, S.; Yu, Z.; Tachibana, Y.; Mishra, A.; Bäuerle, P.; Ohlin, C.A.; Bach, U.; Spiccia, L. Application of the Tris(acetylacetonato)iron(III)/(II) Redox Couple in p-Type Dye-Sensitized Solar Cells. Angew. Chemie Int. Ed. 2015, 54, 3758–3762. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Y.; Zhang, C.; Yuan, J.; Zhao, Y.; Li, Y. A high-performance all-iron non-aqueous redox flow battery. J. Power Sources 2020, 445, 227331. [Google Scholar] [CrossRef]
- Bamgbopa, M.O.; Shao-Horn, Y.; Almheiri, S. The potential of non-aqueous redox flow batteries as fast-charging capable energy storage solutions: Demonstration with an iron–chromium acetylacetonate chemistry. J. Mater. Chem. A 2017, 5, 13457–13468. [Google Scholar] [CrossRef]
- Tachikawa, N.; Haruyama, R.; Yoshii, K.; Serizawa, N.; Katayama, Y. Redox Reaction of Tris(acetylacetonato)iron(III) Complex in an Amide-type Ionic Liquid. Electrochemistry 2018, 86, 32–34. [Google Scholar] [CrossRef] [Green Version]
- Misono, A.; Uchida, Y.; Hidai, M.; Ohsawa, Y. The Oligomerization of Isoprene by Cobalt or Iron Complex Catalysts. Bull. Chem. Soc. Jpn. 1966, 39, 2425–2429. [Google Scholar] [CrossRef] [Green Version]
- Williamson, K.S.; Yoon, T.P. Iron-Catalyzed Aminohydroxylation of Olefins. J. Am. Chem. Soc. 2010, 132, 4570–4571. [Google Scholar] [CrossRef]
- Matsumura, S.; Hlil, A.R.; Lepiller, C.; Gaudet, J.; Guay, D.; Shi, Z.; Holdcroft, S.; Hay, A.S. Stability and Utility of Pyridyl Disulfide Functionality in RAFT and Conventional Radical Polymerizations. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 7207–7224. [Google Scholar] [CrossRef]
- Lübken, D.; Saxarra, M.; Kalesse, M. Tris(acetylacetonato) Iron(III): Recent Developments and Synthetic Applications. Synthesis 2019, 51, 161–177. [Google Scholar] [CrossRef] [Green Version]
- Kadish, K.M.; Van Caemelbecke, E.; D’Souza, F.; Medforth, C.J.; Smith, K.M.; Tabard, A.; Guilard, R. Electrochemistry and Spectroelectrochemistry of.sigma.-Bonded Iron(III) Porphyrins with Nonplanar Porphyrin Rings. Reactions of (OETPP)Fe(R) and (OETPP)FeCl, Where R = C6H5, C6F4H, or C6F5 and OETPP Is the Dianion of 2,3,7,8,12,13,17,18-Octaethyl-5,10,15. Inorg. Chem. 1995, 34, 2984–2989. [Google Scholar] [CrossRef]
- Gumbel, G.; Elias, H. Kinetics and mechanism of ligand substitution in β-diketone complexes of iron(III). Solvolysis controlling the substitution process in alcohol media. Inorg. Chim. Acta 2003, 342, 97–106. [Google Scholar] [CrossRef]
- Heineman, W.R.; Burnett, J.N.; Murray, R.W. Optically Transparent Thin-Layer Electrodes: Studies of lron(ll)-(lll) Acetylacetonate Ligand Exchange Reactions in Acetonitrile. Anal. Chem. 1968, 40, 1970–1973. [Google Scholar] [CrossRef]
- Langon, D.; Cocolios, P.; Kadish, K.M.; Guilard, R. Electrochemistry and Spectroelectrochemistry of σ-Bonded Iron Aryl Porphyrins. 1. Evidence for Reversible Aryl Migration from Iron to Nitrogen of Five-Coordinate Complexes. J. Am. Chem. Soc. 1984, 106, 4472–4478. [Google Scholar] [CrossRef]
- von Eschwege, K.G.; Conradie, J. Iron phenanthrolines: A density functional theory study. Inorg. Chim. Acta 2018, 471, 391–396. [Google Scholar] [CrossRef]
- Ferreira, H.; von Eschwege, K.G.; Conradie, J. Electronic properties of Fe charge transfer complexes—A combined experimental and theoretical approach. Electrochim. Acta 2016, 216, 339–346. [Google Scholar] [CrossRef]
- Conradie, M.M.; Conradie, J. Electrochemical behaviour of Tris(β-diketonato)iron(III) complexes: A DFT and experimental study. Electrochim. Acta 2015, 152, 512–519. [Google Scholar] [CrossRef]
- Conradie, M.M.; van Rooyen, P.H.; Conradie, J. Crystal and electronic structures of tris[4,4,4-Trifluoro-1-(2-X)-1,3-butanedionato]iron(III) isomers (X=thienyl or furyl): An X-ray and computational study. J. Mol. Struct. 2013, 1053, 134–140. [Google Scholar] [CrossRef] [Green Version]
- Conradie, M.M.; van Rooyen, P.H.; Conradie, J. X-ray and electronic structure of tris(benzoylacetonato-κ2O,O′)iron(III). J. Mol. Struct. 2016, 1123, 199–205. [Google Scholar] [CrossRef] [Green Version]
- Conradie, J. DFT Study of bis(1,10-phenanthroline)copper complexes: Molecular and electronic structure, redox and spectroscopic properties and application to Solar Cells. Electrochim. Acta 2022, 418, 140276. [Google Scholar] [CrossRef]
- Baldenebro-Lopez, J.; Flores-Holguin, N.; Castorena-Gonzalez, J.; Almaral-Sanchez, J.; Glossman-Mitnik, D. Theoretical Study of Copper Complexes: Molecular Structure, Properties, and Its Application to Solar Cells. Int. J. Photoenergy 2013, 2013, 613064. [Google Scholar] [CrossRef] [Green Version]
- Tontapha, S.; Uppachai, P.; Amornkitbamrung, V. Fabrication of Functional Materials for Dye-sensitized Solar Cells. Front. Energy Res. 2021, 9, 641983. [Google Scholar] [CrossRef]
- Sharma, K.; Sharma, V.; Sharma, S.S. Dye-Sensitized Solar Cells: Fundamentals and Current Status. Nanoscale Res. Lett. 2018, 13, 381. [Google Scholar] [CrossRef] [PubMed]
- Cahen, D.; Hodes, G.; Grätzel, M.; Guillemoles, J.F.; Riess, I. Nature of Photovoltaic Action in Dye-Sensitized Solar Cells. J. Phys. Chem. B 2000, 104, 2053–2059. [Google Scholar] [CrossRef]
- Benesperi, I.; Michaels, H.; Freitag, M. The researcher’s guide to solid-state dye-sensitized solar cells. J. Mater. Chem. C 2018, 6, 11903–11942. [Google Scholar] [CrossRef] [Green Version]
- Asbury, J.B.; Wang, Y.-Q.; Hao, E.; Ghosh, H.N.; Lian, T. Evidences of hot excited state electron injection from sensitizer molecules to TiO2 nanocrystalline thin films. Res. Chem. Intermed. 2001, 27, 393–406. [Google Scholar] [CrossRef]
- Grätzel, M. Conversion of sunlight to electric power by nanocrystalline dye-sensitized solar cells. J. Photochem. Photobiol. A Chem. 2004, 164, 3–14. [Google Scholar] [CrossRef]
- Pastore, M.; Fantacci, S.; De Angelis, F. Ab initio determination of ground and excited state oxidation potentials of organic chromophores for dye-sensitized solar cells. J. Phys. Chem. C 2010, 114, 22742–22750. [Google Scholar] [CrossRef]
- Grätzel, M. Recent advances in sensitized mesoscopic solar cells. Acc. Chem. Res. 2009, 42, 1788–1798. [Google Scholar] [CrossRef]
- Chaitanya, K.; Ju, X.H.; Heron, B.M. Theoretical study on the light harvesting efficiency of zinc porphyrin sensitizers for DSSCs. RSC Adv. 2014, 4, 26621–26634. [Google Scholar] [CrossRef]
- Chen, C.Y.; Wang, M.; Li, J.Y.; Pootrakulchote, N.; Alibabaei, L.; Ngoc-Le, C.H.; Decoppet, J.D.; Tsai, J.H.; Grätzel, C.; Wu, C.G.; et al. Highly efficient light-harvesting ruthenium sensitizer for thin-film dye-sensitized solar cells. ACS Nano 2009, 3, 3103–3109. [Google Scholar] [CrossRef]
- Luo, J.-H.; Li, Q.-S.; Yang, L.-N.; Sun, Z.-Z.; Li, Z.-S. Theoretical design of porphyrazine derivatives as promising sensitizers for dye-sensitized solar cells. RSC Adv. 2014, 4, 20200–20207. [Google Scholar] [CrossRef]
- Piatkowski, P.; Martin, C.; Di Nunzio, M.R.; Cohen, B.; Pandey, S.; Hayse, S.; Douhal, A. Complete photodynamics of the efficient YD2-o-C8-based solar cell. J. Phys. Chem. C 2014, 118, 29674–29687. [Google Scholar] [CrossRef]
- Arici, M.; Arican, D.; Uǧur, A.L.; Erdoǧmuş, A.; Koca, A. Electrochemical and spectroelectrochemical characterization of newly synthesized manganese, cobalt, iron and copper phthalocyanines. Electrochim. Acta 2013, 87, 554–566. [Google Scholar] [CrossRef]
- Boschloo, G.; Hagfeldt, A. Characteristics of the iodide/triiodide redox mediator in dye-sensitized solar cells. Acc. Chem. Res. 2009, 42, 1819–1826. [Google Scholar] [CrossRef] [PubMed]
- Freitag, M.; Daniel, Q.; Pazoki, M.; Sveinbjörnsson, K.; Zhang, J.; Sun, L.; Hagfeldt, A.; Boschloo, G. High-efficiency dye-sensitized solar cells with molecular copper phenanthroline as solid hole conductor. Energy Environ. Sci. 2015, 8, 2634–2637. [Google Scholar] [CrossRef]
- Yang, W.; Vlachopoulos, N.; Hao, Y.; Hagfeldt, A.; Boschloo, G. Efficient dye regeneration at low driving force achieved in triphenylamine dye LEG4 and TEMPO redox mediator based dye-sensitized solar cells. Phys. Chem. Chem. Phys. 2015, 17, 15868–15875. [Google Scholar] [CrossRef]
- Ellis, H.; Eriksson, S.K.; Feldt, S.M.; Gabrielsson, E.; Lohse, P.W.; Lindblad, R.; Sun, L.; Rensmo, H.; Boschloo, G.; Hagfeldt, A. Linker Unit Modification of Triphenylamine-Based Organic Dyes for Efficient Cobalt Mediated Dye-Sensitized Solar Cells. J. Phys. Chem. C 2013, 117, 21029–21036. [Google Scholar] [CrossRef]
- Higashino, T.; Iiyama, H.; Nimura, S.; Kurumisawa, Y.; Imahori, H. Effect of Ligand Structures of Copper Redox Shuttles on Photovoltaic Performance of Dye-Sensitized Solar Cells. Inorg. Chem. 2020, 59, 452–459. [Google Scholar] [CrossRef]
- Conradie, J. Polypyridyl copper complexes as dye sensitizer and redox mediator for dye-sensitized solar cells. Electrochem. Commun. 2022, 134, 107182. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kin. J. Chem. Phys. 2006, 125, 194101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Stevens, W.J.; Basch, H.; Krauss, M. Compact effective potentials and efficient shared-exponent basis sets for the first- and second-row atoms. J. Chem. Phys. 1984, 81, 6026–6033. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef] [Green Version]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef] [Green Version]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Peterson, K.A.; Woon, D.E.; Dunning, T.H. Benchmark calculations with correlated molecular wave functions. IV. The classical barrier height of the H+H 2 →H 2 +H reaction. J. Chem. Phys. 1994, 100, 7410–7415. [Google Scholar] [CrossRef]
- Wilson, A.K.; van Mourik, T.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. VI. Sextuple zeta correlation consistent basis sets for boron through neon. J. Mol. Struct. THEOCHEM 1996, 388, 339–349. [Google Scholar] [CrossRef]
- Davidson, E.R. Comment on “Comment on Dunning’s correlation-consistent basis sets”. Chem. Phys. Lett. 1996, 260, 514–518. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Wadt, W.R.; Hay, P.J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitale. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Dolg, M.; Wedig, U.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the first row transition elements. J. Chem. Phys. 1987, 86, 866–872. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Skyner, R.E.; Mcdonagh, J.L.; Groom, C.R.; Mourik, T. Van A review of methods for the calculation of solution free energies and the modelling of systems in solution. Phys. Chem. Chem. Phys. 2015, 17, 6174–6191. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Chemcraft—Graphical Software for Visualization of Quantum Chemistry Computations. Available online: http://www.chemcraftprog.com (accessed on 8 May 2022).
- Diaz-Acosta, I.; Baker, J.; Cordes, W.; Pulay, P. Calculated and Experimental Geometries and Infrared Spectra of Metal Tris-Acetylacetonates: Vibrational Spectroscopy as a Probe of Molecular Structure for Ionic Complexes. Part I. J. Phys. Chem. A 2001, 105, 238–244. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | β-Diketone | λA,max,exp | λA,max,calc | λA,max,calc | λA,max,calc | ΔE | ΔZEE | ΔG | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Exp | B3LYP | M06 | CAM-B3LYP | B3LYP | M06 | CAM-B3LYP | B3LYP | M06 | B3LYP | M06 | ||
| 1 | acac | 270 | 260.3 | 254.9 | 255.3 | - | - | - | - | - | - | - |
| 2 | tfaa fac | 271 | 261.9 | 256.0 | 261.0 | 0.004 | 0.017 | 0.005 | 0.000 | 0.005 | 0.000 | 0.001 |
| tfaa mer | 271 | 262.1 | 256.6 | 261.5 | 0.000 | 0.000 | 0.000 | 0.003 | 0.000 | 0.091 | 0.000 | |
| 3 | ba fac | 298 | 303.1 | 301.3 | 282.2 | 0.001 | 0.043 | 0.000 | 0.000 | 0.042 | 0.000 | 0.000 |
| ba mer | 298 | 304.2 | 309.9 | 287.0 | 0.000 | 0.000 | 0.000 | 0.001 | 0.000 | 0.015 | 0.021 | |
| 4 | tfba fac | 304 | 311.0 | 309.8 | 290.6 | 0.008 | 0.051 | 0.008 | 0.004 | 0.034 | 0.001 | 0.000 |
| tfba mer | 304 | 318.6 | 320.2 | 307.8 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.025 | |
| 5 | tffu fac | 333 | 321.0 | 317.0 | 297.8 | 0.000 | 0.010 | 0.000 | 0.002 | 0.008 | 0.007 | 0.141 |
| tffu mer | 333 | 333.0 | 327.1 | 325.5 | 0.000 | 0.000 | 0.001 | 0.000 | 0.000 | 0.000 | 0.000 | |
| 6 | tta fac | 333 | 329.0 | 325.7 | 321.5 | 0.002 | 0.045 | 0.000 | 0.002 | 0.033 | 0.011 | 0.011 |
| tta mer | 333 | 339.2 | 339.8 | 333.1 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | |
| 7 | dbm | 336 | 330.7 | 341.8 | 302.2 | - | - | - | - | - | - | - |
| 8 | bth fac | 361 | 346.6 | 346.6 | 326.8 | 0.000 | 0.043 | 0.000 | 0.004 | 0.039 | 0.039 | 0.036 |
| bth mer | 361 | 347.0 | 351.4 | 327.2 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | |
| 9 | dtm | 376 | 375.8 | 358.7 | 333.7 | - | - | - | - | - | - | - |
| 10 | hfaa | - | 278.3 | 274.0 | 268.1 | - | - | - | - | - | - | - |
| AD a | 7.8 | 11.0 | 18.4 | |||||||||
| MAD b | 3.8 | 4.3 | 11.6 |
| Complex | E/eV | λ/nm | f | from | to | % Contribution | Assignment |
|---|---|---|---|---|---|---|---|
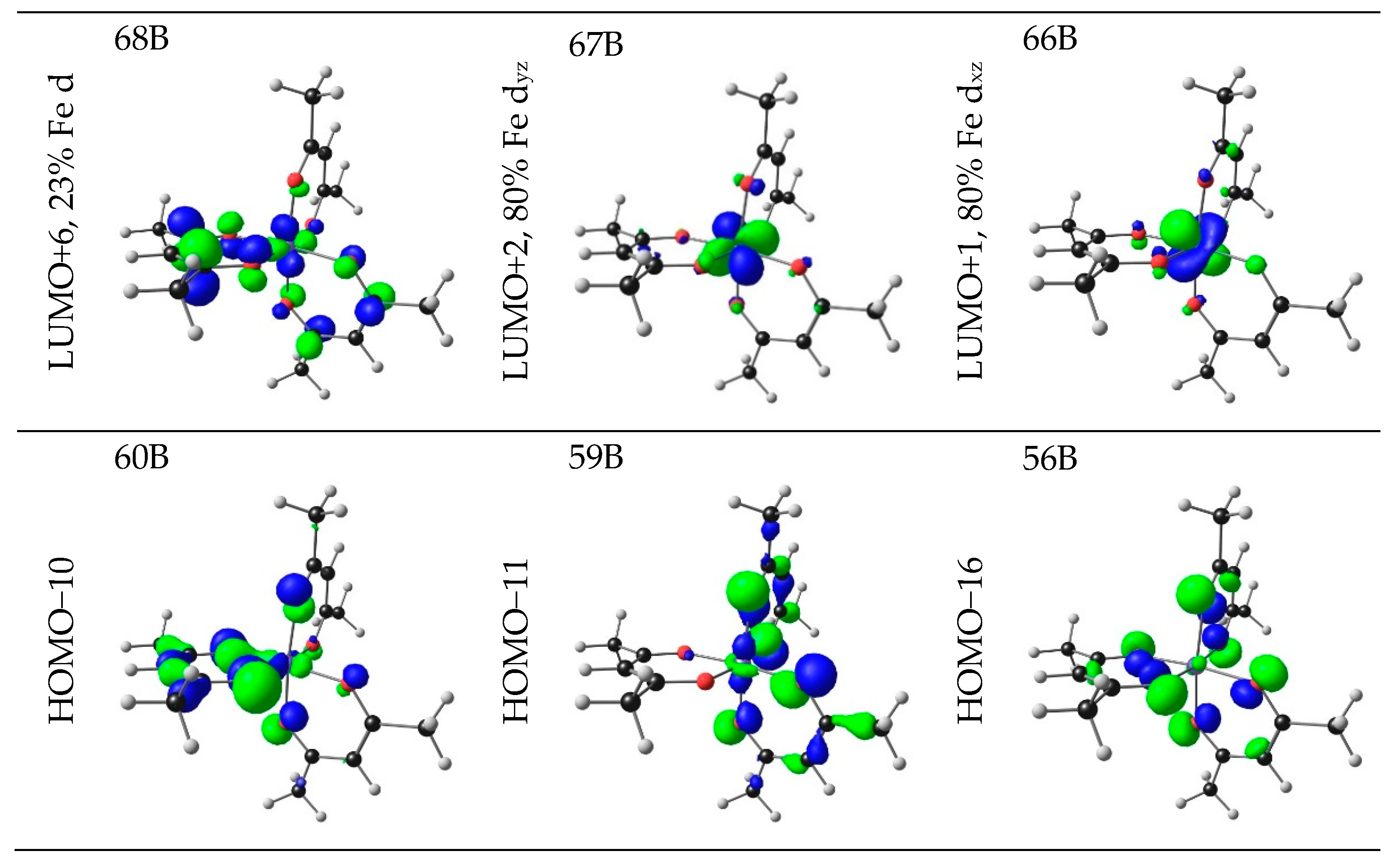
| 1 | 4.76 | 260.34 | 0.2286 | HOMO−16 | LUMO+2 | 6.09 | LMCT |
| HOMO−11 | LUMO+6 | 6.71 | LMCT | ||||
| HOMO−11 | LUMO+9 | 25.49 | LMCT | ||||
| HOMO−10 | LUMO+8 | 25.70 | LMCT | ||||
| 4.76 | 260.33 | 0.2279 | HOMO−16 | LUMO+1 | 6.12 | LMCT | |
| HOMO−11 | LUMO+7 | 6.69 | LMCT | ||||
| HOMO−11 | LUMO+8 | 25.79 | LMCT | ||||
| HOMO−10 | LUMO+6 | 6.75 | LMCT | ||||
| HOMO−10 | LUMO+9 | 25.65 | LMCT | ||||
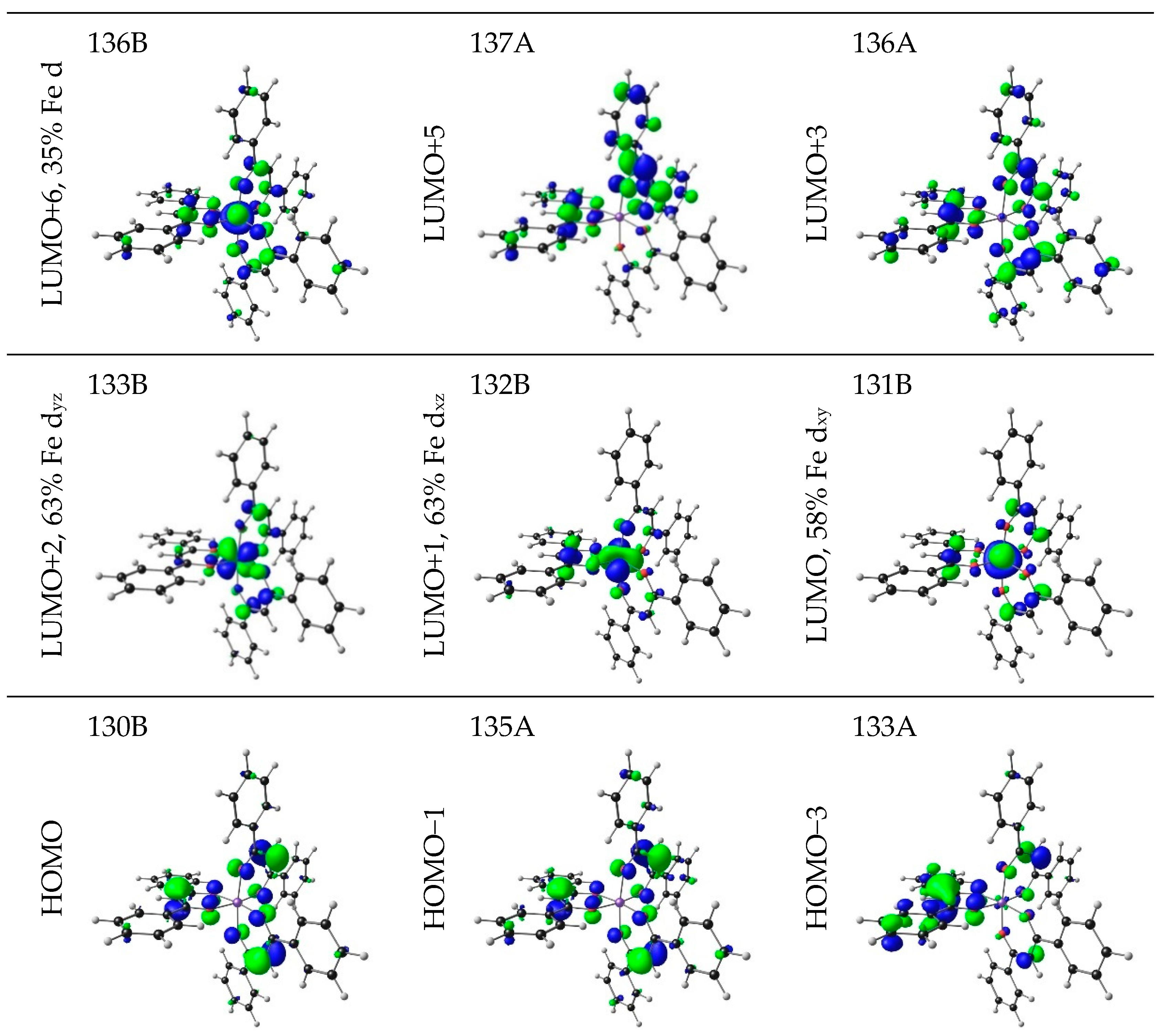
| 7 | 3.75 | 330.68 | 0.8092 | HOMO−3 | LUMO+5 | 3.71 | LLCT |
| HOMO−1 | LUMO+3 | 4.02 | LLCT | ||||
| HOMO−35 | LUMO+2 | 3.61 | LMCT | ||||
| HOMO−34 | LUMO+1 | 3.55 | LMCT | ||||
| HOMO−27 | LUMO | 4.28 | LMCT | ||||
| HOMO−18 | LUMO+1 | 3.62 | LMCT | ||||
| HOMO | LUMO+6 | 20.48 | LMCT | ||||
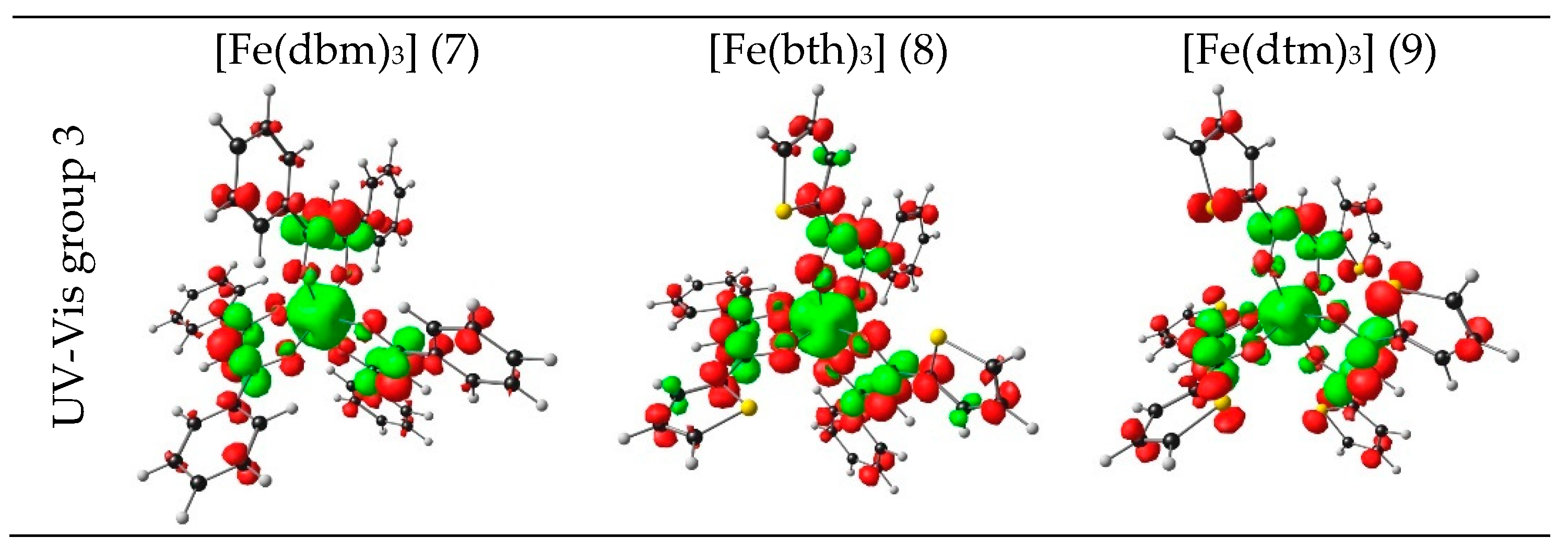
| 9 | 3.30 | 375.76 | 0.5211 | HOMO−26 | LUMO | 17.80 | LMCT |
| HOMO−12 | LUMO | 7.43 | LMCT | ||||
| HOMO−2 | LUMO+6 | 7.30 | LMCT | ||||
| HOMO−2 | LUMO+7 | 6.41 | LMCT | ||||
| HOMO−1 | LUMO+7 | 6.99 | LMCT |
| No | Ligand | λA,max | f | E | LHE | τ | ELUMO | EHOMO | ΔGinject | ΔGregenerate |
|---|---|---|---|---|---|---|---|---|---|---|
| nm | eV | ns | eV | eV | eV | eV | ||||
| 1 | acac | 260.34 | 0.23 | 4.76 | 0.41 | 4 | −3.196 | −6.552 | 2.21 | 1.75 |
| 2 | tfaa fac | 261.86 | 0.30 | 4.73 | 0.49 | 3 | −4.117 | −7.387 | 1.35 | 2.59 |
| tfaa mer | 262.12 | 0.33 | 4.73 | 0.53 | 3 | −4.114 | −7.374 | 1.36 | 2.57 | |
| 3 | ba fac | 303.05 | 0.39 | 4.09 | 0.59 | 4 | −3.263 | −6.463 | 1.63 | 1.66 |
| ba mer | 304.16 | 0.30 | 4.08 | 0.50 | 5 | −3.265 | −6.459 | 1.62 | 1.66 | |
| 4 | tfba fac | 310.95 | 0.53 | 3.99 | 0.71 | 3 | −4.064 | −7.172 | 0.81 | 2.37 |
| tfba mer | 318.63 | 0.49 | 3.89 | 0.67 | 3 | −4.064 | −7.126 | 0.77 | 2.33 | |
| 5 | tffu fac | 320.96 | 0.93 | 3.86 | 0.88 | 2 | −4.075 | −7.048 | 0.82 | 2.25 |
| tffu mer | 332.98 | 0.67 | 3.72 | 0.78 | 2 | −4.080 | −6.972 | 0.75 | 2.17 | |
| 6 | tta fac | 328.98 | 0.90 | 3.77 | 0.88 | 2 | −4.066 | −7.032 | 0.74 | 2.23 |
| tta mer | 339.16 | 0.68 | 3.66 | 0.79 | 3 | −4.065 | −6.965 | 0.69 | 2.16 | |
| 7 | dbm | 330.68 | 0.81 | 3.75 | 0.84 | 2 | −3.330 | −6.373 | 1.38 | 1.57 |
| 8 | bth fac | 346.64 | 0.80 | 3.58 | 0.84 | 2 | −3.394 | −6.312 | 1.26 | 1.51 |
| bth mer | 346.96 | 0.37 | 3.57 | 0.57 | 5 | −3.391 | −6.311 | 1.26 | 1.51 | |
| 9 | dtm | 375.76 | 0.52 | 3.30 | 0.70 | 4 | −3.330 | −6.373 | 0.93 | 1.57 |
| 10 | hfaa | 278.26 | 0.22 | 4.46 | 0.40 | 5 | −5.122 | −8.199 | 0.26 | 3.40 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conradie, M.M. UV-Vis Spectroscopy, Electrochemical and DFT Study of Tris(β-diketonato)iron(III) Complexes with Application in DSSC: Role of Aromatic Thienyl Groups. Molecules 2022, 27, 3743. https://doi.org/10.3390/molecules27123743
Conradie MM. UV-Vis Spectroscopy, Electrochemical and DFT Study of Tris(β-diketonato)iron(III) Complexes with Application in DSSC: Role of Aromatic Thienyl Groups. Molecules. 2022; 27(12):3743. https://doi.org/10.3390/molecules27123743
Chicago/Turabian StyleConradie, Marrigje M. 2022. "UV-Vis Spectroscopy, Electrochemical and DFT Study of Tris(β-diketonato)iron(III) Complexes with Application in DSSC: Role of Aromatic Thienyl Groups" Molecules 27, no. 12: 3743. https://doi.org/10.3390/molecules27123743